
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective hydrogenation of dimethyl terephthalate over a potassium-modified Ni/SiO2 catalyst†
Han Xiao‡
a,
Chao Zhang‡a,
Jiaojiao Zhao b,
Zhaohui Zhengc and
Yuehui Li*b
b,
Zhaohui Zhengc and
Yuehui Li*b
aSchool of Chemical Engineering, Guizhou Minzu University, Guiyang 550025, P. R. China
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou 730000, P. R. China. E-mail: yhli@licp.cas.cn
cCollege of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266000, P. R. China
First published on 31st May 2023
Abstract
Selective hydrogenation of dimethyl terephthalate (DMT) is an ideal way to prepare 1,4-cyclohexane dicarboxylate (DMCD), an important intermediate and monomer. Even though noble metal-based catalysts (e.g., Ru, Pd) have been developed for selective hydrogenation of DMT, the use of non-precious Ni catalysts to achieve high activity and selectivity is still challenging. In this study, we present that only 0.5 wt% of KF by post-impregnated doping can significantly improve the performance of Ni/SiO2 catalysts (83% vs. 96% selectivity; 41% vs. 95% conversion). The selectivity of DMCD can be up to 97%, which is the highest reported over Ni catalysts. The boosting effect of KF modification might be due to higher amounts of Ni(0) species and lower amounts of moderate acidic sites, which are beneficial for selective hydrogenation of phenyl rings over hydrogenolysis of ester groups.
1. Introduction
Dimethyl 1,4-cyclohexane-dicarboxylate (DMCD) is widely applied in the manufacture of coating resin, polyester resin, alkyd resin, and plasticizers.1–3 In industry, DMCD is produced by hydrogenation of DMT using palladium-based heterogeneous catalysts at temperatures ranging from 160 to 180 °C under a high pressure of H2.4 Given the harsh conditions and the use of the noble metal palladium, it is urgent to develop a mild and inexpensive catalytic reaction protocol.In the last decades, various Pd, Ru, or Ni catalysts were developed for DMT hydrogenation to DMCD. Zhang et al. reported a supported Pd/HTC-Al2O3 catalyst giving excellent conversion and selectivity.5 Tan et al. prepared supported Ru5/Alx SBA-15 with 100% selectivity and 93.4% yield (100 °C, 4.14 MPa H2).6 As a non-noble metal case, Ni/Ni(Al)Ox/AlOx catalysts were successfully used with a selectivity of 93.3% and almost complete conversion of DMT.7 A modified skeletal Ni catalyst was produced, yielding DMCD with complete conversion and 92.0% selectivity.8 Despite high selectivity to DMCD, small amounts of byproducts (Scheme 1) were commonly generated with nickel catalysts, whereas almost stoichiometric selectivity could be achieved by using noble metal catalysts.
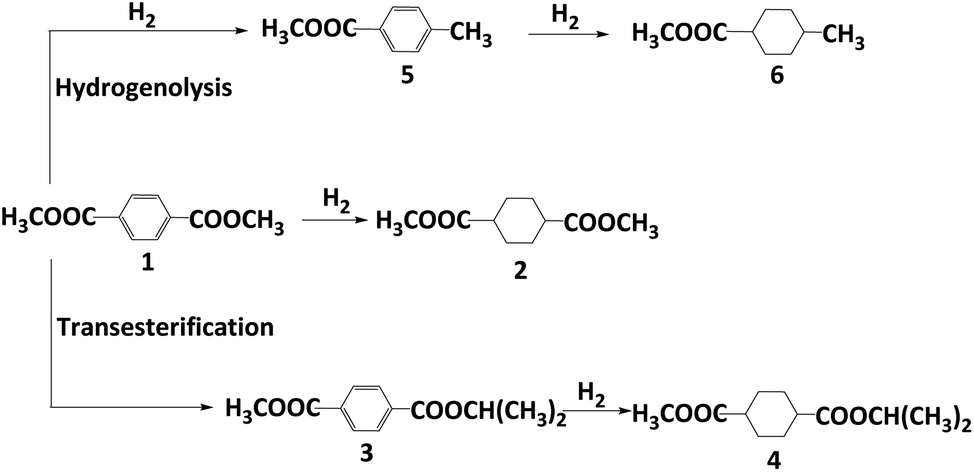 | ||
| Scheme 1 Reaction network of DMT hydrogenation on Ni-based catalysts. | ||
Various strategies have been employed to enhance activity and selectivity, such as utilizing supported bimetallic Ru–Ni/CNT catalysts9 or Re-modified Ru-based catalysts.10 While most studies focus on noble metal catalysts, investigations into modified nickel catalysts for improving catalytic activity and product selectivity are rare. The main by-products of nickel catalysts are due to the hydrogenolysis of the ester group (Scheme 1, products 5 and 6). For example, aryl esters can be activated into tolyl derivatives by homogeneous Ni catalysts, followed by hydrogenolysis using Ni/NHC catalysts.11 Multifunctional Pt/WO3–ZrO2 catalysts promote hydrogenolysis with excellent alkanes selectivity, where the acidity of support is critical.12 Therefore, using an appropriate alkaline promoter might be helpful to inhibit the hydrogenolysis of ester.
In this study, potassium-modified nickel catalysts were prepared using the AE method (ammonia evaporation) and applied to the selective hydrogenation of DMT. H2-TPR results indicate a significant decrease in reduction temperature for K-modified catalysts, which indicates that more metallic nickel species exist compared to pristine Ni/SiO2 under the same conditions. NH3-Temperature Programmed Desorption (NH3-TPD) results show moderate strength acid sites vanished on potassium-modified Ni/SiO2 catalysts, which is likely due to neutralization between K2O and acid sites. Catalysts modified with K-promoter exhibit higher activity and selectivity than pristine Ni/SiO2. Among K-modified catalysts, KF-Ni/SiO2 catalyst is the best catalyst regarding conversion and selectivity (95% conversion and 96% selectivity). The high DMCD selectivity of K-modified catalysts is attributed to lower reduction temperature and smaller acid sites which inhibit hydrogenolysis of ester to alkane.
2. Experimental
2.1 Preparation of catalysts
All catalyst precursors were calcinated at 450 °C for 4 h under ambient conditions. The samples were then reduced in pure H2 flow (30 mL min−1) for 4 h at a certain temperature (400 °C, 500 °C, 550 °C) with a heating rate of 3 °C min. M-Ni/SiO2 catalysts were obtained, where M is the promoter (KVO3, KOH, KF).
3. Results and discussion
3.1 Structural characterization of catalysts
H2-TPR characterization was first carried out to determine the influence of promoters on the reducibility of nickel species on SiO2 support. For the pristine Ni/SiO2 sample (Fig. 1(a)), a broad reduction peak ranging from 400 to 700 °C is assigned to the reduction of nickel silicate hydrate to Ni0. The peak center at approximately 590 °C is much higher than Ni/SiO2 produced by sol–gel14 or wet impregnation (WI).15 The higher reduction temperature indicates that the AE method can enhance the metal–support interaction.16,17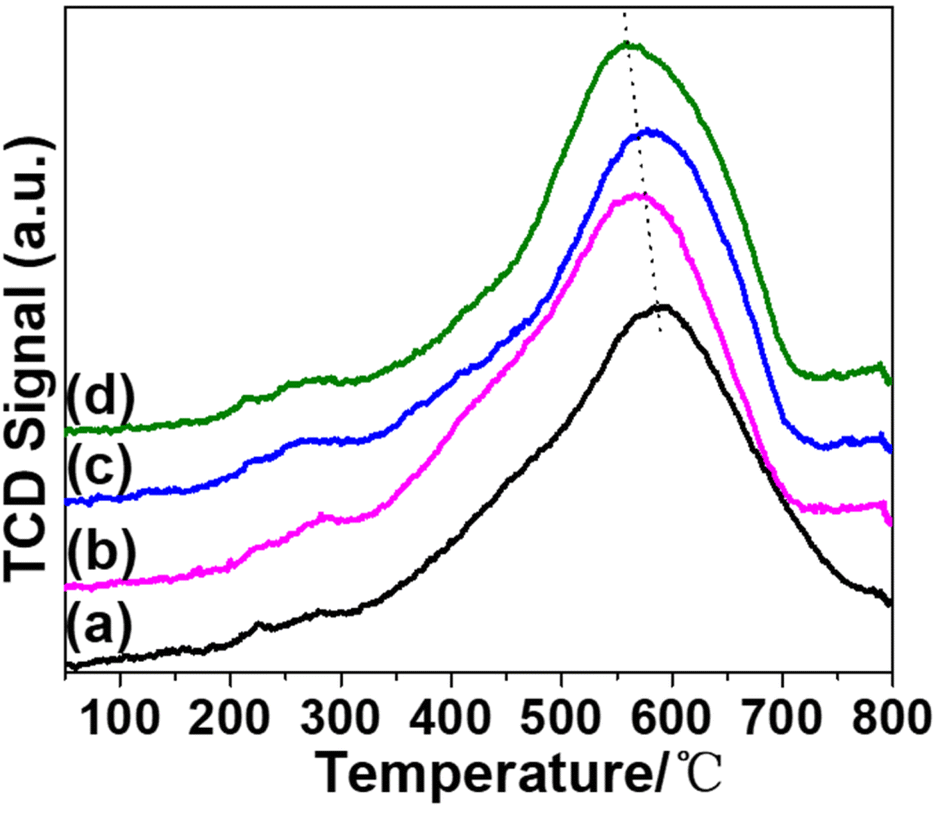 | ||
| Fig. 1 H2-TPR profiles of catalysts and promoters are in the following sequence: none (a); KVO3 (b); KOH (c); KF (d). | ||
As a typical alkali metal hydrogenation dopant, potassium typically hinders catalyst reduction by inhibiting the adsorption and dissociation of hydrogen.18 Chen has reported that K2O inhibits reduction of Ni2+ in Ni/SiO2 catalysts.19 However, in our catalytic system, the reduction temperatures of K-modified catalysts are lower than that of Ni/SiO2. It is reported that the introduction of Mo element can weaken the interaction between Ni2+ species and phyllosilicates.20 Therefore, the introduction of alkaline dopants might weaken interactions between Ni and phyllosilicates, resulting in a reduction temperature higher than nickel oxide but lower than nickel phyllosilicates. On the other hand, the promoting effect of V2O5 and F can facilitate the reduction of nickel phyllosilicate.21,22 The sequence of reduction temperatures is shown as follows: (a) > (c) > (b) > (d). Besides, the reducibility of each catalyst was calculated and listed in Table S1.†
H2-TPD was performed to investigate the effects of potassium on the adsorption properties of hydrogen on metallic Ni (Fig. S1†). All catalysts showed two peaks, the peak at a lower temperature can be attributed to hydrogen chemisorption on metallic Ni. The H2 desorption peak at high temperatures can likely be attributed to the hydrogen chemisorbed on the Ni of nickel phyllosilicates. The dispersion of nickel is calculated and listed in Table S1.†
Fig. 2 depicts X-ray diffraction (XRD) patterns of M-Ni/SiO2 (M = KVO3; KOH; KF) samples following calcination and reduction. For all calcined samples (Fig. 2A), characteristic peaks at 2θ = 34.6° and 60.6° are assigned to (110) and (300) facets of nickel silicate hydrate (JCPDS 20-0790), respectively, which is consistent with previous reports.23 Amorphous SiO2 is responsible for the wide peak centered at 2θ = 22.2° (JCPDS 76-0933).24 No characteristic peak corresponding to NiO indicates that nickel species are primarily nickel phyllosilicates. Although the broad reduction peak starts from 400 °C according to H2-TPR results, no obvious new characteristic peaks can be observed after reduction at 400 °C for 4 hours (Fig. S2†). Increasing the reduction temperature to 500 °C (Fig. S3†), two new peaks at 2θ = 44.5° and 51.8° ascribed to (111) and (200) of Ni0 (JCPDS 04-0850) can be observed.9 Nickel silicate hydrate remains the main species at this temperature. Increasing the reduction temperature to 550 °C, the intensity of the two new peaks increases (Fig. 2B). Although the majority of nickel silicate species are reduced to metallic nickel, some nickel phyllosilicate species (2θ = 34.6° and 60.5°) still exist even after 4 h of reduction in pure H2 at 550 °C. Among the four catalysts, KF-Ni/SiO2 has the weakest peak intensity of nickel silicate species. The crystallite size of the Ni0 nanoparticle is determined using the FWHM of Ni0(111) based on the Scherrer equation (Table 1). The results demonstrate that adding potassium promoter did not alter the peak intensity of nickel particles or appreciably alter their size.
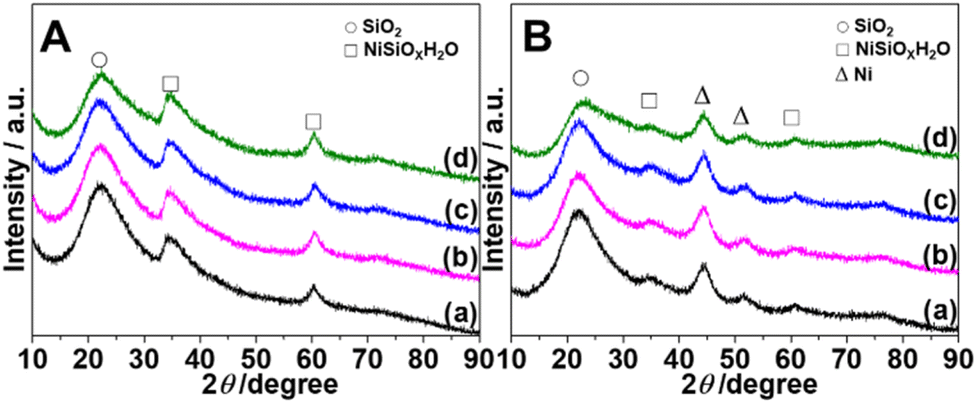 | ||
| Fig. 2 XRD patterns of (A) calcined (450 °C/4 h) and (B) reduced (550 °C/4 h) catalysts. From bottom to top, promoters are in the following sequence: none (a); KVO3 (b); KOH (c); KF (d). | ||
| Catalysts (M-Ni/SiO2)a | Loadingb (wt%) | Crystallite sizec (nm)−1 | SBETd (m2 g−1) | Vpe (cm3 g−1) | Dpf (nm) | |
|---|---|---|---|---|---|---|
| Ni | K | |||||
| a M stands for potassium dopant.b Loadings of Ni and K are determined by ICP-MS.c Calculated by using the Ni (111) reflection at 44.5° based on the Scherrer equation, catalyst was reduced at 550 °C.d BET specific surface area.e Total pore volume.f BJH desorption average pore diameter. | ||||||
| Ni/SiO2 | 13.7 | — | 3.4 | 329 | 0.70 | 8.5 |
| KVO3-Ni/SiO2 | 14.9 | 0.45 | 3.0 | 307 | 0.62 | 8.0 |
| KOH-Ni/SiO2 | 13.0 | 0.44 | 3.3 | 314 | 0.73 | 9.3 |
| KF-Ni/SiO2 | 13.8 | 0.38 | 3.0 | 261 | 0.64 | 8.5 |
The chemical compositions and textural properties of catalysts were listed in Table 1. Nickel ions weakly adsorbed on silica gel were easy to elute during the centrifugal washing process, therefore, the actual nickel loading was slightly lower than the preset value (about 14 wt% vs. 20 wt%). The loading of K is close to the preset values (about 0.4 wt% vs. 0.5 wt%) determined by ICP-MS. BET surface area (SBET), pore volume (Vp), and average pore size (Dp) of catalysts were measured using N2 adsorption–desorption isotherms (Table 1). Both pristine Ni/SiO2 and KOH-modified catalysts exhibit large SBET, Vp, and Dp. Adding potassium promoters significantly affects the physical properties of catalysts. A drop in SBET and Vp indicates that K species have occupied pore channels of support, which is consistent with previous reports.18 KF-Ni/SiO2 catalyst possesses the smallest surface area, volume, and pore size. Except for the fact that K+ occupies part of pore channels, the reaction between F ion and SiO2 can also cause the collapse of the pore structure. This is consistent with the H2-TPR and XRD results, where the weakening of interaction results in lower reduction temperature and fewer amounts of nickel phyllosilicates.
Transmission electron microscopy (TEM) was used to investigate the dispersion and shape of reduced catalysts (Fig. 3). For the Ni/SiO2 catalyst, most of particle sizes range from 2 to 5 nm (Fig. 3A), suggesting an excellent dispersity of Ni nanoparticles on SiO2 support. As magnetic catalysts may cause damage to TEM measurement, KF-Ni/SiO2 sample was chosen to illustrate the influence of potassium promoters on Ni dispersion(Fig. 3B). Compared with Ni/SiO2 catalyst, particle size distribution becomes wider. Even though most particle sizes range from 2 to 5 nm, there are more small (1 to 2 nm) and large (5 to 7 nm) particles in KF-Ni/SiO2. The etching of SiO2 by fluoride ions may lead to the collapse of pore structure, leading to small or large nickel particles. Nevertheless, the average particle sizes of the two catalysts are similar, which is in accordance with previous reports.20 Combined with TEM and XRD results, it can be considered that introducing small amounts of K did not significantly change the size of nickel particles.
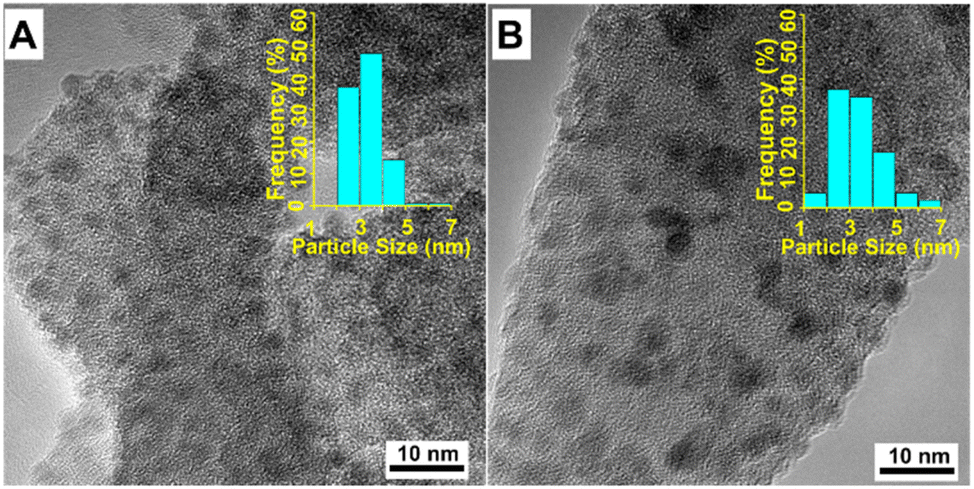 | ||
| Fig. 3 TEM images of Ni/SiO2 (A), and KF-Ni/SiO2 (B). | ||
The surface acidity of potassium-promoted Ni/SiO2 catalysts was evaluated using NH3-Temperature Programmed Desorption (NH3-TPD) (Fig. 4). All catalysts showed a desorption peak attributed to weak acid sites at around 100 °C, which was caused by NH3 adsorption on weak acidic support SiO2. In addition, a broad desorption peak in temperatures ranging from 250 to 350 °C, assigned to moderate-strength acid sites, can be observed (Fig. 4(a)). The nickel phyllosilicate surface or edge of Ni(II) is a primary source of acid sites for AE-prepared pristine Ni/SiO2 catalysts.17,20,25
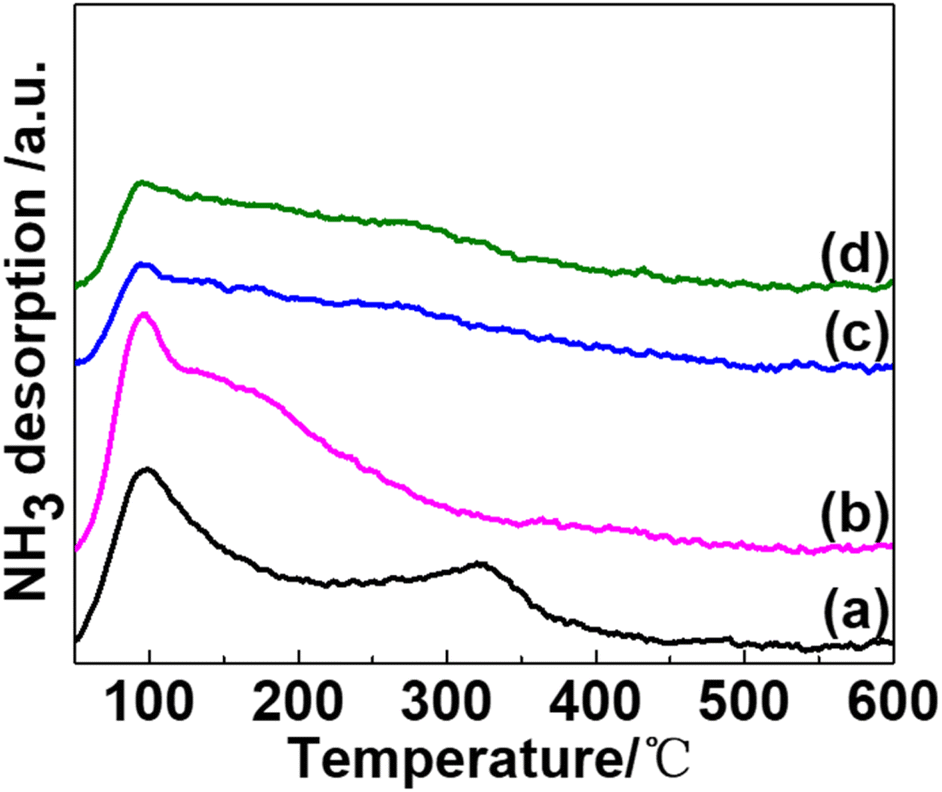 | ||
| Fig. 4 NH3-TPD profiles of catalysts and promoters are in the following sequence: none (a); KVO3 (b); KOH (c); KF (d). | ||
NH3 desorption peaks of K-modified catalysts exhibit significant differences. For KVO3 modified catalyst (Fig. 4(b)), the moderate-strength acid desorption peak vanished, and a shoulder peak ascribed to a weak acid desorption peak, being centered at 150 to 250 °C, was observed. This is due to the combined contribution of K2O and V2O5 oxides, which is formed by calcination of KVO3 in the air. K2O neutralizes moderate-strength acid sites, which is resulted from –OH group in phyllosilicates, and V2O5 produces a new weak acid site desorption peak. For KOH and KF-modified catalysts (Fig. 4(c) and (d)), the desorption peak assigned to the moderate-strength peak vanished, and the desorption peak intensity of weak acid sites decreased. These results indicate that only a 0.5 wt% addition of alkaline potassium dopants can significantly decrease the acidity of catalysts. Although the peak intensity decreased to some extent, the desorption temperature of NH3 on weak acid sites was almost the same among the four catalysts. These results are consistent with previous reports,26,27 where alkali salt could decrease the acidity of Ni/SiO2 catalysts by neutralizing acid sites but did not change the strength distribution of acidity.
The electronic structural information of potassium-modified catalysts was probed using X-ray photoelectron spectra (XPS). According to previous reports, two typical nickel species co-exist in nickel phyllosilicate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ni–PSi, formula: Si2Ni3O5(OH)4 and 2:1 Ni–PSi, formula: Si4Ni3O10(OH)2).13,28 For pristine Ni/SiO2 catalyst (Fig. 5(a)), the spectra were deconvoluted into five peaks centered at around 852.1 eV, 853.3 eV, 856.5 eV, 859.2 eV, and 862.5 eV corresponding to Ni0, Ni2+ of nickel oxide, 1:1 Ni–PSi, 2:1 Ni–Psi, and Ni2+ satellite peak, respectively. For KVO3-Ni/SiO2 catalyst (Fig. 5(b)), a slight shift of 0.2 eV was observed on Ni0 2p3/2 peak. This is due to vanadium species increasing the electron charge density of Ni atoms, consistent with previous reports where V2O5 can effectively improve the dispersion of nickel in Ni/FDU-12 catalyst and alter the electron charge density of nickel.29
:1 Ni–PSi, formula: Si2Ni3O5(OH)4 and 2:1 Ni–PSi, formula: Si4Ni3O10(OH)2).13,28 For pristine Ni/SiO2 catalyst (Fig. 5(a)), the spectra were deconvoluted into five peaks centered at around 852.1 eV, 853.3 eV, 856.5 eV, 859.2 eV, and 862.5 eV corresponding to Ni0, Ni2+ of nickel oxide, 1:1 Ni–PSi, 2:1 Ni–Psi, and Ni2+ satellite peak, respectively. For KVO3-Ni/SiO2 catalyst (Fig. 5(b)), a slight shift of 0.2 eV was observed on Ni0 2p3/2 peak. This is due to vanadium species increasing the electron charge density of Ni atoms, consistent with previous reports where V2O5 can effectively improve the dispersion of nickel in Ni/FDU-12 catalyst and alter the electron charge density of nickel.29
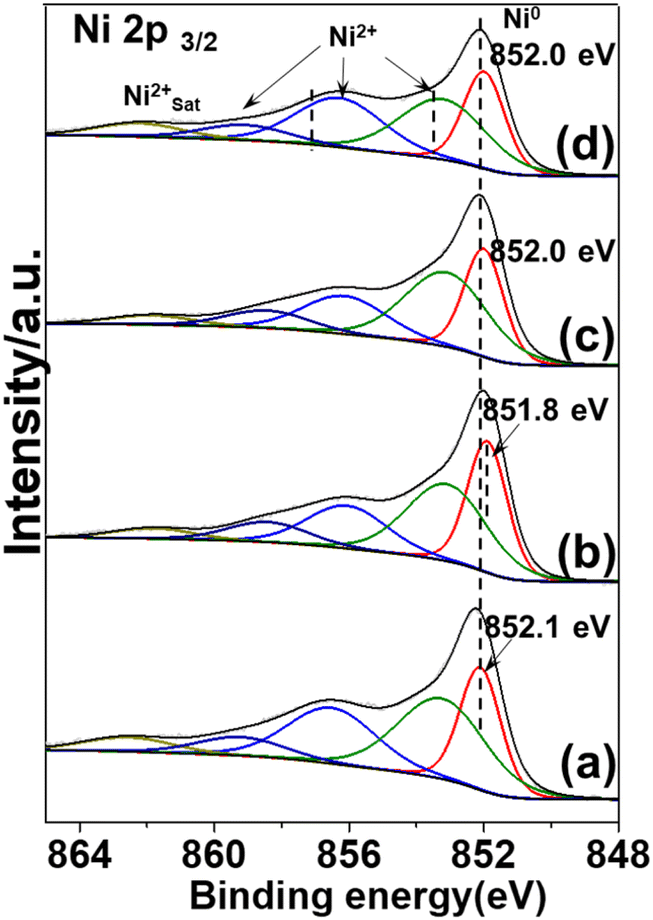 | ||
| Fig. 5 XPS spectra of the reduced catalysts and promoters are in the following sequence: none (a); KVO3 (b); KOH (c); KF (d). | ||
Adding KOH or KF to a pristine Ni/SiO2 catalyst had little effect on the chemical valence state of Ni0 (from 852.1 eV to 852.0 eV) (Fig. 5(c) and (d)). Besides Ni0 species, Ni2+ species attributed to nickel oxide and nickel phyllosilicates species were also observed. This is consistent with XRD results where there were characteristic peaks attributed to nickel phyllosilicates species. The multiple Ni species can also explain the broad reduction peak in H2-TPR results. The deconvoluted area and fraction of different Ni species are listed in Table S2.†
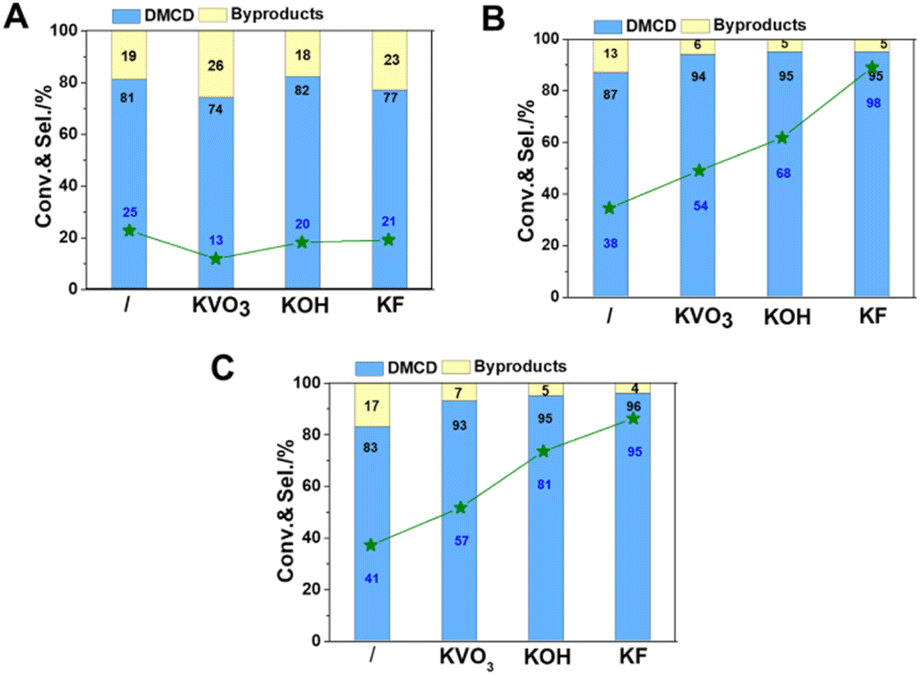 | ||
| Fig. 6 Influence of reduction temperature of catalysts. 400 °C (A); 500 °C (B); 550 °C (C). Reaction conditions: catalyst, 50 mg; DMT, 0.1 g; IPA, 2 mL; the initial pressure of H2, 5 MPa; reaction temperature, 90 °C; reaction time, 4 h. | ||
Considering catalysts reduced at 550 °C have more metallic nickel, they have been chosen as model catalysts to explore the effect of reaction temperature. All catalysts displayed catalytic activity even at a low temperature of 80 °C. Potassium promoters were found to greatly enhance the selectivity of DMCD, with KF-Ni/SiO2 achieving a remarkable 97% selectivity (Fig. 7A). Nickel-based catalysts can catalyze efficiently the hydrogenolysis of the fatty acid ester, and excellent hydrodeoxygenation performance of methyl palmitate to alkanes was acquired due to the abundant acidic sites in modified nickel phyllosilicate catalysts.20 In this work, the introduction of promoters had two key effects: it effectively lowered reduction temperature, thus increasing the proportion of metallic nickel in modified catalysts compared to Ni/SiO2, and neutralized moderate strength acid sites, decreasing hydrogenolysis of ester to alkanes.12 This represents the highest selectivity catalyzed by a non-precious metal catalyst. The conversion of DMT increased as the reaction temperature increased from 80 to 100 °C (Fig. 7B and C), whereas the selectivity of DMCD remained substantially constant. Further increasing the reaction temperature to 110 °C, all catalysts achieved 100% DMT conversion. The main byproduct, methyl 4-methyl-cyclohexane carboxylate (MMCHC) (Scheme 1, product 6), is resulted from the hydrogenation of methyl 4-Methylbenzoate (MMB) (Scheme 1, product 5). Detailed selectivities of the main byproducts are given in Table S4.†
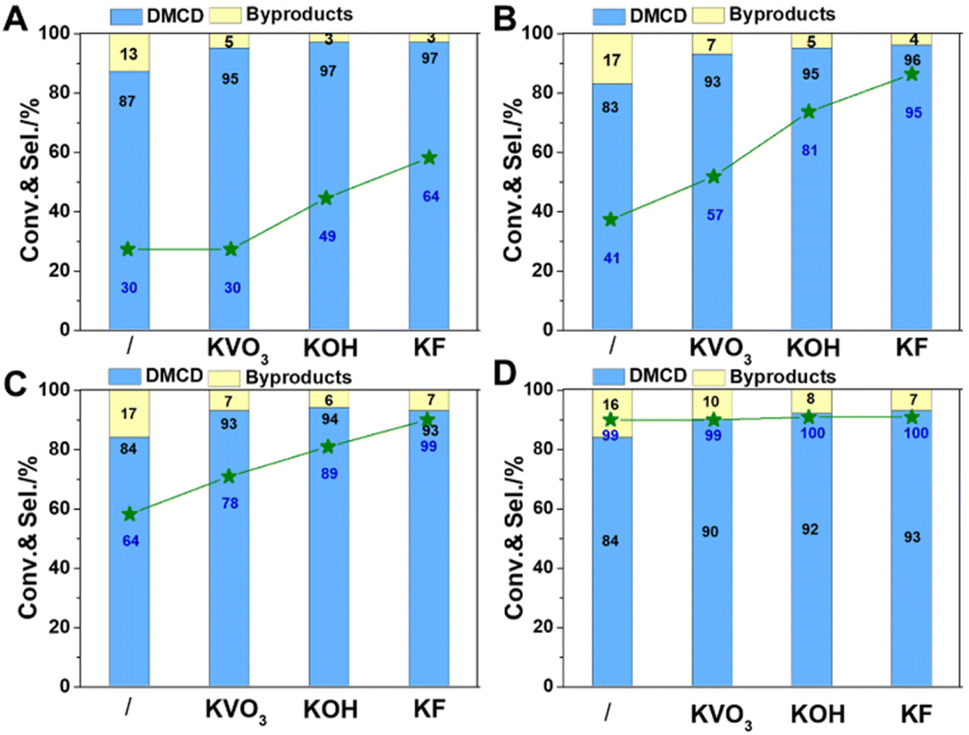 | ||
| Fig. 7 Influence of reaction temperature. 80 °C (A); 90 °C (B); 100 °C (C); 110 °C (D). Reaction conditions: catalyst, 50 mg; DMT, 0.1 g; IPA, 2 mL; the initial pressure of H2, 5 MPa; reaction time, 4 h. | ||
The stability of catalysts is an important consideration when assessing their possible usage in industry. To test the stability of KF-Ni/SiO2, which is an optimal catalyst, it underwent a six-time recycling test (Fig. 8). The catalyst was filtered, washed three times with isopropanol, and directly reused in the next cycle without additional processing or reduction. The DMT conversion was above 98% until the third cycle. After the sixth cycle, it decreased to 89%. The selectivity of DMCD remained consistent throughout six cycles, indicating that the catalyst did not lose its activity or selectivity over time. These results demonstrate that the catalyst exhibited good stability over the course of the recycling test.
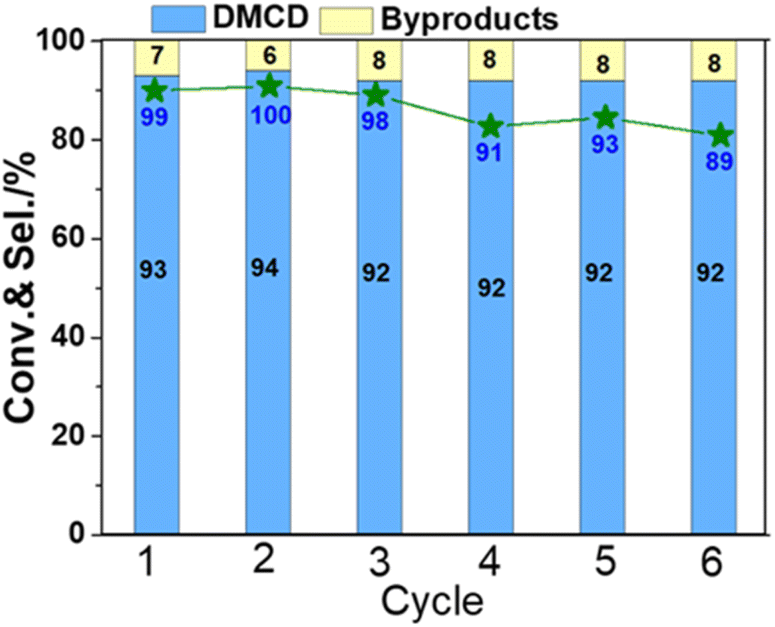 | ||
| Fig. 8 Stability studies for KF-Ni/SiO2 catalyst on DMT conversion and DMCD selectivity. Reaction conditions: catalyst, 50 mg; DMT, 0.1 g; IPA, 2 mL; the initial pressure of H2, 5 MPa; reaction temperature, 100 °C, reaction time, 4 h. | ||
4. Conclusions
In summary, this study developed a potassium-promoted strategy to enhance the selectivity of DMCD during the hydrogenation of DMT. Catalytic performance evaluation showed both conversion and selectivity were significantly improved on K-modified catalysts compared to pristine Ni/SiO2. KF-promoted Ni/SiO2 showed the highest conversion and selectivity within the evaluated range. The remarkable difference in activity and selectivity is due to the lower reduction temperature, which can offer higher amounts of metallic nickel under the same conditions, as well as smaller amounts of acid sites inhibiting the hydrogenolysis of ester. Additionally, the selectivity and reactivity of the KF-promoted catalyst remained consistent throughout six recycling experiments, demonstrating its excellent stability. Overall, the present work provides comprehensive insights into the rational design of nickel catalysts by using potassium modifiers.Additional reference cited within the ESI.†31
Author contributions
Han Xiao: investigation, formal analysis, validation, data curation, writing – original draft. Chao Zhang: investigation, formal analysis, data curation. Jiaojiao Zhao: formal analysis, visualization, writing – review & editing. Zhaohui Zheng: formal analysis, software. Yuehui Li: conceptualization, supervision, resources, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for the financial support from NSFC (21633013, 22102197), funding support from Jiangsu Province Natural Science Foundation (BK20211096), and Guizhou Provincial Science and Technology Projects (ZK[2021]051). Dr Zhao thanks the Science Fund of Shandong Laboratory of Advanced Materials and Green Manufacturing (Yantai, AMGM2021F07).Notes and references
- S. R. Turner, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5847 CrossRef CAS.
- B. J. Sublett and G. W. Connell, US Pat. 5559159, 1996.
- R. R. Ambrose, J. B. O’dwyer, B. K. Johnston, D. P. Zielinski, S. Porter and W. H. Tyger, US Pat. 4859743, 1989 Search PubMed.
- D. Hou, J. Xin, X. Lu, X. Guo, H. Dong, B. Ren and S. Zhang, RSC Adv., 2016, 6, 48737 RSC.
- F. Zhang, J. Chen, P. Chen, Z. Sun and S. Xu, AIChE J., 2012, 58, 1853 CrossRef CAS.
- W. Yu, S. Bhattacharjee, W.-Y. Lu and C.-S. Tan, ACS Sustainable Chem. Eng., 2020, 8, 4058 CrossRef CAS.
- Q. Fan, X. Li, Z. Yang, J. Han, S. Xu and F. Zhang, Chem. Mater., 2016, 28, 6296 CrossRef CAS.
- X. Xu, Z. Rong, W. Du, F. Zheng, Y. Liang, Y. Wang and L. Lv, Fine Chem., 2010, 27, 1239 CAS.
- Y. Huang, Y. Ma, Y. Cheng, L. Wang and X. Li, Ind. Eng. Chem. Res., 2014, 53, 4604 CrossRef CAS.
- E. Qu, J. Luo, X. Di, C. Li and C. Liang, J. Nanosci. Nanotechnol., 2020, 20, 1140 CrossRef CAS PubMed.
- A. Cook, S. Prakash, Y.-L. Zheng and S. G. Newman, J. Am. Chem. Soc., 2020, 142, 8109 CrossRef CAS PubMed.
- K. Yuan, Y. Yamazaki, X. Jin and K. Nozaki, J. Am. Chem. Soc., 2023, 145, 3454 CrossRef CAS PubMed.
- Y. Wu, G. Chang, Y. Zhao and Y. Zhang, Dalton Trans., 2014, 43, 779 RSC.
- F. Ding, Y. Zhang, G. Yuan, K. Wang, I. Dragutan, V. Dragutan, Y. Cui and J. Wu, J. Nanomater., 2015, 16, 60 Search PubMed.
- T. A. Le, J. K. Kang and E. D. Park, Top. Catal., 2018, 61, 1537 CrossRef CAS.
- P. Burattin, M. Che and C. Louis, J. Phys. Chem. B, 2000, 104, 10482 CrossRef CAS.
- Y. W. Wang, Y. J. Zhang, K. J. Wang, L. M. Tan and S. Y. CHEN, J. Fuel Chem. Technol., 2021, 49, 97 CrossRef CAS.
- Y. Yang, H.-W. Xiang, Y.-Y. Xu, L. Bai and Y.-W. Li, Appl. Catal., A, 2004, 266, 181 CrossRef CAS.
- J. Chen, J. Zhang and J. Zhang, React. Kinet. Catal. Lett., 2008, 93, 359 CrossRef CAS.
- (a) B. Peng, Y. Yao, C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 2072 CrossRef CAS PubMed; (b) B. Peng, X. Yuan, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 9400 CrossRef CAS PubMed; (c) L. Yan, X. X. Liu, J. Deng and Y. Fu, Green Chem., 2017, 19, 4600 RSC.
- Y. Zhang, Y. Chen and Q. Liu, Int. J. Hydrogen Energy, 2021, 46, 32003 CrossRef CAS.
- M. Ebrahiminejad and R. Karimzadeh, Mol. Catal., 2022, 517, 112056 CrossRef CAS.
- S. Yuan and L. Zhao, RSC Adv., 2016, 6, 49769 RSC.
- K. Xu, J.-X. Wang, X.-L. Kang and J.-F. Chen, Mater. Lett., 2009, 63, 31 CrossRef CAS.
- F. Dong, G. Ding, H. Zheng, X. Xiang, L. Chen, Y. Zhu and Y. Li, Catal. Sci. Technol., 2016, 6, 767 RSC.
- J. Chang, N. Tsubaki and K. Fujimoto, J. Jpn. Pet. Inst., 2000, 43, 357 CrossRef CAS.
- S. Xie, H. Li, H. Li and J.-F. Deng, Appl. Catal., A, 1999, 189, 45 CrossRef CAS.
- Z. Li, L. Mo, Y. Kathiraser and S. Kawi, ACS Catal., 2014, 4, 1526 CrossRef CAS.
- Z. Tian, H. Yang and Q. Liu, Energy Technol., 2020, 8, 1901270 CrossRef CAS.
- S. Abelló, D. Verboekend, B. Bridier and J. Pérez-Ramírez, J. Catal., 2008, 259, 85 CrossRef.
- F. Wang, C.-M. Li, X.-Y. Zhang, M. Wei, D. G. Evans and X. Duan, J. Catal., 2015, 329, 177 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Catalyst characterization, evaluation of the catalytic performance, Fig. S1–S3, and Tables S1–S3. See DOI: https://doi.org/10.1039/d3ra02223d |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |