
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and photophysical characterization of fluorescent indole nucleoside analogues†
Jacob M. Sawyera,
Kellan T. Passowb and
Daniel A. Harki *ab
*ab
aDepartment of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA. E-mail: daharki@umn.edu
bDepartment of Medicinal Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA
First published on 31st May 2023
Abstract
Fluorescent nucleosides are useful chemical tools for biochemical research and are frequently incorporated into nucleic acids for a variety of applications. The most widely utilized fluorescent nucleoside is 2-aminopurine-2′-deoxyribonucleoside (2APN). However, 2APN is limited by a moderate Stokes shift, molar extinction coefficient, and quantum yield. We recently reported 4-cyanoindole-2′-deoxyribonucleoside (4CIN), which offers superior photophysical characteristics in comparison to 2APN. To further improve upon 4CIN, a focused library of additional analogues combining the structural features of 2APN and 4CIN were synthesized and their photophysical properties were quantified. Nucleosides 2–6 were found to possess diverse photophysical properties with some features superior to 4CIN. In addition, the structure–function relationship data gained from 1–6 can inform the design of next-generation fluorescent indole nucleosides.
Introduction
Isomorphic fluorescent nucleosides are structurally similar to naturally occurring nucleosides and aim to retain the intra- and inter-molecular interactions found with endogenous nucleosides and other molecules. 2-Aminopurine-2′-deoxyribonucleoside (2APN) is the most widely utilized isomorphic fluorescent nucleoside because of its environment sensitive fluorescence along with only minor perturbations to the native B-form DNA structure, which results in only a slight reduction in overall stability of duplex DNA.1–4 Despite its widespread use, 2APN has significant limitations in terms of its photophysical properties such as a moderate Stokes shift, molar extinction coefficient, and quantum yield. The development of fluorescent nucleoside analogues that improve upon the photophysical properties of 2APN has been an area of significant research interest.5–7 Recently, 4-cyanoindole (4CI) was developed into the corresponding fluorescent nucleoside 4-cyanoindole-2′-deoxyribonucleoside (4CIN), which is superior to 2APN in all the aforementioned photophysical properties.8 4CI has been incorporated into a ribonucleoside (4CINr) and a 2′-deoxyribonucleoside-2′-triphosphate (4CINTP),9 and 4CIN has served as a useful probe for DNA8,10 and RNA applications.11 To further optimize 4CIN and interrogate the scope of structural modifications that can contribute to favorable photophysical properties, a library of fluorescent nucleosides combining the structural features of 4CIN and 2APN were synthesized and their photophysical properties characterized (Fig. 1). Nucleoside 1 contains the classical purine heterocycle with the introduction of a nitrile at the 6-position, which is analogous to the 4-position on indole. Nucleoside 2 interrogates whether a 6-amino group can be installed on the 4-cyanoindole (4CI) heterocycle and retain fluorescence properties. In addition, 2 incorporates the amino group found in guanosine, which may be useful in applications involving duplex DNA. Nucleosides 3 and 4 interrogate conversion of the nitrile of 2 to an ester and acid (3 and 4, respectively), whereas 5 and 6 continue that line of investigation to determine if the pendant 6-amino group contributes to favorable photophysical properties. Interestingly, we find that 5 and 6 have higher molar extinction coefficients than 2APN and 4CIN, as well as a higher quantum yield than 2APN.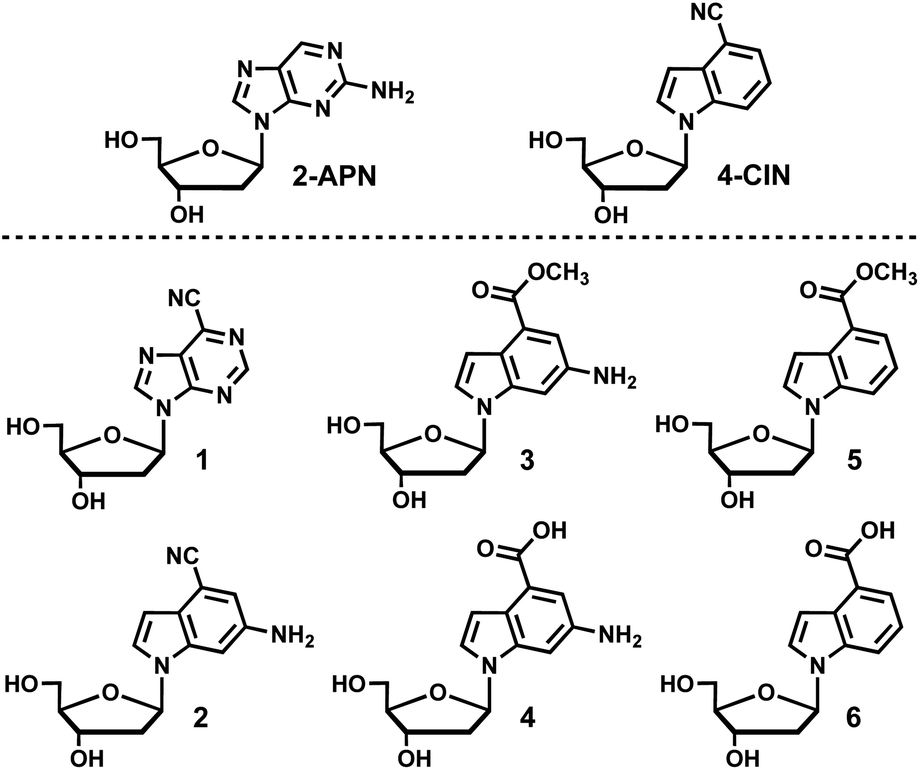 | ||
| Fig. 1 A library of fluorescent nucleosides (1–6) combining the structural features of 2APN and 4CIN. | ||
Results and discussion
Synthesis
The synthesis of 6-cyanopurine-2′-deoxyribonucleoside 1 began with silyl protection of the 3′- and 5′-hydroxyls of 6-chloropurine-2′-deoxyribonucleoside 7 to yield 8. The nitrile was then installed at the 6-position using a palladium-catalyzed cyanation reaction with zinc cyanide to afford 9 in good yield.12 Deprotection of the silyl groups afforded 1 in 36% overall yield (Scheme 1).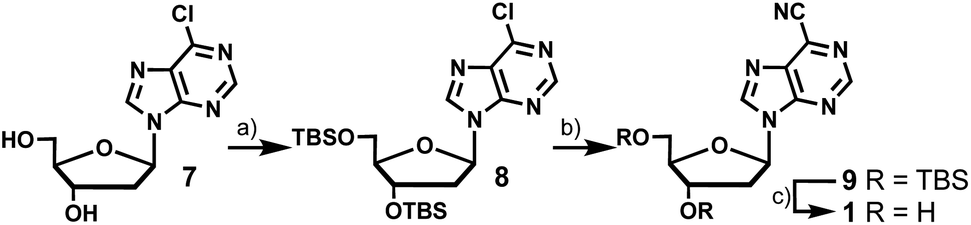 | ||
| Scheme 1 Synthesis of 6-cyanopurine-2′-deoxyribonucleoside (1). Reagents and conditions: (a) TBSCl, imidazole, DMAP, DMF, RT (room temperature) 75%; (b) Zn(CN)2, Pd(PPh3)4, DMF, 90 °C, 71%; (c) TBAF, THF, RT, 67%. | ||
6-Amino-4-cyanoindole-2′-deoxyribonucleoside (2) was prepared from 6-amino-4-cyanoindole (10) (Scheme 2). Di-benzyl protection of the exocyclic amine of 10 resulted in 11 in moderate yield, which provided the coupling partner for established chlorosugar 12 to afford 13 as the β-anomer in good yield. Removal of the N-benzyl and O-toluoyl protecting groups in a 2-step process afforded 2 in an overall yield of 8%.
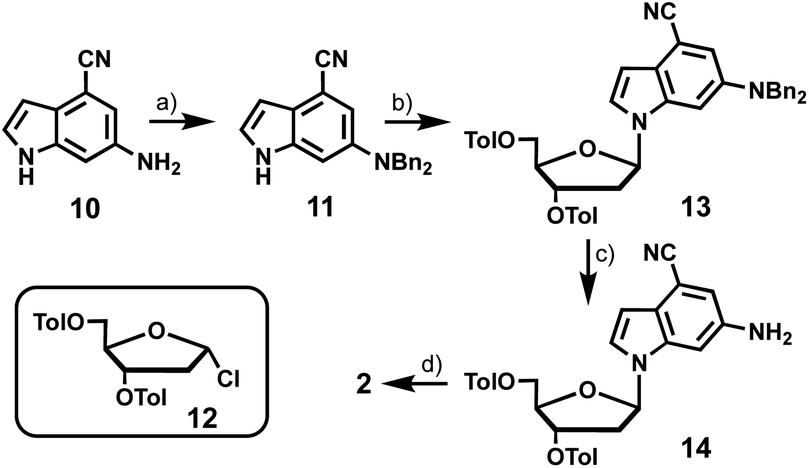 | ||
| Scheme 2 Synthesis of 4-cyano-6-aminoindole-2′-deoxyribonucleoside (2). Reagents and conditions: (a) BnCl, K2CO3, MeOH, reflux, 61%; (b) NaH, MeCN, 30 min, RT; 12, RT, 65%; (c) Pd/C, H2, THF, RT, 38%; (d) K2CO3, MeOH, THF, RT, 50%. | ||
To synthesize the nucleosides bearing 6-amino-4-carboxyindoles (3 and 4) we started with dinitro-toluic acid (15), which underwent a Fischer esterification to generate the carboxylate ester (16, Scheme 3). Ester 16 was then subjected to Leimgruber–Batcho indole synthesis conditions to afford 17.13,14 The subsequent 4 synthesis steps were analogous to those described for the preparation of 2, including benzyl protection of the exocyclic amine of 17, coupling of indole 18 to chlorosugar 12, benzyl deprotection of 19, and saponification of the toluoyl esters of 20 to produce 3 in 6% overall yield. Treatment of 20 directly with aqueous KOH saponified the methyl carboxylate and toluoyl groups yielding 4. However, we wanted to access the methyl carboxylate compound for photophysical testing so this conversion was done over two steps. Employing a weaker base than hydroxide allowed selective removal of the toluoyl groups while avoiding hydrolysis of the methyl carboxylate. Carboxylic acid 4 was then synthesized from 3 by ester saponification with aqueous KOH in 43% yield.
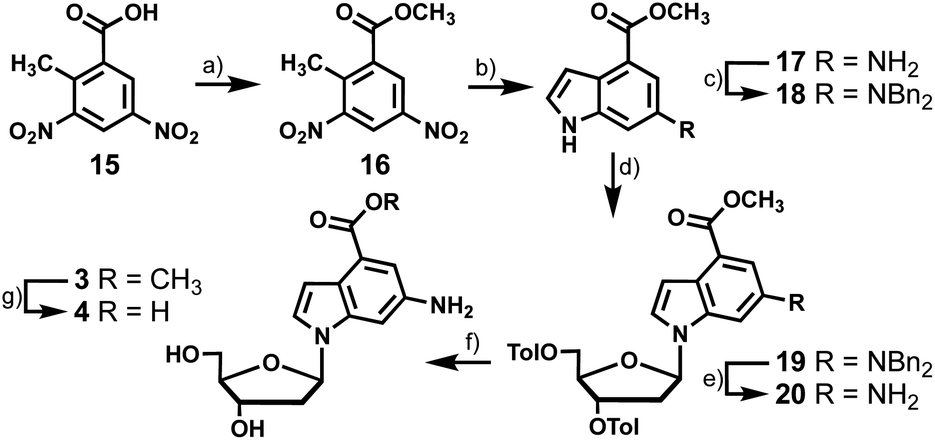 | ||
| Scheme 3 Synthesis of fluorescent nucleosides with 6-amino and 4-carboxylate ester (3) and acid (4) substituents. Reagents and conditions: (a) H2SO4, MeOH, reflux, 98%; (b) DMF-DMA, dioxane, microwave, 90 °C; H2, Pd/C, MeOH, RT, 62%; (c) BnCl, K2CO3, MeOH, reflux, 54%; (d) NaH, MeCN, 30 min, RT; 12, RT, 64%; (e) Pd/C, H2, THF, RT, 55%; (f) K2CO3, MeOH, THF, RT, 51%; (g) KOH, MeOH, THF, 50 °C, 43%. | ||
To access 4-carboxyindole analogues 5 and 6, a simpler synthesis was employed as these indoles lack the exocyclic amine (Scheme 4). Indole 21 was coupled directly to chlorosugar 12 followed by deprotection of the toluoyl groups to yield 5 with an overall yield of 47%. When treated with aqueous KOH, 5 was smoothly converted to the carboxylic acid analogue 6 in 46% yield.
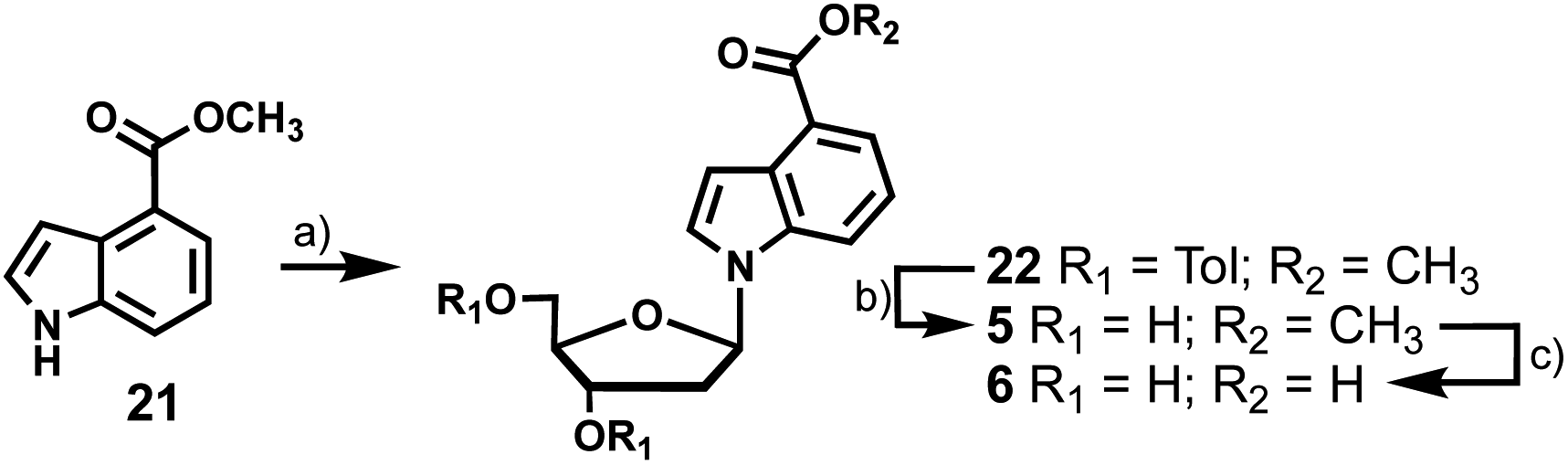 | ||
| Scheme 4 Synthesis of fluorescent indole nucleosides with 4-carboxylate ester (5) and acid (6) substituents. Reagents and conditions: (a) NaH, MeCN, 30 min, RT; 12, RT, 69%; (b) K2CO3, MeOH, THF, RT, 68%; (c) KOH, MeOH, THF, H2O, 50 °C, 46%. | ||
Photophysical characterization of fluorescent nucleosides
The library of fluorescent nucleosides was first characterized by measuring absorption and emission properties (Fig. 2). The nucleosides displayed red-shifted absorption and emission maxima wavelengths in comparison to 2APN and 4CIN, granting high Stokes shifts for each of the analogues with the exception of 1 (Table 1). 4-Cyanopurine-2′-deoxyribonucleoside (1), built on the purine scaffold, was not a fluorescent compound. For this reason, the nucleoside was excluded from further studies. For the compounds with two absorption maxima (2, 3, and 4), the photophysical properties were determined using the wavelength with the highest absorption (represented by λex in Table 1). 2 contains the 4-cyano group of 4CIN and the 6-amino group of 2APN. This compound has a Stokes shift in-between 4CIN (8.5 × 103 cm−1) and 2APN (5.9 × 103 cm−1) at 6.9 × 103 cm−1. Analogues 3 and 4 replace the nitrile at the 4-position with a methyl carboxylate ester (3) and carboxylic acid (4). These functional group changes increased the Stokes shift of both compounds in comparison to 2 granting a Stokes shift of 9.0 × 103 cm−1 and 12.9 × 103 cm−1 for 3 and 4, respectively. Nucleosides 5 and 6 are analogous to 3 and 4, except they lack the 6-amino group. Interestingly, 5 displays a higher Stokes shift than 3 at 10.1 × 103 cm−1. However, in the case of 6, removal of the 6-amino group lowered its Stokes shift in comparison to 4 with a value of 10.1 × 103 cm−1. These data show that the presence of an electron-donating group at the 6-position does not clearly correlate with an effect on the Stokes shift. Regardless, nucleosides 2–6 all have a higher Stokes shift than 2APN and, with the exception of 2, a higher Stokes shift than 4CIN.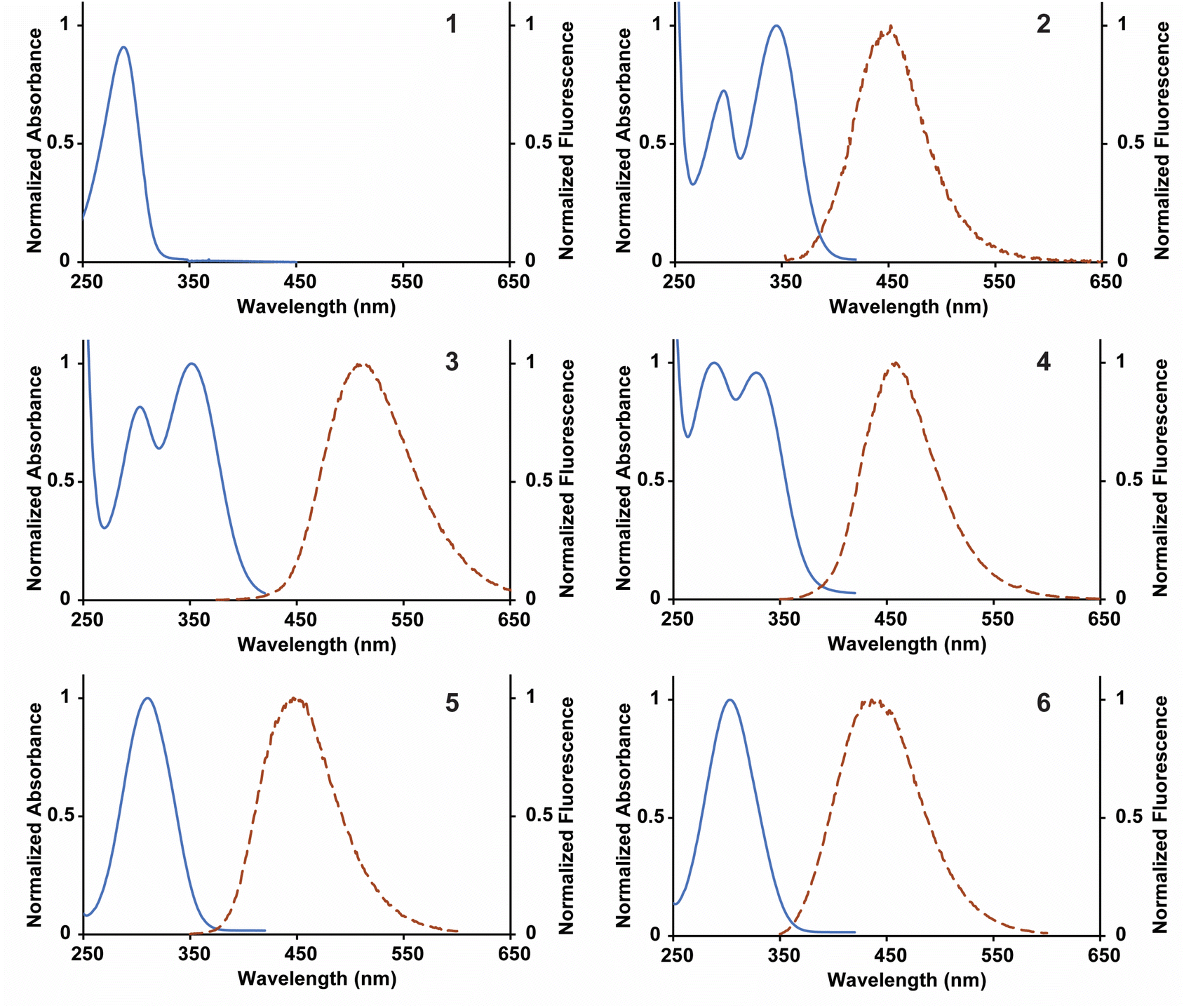 | ||
| Fig. 2 Normalized absorbance and fluorescence spectra for 1–6 collected in distilled and deionized H2O. Fluorescence measurements were obtained by exciting the compounds at their absorbance maximum. | ||
| Nucleoside | λex (nm) | λem (nm) | Stokes shift (cm−1) | ε (M−1 cm−1) | QYb | Brightnessc |
|---|---|---|---|---|---|---|
| a Literature values for 2APN molar extinction coefficient vary.15–17b Calculated relative to QS in aqueous 0.105 M HClO4.c Brightness was calculated by multiplying ε and QY. | ||||||
| 2APN | 303 | 369 | 5.9 × 103 | 5000–6000a | 0.61 ± 0.01 | 3050–3660 |
| 4CIN | 305 | 412 | 8.5 × 103 | 7790 ± 320 | 0.92 ± 0.02 | 7160 |
| 2 | 296, 345 | 452 | 6.9 × 103 | 6430 ± 70 | 0.0067 ± 0.0006 | 40 |
| 3 | 303, 351 | 514 | 9.0 × 103 | 7570 ± 100 | 0.083 ± 0.001 | 630 |
| 4 | 288, 327 | 457 | 12.9 × 103 | 2780 ± 110 | 0.53 ± 0.01 | 1470 |
| 5 | 310 | 451 | 10.1 × 103 | 8910 ± 70 | 0.64 ± 0.01 | 5700 |
| 6 | 303 | 436 | 10.1 × 103 | 8090 ± 50 | 0.84 ± 0.01 | 6800 |
The molar extinction coefficients (ε) of nucleosides 2–6 were determined using Beer's law (Table 1). Following the same trend as was seen with the Stokes shift, 2 has a molar extinction coefficient in between that of 2APN and 4CIN at 6430 ± 70 M−1 cm−1. Changing the functional group at the 4-position from a nitrile to a methyl carboxylate ester increased the molar extinction coefficient of 3 to 7570 ± 100 M−1 cm−1. However, converting the functional group at the 4-position to the carboxylic acid had a detrimental effect on the extinction coefficient, reducing the value for 4 to 2780 ± 110 M−1 cm−1. Nucleosides 5 and 6 both showed an increase in molar extinction coefficients in comparison to 3 and 4 at 8910 ± 70 M−1 cm−1 and 8090 ± 50 M−1 cm−1, respectively. The molar extinction coefficient for all nucleosides, with the exception of 4, were higher than 2APN. 5 and 6 also had higher molar extinction coefficients than 4CIN. These data show that removing the amine at the 6 position grants an increase in the molar extinction coefficient for the fluorophores.
The quantum yield was next determined for 2–6 (Table 1) using an established method relative to a known standard, quinine sulfate (QS).18 Breaking from the trend seen for the previous photophysical properties, 2 did not display a quantum yield in between 2APN and 4CIN, but a lower quantum yield than both at 0.0067 ± 0.0006. Changing from the 4-cyano to the methyl carboxylate ester granted a significant increase in quantum yield for 3 at 0.083 ± 0.001. Converting to the carboxylic acid increased the quantum yield to 0.53 ± 0.01 for 4. The absence of the amine at the 6-position gave the highest quantum yields of the analogues for 5 and 6 at 0.64 ± 0.01 and 0.84 ± 0.01, respectively, which are both higher than 2APN. Analysis of quantum yield follows the same trend as the molar extinction coefficient, whereas removal of the 6-amino group is beneficial.
A useful characteristic for the analysis of fluorophores is the brightness (Φ) because it accounts for both the absorption and fluorescence properties of the fluorophore. Due to significant differences in the quantum yield of analogues 2–6 there was a wide range of corresponding brightness values. Nucleosides 2–4 displayed low brightness values owing to either low quantum yields (2 and 3) or a low extinction coefficient (4). Analogues 5 and 6, which lack the 6-amino substituent, had significantly higher brightness values than 2APN. However, 4CIN still shows the highest brightness of the group.
Conclusions
This study served to combine the structural properties of 4CIN and 2APN into a focused library of fluorescent nucleosides with the goal of identifying compounds with improved photophysical properties and further defining structure–function relationships. Six nucleosides were synthesized and characterized photophysically, and then benchmarked against 2APN and 4CIN. Nucleoside 1, which was built upon the purine scaffold, was not a fluorescent compound. This initial result informed our decision to focus on the indole scaffold. The fluorescent nucleosides bearing the 6-amino substituent (2, 3, and 4) had high Stokes shifts, but overall low brightness. In the case of 2 and 3, the low brightness is attributed to a low quantum yield. Interestingly, for nucleoside 4, the low brightness was due to a low molar extinction coefficient. Conversely, the fluorescent nucleosides lacking the 6-amino substituent, 5 and 6, showed higher Stokes shifts, molar extinction coefficients, and quantum yields in comparison to 2APN. These compounds also had larger Stokes shifts and molar extinction coefficients than 4CIN, although 4CIN has a higher quantum yield. The brightness of 5 and 6 were higher than 2APN, but not 4CIN. Taken together, nucleosides 5 and 6 have improved photophysical properties over 2APN and are viable isomorphic fluorescent nucleosides suitable for future studies. In addition, our data suggest that the presence of an electron donating group at the 6-position of the indole lowers the brightness of fluorescent indole nucleosides, which should inform the design of next-generation compounds with enhanced photophysical properties.Experimental
General information
Reactions were performed in flame-dried glassware under inert atmospheres (nitrogen or argon). Reactions were stirred using Teflon-coated stir bars. Solvents MeCN and CH2Cl2 were dried using an MBraun solvent purification system. Other solvents were purchased as ACS grade (≥99% purity) and used as received. Water for reactions and for photophysical characterization of the fluorescent nucleosides was purified using a MilliQ system (MilliporeSigma). 3,5-Di-O-toluoyl-α-1-chloro-2-deoxy-D-ribofuranose (12) was purchased from Carbosynth. Microwave reactions were performed on a Discover SP Microwave Synthesizer (CEM Corporation). Conditions used for microwave reactions were as follows: 17 bar, 200 watts, and 90 °C with stirring. Silica gel chromatography was performed using Redisep Rf high performance silica gel columns (Teledyne-Isco) on a Combiflash NextGen 300+ instrument (Teledyne-Isco). A Bruker Advance 500 MHz NMR spectrometer was used for the collection of all NMR spectra. NMR spectra were collected at room temperature. High resolution mass spectrometry was performed at the Analytical Biochemistry Core Facility at the University of Minnesota Masonic Cancer Center on an Exactive Plus Orbitrap mass spectrometer (Thermo Scientific).Synthesis
1H NMR (CDCl3, 500 MHz): 8.74 (s, 1H), 8.48 (s, 1H), 6.52 (app t, J = 6.4 Hz, 1H), 4.62 (m, 1H), 4.05 (app q, J = 3.3 Hz, 1H), 3.89 (dd, J = 11.3, 3.7 Hz, 1H), 3.78 (dd, J = 11.3, 2.8 Hz, 1H), 2.64 (m, 1H), 2.49 (m, 1H), 0.92 (s, 9H), 0.90 (s, 9H), 0.11 (s, 6H), 0.09 (s, 6H) δ ppm.
13C NMR (CDCl3, 126 MHz): 151.9, 151.1, 151.0, 143.8, 132.2, 88.2, 84.9, 71.8, 62.7, 41.6, 26.0, 25.7, 18.4, 18.0, −4.6, −4.8, −5.4, −5.5 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C22H40ClN4O3Si2: 499.2322, found: 499.2302.
1H NMR (CDCl3, 500 MHz): 9.04 (s, 1H), 8.67 (s, 1H), 6.55 (app t, J = 6.3 Hz, 1H), 4.63 (app q, J = 3.8 Hz, 1H), 4.06 (app q, J = 3.2 Hz, 1H), 3.90 (dd, J = 11.4, 3.6 Hz, 1H), 3.79 (dd, J = 11.4, 2.7 Hz, 1H), 2.63 (m, 1H), 2.52 (m, 1H), 0.91 (s, 9H), 0.90 (s, 9H), 0.11 (s, 6H), 0.09 (s, 6H) δ ppm.
13C NMR (CDCl3, 126 MHz): 152.5, 152.2, 146.8, 135.6, 130.9, 113.6, 88.4, 85.0, 71.7, 62.6, 53.4, 41.8, 25.9, 25.7, 18.4, 18.0, −4.6, −4.8, −5.4, −5.5 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C23H40N5O3Si2: 490.2664, found: 490.2646.
1H NMR (DMSO-d6, 500 MHz): 9.15 (s, 1H), 9.12 (s, 1H), 6.51 (app t, J = 6.5 Hz, 1H), 5.39 (d, J = 4.2 Hz, 1H), 4.96 (t, J = 5.4 Hz, 1H), 4.46 (m, 1H), 4.09 (app q, J = 5.3 Hz, 1H), 3.91 (app q, J = 4.1 Hz, 1H), 3.63 (m, 1H), 3.55 (m, 1H), 2.79 (m, 1H), 2.41 (m, 1H) δ ppm.
13C NMR (DMSO-d6, 126 MHz): 152.9, 152.7, 149.3, 135.9, 129.4, 114.8, 88.6, 84.7, 70.7, 61.7, 49.1 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C11H12N5O3: 262.0935, found: 262.0919.
1H NMR (MeOD, 500 MHz): 7.29 (m, 8H), 7.22 (m, 2H), 7.18 (d, J = 3.0 Hz, 1H), 6.99 (m, 2H), 4.67 (s, 4H) δ ppm.
13C NMR (MeOD, 126 MHz): 146.3, 140.0, 139.1, 129.6, 128.1, 128.0, 126.7, 123.4, 120.2, 114.5, 102.9, 102.4, 100.3, 56.8 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C23H19N3: 338.1652, found: 338.1631.
1H NMR (MeCN-d3, 500 MHz): 7.98 (d, J = 8.5 Hz, 2H), 7.85 (d, J = 8.5 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.30 (m, 11H), 7.22 (m, 2H), 7.10 (d, J = 2.0 Hz, 1H), 7.04 (d, J = 2.0, 1H), 6.46 (d, J = 3.5 Hz, 1H), 6.27 (dd, J = 5.5, 3.0 Hz, 1H), 5.57 (m, 1H), 4.72 (s, 4H), 4.46 (m, 3H), 2.68 (m, 1H), 2.54 (m, 1H), 2.43 (s, 3H), 2.39 (s, 3H) δ ppm.
13C NMR (MeCN-d3, 126 MHz): 166.8, 166.7, 146.0, 145.5, 145.2, 139.6, 138.5, 130.5, 130.4, 130.2, 130.2, 129.5, 128.0, 128.0, 127.9, 126.0, 123.1, 119.3, 114.8, 103.7, 101.8, 100.7, 86.1, 82.4, 75.9, 65.0, 56.4, 56.3, 37.7, 21.7, 21.6 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C44H39N3O5: 690.2962, found: 690.2928.
1H NMR (MeCN-d3, 500 MHz): 7.91 (d, J = 8.0 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H), 7.24 (m, 5H), 7.00 (s, 1H), 6.80 (s, 1H), 6.39 (d, J = 3.5 Hz, 1H), 6.29 (dd, J = 6.0, 2.5 Hz, 1H), 5.59 (d, J = 7.0 Hz, 1H), 4.50 (app q, J = 7.5 Hz, 1H), 4.43 (m, 2H), 4.15 (s, 2H), 2.75 (m, 1H), 2.60 (m, 1H), 2.34 (s, 3H), 2.32 (s, 3H) δ ppm.
13C NMR (MeCN-d3, 126 MHz): 166.9, 166.8, 145.5, 145.3, 145.0, 138.6, 130.5, 130.4, 130.4, 130.3, 130.2, 128.0, 125.7, 123.6, 119.2, 115.3, 103.8, 102.1, 101.0, 86.2, 82.5, 75.9, 65.0, 37.6, 21.6, 21.6 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C30H27N3O5: 510.2023, found: 510.2017.
1H NMR (MeOD, 500 MHz): 7.45 (d, J = 3.5 Hz, 1H), 7.16 (s, 1H), 6.94 (d, J = 1.5 Hz, 1H), 6.48 (d, J = 3.0 Hz, 1H), 6.31 (dd, J = 6.0, 2.0 Hz, 1H), 4.47 (m, 1H), 3.95 (app q, J = 3.5 Hz, 1H), 3.69 (m, 2H), 2.57 (m, 1H), 2.32 (m, 1H) δ ppm.
13C NMR (MeOD, 126 MHz): 144.8, 139.0, 126.3, 124.7, 119.6, 115.8, 103.5, 102.0, 101.8, 88.3, 86.2, 72.6, 63.5, 40.9 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C14H15N3O3: 274.1186, found: 274.1176.
1H NMR (CDCl3, 500 MHz): 8.85 (d, J = 2.4 Hz, 1H), 8.69 (d, J = 2.4 Hz, 1H), 4.01 (s, 3H), 2.76 (s, 1H) δ ppm.
1H NMR (MeOD, 500 MHz): 7.36 (d, J = 2.0 Hz, 1H), 7.16 (d, J = 3.1 Hz, 1H), 7.04 (d, J = 1.1 Hz, 1H), 6.85 (d, J = 3.1 Hz, 1H), 3.92 (s, 3H) δ ppm.
1H NMR (DMSO-d6, 500 MHz): 10.88 (s, 1H), 7.37 (d, J = 2.3 Hz, 1H), 7.32 (m, 8H), 7.24 (m, 2H), 7.21 (t, J = 2.8 Hz, 1H), 6.90 (d, J = 2.2 Hz, 1H), 6.70 (t, J = 2.6 Hz, 1H), 4.71 (s, 4H), 3.82 (s, 3H) δ ppm.
13C NMR (DMSO-d6, 126 MHz): 167.9, 144.1, 139.5, 138.7, 129.0, 127.2, 127.2, 125.6, 120.9, 119.9, 111.1, 102.2, 101.0, 55.6, 52.0 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C24H23N2O2: 371.1754, found: 371.1713.
1H NMR (MeCN-d3, 500 MHz): 8.02 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 2.0 Hz, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.32 (m, 10H), 7.28 (d, J = 2.0 Hz, 1H), 7.24 (t, J = 6.5 Hz, 2H), 7.11 (d, J = 2.5 Hz, 1H), 6.88 (d, J = 3.0 Hz, 1H), 6.32 (dd, J = 8.5, 5.5 Hz, 1H), 5.60 (d, J = 6.0 Hz, 1H), 4.76 (s, 4H), 4.53 (m, 1H), 4.47 (m, 2H), 3.87 (s, 3H), 2.72 (m, 1H), 2.56 (m, 1H), 2.46 (s, 3H), 2.43 (s, 3H) δ ppm.
13C NMR (MeCN-d3, 126 MHz): 168.4, 166.9, 166.7, 145.7, 145.5, 145.2, 140.3, 140.1, 130.5, 130.4, 130.2, 129.5, 128.1, 128.0, 127.9, 125.1, 122.7, 121.5, 113.1, 104.7, 103.2, 86.0, 82.3, 76.0, 65.1, 56.5, 52.2, 37.6, 21.7, 21.6 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C45H42N2O7: 723.3065, found: 723.2990.
1H NMR (MeOD, 500 MHz): 8.00 (d, J = 10.0 Hz, 2H), 7.90 (d, J = 10.0 Hz, 2H), 7.37 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 3.5 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 1.5 Hz, 1H), 6.90 (d, J = 3.5 Hz, 1H), 6.47 (dd, J = 6.0, 2.5 Hz, 1H), 5.75 (m, 1H), 4.6 (m, 3H), 3.92 (s, 3H), 2.94 (m, 1H), 2.70 (m, 1H), 2.44 (s, 3H), 2.41 (s, 3H) δ ppm.
13C NMR (MeOD, 126 MHz): 169.5, 167.8, 167.6, 145.8, 145.5, 143.9, 139.9, 130.8, 130.7, 130.3, 130.3, 128.2, 128.2, 125.0, 123.4, 122.8, 115.0, 105.2, 102.5, 86.6, 82.9, 76.5, 65.4, 52.2, 37.9, 21.7, 21.6 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C31H30N2O7: 543.2126, found: 543.2071.
1H NMR (MeOD, 500 MHz): 7.37 (s, 1H), 7.36 (s, 1H), 7.15 (d, J = 2.0 Hz, 1H), 6.92 (d, J = 3.5 Hz, 1H), 6.33 (dd, J = 6.5, 1.5 Hz, 1H), 4.47 (m, 1H), 3.94 (app q, J = 4.0 Hz, 1H), 3.92 (s, 3H), 3.69 (m, 2H), 2.58 (m, 1H), 2.30 (m, 1H) δ ppm.
13C NMR (MeOD, 126 MHz): 169.6, 143.6, 139.8, 125.4, 123.4, 122.6, 114.8, 104.8, 102.4, 88.1, 86.0, 72.7, 63.6, 52.1, 40.7 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C15H18N2O5: 307.1288, found: 307.1273.
1H NMR (MeOD, 500 MHz): 7.24 (d, J = 3.5 Hz, 1H), 7.20 (d, J = 2.0 Hz, 1H), 6.99 (m, 2H), 6.32 (dd, J = 6.0, 2.0 Hz, 1H), 4.46 (m, 1H), 3.92 (app q, J = 4.0 Hz, 1H), 3.67 (m, 2H), 2.59 (m, 1H), 2.28 (m, 1H) δ ppm.
13C NMR (MeOD, 126 MHz): 176.9, 143.0, 139.6, 132.2, 123.5, 123.5, 113.9, 105.7, 99.5, 87.9, 86.0, 72.7, 63.7, 40.6 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C14H16N2O5: 293.1132, found: 293.1130.
1H NMR (MeOD, 500 MHz): 8.01 (d, J = 8.0 Hz, 2H), 7.89 (m, 3H), 7.83 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 3.0 Hz, 1H), 7.34 (d, J = 8.5 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 7.16 (t, J = 7.5 Hz, 1H), 7.08 (d, J = 3.0 Hz, 1H), 6.61 (dd, J = 5.5, 2.5 Hz, 1H), 5.78 (m, 1H), 4.63 (m, 3H), 3.95 (s, 3H), 2.99 (m, 1H), 2.75 (m, 1H), 2.44 (s, 3H), 2.42 (s, 3H) δ ppm.
13C NMR (MeOD, 126 MHz): 169.6, 167.9, 167.8, 145.9, 145.7, 138.5, 131.0, 130.9, 130.5, 130.5, 130.4, 128.4, 128.3, 127.9, 124.8, 122.8, 122.4, 116.7, 105.4, 87.1, 83.3, 76.5, 65.4, 52.4, 38.4, 21.8, 21.8 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C31H29NO7: 528.2017, found: 528.1993.
1H NMR (MeOD, 500 MHz): 7.84 (d, J = 7.5 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.65 (d, J = 4.0 Hz, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.12 (d, J = 3.5 Hz, 1H), 6.49 (dd, J = 6.0, 1.5 Hz, 1H), 4.50 (m, 1H), 3.96 (m, 4H), 3.70 (m, 2H), 2.62 (m, 1H), 2.37 (m, 1H) δ ppm.
13C NMR (MeOD, 126 MHz): 169.6, 138.5, 130.0, 127.9, 124.5, 122.5, 122.0, 116.1, 104.9, 88.3, 86.2, 72.7, 63.5, 52.2, 41.0 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C15H17NO5: 292.1179, found: 292.1173.
1H NMR (MeOD, 500 MHz): 7.85 (d, J = 7.0 Hz, 1H), 7.78 (d, J = 8.5 Hz, 1H) 7.61 (d, J = 3.5 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 7.14 (d, J = 3.5 Hz, 1H), 6.48 (dd, J = 6.0, 2.0 Hz, 1H), 4.49 (m, 1H), 3.98 (app q, J = 4.0 Hz, 1H), 3.70 (m, 2H), 2.61 (m, 1H), 2.36 (m, 1H) δ ppm.
13C NMR (MeOD, 126 MHz): 171.2, 138.4, 130.3, 127.6, 124.7, 123.3, 122.0, 115.8, 105.1, 88.2, 86.2, 72.7, 63.5, 41.0 δ ppm.
HRMS-ESI+: (m/z) calcd [M + H+] for C14H15NO5: 278.1023, found: 278.1022.
UV absorption spectroscopy measurements
Absorbance measurements were collected on an Agilent Cary 100 UV-vis spectrometer (Simple Reads Software, spectral bandwidth 1.5 nm). Samples were dissolved in MilliQ water at 0.10 to 0.35 mM. Readings were taken at room temperature. 1 cm path length quartz cuvettes were used for all readings (1.16 mL volume, Starna Cells, Inc.).Steady state fluorescence measurements
Steady state fluorescence measurements were collected on a Varian Cary Eclipse fluorescence spectrometer. Samples were dissolved in MilliQ water (apart from quinine sulfate in 0.105 M aq. HClO4) at concentrations ranging from 0.10 to 0.35 mM. Readings were taken at room temperature with an excitation slit of 2.5 nm and an emission slit of 5.0 nm. 1 cm path length quartz cuvettes were used for all readings (1.16 mL volume, Starna Cells, Inc.).Molar extinction coefficient determinations
A known mass of each sample was measured in a dry glass vial. To this sample was added a known volume of MilliQ water so that the concentration of each sample was known. Serial dilutions were performed so there were 6 samples of known concentration. Absorbance measurements were taken using a UV-vis at the absorbance maximum for each compound (all absorbances in the range of 0.1–1.0). 1 cm path length quartz cuvettes were used for all readings (1.16 mL volume, Starna Cells, Inc.). Beer's law plots were constructed where the slope of the best fit line for plots of absorbance vs. concentration gives the molar extinction coefficient in M−1 cm−1. This process was repeated 3 times for each sample. The molar extinction coefficients were averaged, and standard deviation was calculated using Microsoft Excel.Quantum yield determinations
Quantum yield calculations were determined using the method described by Würth, et al.18 with quinine sulfate in 0.105 M aq. HClO4 used as the standard. Samples were dissolved in MilliQ water and placed in 1 cm path length quartz cuvettes (1.16 mL volume, Starna Cells, Inc.). Dilutions of the sample were performed until the absorbance measurement of the solution was near 0.05 (0.03–0.06 was considered acceptable). The absorbance of the standard (quinine sulfate dissolved in 0.105 M aq. HClO4) was made to match the absorbance of the standard solution (error of 0.001 was considered acceptable). All readings were collected on an Agilent Cary 100 UV-vis spectrometer (Simple Reads Software, spectral bandwidth 1.5 nm). Fluorescence measurements were collected of the sample and the standard using the excitation wavelength of the maximum absorbance for the sample. The area under the curve of the sample and standard was calculated using the fluorometer software. All fluorescence measurements were taken using a Varian Cary Eclipse fluorescence spectrometer (excitation slit 1.5 nm, emission slit 10 nm). Quantum yield was calculated using the following equations:| f = 1 − 10−A(λex) | (1) |
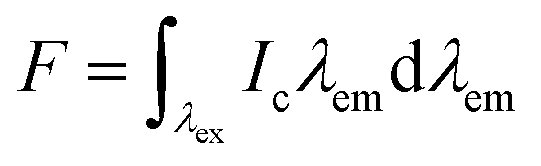 | (2) |
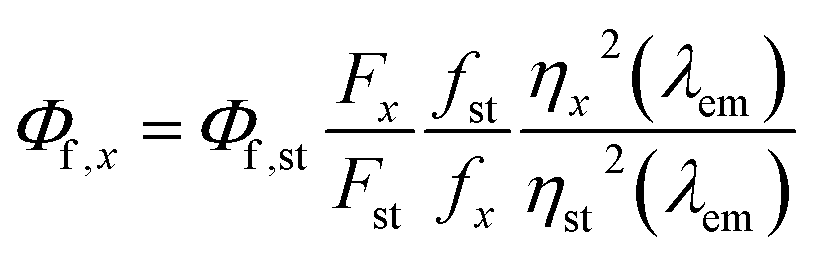 | (3) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding from the National Institutes of Health P01-CA234228 and R01-GM110129. Mass spectrometry was performed at the University of Minnesota, Masonic Cancer Center, Analytical Biochemistry Core Facility, which is supported by P30-CA77598.References
- S. M. Law, R. Eritja, M. F. Goodman and K. J. Breslauer, Biochemistry, 1996, 35, 12329–12337 CrossRef CAS PubMed.
- D. Xu, K. O. Evans and T. M. Nordlund, Biochemistry, 1994, 33, 9592–9599 CrossRef CAS PubMed.
- S. P. Hancock, T. Ghane, D. Cascio, R. Rohs, R. Di Felice and R. C. Johnson, Nucleic Acids Res., 2013, 41, 6750–6760 CrossRef CAS PubMed.
- A. Dallmann, L. Dehmel, T. Peters, C. Mügge, C. Griesinger, J. Tuma and N. P. Ernsting, Angew. Chem., Int. Ed., 2010, 49, 5989–5992 CrossRef CAS PubMed.
- D. Dziuba, P. Didier, S. Ciaco, A. Barth, C. A. M. Seidel and Y. Mély, Chem. Soc. Rev., 2021, 50, 7062–7107 RSC.
- D. Dziuba, Methods Appl. Fluoresc., 2022, 10, 044001 CrossRef PubMed.
- A. A. Tanpure, M. G. Pawar and S. G. Srivatsan, Isr. J. Chem., 2013, 53, 366–378 CrossRef CAS.
- K. T. Passow and D. A. Harki, Org. Lett., 2018, 20, 4310–4313 CrossRef CAS PubMed.
- K. T. Passow, N. M. Antczak, S. J. Sturla and D. A. Harki, Curr. Protoc. Nucleic Acid Chem., 2020, 80, e101 CAS.
- I. A. Ahmed, A. Acharyya, C. M. Eng, J. M. Rodgers, W. F. DeGrado, H. Jo and F. Gai, Molecules, 2019, 24, 602–613 CrossRef PubMed.
- C. Steinmetzger, C. Bäuerlein and C. Höbartner, Angew. Chem., Int. Ed., 2020, 59, 6760–6764 CrossRef CAS PubMed.
- L. L. Gundersen, Acta Chem. Scand., 1996, 50, 58–63 CrossRef CAS.
- A. Hall, S. H. Brown, I. P. Chessell, A. Chowdhury, N. M. Clayton, T. Coleman, G. M. P. Giblin, B. Hammond, M. P. Healy, M. R. Johnson, A. Metcalf, A. D. Michel, A. Naylor, R. Novelli, D. J. Spalding, J. Sweeting and L. Winyard, Bioorg. Med. Chem. Lett., 2007, 17, 916–920 CrossRef CAS PubMed.
- A. D. Batcho and W. Leimgruber, US Pat., 3,976,639, 1976 Search PubMed.
- J. Smagowicz and K. L. Wierzchowski, J. Lumin., 1974, 8, 210–232 CrossRef CAS.
- A. Mikhaylov, S. de Reguardati, J. Pahapill, P. R. Callis, B. Kohler and A. Rebane, Biomed. Opt. Express, 2018, 9, 447–452 CrossRef CAS PubMed.
- A. Albert and H. Taguchi, J. Chem. Soc., Perkin Trans. 2, 1973, 1101–1103 RSC.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Nat. Protoc., 2013, 8, 1535–1550 CrossRef PubMed.
- N. S. Girgis, H. B. Cottam and R. K. Robins, J. Heterocycl. Chem., 1988, 25, 361–366 CrossRef CAS.
- Z. Kazimierczuk, H. B. Cottam, G. R. Revankar and R. K. Robins, J. Am. Chem. Soc., 1984, 106, 6379–6382 CrossRef CAS.
- S. S. Bag, S. K. Das and H. Gogoi, Tetrahedron, 2018, 74, 2218–2229 CrossRef CAS.
- E. Fischer and A. Speier, Chem. Ber., 1895, 28, 3252–3258 CrossRef CAS.
- M. Hartmann, J. Huber, J. S. Kramer, J. Heering, L. Pietsch, H. Stark, D. Odadzic, I. Bischoff, R. Fürst, M. Schröder, M. Akutsu, A. Chaikuad, V. Dötsch, S. Knapp, R. M. Biondi, V. V. Rogov and E. Proschak, J. Med. Chem., 2021, 64, 3720–3746 CrossRef CAS PubMed.
- R. S. Coleman, Y. Dong and J. C. Arthur, Tetrahedron Lett., 1993, 34, 6867–6870 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03457g |
| This journal is © The Royal Society of Chemistry 2023 |