
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron-based octahedral dication experimentally detected: DFT surface confirms its availability†
Willi Keller*a,
Menyhárt B. Sárosi‡
b,
Jindřich Fanfrlík c,
Michal Strakac,
Josef Holubd,
Michael L. McKeee and
Drahomír Hnyk*d
c,
Michal Strakac,
Josef Holubd,
Michael L. McKeee and
Drahomír Hnyk*d
aInstitut für Chemie, Universität Hohenheim, Garbenstrasse 30, D-70599 Stuttgart, Germany. E-mail: willi.keller@uni-hohenheim.de
bWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstrasse 2, 04103 Leipzig, Germany
cInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, CZ-166 10 Praha 6, Czech Republic
dInstitute of Inorganic Chemistry of the Czech Academy of Sciences, CZ-250 68 Husinec-Řež, Czech Republic. E-mail: hnyk@iic.cas.cz
eDepartment of Chemistry and Biochemistry, Auburn University, Auburn, AL 36849, USA
First published on 28th June 2023
Abstract
Borane and heteroborane clusters have been known as neutral or anionic species. In contrast to them, several ten-vertex monocationic nido and closo dicarbaborane-based systems have recently emerged from the reaction of the parent bicapped-square antiprismatic dicarbaboranes with N-heterocyclic carbenes followed by the protonization of the corresponding nido intermediates. The expansion of these efforts has afforded the very first closo-dicationic octahedral phosphahexaborane along with new closo-monocationic pnictogenahexaboranes of the same shapes. All are the products of the one-pot procedure that consists in the reaction of the same carbenes with the parent closo-1,2-Pn2B4Br4 (Pn = As, P). Whereas in the case of phosphorus such a monocation appears to be a mixture of stable intermediates, and arsenahexaboranyl monocation has occurred as the final product, all of them without using any subsequent reaction. The well-established DFT/ZORA/NMR approach has unambiguously confirmed the existence of these species in solution, and computed electrostatic potentials have revealed the delocalization of the positive charge in these monocations and in the very first dication, namely within the octahedral shapes in both cases.
Introduction
N-Heterocyclic carbenes (NHCs, see Scheme 1) have become very frequently employed agents in chemical sciences. These soft acids comprise a large variety of stereoelectronic properties that are based on a myriad of combinations of five-membered N-heterocyclic rings, such as imidazole, 1,2,4,-triazole etc., and ligands on one or more nitrogen atoms, with at least one of them being a neighbor of a divalent carbon atom.1 The ability to donate a π-electron from an occupied pz orbital of nitrogen to an empty pz orbital (with both orbitals being perpendicular to the plane of the pentagonal C3N2 ring in terms of adopting natural chemical language of molecular orbitals and a coordinate system with the z-axis perpendicular to the plane of the page) of the divalent carbon makes NHCs efficient electron donors.2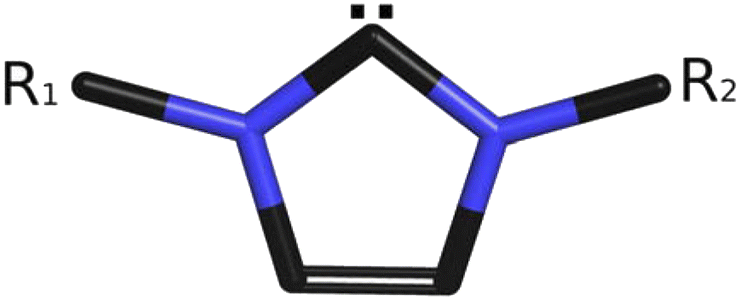 | ||
| Scheme 1 An archetype of N-heterocyclic carbenes (NHCs) (R1, R2 are different or identical aromatic or aliphatic substituents). | ||
In contrast, boron and heteroborane clusters have been known for the existence of multicenter bonding schemes for decades.3 Such electron imbalance results in various molecular shapes as a consequence of multicenter bonding,4 with the three-center two-electron (3c-2e) bond being the most prominent. The number of skeletal electrons within a cluster defines the corresponding electron count and the clusters are classified accordingly. The most stable structural motifs among them are the closo-heteroboranes of icosahedral, bicapped square-antiprismatic and octahedral shapes,5 with icosahedral heteroboranes being experimentally recognized as slight electron acceptors.6 They exist as neutral or anionic species (for the molecular shapes exemplified by the parent closo-boranes, see Scheme 2).
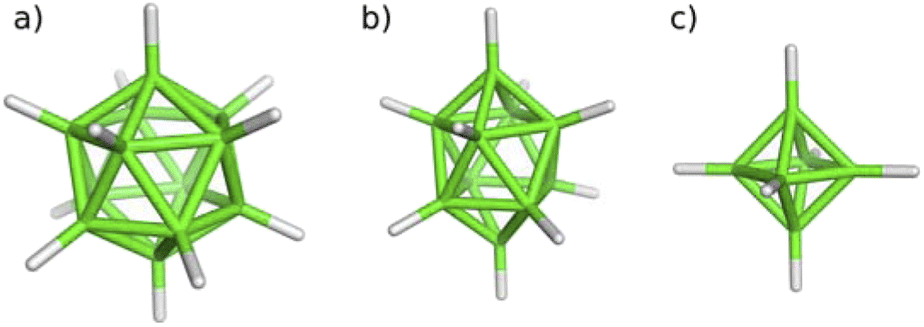 | ||
| Scheme 2 (a) Icosahedral, (b) bicapped square-antiprismatic and (c) octahedral closo-boranes. | ||
The corresponding cations of such shapes are unknown, although some predictive computational studies have been performed and their existence could not be ruled out. These assumptions are applicable to icosahedral, bicapped square-antiprismatic and octahedral tricarba monocations C3Bn−3Hn+ (n = 12, 10, 6).7,8
Mutual reactions of neutral NHCs with neutral heteroboranes might offer options for producing unusual motifs as a whole because both reaction components are far from being conventional. Indeed, the few attempts that have already been made have offered very interesting conclusions.
Therefore, icosahedral closo-carboranes9 and closo-thiaboranes10 underwent reactions with NHCs (with both bulky R's represented by 2,6-iPr2C6H3, abbreviated as Idip) and both twelve-vertex species (for the corresponding shapes, see Scheme 2) were reduced to the so-called nido structural patterns (either neutral or monoanionic) with either twelve or eleven vertices (see Scheme 3). The same kind of N-heterocyclic carbene was used for attacking all three available bicapped-square antiprismatic closo-1,N-C2B8H10 (N = 2, 6, 10).
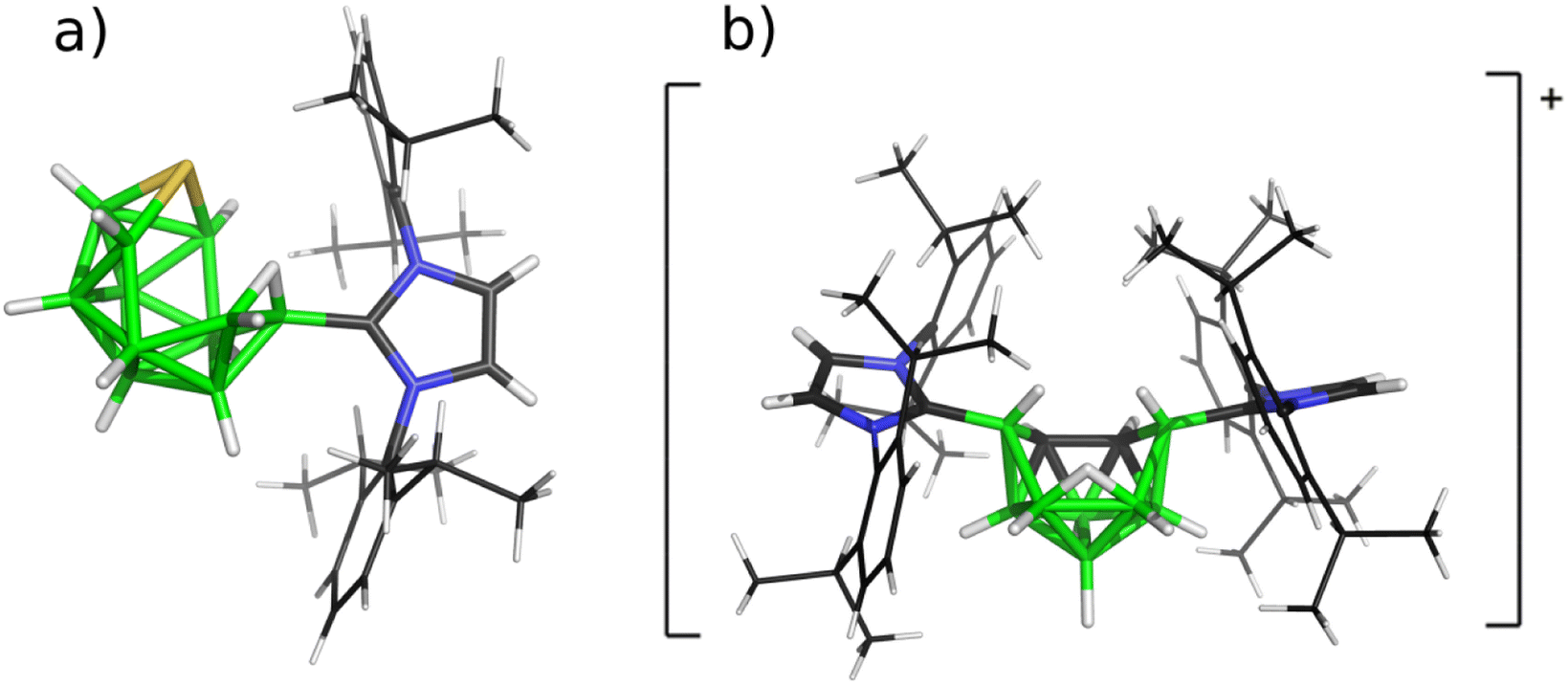 | ||
| Scheme 3 Molecular diagrams of the products of the reactions of (a) twelve- and (b) ten-vertex closo-heteroboranes with Idip to obtain SB11H11-Idip and [6,9-Idip2-C2B8H11]+Cl− based on their crystal structures published in ref. 10 and 11, respectively. | ||
These attempts provided mono- and/or bis-carbene adducts that underwent protonation, surprisingly resulting in nido-shaped monocations. Interestingly enough, the bicapped-square antiprismatic shape in the cationic features obtained is retained only in the case of (2-Idip-closo-1,10-C2B8H9)+ (ref. 11) (for one of these cations, see Scheme 3).
The chemistry of octahedral closo-heteroboranes has so far received less attention than their two larger parent congeners. Apart from carboranes of these shapes,5 the only others known include the perhalogenated octahedral pnictogenahexaboranes closo-1,2-Pn2B4X4 (Pn = P, As, X = Cl, Br)12 (see Scheme 4) and the chalcogenahexaboranes closo-EB5Cl5 (E = Se, Te)13 (for the octahedral molecular shape, see Scheme 2c), from which closo-1,2-P2B4Br4 and closo-1,2-As2B4Br4 are prone to react with an O-base such as THF by successive insertion into two B–Br bonds with different NMR properties with respect to the parent octahedra.14 The reactions of closo perhalogenated heteroboranes with NHC are even rarer and should facilitate the formation of cations by the elimination of halide.
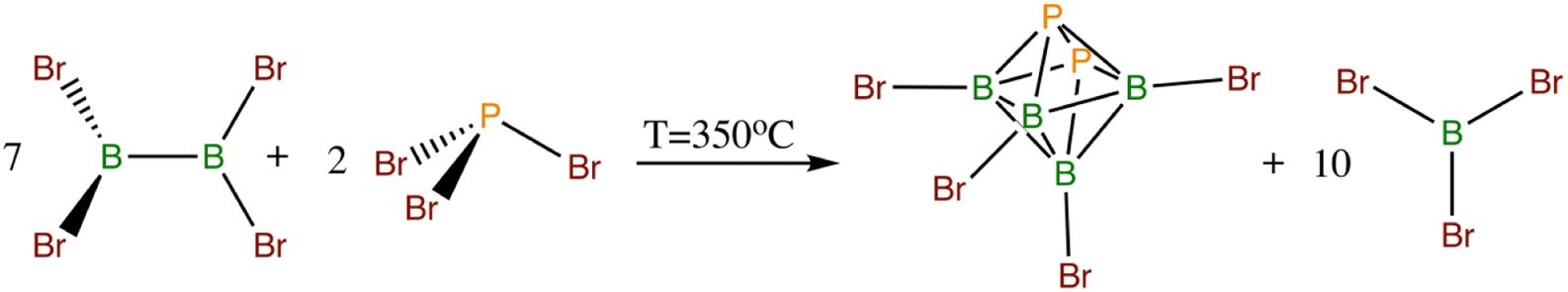 | ||
| Scheme 4 Thermodynamically-driven formation of the octahedral closo-1,2-P2B4Br4. The arsenic analog originates in the same manner as in the case of PBr3. | ||
However, Braunschweig et al. have recently investigated the Lewis acid–base reaction of the classically B–B bonded tetrabromo diborane (4) B2Br4 with two equivalents of Idip, producing a diadduct as an intermediate, which is finally converted with sodium naphthalenide to the first reported bis(NHC)-stabilized triple-bonded diboryne;15 the stabilization effect of the carbene moieties has already been examined as well.16
In order to gain a deeper insight into the behavior of polyhedral octahedral bromoboranes towards NHC, we now examined the reaction of Pn2B4Br4 with Idip and selected CH2Cl2 as a slightly polar solvent with the dipole moment of ca 1.6 D. The reactivity of Pn2B4Br4 towards THF14 (with the same magnitude of the dipole moment as that measured for CH2Cl2) and the tendency of the Pn–Pn vector presented in a heteroborane cluster to form a co-crystal with the π-electron system, as exemplified by toluene,17 of a 2D aromatic system prompted us to avoid aromatic compounds as solvents in the first experiments.
Results and discussion
1. NMR and structural aspects
The reaction of P2B4Br4 with two equivalents of Idip in CH2Cl2 proceeds successively and starts already during heating, but it does not lead to a direct adduct formation as in the case described in ref. 15. It is very likely that Idip induces such a reaction with P2B4Br4 by attacking a phosphorus atom first (P(1), due to the presence of a σ-hole on it14), thereby enabling the attack18 on the second cluster atom with the second Idip, i.e. on B(6), with the whole process being the result of the antipodal coupling19 of these two cage atoms. At first, the 11B NMR spectrum shows three signals at −2.6 ppm, 10.2 ppm and 15.8 ppm in the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio. The two signals in 31P NMR at −123 ppm and −56 ppm are split into doublets due to 1J(31P–31P). In analogy to the reaction with THF, the equivalency of the P-atoms is obviously detached by the substitution on the antipodally coupled pair of B(6) and P(1) in the very first intermediate 1,6-Idip2-P2B4Br3+ (1b), from which another intermediate, i.e. 4-Idip-P2B4Br3+ (1a) originates (see below for the rationalization of the beginning of this reaction). The coupling constant of |217| Hz is much larger than that observed in the reaction with THF (|143| Hz). The examination of the reaction pathways below explains the formation of the final product, i.e. 3,5-Idip2-P2B4Br22+ (3). Its 11B NMR spectrum shows two signals at −1.2 ppm and 17.4 ppm in the 1:1 ratio and one signal in 31P NMR at −144.1 ppm. Neither the monocationic intermediates nor the dication show a cross–peak correlation in the 11B11B COSY NMR spectrum, which may be the result of increased electron deficiency within the cluster framework due to cation formation and is fully consistent with an earlier observation.11
:1:1 ratio. The two signals in 31P NMR at −123 ppm and −56 ppm are split into doublets due to 1J(31P–31P). In analogy to the reaction with THF, the equivalency of the P-atoms is obviously detached by the substitution on the antipodally coupled pair of B(6) and P(1) in the very first intermediate 1,6-Idip2-P2B4Br3+ (1b), from which another intermediate, i.e. 4-Idip-P2B4Br3+ (1a) originates (see below for the rationalization of the beginning of this reaction). The coupling constant of |217| Hz is much larger than that observed in the reaction with THF (|143| Hz). The examination of the reaction pathways below explains the formation of the final product, i.e. 3,5-Idip2-P2B4Br22+ (3). Its 11B NMR spectrum shows two signals at −1.2 ppm and 17.4 ppm in the 1:1 ratio and one signal in 31P NMR at −144.1 ppm. Neither the monocationic intermediates nor the dication show a cross–peak correlation in the 11B11B COSY NMR spectrum, which may be the result of increased electron deficiency within the cluster framework due to cation formation and is fully consistent with an earlier observation.11
The corresponding reaction of As2B4Br4 with Idip is quite retarded in comparison with P2B4Br4, which is analogous to its reactivity with THF.14 Therefore, the interpretation of 11B NMR spectra is less convincing and some signals could be beneath both signals of the starting As2B4Br4, the latter being dominant. The 11B NMR spectrum of the monocationic 4-Idip-As2B4Br3+ (2), obviously detected as the final product, shows three signals at 7.2 ppm (B(3,5)), 21.5 ppm (B(4)) and 23.5 ppm (B(6)), with the signal at 7.2 ppm being superimposed by B(3,5) of the starting As2B4Br4. Further reaction could not be studied unambiguously due to decomposition processes.
The resulting cations were not susceptible to electron ionization (EI) mass spectroscopy. Instead, albeit only in low intensity, the phosphaborane dication containing two Idip moieties was detected as the monoanionic adduct [P2B4Br2(Idip)2(HCl)(2Br−Cl−)]− at m/z = 1273 when the reaction solution in methylene chloride had been subjected to electrospray ionization in negative mode. Such anionic adduct-ion formation from either neutral20 or mono- and even dicationic21 analytes as in the current study with components of the eluent is a frequently observed phenomenon in mass spectra, originating from the electrospray ionization process.22 In these experiments, the halide ions were produced in situ from halogenated solvents.23,24 This process is known to be poorly understood. For further experimental details, including decisive spectroscopic measurements, see the ESI.†
Since the influence of spin–orbit coupling in 11B NMR computations is known,14 we have applied the DFT/ZORA/NMR structural tool, which includes spin–orbit coupling within B–Br bonds,14 in the following structural analyses together with MS spectroscopy. The joint experimental/computational study of the reactions of closo-1,2-Pn2B4Br4 (Pn = P, As) provided a very good ground for believing15 that these approaches are also relevant to the current study. At first, we used the simplified H-NHC in shielding-tensor computations, in which 2,6-iPr2C6H3 is replaced by hydrogen, because this simplification performed well for the cations based on bicapped square-antiprismatic carboranes and helped to reveal their molecular shapes in terms of the ab initio/GIAO/NMR approach.11 The final geometry optimizations of the obtained mono- and dications, in which Idip had been identified, also included the dispersion correction D3(BJ), suggested by Grimme,25 because its use is justified by the possible interactions of the benzene rings.26 On that basis, the formed monocationic 4-Idip-closo-1,2-P2B4Br3+ (1a), 1,6-Idip2-closo-1,2-P2B4Br3+ (1b) and 4-Idip-closo-1,2-As2B4Br3+ (2) together with dicationic 3,5-Idip2-closo-1,2-P2B4Br22+ (3) structural assemblies were subjected to structural studies using the above joint computational/experimental NMR approach (for the final products, see Fig. 1 and also Table 1). Table 1 reveals that the average values of the computed chemical shifts of 1a and 1b provide very good agreement with the experimental values in CH2Cl2 solution in the first steps, enabling the reaction to take place. Conceivably, if the average values of the computed 31P and 11B chemical shifts agree well with the experimental values, the ratio of 1a and 1b can be considered as 1:1. This finding is in general agreement with the examined reaction pathways given below. When the charge of 3,5-Idip2-closo-1,2-P2B4Br2 was postulated to be zero, the P2B4 core opened and the geometry optimization did not converge. Furthermore, the 4,6-Idip2-closo-1,2-P2B4Br22+ structural alternative could be ruled out due to the considerable steric hindrance of two Idips (for NMR consequences, see also Table 1). Note that the agreement between the computed and experimental 31P and 11B NMR chemical shifts in the dication 3 is remarkable, i.e. the internal coordinates derived at the B3LYP/6-311+G(2d,p)+D3(BJ) level provide a good basis for believing that they are a very good representation of its molecular geometry in solution.
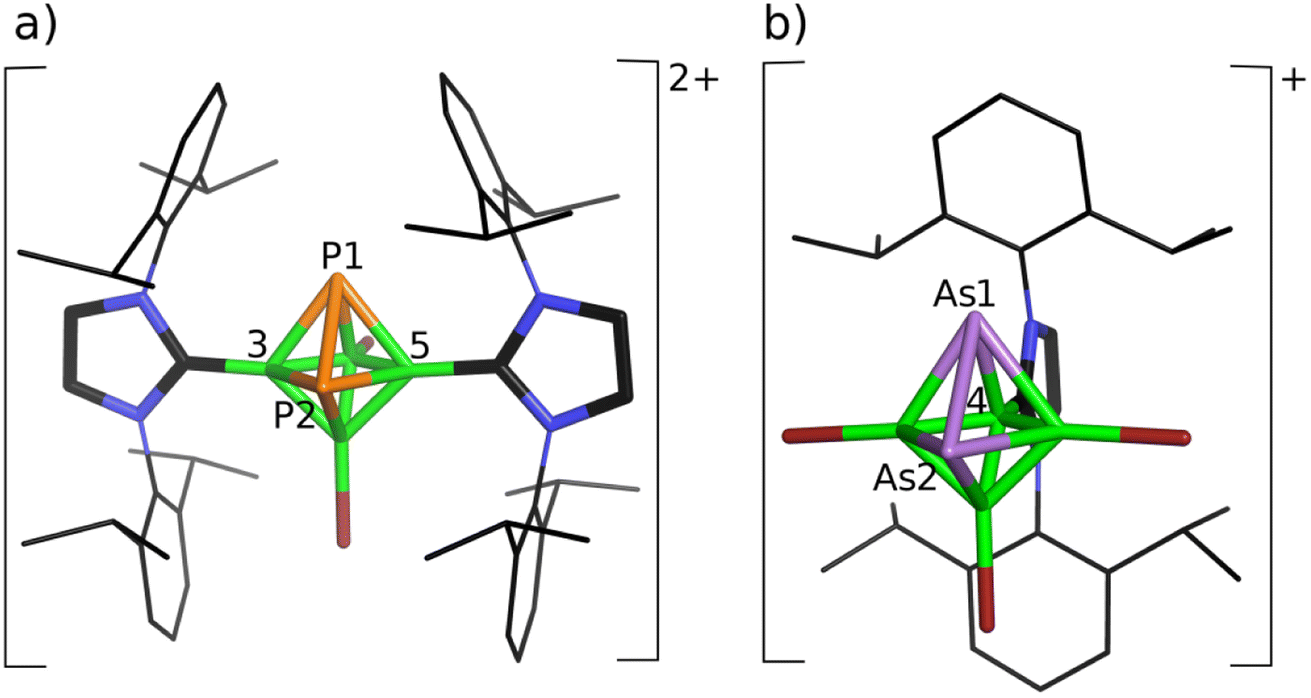 | ||
| Fig. 1 Molecular diagrams of the final products of the reaction of Pn2B4Br4 with Idip: (a) [3,5-Idip2-closo-Pn2B4Br2]2+Br22− and (b) [4-Idip-closo-As2B4Br3]+Br−. Hydrogen atoms are omitted for clarity. For monocationic phosphaintermediates, see the reaction pathways below. | ||
| a ZORA-SO-PBE0/TZ2P//B3LYP/6-311+G(2d,p)+D3(BJ).b For [3-(H-NHC)-1,2-P2B4Br3]+ Br−, these values are computed to be −148 ppm for P(1) and −138 ppm for P(2).c The average values according to the symmetry of the P2B4 core.d For [4,6-(H-NHC)2-1,2-P2B4Br2]2+ 2Br−, these values are computed to be −21 ppm.e For [3-(H-NHC)-1,2-As2B4Br3]+ Br−, this value is computed to be −5.7 ppm.f After heating in CH2Cl2 to 50 °C for 10 min.g After one day at −10 °C in THF, this value is measured to be 144.1 ppm.h Below B(3,5) of the starting As2B4Br4.i The average values of 1a and 1b. | ||||||
|---|---|---|---|---|---|---|
| [4-Idip-1,2-P2B4Br3]+ Br−, 1a | ||||||
| P(1) | P(2) | B(3) | B(4) | B(5) | B(6) | |
| ZORA | −33 | −157 | 0.8 | 15.5 | 0.8 | 17.5 |
|
||||||
| [1,6-Idip2-1,2-P2B4Br3]+ Br−, 1b | ||||||
| ZORA | −81 | −108 | −6.5 | 3.5 | −6.5 | 12.7 |
| (1a + 1b)/2i | −57 | −133 | −3.7 | 9.5 | −3.7 | 15.1 |
| Exp.f | −56.0 | −122.7 | −2.6 | 10.2 | −2.6 | 15.8 |
|
||||||
| [4-(H-NHC)-1,2-P2B4Br3]+ Br− | ||||||
| P(1) | P(2) | B(3) | B(4) | B(5) | B(6) | |
| ZORA | −22b | −151b | 2.9 | 15.2 | 2.9 | 19.0 |
|
||||||
| [3,5-Idip2-1,2-P2B4Br2]2+ 2Br−, 3 | ||||||
| P(1) | P(2) | B(3) | B(4) | B(5) | B(6) | |
| ZORAc | −143 | −143 | −1.2 | 17.4 | −1.2 | 17.4 |
| Exp.g | −145.7 | −145.7 | −1.2 | 17.4 | −1.2 | 17.4 |
|
||||||
| [3,5-(H-NHC)2-1,2-P2B4Br2]2+ 2Br− | ||||||
| P(1) | P(2) | B(3) | B(4) | B(5) | B(6) | |
| ZORA | −150d | −150d | −0.3 | 22.2 | −0.3 | 22.2 |
| [4-Idip-1,2-As2B4Br3]+ Br−, 2 | ||||||
| As(1) | As(2) | B(3) | B(4) | B(5) | B(6) | |
| ZORA | 7.4 | 20.1 | 7.4 | 20.4 | ||
| Exp.f | 7.2 | 21.5 | 7.2 | 23.5 | ||
|
||||||
| [4-(H-NHC)-1,2-As2B4Br3]+ Br− | ||||||
| As(1) | As(2) | B(3)e | B(4) | B(5) | B(6) | |
| ZORA | 7.2 | 20.4 | 7.2 | 21.7 | ||
Table 1 further outlines the influence of various NHCs on the magnetic shielding of the cations and, therefore, on the electronic structures of the given motifs. Although most of the computed 31P and 11B NMR chemical shifts of 1a, 2 and 3 do not differ much from those based on the structures in which Idip is replaced by a simple H-NHC, which worked quite well in ref. 11, some of them exhibit larger discrepancies (1b was not considered with H-NHC). This observation might be ascribed to the influence of the shielding cones27 connected with the benzene rings (for an example, see Fig. 2). Specifically, the 31P NMR chemical shifts of the P atoms in 1a are slightly shielded with respect to the same P atoms in 4-(H-NHC)-closo-1,2-P2B4Br3+. On the contrary, the δ(11B) NMR of B(6) in 3 is deshielded in relation to the same atom in 3,5-(H-NHC)2-closo-1,2-P2B4Br22+. Since the electron count (the number of skeletal electron pairs (SEPs) for the cluster with n vertices)28 of both parent pnictogenahexaboranes is equal, namely 6 + 1 = 7, such a number is equal to 6 + 0.5 for 1a, 1b, 2 and to 6 for 3. In other words, the number of SEPs in 3 is the same as in the six-vertex hypercloso29 hexaborane. Fig. 3 and 4 exemplify the bonding and electronic properties of 3; those of 1 and 2 follow the same patterns (using the optimized structure with the simple H-NHC for the intrinsic-bond orbital (IBO) analyses is justified on the basis of the NMR considerations given above that take the same H-NHC-based optimized structure into account). The bonding properties of the cations within the so-called IBO scheme30 do not differ appreciably from those examined in the parent closo-pnictogenahexaboranes. Conceivably, the whole molecular surface of 3 has a positive electrostatic potential (ESP). The most positive ESP molecular surface (over 113 kcal mol−1, the dark-blue area in Fig. 4) of 3 is on the imidazole ring and also on the octahedral cage. The least positive ESP molecular surface (below 88 kcal mol−1, the green area in Fig. 4) is on the isopropyl moieties.
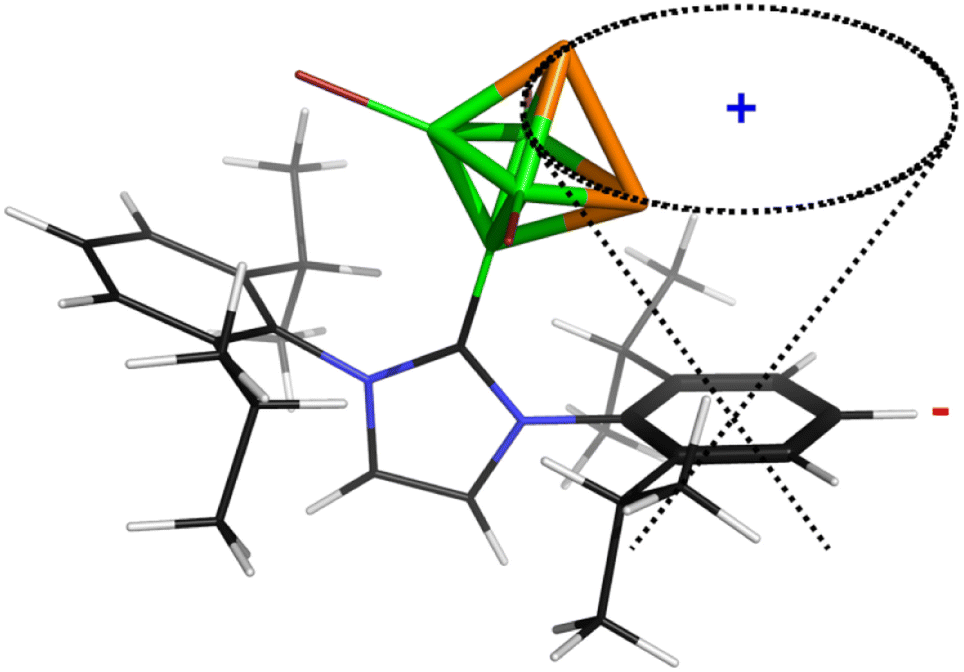 | ||
| Fig. 2 The shielding cone in 1a (blue sign: shielding area, red sign: deshielding area). The second cone, associated with the other side of the aromatic ring, is omitted for clarity. | ||
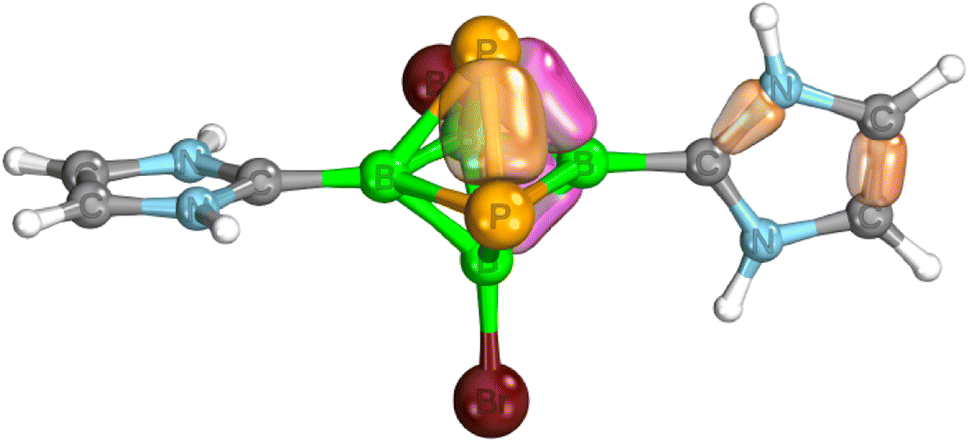 | ||
| Fig. 3 Examples of IBOs for a simplified model of 3 with hydrogens instead of 2,6-(iPr)2-C6H3 in the carbene moieties – see the text. Color coding: yellow – 2c-2e, pink – 3c-2e. | ||
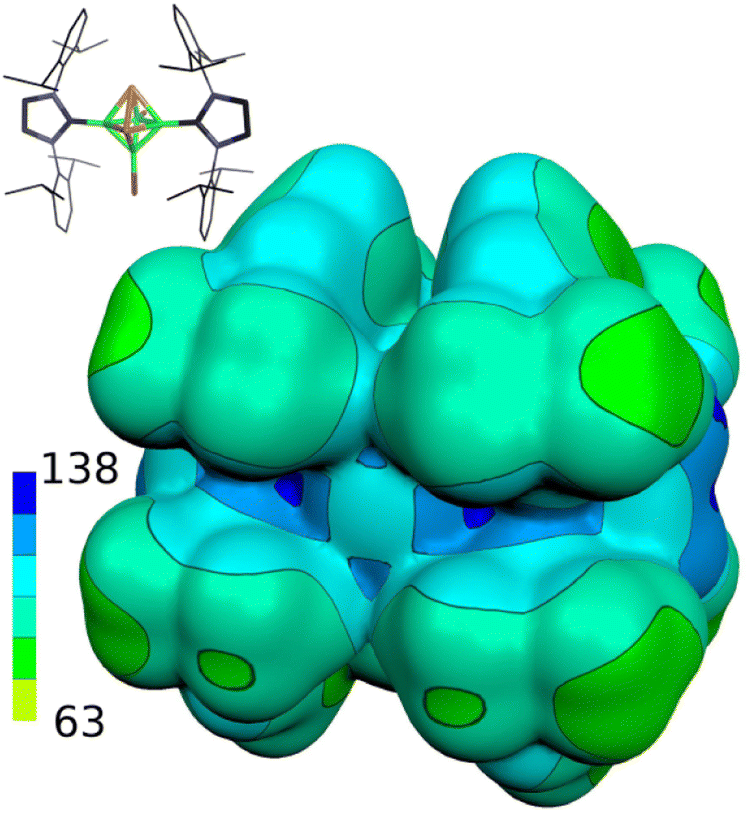 | ||
| Fig. 4 The ESP molecular isosurface of 3. The color range of the ESP in kcal mol−1. | ||
2. Searching the reaction pathways
Since we have to deal with the cations as the final product(s), we have computed reaction profiles by employing the SMD implicit solvent model of dichloromethane for each of the stationary points associated with the pathways below. The necessity of employing dispersion correction has already been pointed out. Using a smaller basis set for the single-point computations in the pathway search (e.g. B3LYP/6-31+G(d)+D3(BJ), i.e. the DZ basis set) instead of B3LYP/6-311+G(2d,p)+D3(BJ) has resulted in higher relative energies of some stationary points, with their high energy excess being in contradiction with the results achieved experimentally. This is particularly true when Br− is taken into account. Moreover, with the mentioned DZ basis set, the computed basis set superposition error (BSSE)31 has been found to be much higher than with the 6-311+G(2d,p) basis set of the TZ type, which further supports the use of the TZ basis set. In order to illustrate the pathway more clearly, we opted for showing the reactions with ID, i.e. with hydrogens instead of very bulky iPr groups. Tables 2 and 3 justify such an assumption. At first, we started searching the reaction pathway with one ID (or Idip). However, the elimination procedure of Br− was connected with a very high barrier of 69.2 kcal mol−1. Therefore, we did follow the experimental procedure described earlier but started the reaction with the formation of 1,6-Idip2-closo-1,2-P2B4Br3+, from which 4-Idip-closo-1,2-P2B4Br3+ originates. The latter procedure takes place through one transition state (TS1) and two elimination procedures as indicated in Fig. 5 (PES1). Table 2 offers energetic criteria for this pathway with both ID and Idip participations. In essence, there are two TS1s, namely TS1-a and TS1-b, both of which possess a transition vector where the ligand moves toward/away from a boron vertex. Table 2 provides their energy differences, whereas the values of the corresponding imaginary frequencies are available in Table S1.† Consequently, there are two monocationic 4-Idip-closo-1,2-P2B4Br3+, differing in the positions of the Idip ligands which are not distinguishable by means of experimental NMR (see also below).| Species | PG | L = ID | L = Idip | ||
|---|---|---|---|---|---|
| ΔH (298 K) | ΔG (298 K, CH2Cl2) | ΔH (298 K) | ΔG (298 K, CH2Cl2) | ||
| a ΔH (298 K) and ΔG (298 K, CH2Cl2) at the B3LYP/6-311+G(2d,p)+D3(BJ)//B3LYP/6-31+G(d)+D3(BJ) level of theory. The free energy of solvation in dichloromethane (kcal mol−1) using the SMD solvation model. All energies in kcal mol−1 relative to 1,2-L2-P2B4Br4 and the appropriate number of Br− anions. | |||||
| P2B4Br4 + 2L | 0.0 | 0.0 | 0.0 | 0.0 | |
| 1-L-P2B4Br4 + L | Cs | −20.6 | −6.8 | −24.7 | −7.0 |
| 1,2-L2-P2B4Br4-a | C2v | −13.3 | 10.3 | −20.2 | 16.5 |
| 1,2-L2-P2B4Br4-b | C1/Cs | −19.3 | 6.1 | −25.2 | 7.7 |
| TS1-a | C1 | −9.6 | 19.7 | −5.1 | 33.8 |
| TS1-b | C1 | −10.9 | 28.0 | ||
| 1,6-L2-P2B4Br4 | C1 | −32.9 | −3.3 | −42.3 | −4.5 |
| 1,6-L2-P2B4Br3+ | C1 | −42.8 | −22.6 | −50.6 | −20.5 |
| 4-L-P2B4Br3-a+ | Cs | −15.8 | −8.8 | −26.3 | −16.8 |
| 4-L-P2B4Br3-b+ | Cs | −20.1 | −14.1 | −24.3 | −14.6 |
| Species | PG | L = ID | L = IDip | ||
|---|---|---|---|---|---|
| ΔH (298 K) | ΔG (298 K, CH2Cl2) | ΔH (298 K) | ΔG (298 K, CH2Cl2) | ||
| a ΔH (298 K) and ΔG (298 K, CH2Cl2) at the B3LYP/6-311+G(2d,p)+D3(BJ)//B3LYP/6-31+G(d)+D3(BJ) level. The free energy of solvation in dichloromethane (kcal mol−1) using the SMD solvation model. All energies in kcal mol−1 relative to 1,2-ID2-P2B4Br4 and the appropriate number of Br− anions.b In the case of L = Idip, there are two candidates for the intermediate (INT). They differ in the orientation of one of the Idip ligands. Only INT-a is shown. | |||||
| P2B4Br4 + 2L | 0.0 | 0.0 | 0.0 | 0.0 | |
| 1-L-P2B4Br4 + L | Cs | −20.6 | −6.8 | −24.7 | −7.0 |
| 1,2-L2-P2B4Br4-a | C2v | −13.3 | 10.3 | −20.2 | 16.5 |
| 1,2-L2-P2B4Br4-b | C1/Cs | −19.3 | 6.1 | −25.8 | 7.7 |
| TS2 | C1 | −18.7 | 17.3 | −15.2 | 22.2 |
| 1,3-L2-P2B4Br4-a | C1 | −44.3 | −16.4 | −45.5 | −8.0 |
| 1,3-L2-P2B4Br4-b | C1 | −42.8 | −16.7 | ||
| 1,3-ID2-P2B4Br3+ | C1 | −44.8 | −24.8 | −42.9 | −15.0 |
| TS3(+) | C1 | −16.4 | 3.1 | −25.2 | 2.1 |
| 3,5-L2-P2B4Br3+ | C1 | −49.0 | −28.6 | −50.4 | −21.4 |
| 3,5-L2-P2B4Br22+ | C2 | −40.6 | −30.4 | −62.7 | −41.2 |
| [3,5-L2-P2B4Br2][Br]2 | C2 | −71.6 | −43.9 | −72.2 | −38.2 |
| Rearrangement | Rearrangement | ||||
| 1,3-L2-P2B4Br3+ | C1 | −44.8 | −24.8 | −42.9 | −15.0 |
| TS4(+) | C1 | −15.5 | 4.6 | −26.7 | 0.3 |
| INT-a(+)b | C1 | −26.4 | −8.5 | −31.6 | −3.9 |
| INT-b(+)b | C1 | −29.1 | −1.0 | ||
| TS5(+) | C1 | −18.8 | 0.5 | 23.3 | 6.7 |
| 1,6-L2-P2B4Br3+ | Cs | −42.8 | −22.6 | −50.6 | −20.5 |
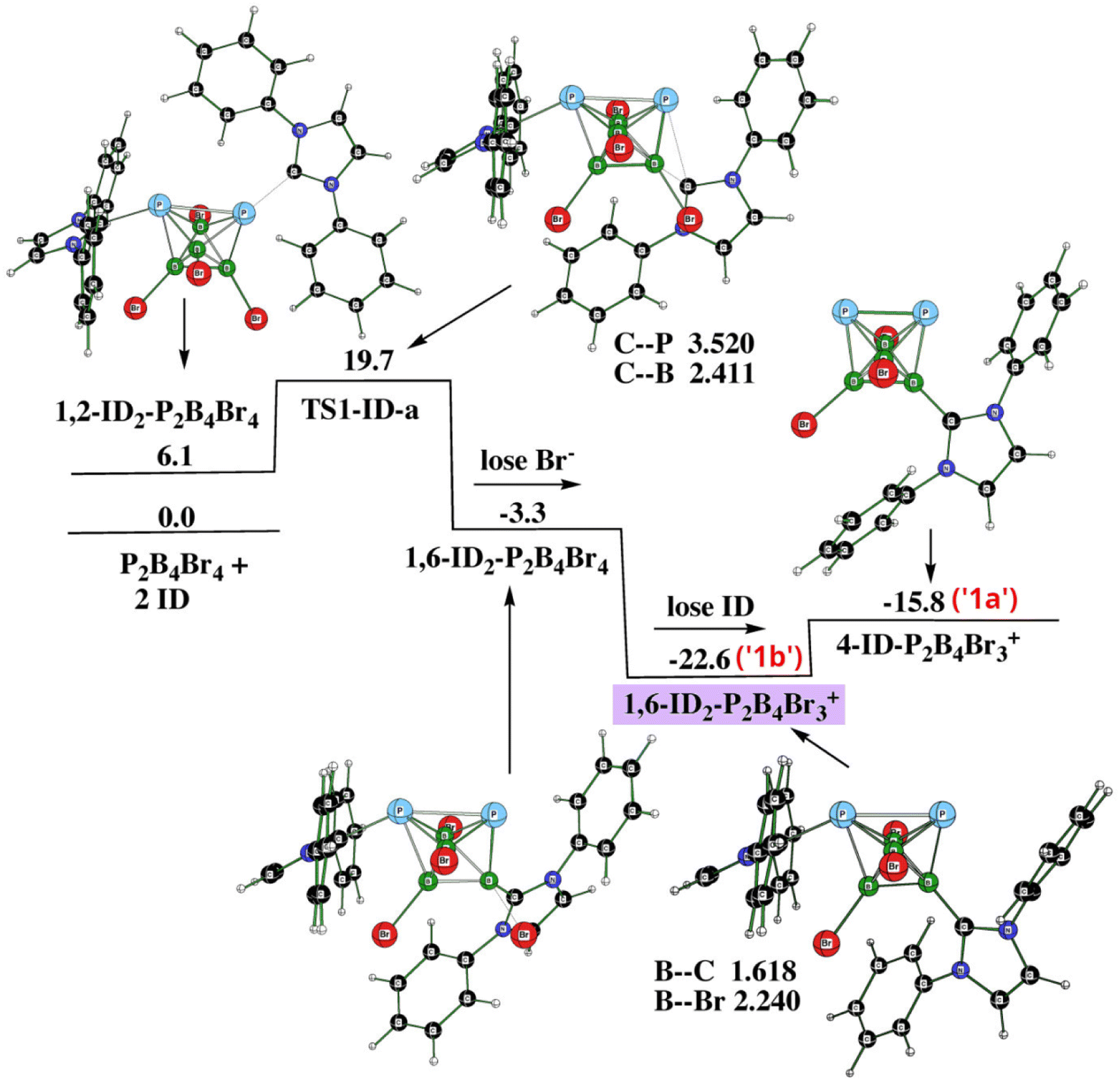 | ||
| Fig. 5 The potential energy surface PES1 with the relative Gibbs free energies (kcal mol−1) of the individual stationary points of the reaction of closo-1,2-P2B4Br4 with ID as computed at the SMD(CH2Cl2)/B3LYP/6-311+G(2d,p)+D3(BJ)//B3LYP/6-31+G(d)+D3(BJ) level of theory (for the methodology description, see computational details in ESI†). The energies are given with respect to closo-1,2-P2B4Br4 + 2ID. | ||
Fig. 6 (PES2) shows the entire procedure for obtaining 3. It requires two transition states as well as two elimination reactions, with the latter being analogous to the formation of the monocation(s). Note that the cationic structure 1,3-ID2-1,2-P2B4Br3+ (yellow) acts as one of the two decisive stationary points assuring the formation of the dication from the monocation; the other one is 1,6-ID2-1,2-P2B4Br3+ (violet) as depicted in Fig. 5. Table 3 gives all energy details.
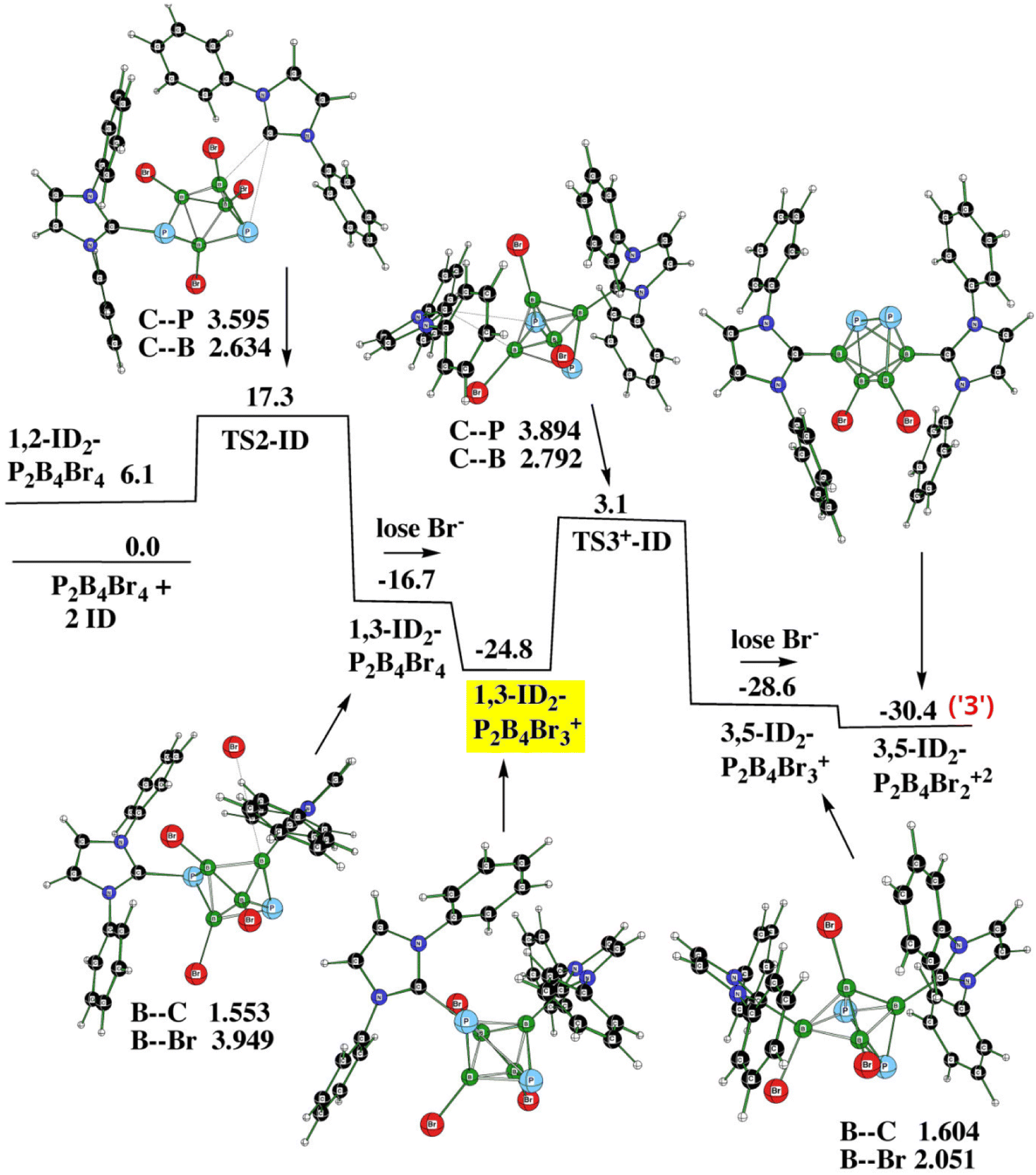 | ||
| Fig. 6 The potential energy surface PES2 with the relative Gibbs free energies (kcal mol−1) of the individual stationary points of the reaction of closo-1,2-P2B4Br4 with ID to form 3 as computed at the SMD(CH2Cl2)/B3LYP/6-311+G(2d,p)+ D3(BJ)//B3LYP/6-31+G(d)+D3(BJ) level of theory (for the methodology description, see computational details in ESI†). The energies are given with respect to closo-1,2-P2B4Br4 + 2ID. | ||
Fig. 7 (PES3) illustrates the connection of PES1 and PES2 through 1,6-ID2-1,2-P2B4Br3+ (violet in PES1) and 1,3-ID2-1,2-P2B4Br3+ (yellow in PES2), which reflects the experimental observation that the formation of monocationic and dicationic species occurs simultaneously. Such an arrangement of these two cations proceeds again through transition states and one intermediate (INT) that adopts the shape of pentagonal pyramide, a process observed earlier for the two isomers of closo-C2B4H6.32
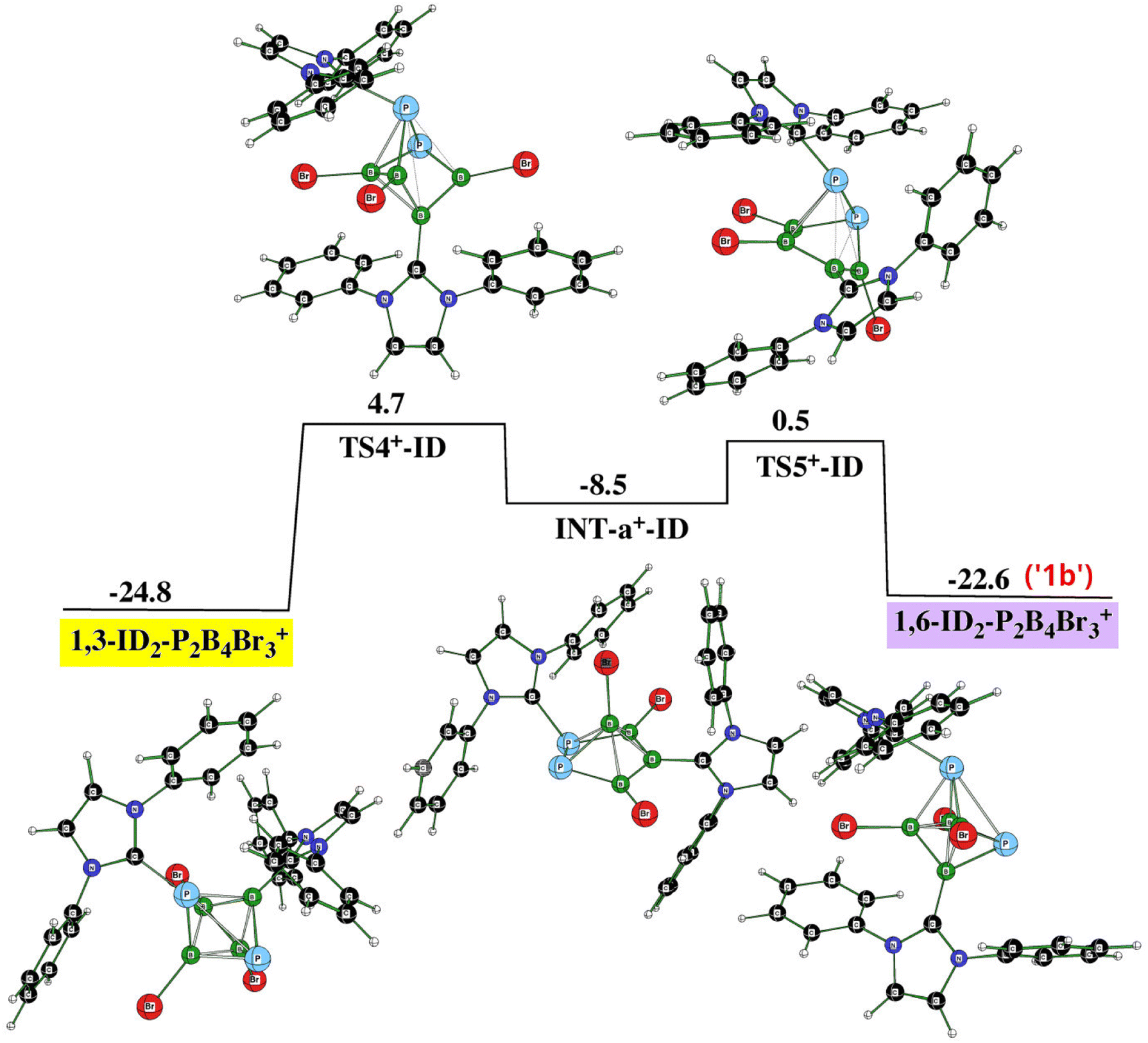 | ||
| Fig. 7 The potential energy surface PES3 with the relative Gibbs free energies (kcal mol−1) of the individual stationary points of the rearrangement between 1,6-ID2-1,2-P2B4Br3+ and 1,3-ID2-1,2-P2B4Br3+ as computed at the SMD(CH2Cl2)/B3LYP/6-311+G(2d,p)+D3(BJ)//B3LYP/6-31+G(d)+D3(BJ) level of theory (for the methodology description, see computational details in ESI†). The energies are given with respect to closo-1,2-P2B4Br4 + 2ID. | ||
Fig. 8 offers the molecular diagram of [3,5-IDip2-P2B4Br2][Br]2, i.e. the dication 3 in the form of an ionic pair with bromide anions. Their comparison in terms of electronic energy for [3,5-Idip2-P2B4Br2][Br]2 and 3,5-Idip2-P2B4Br22+ + 2Br− at the B3LYP/6-311+G(2d,p)+D3(BJ) and B3LYP/6-31+G(d)+ D3(BJ) levels shows that the binding energy of two Br− anions is 34.2 kcal mol−1 smaller in the case of the larger basis set. This supports the idea that there is a large BSSE (see above) at the B3LYP/6-31+ G(d)+D3(BJ) level for [3,5-Idip2-P2B4Br2][Br]2, which further disqualifies the use of the smaller basis set for single-point computations. Therefore, it is possible that the [3,5-IDip2-P2B4Br2][Br]2 complex does not exist in CH2Cl2 solution or there could be an equilibrium with the dication itself and the two bromide anions in it. Indeed, the computed 31P and 11B NMR chemical shifts of the ionic pair [3][Br]2 differ from those computed for the dication itself by less than 1.5 ppm, which gives a sort of preference to the latter equilibrium. Moreover, the formation of an adduct Idip⋯HBr cannot be ruled as also indicated in ref. 10, 11, and 15.
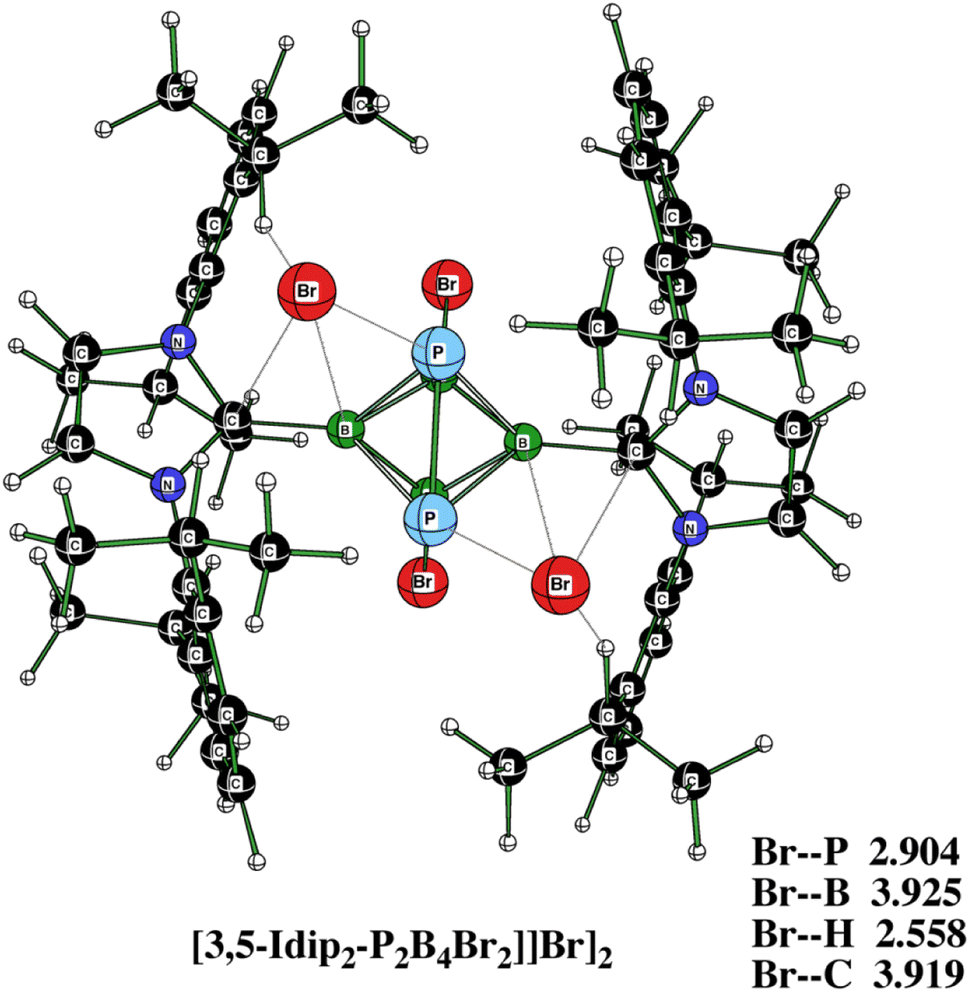 | ||
| Fig. 8 The molecular diagram of [3,5-Idip2-P2B4Br2][Br]2 at B3LYP/6-31+G(d) + D3(BJ), 3[Br]2. | ||
Obviously, 4-Idip-closo-As2B4Br3+ (2), originates in the same manner as the phosphorus analogue.
As already indicated, 1,2-P2B4Br4 easily reacts with THF to provide 4-OBuBr-1,2-P2B4Br3 (4-OBuBr-PB).14 Guided by these findings, we have also examined the reaction 1,2-P2B4Br4 with ID (ID instead of Idip for the reasons given above) in the presence of THF instead of CH2Cl2. According to the HOMO–LUMO concept (LUMO is connected with the electron acceptor diphosphahexaborane),2 the HOMO–LUMO gap for the pair P2B4Br4 + Idip at HF/6-311+G(2d,p) is about 51.3 kcal mol−1 more favorable than that for the pair P2B4Br4 + THF, which means that the corresponding initial mutual contact is more affordable for the first pair. When comparing the P2B4Br4/Idip and 4-OBuBr-PB/Idip FMO pairs, the former is about 4.9 kcal mol−1 more favorable than the latter as computed at the same level. The same applies to As2B4Br4. In other words, the reaction of Pn2B4Br4 with Idip should be preferred over that with THF, i.e. the latter acts as a solvent as CH2Cl2 does when 1,2-P2B4Br4 and Idip are present together.
In order to examine this conclusion computationally, we tried to find the transformation of “THF-based” 4-OBuBr-1,2-P2B4Br3 with Idip to 4-Idip-1,2-P2B4Br3+ as a competitive reaction with respect to that illustrated in Fig. 5. As can be seen from Fig. 9, such a reaction is highly endothermic, which is in line with the conclusion already postulated in terms of the above HOMO/LUMO approach. The only “exothermic” part of this pathway is the formation of the intermediate 4-ID-4-OBuBr-1,2-P2B4Br3, which lies 0.8 kcal mol−1 below the starting motifs of this reaction. Note that the attack of ID on the P atom antipodally coupled with B(4) (to which OBuBr is bonded) is 7.6 kcal mol−1 less favorable than the current situation, in which the first intermediate, 1-ID-3-OBuBr-PB, is formed (see Fig. 9). The diamagnetic shielding of this P atom caused by an increase of electron density on the P atom,14 which makes the attack of the electron pair of ID on this pnictogen energetically more demanding. In summary, 1,2-P2B4Br4 provides 3 also in the presence of THF (see Fig. S5 and S6 in ESI†). Interestingly enough, these figures reveal the change of the 1a:1b ratio in the course of the reaction; their overall ten resonances are subject to mutual “mixing”.
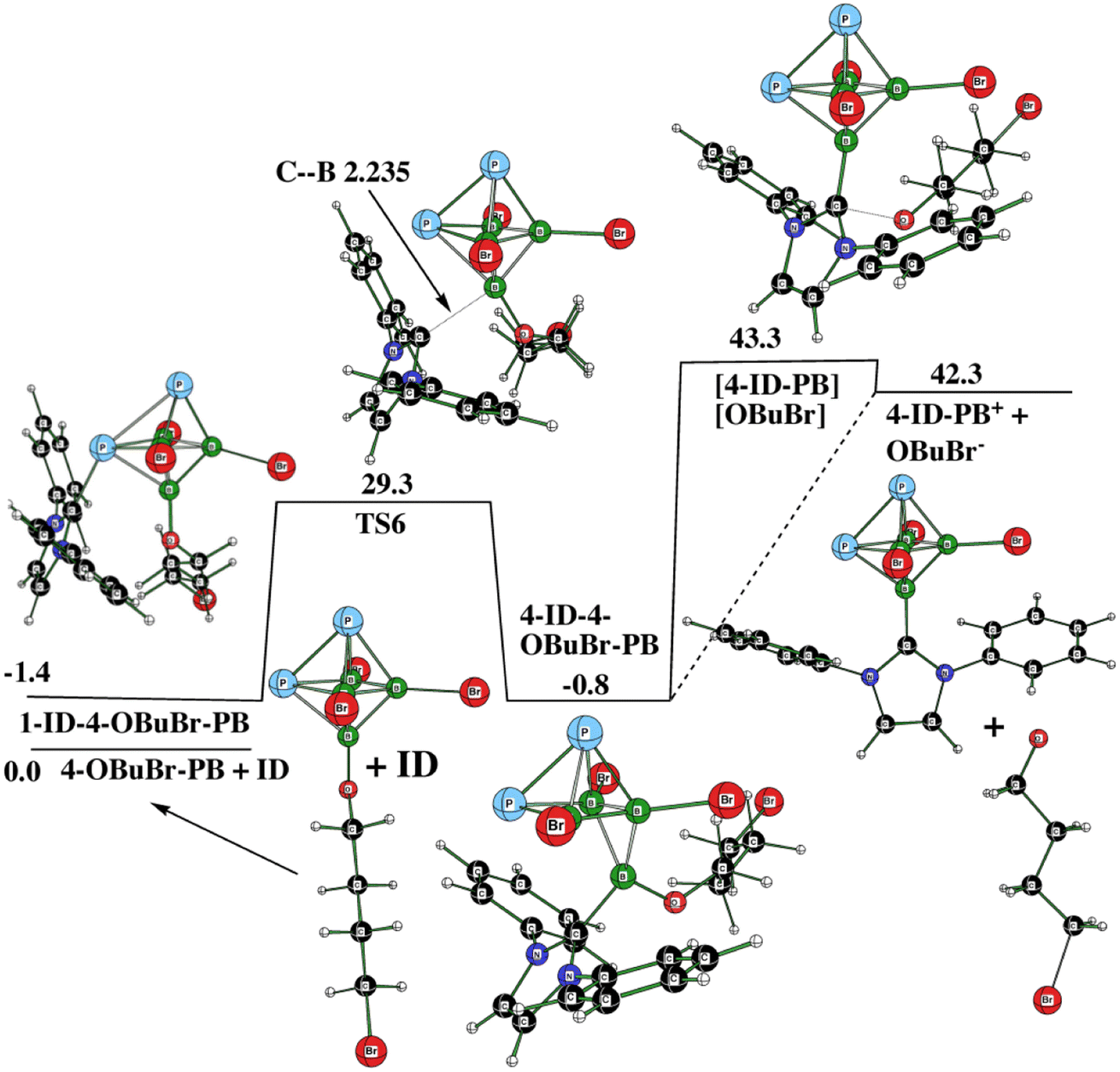 | ||
| Fig. 9 The potential energy surface PES4 with the relative Gibbs free energies (kcal mol−1) of the individual stationary points of the endothermic transformation of 1-ID-3-OBuBr-1,2-P2B4Br3 + THF to form 4-ID-P2B4Br3+ as computed at the SMD(THF)/B3LYP/6-311+G(2d,p)+D3(BJ) level of theory (for the methodology description, see computational details in ESI†). | ||
Conclusions
The unique dication 3,5-Idip2-1,2-P2B4Br22+ and the monocations 1,6-Idip2-closo-P2B4Br3+ and 4-Idip-closo-Pn2B4Br3+ (Pn = P and As, for P as intermediates), all of them adopting octahedral shapes, have been afforded. Computations of the electrostatic potentials have revealed that their positive charges are delocalized over all the moieties including the octahedrons. Their existence and molecular structures in solution have been derived through the application of the well-established joint experimental/computational DFT/ZORA/NMR approach. The agreement between the computed and experimental 31P and 11B chemical shifts is remarkable in the case of the dication and more than satisfactory for the mixture of the monocations 1,6-Idip2-closo-P2B4Br3+ and 4-Idip-closo-P2B4Br3+, appearing as intermediates together with 4-IDip-closo-As2B4Br3+. This confirms the reliability of their molecular geometries in CH2Cl2 or THF solutions. In theory, the substitution of a terminal atom bonded to boron with N-heterocyclic carbene results in the formation of a cation in any neutral closo heteroborane. These efforts open new possibilities in boron-cluster chemistry, so far mainly limited by the existence of neutral or anionic cages of various shapes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Czech Science Foundation (Project No. 22-03945S).Notes and references
- See e.g. H. A. Huynh, Chem. Rev., 2018, 118, 9457 CrossRef CAS PubMed.
- For example I. Fleming, Frontier Orbitals and Organic Chemical Reactions, John Wiley and Sons, Chichester, Sussex, UK, 1976 Search PubMed.
- D. Hnyk and D. A. Wann, Boron: the Fifth Element Challenges and Advances in Computational Chemistry and Physics, ed. D.Hnyk, M. L.McKee, 2016, vol. 20, ch. 2 and the extensive references therein Search PubMed.
- P. Melichar, D. Hnyk and J. Fanfrlík, Phys. Chem. Chem. Phys., 2018, 20, 4666 RSC and the references therein..
- P. v. R. Schleyer, K. Najafian and A. M. Mebel, Inorg. Chem., 1998, 37, 6765 CrossRef CAS PubMed.
- D. Hnyk, V. Všetečka, L. Drož and O. Exner, Collect. Czech. Chem. Commun., 2001, 66, 1375 CrossRef CAS.
- D. Hnyk and E. G. Jayasree, J. Comput. Chem., 2013, 34, 656 CrossRef CAS PubMed.
- D. S. Perumalla, S. Ghorai, T. Pal, D. Hnyk, J. Holub and E. D. Jemmis, J. Comput. Chem., 2023, 44, 256 CrossRef CAS PubMed.
- C. E. Willans, C. A. Kilner and M. A. Fox, Chem.–Eur. J., 2010, 16, 10644 CrossRef CAS PubMed.
- J. Vrána, J. Holub, Z. Růžičková, J. Fanfrlík, D. Hnyk and A. Růžička, Inorg. Chem., 2019, 58, 2471 CrossRef PubMed.
- J. Vrána, J. Holub, M. A. Samsonov, Z. Růžičková, J. Cvačka, M. L. McKee, J. Fanfrlík, D. Hnyk and A. Růžička, Nat. Commun., 2021, 12, 4971 CrossRef PubMed.
- (a) W. Keller, L. G. Sneddon, W. Einholz and A. Gemmler, Chem. Ber., 1992, 125, 2343 CrossRef CAS; (b) W. Keller, W. Einholz, D. Rudolph and T. Schleid, Z. Anorg. Allg. Chem., 2017, 643, 664 CrossRef CAS.
- (a) W. Keller, M. Hofmann and Z. Anorg, Allg. J. Chem., 2017, 643, 729 CrossRef CAS; (b) W. Keller, J. Ballmann, M. B. Sárosi, J. Fanfrlík and D. Hnyk, Angew. Chem., Int. Ed., 2023, 62, e2022119018 Search PubMed.
- W. Keller, M. Hofmann, M. B. Sárosi, J. Fanfrlík and D. Hnyk, Inorg. Chem., 2022, 61, 16565 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurts, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef CAS PubMed.
- R. Köppe and H. Schnöckel, Chem. Sci., 2015, 6, 1199 RSC.
- J. Fanfrlík and D. Hnyk, Crystals, 2018, 8, 390 CrossRef the P-P vector forms co-crystals with π-electron systems exemplified by toluene..
- J. P. Waters, T. A. Everitt, K. Myers and J. M. Goicoechea, Chem. Sci., 2016, 7, 6981 RSC.
- (a) S. Heřmánek, D. Hnyk and Z. Havlas, J. Chem. Soc., Chem. Commun., 1989, 1859 RSC; (b) M. Bühl, P. v. R. Schleyer, Z. Havlas, D. Hnyk and S. Heřmánek, Inorg. Chem., 1991, 30, 3107 CrossRef.
- A. Kruve and K. Kaupmees, J. Am. Soc. Mass Spectrom., 2017, 28, 887 CrossRef CAS PubMed.
- C. Xu, H. Guo, Z. S. Breitbach and D. W. Armstrong, Anal. Chem., 2014, 86, 2665 CrossRef CAS PubMed.
- A. Kruve and K. Kaupmees, J. Am. Soc. Mass Spectrom., 2017, 28, 887 CrossRef CAS PubMed.
- L. Nagy, T. Nagy, G. Deák, Á. Kuki, M. Purgel, M. Narmandakh, B. Iván, M. Zsuga and S. Kéki, J. Am. Soc. Mass Spectrom., 2016, 27, 432 CrossRef CAS PubMed.
- Anionic adducts of halogenated closo-heteroboranes, such as [P2B4Br5]−, [P2B4Br4(OBuBr)]− or [P4B8Br7]−, were formed in ESI-mass spectroscopy from solutions of neutral closo-P2B4Br4, closo-P2B4Br3ObuBr or conjuncto-(P2B4Br3)2 in methylene chloride.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Maué, P. H. Strebert, D. Bernhard, S. Rösel, P. R. Schreiner and M. Gerhards, Angew. Chem., Int. Ed., 2021, 60, 11305 CrossRef PubMed.
- M. Baranac-Stojanovic, RSC Adv., 2014, 4, 308 RSC.
- See e.g. K. Wade, Electron Deficient Compounds, Nelson, London, 1971 Search PubMed.
- For hypercloso boron halides, see e.g. J. A. Morrison, Chem. Rev., 1991, 91, 35 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834 CrossRef CAS PubMed.
- M. Gray, P. E. Bowling and J. M. Herbert, J. Chem. Theory Comput., 2022, 18, 6742 CrossRef CAS PubMed.
- For the rearrangement in octahedral cages, see: M. L. Kee, J. Am. Chem. Soc., 1988, 110, 5317 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Descriptions of experiments, 11B and 31P NMR spectra, ESI-MS spectra, energy characterizations of all the stationary points found, Cartesian geometries of all the detectable species. See DOI: https://doi.org/10.1039/d3ra03665k |
| ‡ Present address: Zentrum für Nanosystemchemie, Universität Würzburg, Theodor-Boveri-Weg, 97074 Würzburg, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |