
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient method to access spiro pseudoindoxyl ketones: evaluation of indoxyl and their N-benzylated derivatives for inhibition of the activity of monoamine oxidases†
Karuppaiah Perumala,
Jiseong Leeb,
Sesuraj Babiola Annes a,
Subburethinam Ramesh*a,
T. M. Rangarajanc,
Bijo Mathewd and
Hoon Kim*b
a,
Subburethinam Ramesh*a,
T. M. Rangarajanc,
Bijo Mathewd and
Hoon Kim*b
aDepartment of Chemistry, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur 613 401, Tamil Nadu, India. E-mail: rameshsbdu@gmail.com
bDepartment of Pharmacy, Research Institute of Life Pharmaceutical Sciences, Sunchon National University, Suncheon 57922, Republic of Korea. E-mail: hoon@sunchon.ac.kr
cDepartment of Chemistry, Sri Venkateswara College, University of Delhi, New Delhi, India
dDepartment of Pharmaceutical Chemistry, Amrita School of Pharmacy, Amrita Vishwa Vidyapeetham, AIMS Health Sciences Campus, Kochi 682 041, India
First published on 22nd August 2023
Abstract
A simple, metal-free approach was developed to obtain novel pseudoindoxyl derivatives. The reaction was mediated by tBuOK on tetrahydrocarbazole 8 in dimethyl sulfoxide (DMSO) at room temperature through the hydroxylation of the indole double bond and a subsequent pinacol-type rearrangement. Spiro pseudoindoxyl compounds and their N-benzylated derivatives were assessed for their inhibitory activities against monoamine oxidase (MAO) enzymes. Based on half-maximal inhibitory concentration (IC50) values, 13 compounds were found to have higher inhibitory activity against MAO-B than against MAO-A. With regard to MAO-B inhibition, 11f showed the best inhibitory activity, with an IC50 value of 1.44 μM, followed by 11h (IC50 = 1.60 μM), 11j (IC50 = 2.78 μM), 11d (IC50 = 2.81 μM), and 11i (IC50 = 3.02 μM). Compound 11f was a competitive inhibitor with a Ki value of 0.51 ± 0.023 μM. In a reversibility experiment using dialysis, 11f showed effective recovery of MAO-B inhibition similar to that of safinamide. These experiments suggested that 11f was a potent, reversible, and competitive inhibitor of MAO-B activity.
Introduction
Many alkaloids and biologically active substances have spiro core heterocyclic structures.1 Indole is an important heterocyclic core found in many natural products. The pseudoindoxyl sub-structural motif is a unique subgroup of the class of oxygenated indoles among the many alkaloids.1 The pesudoindoxyl structure (especially one with a spiro quaternary C2 carbon center) is an essential skeleton of indole alkaloids. The pseudoindoxyl alkaloids are an excellent example of a 2,2-disubstitution of a pseudoindoxyl compound, which is a preferred entity because it is a vital part of many important chemical compounds.2 Examples of characteristic natural products with different biological activities in these spiro pseudoindoxyl compounds include duocarmycin c1/b2, brevianamide A (4), (+)-aristotelone (3), and diketopiperazine (7a) (Fig. 1).3 | ||
| Fig. 1 Examples of important pseudoindoxyl derivatives. | ||
Moreover, the extracts of leaves of Mitragyna speciosa and flowering plants of the Rubiaceae family contain alkaloids with diverse structural motifs such as indole, indolenine, and spiro pseudoindoxyl. The natural product mitragynine undergoes rearrangement into mitragynine pseudoinoxyl after oxidation at an indole site to form 7-hydroxymitragynine, and all of them show powerful pain-relieving (or analgesic) effects and opioid activity. The mitragynine pseudoindoxyl acts non-selectively on mu- and delta-opioid receptors.2h,j These opioids have shown neuroprotective effects against hypoxia–ischemia injury and have a modulatory effect on the regulation of neurotransmitter chemicals. Recently, the importance of opioids receptors in modulation of more complicated neurodegenerative diseases (e.g., Alzheimer's disease (AD)) has been shown.2j,k The biological activity of these alkaloids has yet to be explored. Because of the affinity of these compounds with opioid receptors, we were interested in evaluating the role of the pseudoindoxyl core in neurodegenerative diseases by inhibiting some of their target enzymes, such as monoamine oxidases (MAOs).
Furthermore, in recent years, these compounds have been demonstrated to have fascinating roles in terms of bioactivity, materials research, and optoelectronic research (Fig. 1).4–7 For example, the lipid droplets in the 3T3L1 cell line and fat deposits in zebrafish were strained using these pseudoindoxyl derivatives without apparent toxicity up to 100 mM.4 Alkyne-linked 2,2-disubstituted pseudoindoxyl oligomers have been identified as extended beta-strand mimics.5 Four organic dye compounds generated from pseudoindoxylyl showed excellent photophysical characteristics.6 A novel family of pseudoindoxyl compounds with a useful two-photon characteristic has been used for bioimaging and sensing studies.7
Therefore, the methodology in accessing the pseudoindoxyl core is of high interest and in demand. Significant effort has been invested in designing these crucial structures,8 such as 8-desbromohinckdentine A.1 A typical procedure involves the two-step process of hydroxylation using oxidants such as DMDO, mCPBA, HNO2, and oxone, followed by a reaction in which the hydroxyl-imine is rearranged to produce a spiro compound (2,2-disubstituted pseudoindoxyl) using a strong acid or base8a–k (Scheme 1, eqn (a)). Several methods have been reported for accessing the pseudoindoxyl core. Glorius et al. described a highly enantioselective NHC-catalyzed annulation method for synthesizing pseudoindoxyl derivatives from unsaturated aldehydes and aurones (Scheme 1, eqn (b)).9a In contrast, Sorensen described a two-step process for the Houben–Hoesch cyclization and Ugi reaction utilizing imines and isocyanides (Scheme 1, eqn (c)).9b Allenamides were activated by Rossi et al. using a gold catalyst, and the resultant furan ring-opening reaction led to the production of pseudoindoxyl derivatives (Scheme 1, eqn (d)).9d Xiao et al. reported on visible-light hydroxylation and cyclization catalyzed by Ru(bpy)3Cl2·6H2O.9f Stepwise oxidation followed by a rearrangement reaction, stoichiometric oxidants,8 as well as the need for transition metals,9 expensive metal photocatalysts,9f and high reaction temperatures are the drawbacks of these methodologies.8,9 As a result, alternative synthetic routes to the spiro-pseudoindoxyl structure must be developed using a mild and efficient approach.
 | ||
| Scheme 1 Synthetic approaches for spiro pseudoindoxyl derivatives. | ||
MAOs are necessary for the management of brain function. MAOs oxidize monoamines to produce ammonia, aldehydes, and hydrogen peroxide as byproducts. Excessive production of these byproducts of monoamine metabolism generate free radicals, which lead to apoptosis and give rise to several neurodegenerative disorders, such as AD and Parkinson's disease (PD).10,11 MAO inhibitors are employed as co-adjuvants to treat neurodegenerative illnesses (selective inhibitors of MAO-B) or antidepressants (selective inhibitors of MAO-A). Such studies may be of significant interest because of the crucial role of MAOs in regulating brain processes, mood, cognitive activity, and monoamine catabolism.12,13 The US Food and Drug Administration-approved MAO-B inhibitor selegiline binds irreversibly to the enzyme target.14 This action can lead to target disruption, a prolonged duration of action, and poor pharmacokinetic profiles.15–17 The lack of alternatives, combined with the limitations of existing drugs, has led to a new trend in the design and development of molecules in the reversible-binding mode. Many research teams have explored the potential of MAO-B inhibition of indole derivatives and a bicyclic heteroaromatic nucleus.18–20 However, the role of the natural products spiro pseudoindoxyls in neurodegenerative diseases have not been explored. We were able to develop a simple and straightforward methodology for accessing the pseudoindoxyl core of natural products. Hence, we were interested in evaluating the MAO inhibitory effects of the pseudoindoxyl motif and its N-benzyl derivatives.
Results and discussion
Chemistry
The development of cheaper and more environmentally friendly synthetic-chemical approaches is highly desirable. In this context, we developed a metal-free, base-promoted synthesis of heterocyclic spiro compounds through 3-hydroxylation of tetrahydro carbazole. This novel approach incorporates a metal-free, one-pot synthesis under mild reaction conditions with a cheap base (tBuOK). Tetrahydrocarbazole (THC) was used as a precursor for synthesizing spiro ketones, which were obtained readily using Fisher indole synthesis. The MAO-inhibitory profiles of these novel synthesized spiro-based indole derivatives were evaluated (Scheme 1, eqn (e)).Tetrahydrocarbazole THC (8a)21 (1 equiv), NaOH (1.5 equivalent), and a solvent combination (water–DMSO) were used for the initial optimization study, and the final product 10a was obtained at 40% yield without formation of the intermediate 9a. The structure of compound 10a was determined unambiguously by 1H, 13C, and high-resolution mass spectrometry (HRMS) (ESI†). The aliphatic methylene (–CH2) protons shifted from their original positions in compound 8a owing to formation of a spiro ring in compound 10a. The chemical shift was most likely caused by the more strained structure and presence of a carbonyl group at C3 in compound 10a, which caused the chemically equivalent –CH2 protons to become non-equivalent 2.11–2.05 (2H, m), 2.00–1.93 (2H, m), 1.88–1.83 (2H, m), and 1.75–1.70 (2H, m). The broad singlet at 5.00 ppm indicated the presence of a free-NH group. The deshielding behavior exhibited by the aromatic ring appeared as one doublet, one triplet, and one multiplet at the corresponding frequencies of 7.61, 7.43, and ∼6.8 ppm, respectively. The overlap of a doublet and a triplet caused the appearance of a multiplet. The signal at 205.20 ppm in the 13C nuclear magnetic resonance (NMR) spectrum of CDCl3 at 75 MHz demonstrated the presence of ketone functionality. The less intense peaks at 159.96, 120.58, and 74.64 ppm were responsible for quaternary carbons in 10a. The peak at 74.64 ppm was highly significant and revealed a newly formed quaternary spiro carbon. Aromatic deshielded carbons were represented by peaks at 136.89, 124.54, 118.65, and 112.22 ppm. Aliphatic methylene carbons were indicated by peaks at 38.01 and 25.41°. HRMS (m/z) analysis verified the mass of 10a. Further optimization of reaction conditions using a water–DMF solvent mixture reduced the yield to 38% (Table 1, entries 1 and 2). Use of a K2CO3 base (Table 1, entries 3 and 4), with or without an inert atmosphere, did not significantly affect the yield of the spiro compound 10a. The yield did not increase considerably upon addition of Cs2CO3 (Table 1, entry 5). The base t-BuOK facilitated the reaction in an N2 environment, improving the yield of spiro compound 10a significantly (68%) in 24 h (Table 1, entry 6). With this successful attempt, reactions were also carried out under air and an oxygen-containing atmosphere, and afforded 32% and 69% yields, respectively, of the spiro compound 10a (Table 1, entries 7 and 8). The results stated above indicated that the base was quenched in open air owing to moisture and that the presence of oxygen was crucial for the reaction because the reaction was completed within 2.5 h in an oxygen balloon. We repeated the reaction in the absence of light to establish the importance of visible light, but a discernible response was not observed (Table 1, entry 9). The conversion of the starting material THC 8a was affected by reducing the reaction time to 1 h (Table 1, entry 10). While heating the reaction mixture with DMF at 50 °C, several spots in thin-layer chromatography (TLC) were seen (Table 1, entry 11). The optimized conditions for the synthesis of spiro pseudoindoxyl compound 10a were, therefore, set at 1 equiv. of THC 8a in DMSO solvent and 1.5 equiv. of t-BuOK under an O2 atmosphere for 2.5 h (Table 1, entry 8).
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yieldb (%) |
| a Unless noted otherwise, base 1.5 equiv., DMSO 1 mL, 24 h, N2 atmosphere.b Isolated yield.c Open air.d Under O2 atm, 2.5 h.e Under dark condition.f 1 h.g 50 °C, 2 h. | |||
| 1c | NaOH (1.5 equiv.) | H2O + DMSO | 40 |
| 2c | NaOH (1.5 equiv.) | H2O + DMF | 38 |
| 3c | K2CO3 (1.5 equiv.) | DMSO | 40 |
| 4 | K2CO3 (1.5 equiv.) | DMSO | 47 |
| 5 | Cs2CO3 (1.5 equiv.) | DMSO | 52 |
| 6 | t-BuOK (1.5 equiv.) | DMSO | 68 |
| 7c | t-BuOK (1.5 equiv.) | DMSO | 32 |
| 8d | t-BuOK (1.5 equiv.) | DMSO | 69 |
| 9e | t-BuOK (1.5 equiv.) | DMSO | 60 |
| 10f | t-BuOK (1.5 equiv.) | DMSO | 41 |
| 11g | t-BuOK (1.5 equiv.) | DMF | 36 |

The scope and limitations of synthesizing spiro pseudoindoxyl derivatives were examined under ideal conditions (Scheme 2). Spiro pseudoindoxyl derivatives 10a–10j were synthesized using various amounts of THC (8a–8j),21 and the isolated products were characterized by NMR and HRMS. The yield of product 10b was not affected significantly by the electron-donating group at the R1 position of THC (8b). The compounds with chloro- and bromo-substitution at the R1 position (8c and 8d) produced the corresponding spiro compounds (10c and 10d) in better yields than the fluoro-substituted THC 8e, among the halogen groups (8c–8e) in the R1 position of THC. The yield of compound 10f was reduced marginally, whereas the yield of the electron-withdrawing bromo-substituted compound 10g was lower than that of compound 10f. The methyl substitution at R3 had little effect on the yield of product 10h, whereas the bromo substitution at R3 had a considerable effect on the yield of product 10i. The disubstituted compound (8j) yielded spiro compound 10j in modest yield.
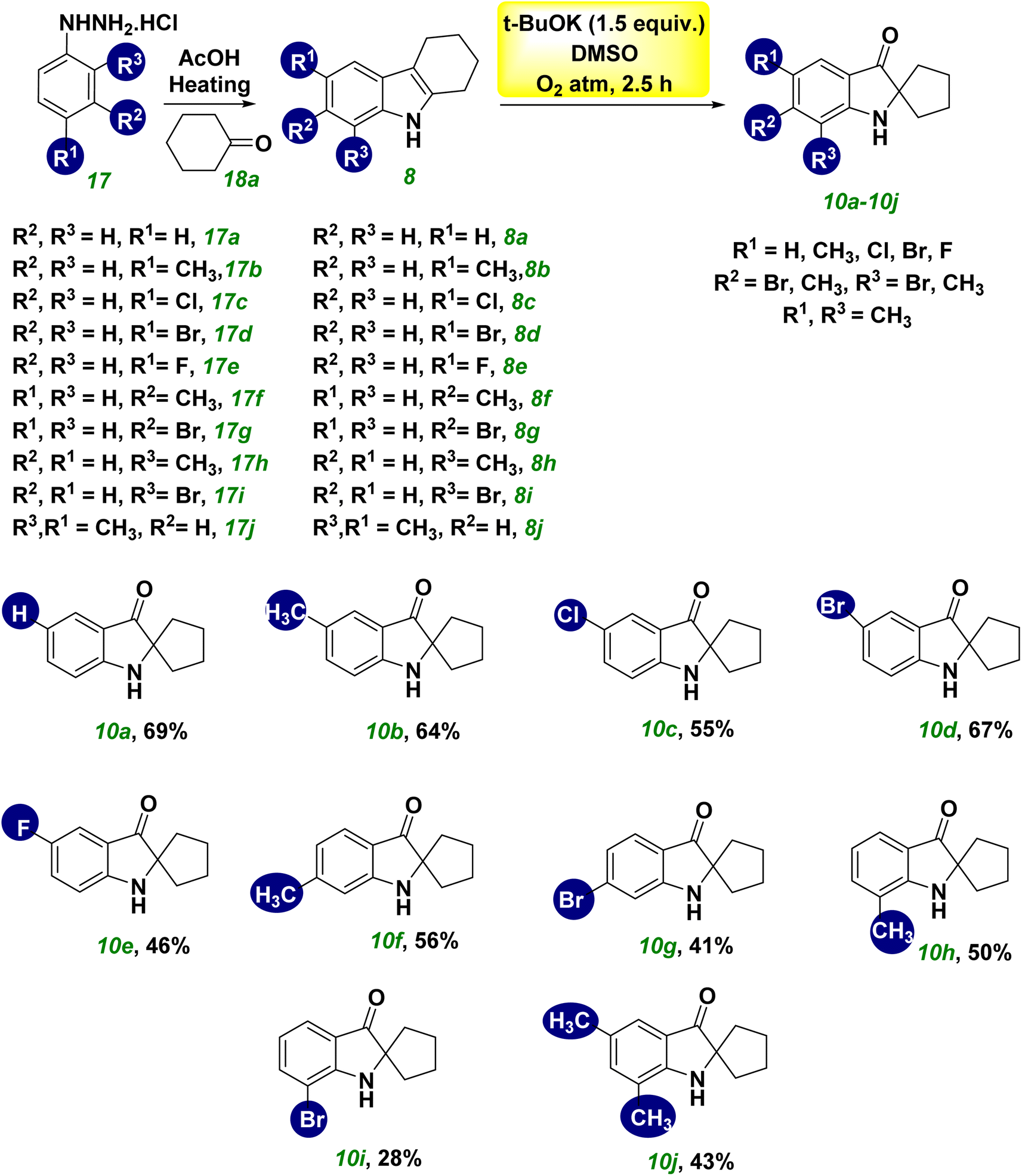 | ||
| Scheme 2 Scope of the synthesis of spiro pseudoindoxyl derivatives 10a–10j. | ||
Next, we planned to benzylate the nitrogen group of spiro pseudoindoxyl compound 10a. When benzyl bromide 19a was used to treat a mixture of spiro pseudoindoxy compounds 10a and DMF in the presence of NaH and nitrogen gas, a good-to-outstanding yield of N-benzylated spiro pseudoindoxyl compounds 11a was obtained (Scheme 3). By adjusting benzyl bromides (19b–19g) and spiro pseudoindoxyl compounds10b–10e, the scope of the reaction was determined (11b–11k). Benzylation of spiro compound 10 with an electron-donating compound (–CH3, 10b), electron-withdrawing compounds –Cl (10c), –Br (10d), –F (10e), and unsubstituted compound 10a afforded average-to-high yields of benzylated compounds 11a–11e. Compared with electron-donating and other halogen substituents, –F affected the benzylation pathway because it could reduce the nucleophilicity of the ring. We also synthesized several additional derivatives utilizing various benzyl bromides 19b–19g that were tested. Except for the –CN group 19d, which had a strong withdrawing character, all substitutes for 19 afforded good yields (11f, 11g, 11i, 11j); this was because 19d reduced the stability of the phenylmethylium ion during the benzylation reaction. Owing to the ortho effect, the yield of product 11k decreased when the benzyl ring (o-Br, 19g) was ortho-substituted. A probable mechanistic route for the metal-free hydroxylation cascade reaction based on the substituent impact and reactivity of the substrate was postulated. Following that, we attempted a gram-scale synthesis with compound 8a under standard reaction conditions, which yielded 69% of the product 10a (eqn (1), Scheme 4). Under standard reaction conditions of the benzylation reaction, compounds 10a and benzyl bromide provided compound 11a in an excellent yield of 91% (eqn (2), Scheme 4).
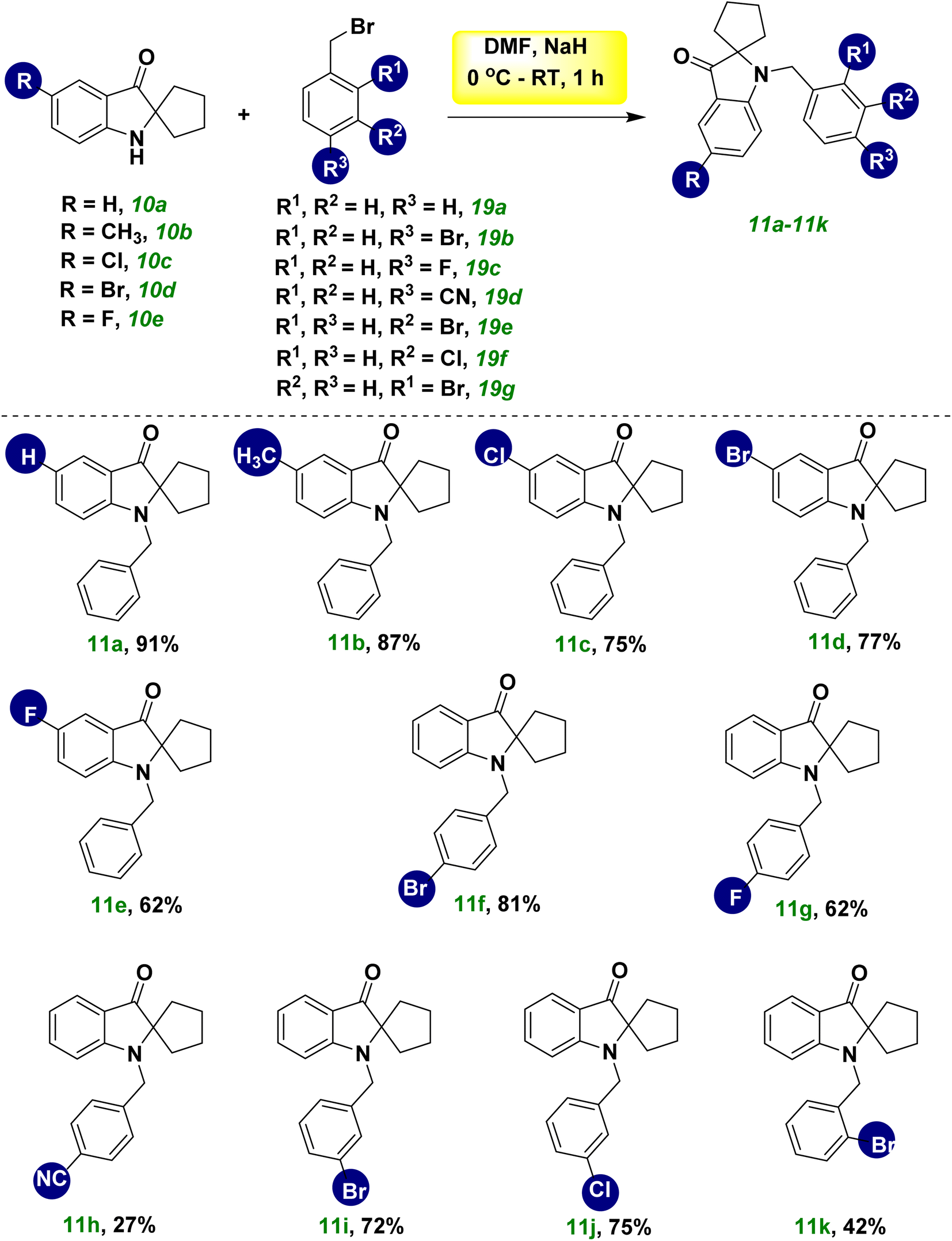 | ||
| Scheme 3 Synthesis of various benzylated-spiro pseudoindoxyl derivatives 11a–11k. | ||
 | ||
| Scheme 4 Gram-scale synthesis. | ||
We undertook some control experiments to ascertain the reaction mechanism. When we carried out a reaction with 17 under optimized conditions, the expected product 17a was not obtained. This result indicated the importance of the secondary cyclic amine system compared with the tertiary amine (eqn (1), Scheme 5). To ascertain if any radical path was involved in the reaction conditions, C–H hydroxylation was carried out with the radical quencher TEMPO (2 equivalent). However, we did not find a decrement in the yield because 69% of the product was obtained (eqn (2), Scheme 5). Compound 8a was treated with 19 under standard conditions, but product 11a was not obtained. This result clearly indicated that the deprotonation followed by an SN2 reaction with 19a was faster than the hydroxylation reaction because 11a product was not formed (eqn (3), Scheme 5).
 | ||
| Scheme 5 Control experiment. | ||
Based on the substrate scope, control experiments, and the literature,28 we proposed a reaction mechanism for formation of the spiro ring (Scheme 6). For the initial formal hydroxylation of THC 8a using DMSO in the presence of t-BuOk, the base removed a proton from THC 8a and generated anion 9a. This anion was stabilized by the enamine system, and attacked oxygen molecules. Subsequently, the newly formed superoxide anion 9c abstracted a proton from 8a and generated 9d. Further transformation of 9d into 9e facilitated an intra-molecular pinacol-type rearrangement for formation of the 9f intermediate. Upon proton transfer on 9f, compound 10a was obtained readily. A benzylation reaction on 10a followed deprotonation of 10a by NaH as well as a SN2 nucleophilic reaction with 19a, and resulted in the product 11a.
 | ||
| Scheme 6 Mechanistic pathway for spiro-ring formation and benzylation reaction. | ||
Biochemistry
| Compound | Residual activities at 10b μM (%) | IC50 (μM) | SI (MAO) | ||||
|---|---|---|---|---|---|---|---|
| MAO-A | MAO-B | AChE | BChE | MAO-A | MAO-B | ||
| a SI, selectivity index = IC50 (MAO-A)/IC50(MAO-B).b A negative value means that the slope of the activity in the presence of the inhibitor was lower than that in the absence of the inhibitor (i.e., control) in the continuous assay. | |||||||
| 10a | 66.86 ± 2.47 | 57.04 ± 2.99 | 76.29 ± 6.64 | 73.90 ± 4.90 | 20.78 ± 2.89 | 15.56 ± 2.33 | 1.34 |
| 10b | 72.15 ± 3.58 | 82.16 ± 9.06 | 83.39 ± 1.93 | 73.82 ± 7.10 | >40 | >40 | — |
| 10c | 79.09 ± 6.43 | 78.03 ± 5.36 | 100.47 ± 5.23 | 73.59 ± 4.50 | 20.17 ± 3.78 | 23.62 ± 3.89 | 0.85 |
| 10d | 75.45 ± 1.29 | 66.95 ± 1.20 | 60.87 ± 4.42 | 76.34 ± 3.15 | 20.59 ± 0.49 | 16.54 ± 2.01 | 1.24 |
| 10e | 68.67 ± 1.16 | 67.75 ± 0.76 | 80.16 ± 1.17 | 73.96 ± 2.95 | 30.68 ± 5.72 | 26.11 ± 4.15 | 1.18 |
| 11a | 80.67 ± 0.94 | 61.27 ± 4.98 | 68.31 ± 0.51 | 75.90 ± 1.27 | >40 | 12.20 ± 1.02 | 3.28 |
| 11b | 79.56 ± 5.03 | 26.12 ± 2.75 | 80.92 ± 5.07 | 65.10 ± 0.74 | >40 | 5.99 ± 0.36 | 6.68 |
| 11c | 65.96 ± 0.11 | 28.99 ± 4.10 | 92.81 ± 8.99 | 59.41 ± 5.45 | 19.19 ± 0.048 | 5.76 ± 1.24 | 3.33 |
| 11d | 62.00 ± 0.94 | −16.24 ± 2.34 | 81.90 ± 1.22 | 71.19 ± 4.17 | 14.04 ± 0.96 | 2.81 ± 0.71 | 5.00 |
| 11e | 81.33 ± 2.56 | 43.56 ± 5.39 | 99.10 ± 14.24 | 52.80 ± 3.25 | 31.06 ± 4.05 | 9.33 ± 1.82 | 3.33 |
| 11f | 67.91 ± 6.53 | 5.22 ± 1.06 | 68.95 ± 0.39 | 72.06 ± 6.88 | 17.15 ± 1.14 | 1.44 ± 0.21 | 11.91 |
| 11g | 73.81 ± 6.06 | 56.98 ± 0.00 | 75.50 ± 3.71 | 83.65 ± 1.02 | 24.17 ± 0.41 | 12.59 ± 1.41 | 1.92 |
| 11h | 63.51 ± 5.73 | 2.19 ± 0.95 | 54.58 ± 5.23 | 88.43 ± 0.89 | 22.42 ± 0.68 | 1.60 ± 0.46 | 14.01 |
| 11i | 24.12 ± 2.50 | 11.00 ± 0.67 | 68.56 ± 2.92 | 69.27 ± 3.68 | 5.58 ± 0.39 | 3.02 ± 0.39 | 1.85 |
| 11j | 45.24 ± 3.37 | −3.28 ± 2.80 | 70.21 ± 5.56 | 72.36 ± 9.17 | 8.95 ± 0.098 | 2.78 ± 0.60 | 3.22 |
| 11k | 41.94 ± 4.56 | 60.20 ± 1.44 | 68.03 ± 3.68 | 51.41 ± 0.53 | 9.01 ± 0.36 | 14.31 ± 1.45 | 0.63 |
| Toloxatone | — | — | — | — | 1.646 ± 0.094 | >40 | <0.041 |
| Safinamide | — | — | — | — | >40.0 | 0.019 ± 0.0019 | >2105.26 |
| Clorgyline | — | — | — | — | 0.0079 ± 0.00094 | 2.43 ± 0.71 | 0.0033 |
| Pargyline | — | — | — | — | 2.15 ± 0.23 | 0.11 ± 0.011 | 19.55 |
Spiro-compounds were classified into two types. Modified isatin compounds were divided into two groups based on the absence (10a–10e) or presence (11a–11k) of a separate benzyl ring. In most cases, compounds with a benzyl ring showed greater inhibition of MAO-B activity. If the substituent was present at the meta- or para-sites of the benzyl ring (11f–11k), low IC50 values were observed, except for 11g. In addition, if the halogen substituent was bound to the isatin ring, good inhibitory activity was observed in the order –Br (11d) > –Cl (11c) > –F (11e). When comparing compounds in which -Br was bonded to the N-benzyl ring, compounds 11d, 11f, and 11i, except for 11k, showed good inhibition of MAO-B activity.
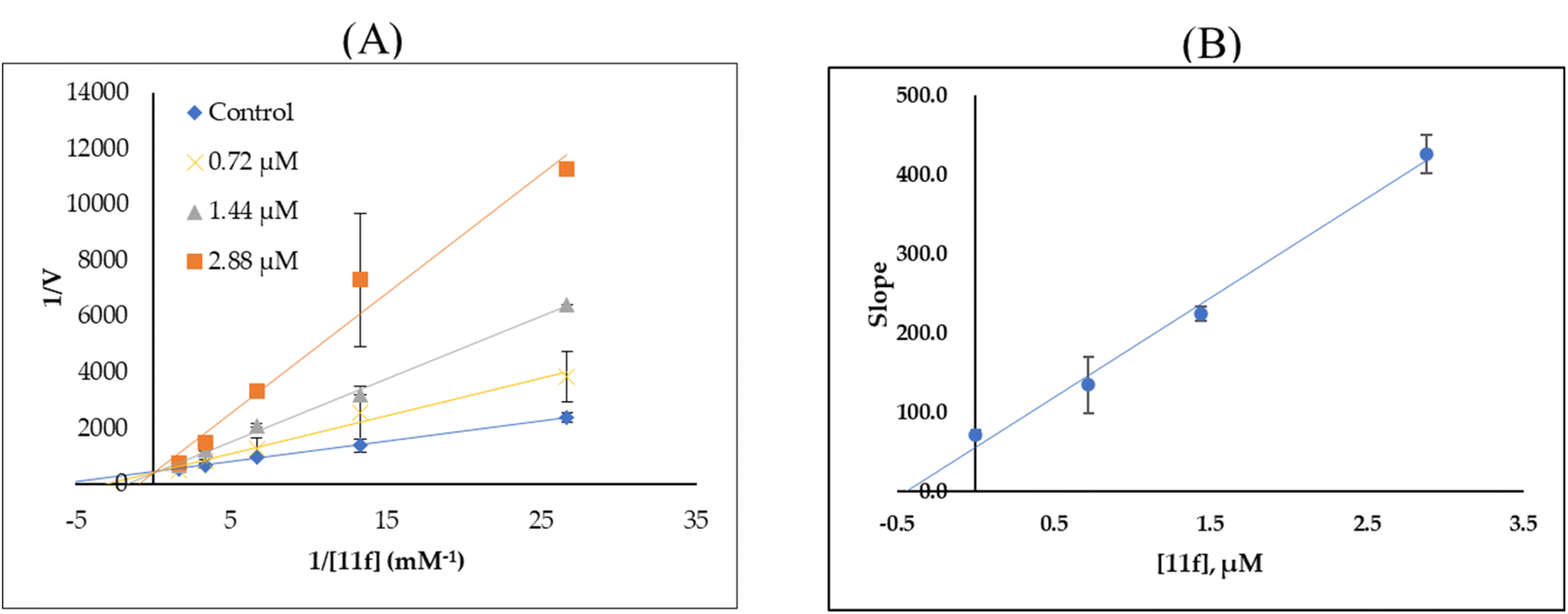 | ||
| Fig. 2 Lineweaver–Burk plots for MAO-B inhibition by 11f (A) and secondary plots (B) of the slope vs. inhibitor concentration. | ||
 | ||
| Fig. 3 Recoveries of MAO-B inhibition by 11f using dialysis experiments. The concentration of compounds used was 2-times their IC50 value. | ||
Conclusions
We devised a novel, metal-free protocol for easy access to spiro pseudoindoxyl ketones. The reaction proceeded in one pot at room temperature with a cheap t-BuOK base under an oxygen atmosphere in DMSO. The methodology afforded products with different substituted THC values in moderate-to-good yields. The electron-releasing group on the benzene ring afforded products with good yields, whereas electron-withdrawing substituents, such as F and 10e, generated moderate yields. To evaluate the cascade process, several 2-spirocyclo-3-oxindoles were synthesized. The reaction progressed through hydroxylation of the carbazole double bond and a subsequent intramolecular pinacol–pinacolone-type rearrangement. Then, the spiro compounds were transformed into their N-benzylated derivatives, and both series were evaluated for MAO inhibition. The non-benzylated spiro derivatives 10a–10e did not exhibit good inhibition of MAO activity. In contrast, all N-benzylated spiro derivatives exhibited good inhibition of MAO-B activity (IC50 = 1.60–9.33 μM), except for 11a, 11g, and 11k. Among them, compounds 11f and 11h exhibited the best inhibitory effects, with the IC50 value of 1.44 μM and 1.60 μM, respectively. These interesting natural product-based spiro pseudoindoxyl ketone core derivatives may be attractive for further exploration of biological activity because of the simple protocol needed to obtain them.Experimental section
General information
All reactions were undertaken in oven-dried round-bottom flasks and vials. Analytical TLC was done using F254 silica gel-precoated plates (Merck 60; Merck, Darmstadt, Germany). 1H NMR spectra were recorded using a NMR spectrometer (600 MHz; JEOL, Tokyo, Japan) and a spectrometer (300 MHz; Bruker, Ettlingen, Germany). 13C NMR spectra were recorded using a NMR spectrometer (150 MHz; JEOL) and a spectrometer (100 or 125 MHz (Bruker) using CDCl3. 1H NMR spectra are reported in parts per million (ppm) downfield of the internal standard (tetramethylsilane). The multiplicities are denoted as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or dd (doublet of doublets). The coupling constants (J) are reported in hertz (Hz). HRMS was undertaken using a quadrupole time of flight-micro mass spectrometer. Melting points were determined using a capillary melting-point apparatus and are uncorrected.Materials and methods
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. R. sincerely thanks SERB, Government of India, New Delhi, for financial support under the SERB-Research Scientist Program (SB/SRS/2022-23/78/CS) and thanks SASTRA Deemed University for a TRR research grant. The authors gratefully acknowledge the DST-FIST grant (SR/FST/CS-I/2018/62) to SCBT and SASTRA Deemed University for providing the NMR facility. K. P. gratefully acknowledges Teaching Assistantship from SASTRA Deemed University, Thanjavur, India.References
-
(a) P. S. Steyn, Tetrahedron Lett., 1971, 12, 3331 CrossRef
; (b) G. Laus, Phytother. Res., 2004, 18, 259 CrossRef CAS PubMed
-
(a) A. J. Hutchison and Y. Kishi, Stereospecific total synthesis of dl- austamide, J. Am. Chem. Soc., 1979, 101, 6786–6788 CrossRef CAS
-
(a) R. M. Williams and R. J. Cox, Acc. Chem. Res., 2003, 36, 127–139 CrossRef CAS PubMed
-
(a) J. H. Lee, J. H. So, J. H. Jeon, E. B. Choi, Y. R. Lee, Y. T. Chang, C. H. Kim, M. A. Bae and J. H. Ahn, Chem. Commun., 2011, 47, 7500–7502 RSC
- P. N. Wyrembak and A. D. Hamilton, J. Am. Chem. Soc., 2009, 131, 4566–4567 CrossRef CAS PubMed
- S. Matsumoto, D. Samata, M. Akazome and K. Ogura, Tetrahedron Lett., 2009, 50, 111–114 CrossRef CAS
- H. Chen, H. Shang, Y. Liu, R. Guo and W. Lin, Adv. Funct. Mater., 2016, 26, 8128–8136 CrossRef CAS
-
(a) E. F. Khusnutdinova, O. B. Kazakova, A. N. Lobov, O. S. Kukovinets, K. Y. Suponitsky, C. B. Meyers and M. N. Prichard, Org. Biomol. Chem., 2019, 17, 585–597 RSC
-
(a) C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232–10236 CrossRef CAS PubMed
- Bhawna, A. Kumar, M. Bhatia, A. Kapoor, P. Kumar and S. Kumar, Eur. J. Med. Chem., 2022, 242, 114655 CrossRef CAS PubMed
- S. Kumar, A. S. Nair, V. Bhashkar, S. T. Sudevan, V. P. Koyiparambath, A. Khames, M. A. Abdelgawad and B. Mathew, ACS Omega, 2021, 6, 23399–23411 CrossRef CAS PubMed
- P. Guglielmi, S. Carradori, I. D'Agostino, C. Campestre and J. P. Petzer, Expert Opin. Ther. Pat., 2022, 32, 849–883 CrossRef CAS PubMed
- R. K. P. Tripathi and S. R. Ayyannan, Med. Res. Rev., 2019, 39, 1603–1706 CrossRef CAS PubMed
- Y. Y. Tan, P. Jenner and S. D. Chen, J. Parkinson's Dis., 2022, 12, 477–493 CAS
- M. Alborghetti and F. Nicoletti, Curr. Neuropharmacol., 2019, 17, 861–873 CrossRef CAS PubMed
- C. D. Binde, I. F. Tvete, J. I. Gåsemyr, B. Natvig and M. Klemp, Eur. J. Clin. Pharmacol., 2020, 76, 1731–1743 CrossRef CAS PubMed
- P. Riederer and T. Müller, J. Neural Transm., 2018, 125, 1751–1757 CrossRef CAS PubMed
- N. T. Tzvetkov, S. Hinz, P. Küppers, M. Gastreich and C. E. Müller, J. Med. Chem., 2014, 57, 6679–6703 CrossRef CAS PubMed
- A. Elkamhawy, S. Paik, H. J. Kim, J. H. Park, A. M. Londhe, K. Lee, A. N. Pae, K. D. Park and E. J. Roh, J. Enzyme Inhib. Med. Chem., 2020, 35, 1568–1580 CrossRef CAS PubMed
- R. Sasidharan, S. L. Manju, G. Uçar, I. Baysal and B. Mathew B, Arch. Pharm., 2016, 349, 627–637 CrossRef CAS PubMed
-
(a) C. U. Rogers and B. B. Corson, J. Am. Chem. Soc., 1947, 69, 2910–2911 CrossRef CAS
- J. M. Oh, T. M. Rangarajan, R. Chaudhary, R. P. Singh, M. Singh, R. P. Singh, A. R. Tondo, N. Gambacorta, O. Nicolotti, B. Mathew and H. Kim, Molecules, 2020, 25, 2356 CrossRef CAS PubMed
- J. M. Oh, Y. Kang, J. H. Hwang, J. H. Park, W. H. Shin, S. K. Mun, J. U. Lee, S. T. Yee and H. Kim, Int. J. Biol. Macromol., 2022, 217, 910–921 CrossRef CAS PubMed
- S. C. Baek, M. H. Park, H. W. Ryu, J. P. Lee, M. G. Kang, D. Park, C. M. Park, S. R. Oh and H. Kim, Bioorg. Chem., 2019, 83, 317–325 CrossRef CAS PubMed
- S. C. Baek, H. W. Lee, H. W. Ryu, M. G. Kang, D. Park, S. H. Kim, M. L. Cho, S. R. Oh and H. Kim, Bioorg. Med. Chem. Lett., 2018, 28, 584–588 CrossRef CAS PubMed
- J. M. Oh, H. J. Jang, W. J. Kim, M. G. Kang, S. C. Baek, J. P. Lee, D. Park, S. R. Oh and H. Kim, Int. J. Biol. Macromol., 2020, 151, 441–448 CrossRef CAS PubMed
- H. W. Lee, H. W. Ryu, M. G. Kang, D. Park, S. R. Oh and H. Kim, Bioorg. Med. Chem. Lett., 2016, 26, 4714–4719 CrossRef CAS PubMed
- B. Moreshwar, S. Yogesh, M. Shreyas, H. Anirban and G. Boopathy, Org. Lett., 2017, 19, 3628–3631 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} NMR and HRMS spectra of all compounds. See DOI: https://doi.org/10.1039/d3ra03641c |
| This journal is © The Royal Society of Chemistry 2023 |