
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon black supported Ag nanoparticles in zero-gap CO2 electrolysis to CO enabling high mass activity†
Khaled Seteiz‡
 ab,
Josephine N. Häberlein‡ab,
Philipp A. Heizmannab,
Joey Dischab and
Severin Vierrath*abc
ab,
Josephine N. Häberlein‡ab,
Philipp A. Heizmannab,
Joey Dischab and
Severin Vierrath*abc
aElectrochemical Energy Systems, IMTEK – Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: severin.vierrath@imtek.uni-freiburg.de
bUniversity of Freiburg, Institute and FIT – Freiburg Center for Interactive Materials and Bioinspired Technologies, Georges-Köhler-Allee 105, 79110 Freiburg, Germany
cHahn-Schickard, Georges-Koehler-Allee 103, 79110 Freiburg, Germany
First published on 21st June 2023
Abstract
In this study Ag nanoparticles supported on carbon black (Ag/C) were studied as catalysts for the electrochemical reduction of CO2 to CO. The nanoparticles were synthesized on three carbon supports, namely Super P, Vulcan and Ketjenblack with surface areas from 50 to 800 m2 g−1 using cysteamine as a linker as proposed by Kim et al., J. Am. Chem. Soc., 2015, 137, 13844. Gas diffusion electrodes were fabricated with all three Ag/Cs and then characterized in a zero-gap electrolyzer. All three supported catalysts achieve high voltage efficiencies, mass activities, and faradaic efficiencies above 80% up to 200 mA cm−2 with Ag loadings of ∼0.07 mg cm−2. Using an IrO2 anode, a partial CO current density of 196 mA cm−2 at 2.95 V and a mass activity of 3920 mA mg−1 at a cell voltage of 3.2 V was achieved. When changing the electrolyte from 0.1 M KOH to 0.1 M CsOH, it is possible to achieve 90% FECO at 300 mA cm−2. This results in a mass activity up to 5400 mA mg−1. Moreover, long-term tests at 300 mA cm−2 with 0.1 M CsOH resulted in FECO remaining above 80% over 11 h. The electrochemical performance did not show a dependence on the carbon support, indicating that mass transport is limiting the cathode, rather than catalyst kinetics. It is worth noting that this may only apply to electrodes with PTFE binders as used in this study, and electrodes with ionomer binders may show a dependence on the catalyst support.
1. Introduction
To achieve global net-zero CO2 emission targets, we must defossilize all sectors that rely on carbon energy and fossil feedstock.1 Electrochemical reduction of CO2 is a very promising approach to defossilize the chemical industry and create a closed carbon loop.2,3 This technology enables the production of chemical feedstocks using captured CO2, water, and renewable energy. A wide range of cell designs and target products with carbon skeletons Cx, usually ranging from x = 1–3 (e.g., CO, formic acid, ethanol, n-propyl alcohol, and ethylene) are the subject of current research.4–7 The simplest product of electrochemical CO2 reduction is CO, which is a widely used feedstock for a wide range of industries.8 Hence, this work focuses on the production of CO via electrochemical CO2 reduction in a zero-gap electrolyzer cell. In this setup, the CO2 reduction catalyst is deposited onto a gas diffusion medium to form the so-called gas diffusion electrode (GDE). During operation the GDE is fed with humidified CO2, which is being reduced at the catalyst in the presence of water, forming CO and hydroxide anions (OH−).Metal-based catalysts such as copper (Cu), gold (Au), silver (Ag), and zinc (Zn) are most commonly used to facilitate this reduction reaction. Besides catalytic activity also the selectivity plays a key role, especially towards the strongly competing hydrogen evolution reaction. Among these metal-based catalysts, Au has the highest selectivity for the production of CO, followed by Ag, Cu, and Zn.9 This is because Au and Ag have the optimal binding energy for the intermediate CO species, i.e. the intermediate CO species are strongly bound to the catalyst surface, preventing them from undergoing further reduction to other products.10
According to a study by Lim et al., density functional theory (DFT) calculations suggest that the required overpotential for the electroreduction of CO2 to CO (CO2RR) can be significantly reduced (∼0.5 V) by doping Ag-based catalysts with sulfur (S) or arsenic (As).11 They proposed that the enhanced performance of the Ag–S or Ag–As catalysts is due to the introduction of covalent bonds on the Ag surface. The theoretical conjecture proposed by Lim et al. was subsequently confirmed by experimental work conducted by Kim et al. In their study, the researchers synthesized S doped Ag/C catalysts with varying Ag nanoparticle sizes using a simple one-pot synthesis with cysteamine as an anchoring agent.12 This approach has the advantage that it allows for the direct growth of immobilized Ag nanoparticles on a carbon support. The use of an anchoring agent, such as cysteamine, enables the formation of nucleation sites that promote the growth of monodispersed and homogeneous Ag nanoparticles, maximizing the surface area of the catalyst. Additionally, the anchoring agent is thought to change the electronic structure of the active sites on the Ag nanoparticles, as indicated by X-ray photoelectron spectroscopy and calculated by DFT. This is believed to result in more favorable binding energies between the intermediate CO species and the Ag nanoparticles, leading to a reduced overpotential for the reaction.12 They found that Ag nanoparticles with an average diameter of about 5 nm exhibit the highest CO2 reduction reaction activity with faradaic efficiencies up to 84.4% at −0.75 V (vs. RHE) in H-cells.
In addition to the work by Kim et al.,12 other researchers have also studied the effect of nanoparticle size on the activity and selectivity of Ag-based catalysts for CO2RR. For example, Deng et al. demonstrated that the CO2RR activity of Ag nanoparticles is mainly dominated by Ag(100) sites, with sub-5 nm Ag nanoparticles supported on highly oriented pyrolytic graphite being the optimal size for these catalysts.13 Similarly, Liu et al. synthesized Ag nanocubes that are fully enclosed by energetically favorable Ag(100) facets, which have a lower d-band center (−3.118 eV) than Ag(111) and edge sites (−3.646 eV and −3.586 eV, respectively).14 These studies highlight the importance of the size and surface structure of Ag nanoparticles for the performance of Ag-based catalysts in CO2RR. These three studies focused on testing the Ag nanoparticles in H-cells. Electrolysis carried out in H-type cells are susceptible to reach mass-transport limitations resulting in low current densities below 100 mA cm−2.15 Moreover, the dissolved CO2 reacts with OH− to form carbonates in basic electrolytes.16,17 The consequence of that is the loss of CO2 decreasing CO2RR activity due to change of the pH value towards a more acidic environment, which affects the catalyst.18,19 Thus, H-type cells are not suitable for use in industrial conditions. In state-of-the-art CO2 electrolysis to CO non-supported Ag NPs are commonly used.20 However, supported catalysts can be more cost-effective compared to unsupported catalysts because the support material reduces the amount of catalytic material needed. Furthermore, the carbon support introduces an additional degree of freedom in the design, e.g. to ensure that reaction sites are well dispersed.
In this work, the synthesis of S-doped Ag/C catalysts developed by Kim et al.12 was adapted and applied in a zero-gap cell with a gas-fed cathode to show the potential in an industry relevant setup. This cell design enables high mass activities with low Ag loadings. Furthermore, an optimized synthesis was applied to various carbon supports in order to investigate the influence of the carbon surface morphology and Ag nanoparticle position on CO2RR activity.
2. Experimental
2.1 Materials
All reagents and solvents used in this study were purchased from commercial sources. Unless otherwise noted, these chemicals were used without further pretreatment or purification. For the Ag/C synthesis, silver nitrate (AgNO3, Sigma-Aldrich, 99.0%) was used as Ag precursor and cysteamine (Sigma-Aldrich) as anchoring agent. Carbon black Super P® conductive (Alfa Aesar, 99+%), carbon black Vulcan (Fuel cell store, Vulcan XC-72-R) and carbon black Ketjen (Nouryon, Ketjenblack EC-300J) were used as carbon support materials. Ethylene glycol (EG, Thermo Fisher, 99%), isopropyl alcohol (IPA, Carl Roth, >99.5%), methanol (MeOH, Carl Roth >99.5%) were used as solvents. Besides the synthesized Ag/C, commercial Ag/C (FC catalysts, 40% and 80% Ag on Vulcan) were used as cathode catalysts. Nickel iron oxide nanopowder (NiFe2O4, US Research Nanomaterials Inc, 99.99%) and iridium oxide nanopoweder (IrO2, Alfa Aesar, Premion 99.99%) were used as anode catalyst, respectively. Polytetrafluoroethylene (PTFE, Sigma-Aldrich, 60 wt% dispersion in H2O) was used as cathode catalyst layer binder, Aemion+ (Ionomer Innovations Inc., Aemion™ AP2-HNN8-00-X) as ionomer and zirconium oxide balls (ZrO2, Retsch, 5 mm) for grinding anode catalysts. Gas diffusion layer (GDL, Freudenberg carbon paper H23C6), Ni Felt (Bekaert, 200 μm), anion exchange membrane (AEM, Ionomer Innovations Inc., Aemion™ AF2-HNN8-50-X) and KOH (Sigma-Aldrich, pellets, > 85%) were used for cell testing. Lead(II) acetate (Pb(ac)2, Sigma-Aldrich, ≥99.99%), sodium perchlorate (NaClO4, Sigma-Aldrich, 98%), perchloric acid (HClO4, Sigma-Aldrich, 70%) and sulfuric acid (H2SO4, Sigma-Aldrich, 98%) were used for surface area characterization. Cesium hydroxide (CsOH, Sigma-Aldrich, 99.95%) was used for long-term tests.2.2 Ag/C synthesis
The synthesis of Ag/C was adapted and scaled up to different carbon supports as described by Kim et al.12 A simplified illustration of the synthesis can be seen in ESI Fig. S1.† First, carbon support (200 mg) in ethylene glycol (100 mL) was ultrasonicated (Bandelin Sonorex RK) for 30 min. Cysteamine (10 mg) was added to the carbon support solution and ultrasonicated for another 30 min. Then the mixture was added to a preheated (50 °C) solution of silver nitrate (200 mg) in ethylene glycol (100 mL), kept for 10 min at 50 °C before being heating up to 180 °C under reflux. The heating at 180 °C was maintained for 60 min. After cooling below 30 °C, the solution was washed with isopropyl alcohol, filtered and dried overnight in an oven at 80 °C. The final Ag/C catalysts were obtained as a black powder and yielded a mass between 200–300 mg, respectively.2.3 Electrode preparation
Cathode GDEs were prepared by sonicating 100 mg of the respective Ag/C catalyst with 0.67 g of a 5 wt% PTFE dispersion (in H2O) in 7 mL H2O and 7 mL isopropyl alcohol for 30 minutes. The ink was then spray-coated onto the GDL with an ultrasonic spray coater (SNR 300, Sonocell). The hotplate below the GDEs was heated to 40 °C to accelerate solvent evaporation during ultrasonic spray-coating. Low Ag loadings with 0.05 mgAg cm−2 and 0.07 mgAg cm−2 for cathode Ag/C GDE's were obtained (Fig. S2†).The anode ink was obtained by dispersing 600 mg NiFe2O4 or IrO2 in 1.8 mL H2O and 1.8 g of a 2.5 wt% Aemion+-solution (in MeOH/H2O 10/1 w/w). In addition, ZrO2 grinding balls were added. After 2 days on a roll mixer (IKA, Roller 10) the ink was casted with a bar coater (Mayer rod, 150 μm wet film thickness) onto the AEM to obtain the half catalyst coated membrane (HCCM) (Fig. S3†). 200 μm thick Ni-felts were used as anode electrode and cleaned by sonication in acetone, isopropyl alcohol and DI-H2O, consecutively.
2.4 Material characterization
2D analysis. The nucleation and growth behavior of the Ag nanoparticles may vary slightly depending on the carbon primary particle due to synthetically based chemical and structural inhomogeneities in the carbon support. For this reason, 2000 nanoparticles on at least 5 different Ag/C particles were considered to allow sufficient quantitative comparisons between all samples. All individual Ag nanoparticles were measured via ImageJ 1.53c.21
3D analysis. The acquired tilt series image pairs were aligned by cross-correlation using Inspect3D. By tracking individual Ag nanoparticles on the sample, this cross-correlation was further refined (see ESI videos 00 and 01† for an exemplary comparison before and after image alignment). The micrographs were then binned by a factor of 2, followed by reconstruction of the tomogram by using an OSEM algorithm with 20 iterations.22 This provided sufficient tomogram quality to distinguish Ag nanoparticles sitting on the exterior surface of the carbon support from nanoparticles inside the support. The tomograms were segmented in ImageJ iteratively using the Trainable Weka Segmentation plugin and visualized with the open source tomography platform TomViz.23,24 Due to methodological (e.g., missing-wedge artifacts due to limited tilt range) and experimentally introduced artifacts (e.g., possible slight intraparticle movements throughout the acquisition), only the position of the nanoparticles but no other morphological aspects were analyzed, also because the direct evaluation of particle diameters from the raw 2D TEM micrographs in our opinion is more precise due to the not required intermediate processing steps and statistically more robust ascribable to the increased number of considered Ag/C parent particles.
2.5 Electrochemical measurements
| UiR corrected = Ucell − i·HFRi. |
Herein, nx denotes the measured mole fraction of the product of interest as quantified by GC, F is the Faraday constant (96
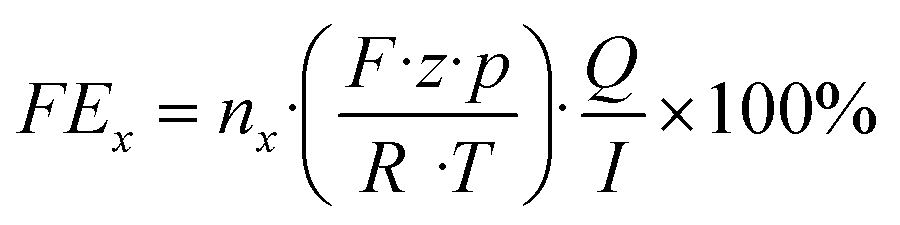
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), R the ideal gas constant (8.314 J K−1 mol−1), Q the volumetric flow rate, T the temperature, z the charge transfer number, p the pressure and I the total current. By multiplying the product-specific Faraday efficiency with the total current density, the product-specific partial current density ix can be obtained.
485 C mol−1), R the ideal gas constant (8.314 J K−1 mol−1), Q the volumetric flow rate, T the temperature, z the charge transfer number, p the pressure and I the total current. By multiplying the product-specific Faraday efficiency with the total current density, the product-specific partial current density ix can be obtained.3. Results & discussion
The influence of carbon support was investigated by applying the synthesis method described in the experimental section to three different common and widely used carbon supports: Ketjenblack with a high surface area of approximately ∼800 m2 g−1, Vulcan with a medium surface area of ∼200–250 m2 g−1 and carbon black Super P with a low surface area of ∼50 m2 g−1.28–31 TEM micrographs show that the carbon supports are composed of primary particles with sizes in the range of 20–60 nm, which then form larger aggregates with sizes of approximately 250–1000 nm (Fig. S6–S8†). The micrographs further indicate that the primary particles of the carbon supports consist of a shell of graphitic carbon and a core of amorphous carbon. The greatly increased surface area of the Ketjenblack sample presumably is due to the degree of hollowing of the carbon support rather than the primary particle sizes. In literature, pore volumes of ∼2 m3 g−1 are reported for Ketjenblack, compared to ∼0.3 m3 g−1 for Super P (see the ESI† for a detailed characterization of the carbon supports).32Representative TEM micrographs of all synthesized Ag/Cs can be seen in Fig. 1. No significant change in carbon support morphology due to nanoparticle synthesis was observed. For the three carbon substrates shown, spherical Ag nanoparticles are homogeneously distributed over the entire carbon support surface. The average nanoparticle diameter was found to be inversely correlated to the surface area: Ag/CSuper P (5.3 nm ± 1.5 nm) > Ag/CVulcan (4.7 nm ± 1.5 nm) > Ag/CKetjen (4.4 nm ± 1.3 nm) (Fig. S14–S16†). This is potentially a consequence of the sharp increase in surface area from Super P to Ketjenblack with concomitant increase in the number of Ag crystallization nuclei and the decrease of probability of agglomeration of individual nanoparticles. Although a strong dependence of nanoparticle size and CO2 activity has been reported, the large overlap of the histograms (Fig. S17†) and the marginal difference of the average particle diameter from 4.4 nm to 5.3 nm allows comparing the three samples.13 The catalyst loadings were 31 wt% for Ag/CSuper P < 33 wt% for Ag/CVulcan < 37 wt% for Ag/CKetjen as determined by TGA (Fig. S4†). Again this trend could be explained by the increasing surface area and thus nuclei and deposition using the same synthesis parameters for all three carbons.
 | ||
| Fig. 1 Representative TEM micrographs of the synthesized Ag/C samples. Red: Ag/CSuper P, green: Ag/CVulcan, blue: Ag/CKetjen. | ||
As reported for fuel cells, the position of the catalyst on the carbon support can have a significant impact on the performance. In general, nanoparticle catalysts on the surface of the carbon support have better access to reactants leading to higher reaction rates. However, an adverse effect can be observed for fuel cells, when catalyst particles on the carbon surface are rendered inactive by adsorbing functional groups of the ion-conductive binder.33 As a consequence, electrodes in fuel cells are optimized depending on the desired activity and reactant accessibility by choosing a carbon support with the right surface area. Typically, low surface area carbons lead to particles being dispersed on the outer surface of the carbon, while higher surface carbon features mesopores that contain a large fraction of the catalyst particles.34
To determine the position 3D STEM tilt series were recorded (Fig. 2a–c) and 3D volumes were reconstructed (Fig. 2d–f, ESI videos 02, 03, 04 and Fig. S18†). As can be seen in Fig. 2, the proportion of internal particles increases from ∼10% for Super P to 20% for Vulcan and ∼65% for Ketjenblack. This finding is consistent with studies on similar Pt/C catalysts, and is likely due to the higher porosity of the Ketjenblack carbon support. For example, Padgett et al. reported an interior fraction of around 0.2 for 10 wt% Pt/Vulcan, and Sneed et al. reported an interior fraction of around 0.7 for 20 wt% Pt/Ketjenblack.29,35 (see the ESI† for a detailed discussion of the TEM data).
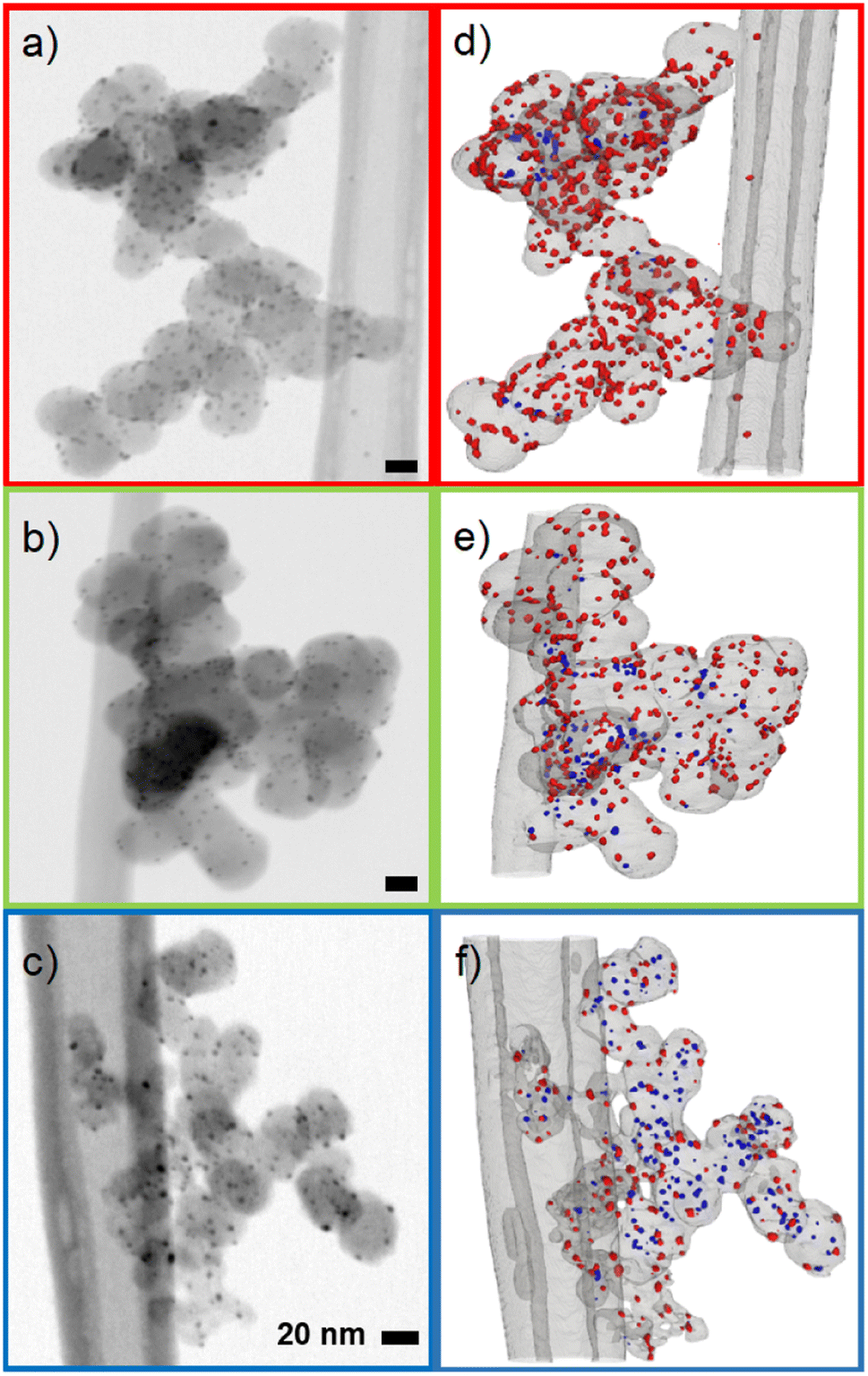 | ||
| Fig. 2 Electron tomography of Ag synthesized on three carbon blacks: Ag on Super P (red), Ag on Vulcan (green), Ag on Ketjenblack (blue). (a–c): Representative BF STEM micrographs of the Ag/C samples. (d–f): Segmented volumes showing interior (blue) and exterior Ag nanoparticles (red). Note that the segmented volumes contain depth and thus the 2D vs. 3D projections do not match exactly in this representation. | ||
It is difficult to determine the precise fraction of nanoparticles that are inside or outside the carbon pores based on 2D TEM micrographs. However, some nanoparticles can be assigned to the outside of the carbon support if they are clearly visible outside the projection of the carbon primary particle, as shown by Harzer et al.36 In turn, if a nanoparticle is totally encompassed by the carbon particle's projection, it cannot be distinguished whether it is on the surface or in a carbon pore. From this derivation, a qualitative decrease in exterior particles in the order Super P > Vulcan > Ketjenblack was observed. To better estimate the fraction of interior and exterior nanoparticles, 3D STEM tilt series were recorded and 3D volumes were reconstructed (see ESI videos 02, 03, 04 and Fig. S18†). We make no claim to perfect reconstructions, as tilt series based electron tomography has method-based problems (e.g., the missing wedge problem introducing elongation and ghost tail artifacts) and the samples themselves are difficult to investigate (e.g., limited radiation dose due to rapid degradation of the carbon species).37 For an in-depth analysis of the issues involved in STEM tomography of metal/carbon based catalysts, the reader is referred to the highly recommended paper by Padgett et al.34 Nevertheless, the position of an individual nanoparticle can already be estimated much more accurately from the raw tilt series than with single projection 2D micrographs (Fig. S19 and ESI video 05†), and the reconstructed volumes obtained allow sufficient discrimination between interior and exterior nanoparticles.
Raman spectroscopy was used to identify the Ag–S bond formation in the Ag/C catalysts and potential changes in the graphitic structure of the various carbon supports. Sadovnikov et al. investigated nanostructured Ag sulfide using Raman spectroscopy and found a series of vibrations in the range of 90–260 cm−1 caused by Ag–S bonds.38 The Raman spectra of our synthesized Ag/C catalysts also show Raman bands at 90–220 cm−1, which indicates the formation of Ag–S bonds (Fig. S20†). This is further confirmed by the absence of near field Raman bands in the case of the various carbon supports and the commercial Ag/CVulcan catalysts. In addition, the Ketjenblack samples contain more structural disorder and defects, as indicated by the highest D to G band ratios (Fig. S21†). The following trend can be observed when comparing carbon blacks: Ketjenblack has the highest defect concentration (ID/IG ratio of 1.18), followed by Vulcan (ratio of 1.08), and then Super P (ratio of 0.85). This is due in part to Ketjenblack having a higher BET surface area, which can result in a larger number of surface defects. This increase in surface defects can contribute to an increase in the intensity of the D band in the Raman spectra. The ID/IG ratios for Ketjenblack, Vulcan and Super P are consistent with the ratios reported in the literature.39,40 It's interesting to note that Ag supported catalysts demonstrate higher D to G band ratios compared to the respective carbon black. The higher ratio can be attributed to the presence of Ag NPs on the surface of the carbon black, inducing more structural disorder, which in turn increases the intensity of the D band in Raman spectra. Note that a low laser power of 2 mW was used for these measurements due to the fragile nature of the carbon supports.
The ECSA was evaluated using Pb UPD (Fig. S22†). The pristine GDL only showed bulk Pb deposition starting at −0.6 V vs. SCE and more negative potentials, indicating no contribution to the active surface area. However, Pb UPD and Pb monolayer desorption on Ag could be obtained with the Ag/C catalysts. The ECSA was estimated by the Pb-UPD desorption peak.26 The ECSA of the Ag/C catalysts showed a slight trend in the order Ag/CKetjen > Ag/CVulcan > Ag/CSuper P, which can be explained with their increasing average particle size of 4.4 nm, 4.7 nm, and 5.3 nm (Fig. S14–S16†).
Additionally, EDLC measurements were conducted (Fig. S23†). CVs in the non-faradaic region from 0.2–0.3 V vs. SCE were recorded, and the charging current was plotted against the scan rate. The slope of the regression line represents the EDLC value. The EDLC of the pristine GDL was negligible due to its low wettability properties with a water contact angle of 144°, resulting in no significant formation of a double-layer (Fig. S24†). The EDLC values were calculated as 18.7 F g−1 for Ag/CSuper P, 43.9 F g−1 for Ag/CVulcan, and 103.6 F g−1 for Ag/CKetjen. The increase in EDLC is the result of the increasing carbon surface area and increased wettability from Ag/CSuper P (water contact angle of 127°) to Ag/CKetjen (water contact angle of 103°, Fig. S24†). Tashima et al. showed that a highly porous Ketjenblack (BET = 1445 m2 g−1) had the highest EDLC value of 59.2 F g−1 compared to acetylene black with a small BET of 66 m2 g−1.41 Therefore, the significant change in EDLC for the Ag/C catalysts is mainly due to the carbon support and should not be considered a measure for active area of (supported) catalysts. In contrast, Pb-UPD selectively measures the surface of Ag.
The performance of the three carbon supports with Ag catalysts was assessed using a zero-gap electrolyzer. Ag/CKetjen, Ag/CVulcan, and Ag/CSuper P gas diffusion electrodes were assembled with an Aemion+ 50 μm membrane and a NiFe2O4 anode. A PTFE particle dispersion was used as a cathode catalyst binder to minimize the influence of interaction between binder and catalyst particles. The PTFE particles increase the hydrophobicity in the catalyst layer, potentially helping to mitigate electrode flooding.42 Fig. 3a shows the cell voltage dependent on current density for cells containing Ag/CKetjen, Ag/CVulcan, and Ag/CSuper P with no significant trend. For instance, at 100 mA cm−2 cell voltages were between 2.95 V and 3 V for Ag/C catalysts, respectively. When changing the anode catalyst to IrO2 the cell voltage was reduced by 300 mV to 2.65 V with Ag/CSuper P as cathode catalyst due to the higher OER activity of IrO2 compared to NiFe2O4.43,44 Fig. 3b shows the high-frequency resistance (HFR) with a trend in the order of Ag/CSuper P < Ag/CKetjen < Ag/CVulcan, resulting in slightly lower cell voltages of Ag/CSuper P with 3.58 V compared to Ag/CVulcan with 3.65 V at high current density (300 mA cm−2). The cell voltage and resistance measurements were repeated showing negligible deviation with cell voltages being between 2.95 V at and 3 V at 100 mA cm−2 confirming reproducibility (Fig. S25†). Overall, the results showed that the carbon support does not have a significant effect on the performance of the Ag/C catalysts. Furthermore, we conducted cell tests using Super P as the carbon support for the cathode catalyst in combination with an IrO2 anode (Fig. S26†). The cell demonstrated a maximum jCO of 0.84 mA cm−2 when a current density of 100 mA cm−2 was applied at 3.78 V. These results suggest that the carbon support does not exhibit any significant catalytic activity for the reduction of CO2 to CO. Therefore, the CO mass activity can be predominantly attributed to the presence of Ag on the Ag/C catalysts.
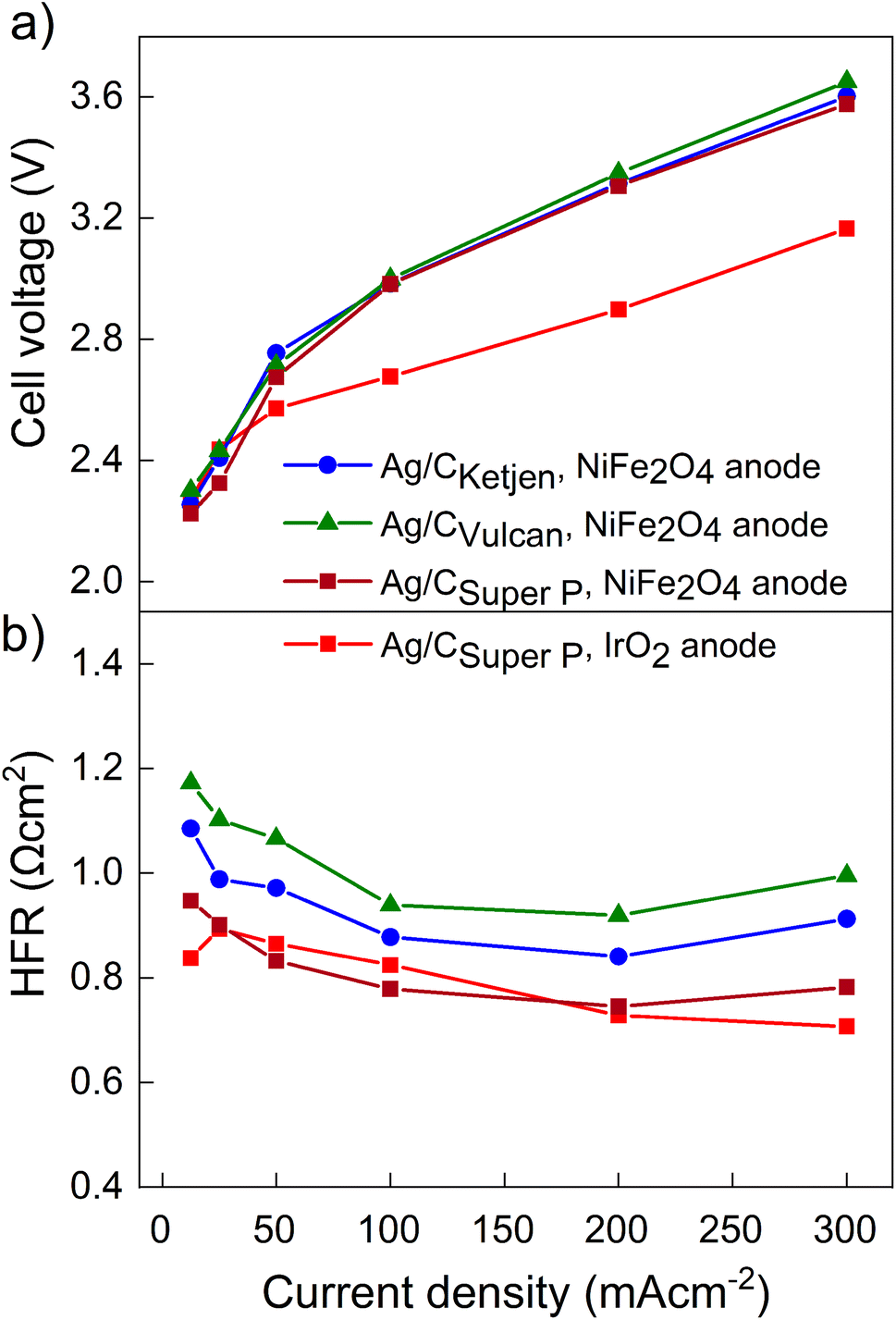 | ||
| Fig. 3 (a) Cell voltages and (b) high frequency resistances of Ag/CKetjen, Ag/CVulcan and Ag/CSuper P catalysts in 0.1 M KOH at ambient temperature. 25% PTFE is used as cathode catalyst binder and NiFe2O4 as anode catalyst. Only for second Super P sample IrO2 was used as anode catalyst. | ||
In AEM zero-gap CO2 electrolysis, cross-over of CO2 gas from the cathode to the anode is always present, altering the pH of the anolyte. This reduces the overall performance of the cell. Janáky et al. found that to conduct long-term studies, the anolyte must either be frequently replenished to maintain a high alkaline pH or an anode catalyst that can withstand OER at close to neutral pH must be used.45 However, in our study the pH change during the measurements was small due to the short measurement duration.
Fig. 4a shows the iR-corrected cell voltage, i.e. without ohmic contributions from e.g. the membrane, over CO partial current density. Similar partial current densities jCO of 178 mA cm−2 at 3.3 V were obtained for all Ag/C catalysts with NiFe2O4. The faradaic efficiency FECO for the Ag/C catalysts remained above 80% up to 200 mA cm−2, while it drops to 59% at 300 mA cm−2 (Fig. S27†). The cells reproduced using Ag/C catalysts maintained FECO above 80% up to 200 mA cm−2. Again these results indicate that the type of carbon support on the Ag/C catalyst does not have a significant effect on the electroreduction of CO2 to CO in our system. This is likely because the Ag/C catalysts have similar particle size distributions with average diameters of around 5 nm. It further indicates that the electrode binder (PTFE) does not poison or block the active catalyst particles. This is an indicator that the peak performance is not limited by kinetics from the catalyst but by other mass transport limitations that affect all supported catalysts similarly. Cell performance with IrO2 on the anode side, however, lead to a maximum jCO of 196 mA cm−2 at 2.95 V with FECO remaining over 90% up to 200 mA cm−2. In contrast, with commercial Ag/CVulcan catalysts we were not able to produce current densities greater than 30 mA cm−2 (Fig. S28†). This poor performance is likely due to the large, unbound Ag nanoparticles with low surface-to-volume ratios (Fig. S29 and S30†).
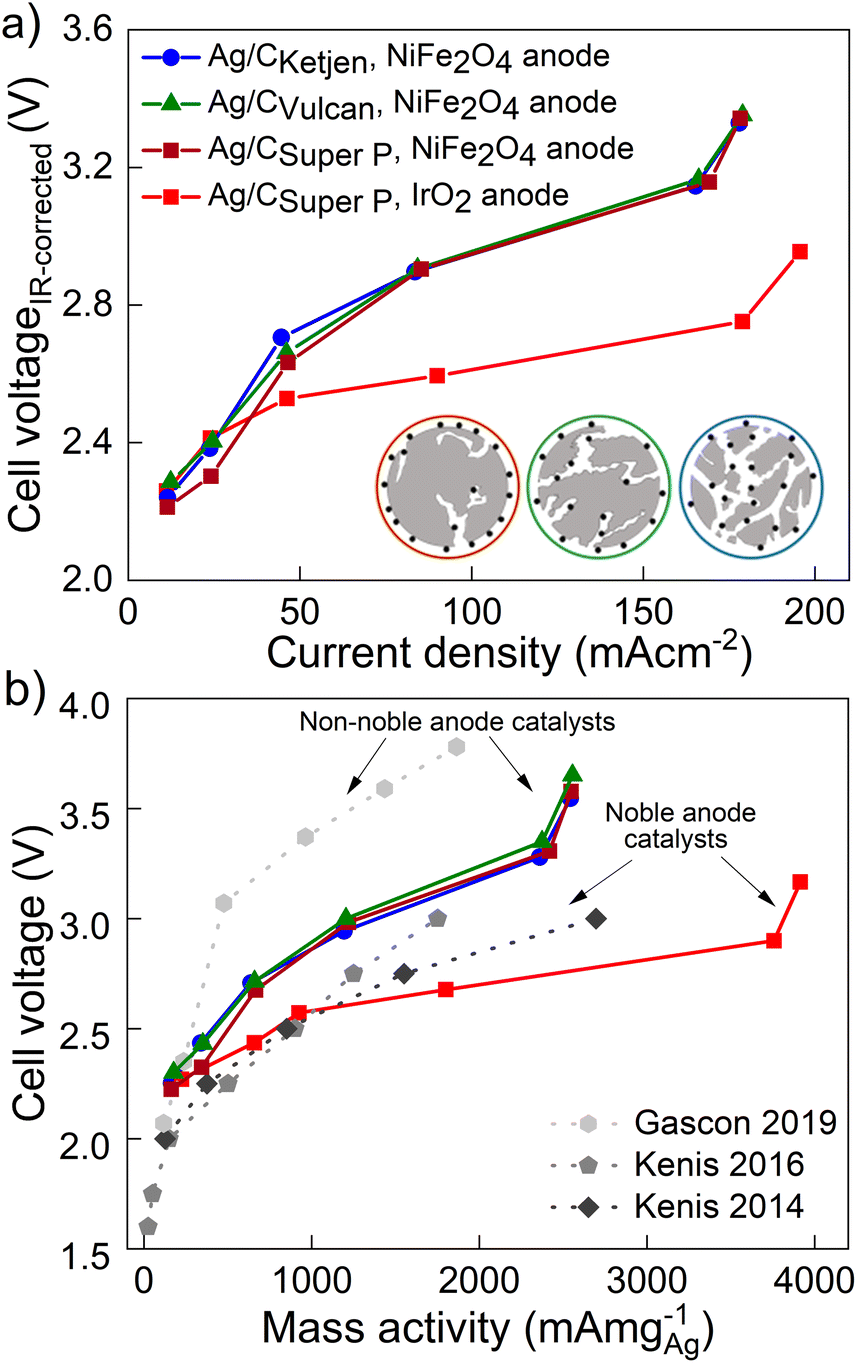 | ||
| Fig. 4 (a) iR-Corrected cell voltage over CO partial current density for Ag/CKetjen, Ag/CVulcan and Ag/CSuper P catalysts in 0.1 M KOH at ambient temperature. (b) CO partial current density relative to Ag loading of selected supported Ag catalysts achieving high mass activities.46–48 | ||
The Ag loading on the cathode GDEs with the synthesized Ag/C catalysts was determined to be 0.07 mgAg cm−2 by XRF (Fig. S2†), except for the Ag/CSuper P GDE with IrO2 on the anode side (0.05 mgAg cm−2).
The performances of the various catalysts normalized by the mass of Ag are compared in Fig. 4b. The Ag/C catalysts presented here demonstrate high mass activities amongst reported literature for carbon supported Ag catalysts (see Table 1): 2543 mA mg−1 for Ag/CKetjen, 2556 mA mg−1 for Ag/CVulcan and 2547 mA mg−1 for Ag/CSuper P with NiFe2O4 anodes. Gascon and coworkers achieved a high mass activity of 1864 mA mg−1 at 3.78 V with porous carbon supported Ag (Ag/MPL).46 This high cell voltage is comparable to our cell voltages (3.54–3.65 V) of the peak mass activity and probably characteristic for non-noble Ni anode catalysts. In contrast, Kenis and coworkers achieved remarkable mass activities up to 2696 mA mg−1 at 3 V with supported Ag catalysts and noble anode catalysts.47–49 For comparison, our Ag/CSuper P GDE with noble IrO2 catalyst exhibited an outstanding mass activity of 3920 mA cm−2 with 0.1 M KOH at 3.16 V surpassing the reported literature data on supported Ag and Au catalysts as summarized in Table 1.
| Supported Ag/Au catalyst | Metal loading (mg cm−2) | Metal NP size (nm) | Anode catalyst | Electrolyte | jCO, max (mA cm−2) | Cell voltage at jCO, max (V) | Mass activity (mA mg−1) | Reference |
|---|---|---|---|---|---|---|---|---|
| Ag/CKetjen | 0.05 | 4.4 ± 1.3 | IrO2 | 0.1 M CsOH | 270 | 3.34 | 5400 | This work |
| Ag/CSuper P | 0.05 | 5.3 ± 1.5 | IrO2 | 0.1 M CsOH | 261 | 3.38 | 5220 | This work |
| Ag/CSuper P | 0.05 | 5.3 ± 1.5 | IrO2 | 0.1 M KOH | 196 | 3.16 | 3920 | This work |
| Ag/CKetjen | 0.07 | 4.4 ± 1.3 | NiFe2O4 | 0.1 M KOH | 177.9 | 3.55 | 2543 | This work |
| Ag/CVulcan | 0.07 | 4.7 ± 1.5 | NiFe2O4 | 0.1 M KOH | 178.9 | 3.65 | 2556 | This work |
| Ag/CSuper P | 0.07 | 5.3 ± 1.5 | NiFe2O4 | 0.1 M KOH | 178.3 | 3.58 | 2547 | This work |
| 5 wt% Ag/TiO2 | 0.017 | 70 | Pt | 1 M KOH | 45.6 | 3 | 2696 | Kenis 2014 (ref. 47) |
| Ag/MPL-3C | 0.20 | 25–30 | Ni mesh | 0.1 M KHCO3 | 385 | 3.8 | 1864 | Gascon 2019 (ref. 46) |
| AgNP/MWCNT | 0.20 | — | IrO2 | 1 M KOH | 350 | 3 | 1750 | Kenis 2016 (ref. 48) |
| Au25(SR)18/CB | 0.00096 | 1.4 ± 0.4 | Pt | 0.1 M KHCO3 | 1.59 ± 0.16 | 2.73 | 1656 ± 163 | Jin 2015 (ref. 59) |
| Au24NHC/Carbon | 0.066 | — | IrO2 | 0.1 M KHCO3 | 90 | 3.2 | 1360 | Crudden 2022 (ref. 60) |
| Au/C | 0.4 | 11 | IrO2 | H2O | 500 | 3 | 1250 | Zhuang 2019 (ref. 61) |
| Au/CN | 0.2 | 2–3 | RuIrOx | 2 M KOH | 180 | 3 | 900 | Wu 2022 (ref. 62) |
Long-term tests with Ag/CSuper P and Ag/CKetjen were conducted as shown in Fig. 5. In the first long-term measurement with Ag/CSuper P, 0.05 mgAg cm−2 in 0.1 M KOH at 100 mA cm−2 for 11 h (shown in Fig. 5a), the FECO remained above 80% for 2 hours, but then dropped to 60%. This decrease in FECO was likely due to the accumulation of potassium carbonates on the cathode side, which can lower the kinetics towards CO2 to CO reduction and result in reduced current densities.45 To address this issue, regular cathode flushs with 50 μL 0.1 M CsOH were used to remove any accumulated carbonates on the surface of the cathode, which successfully restored FECO to above 80%.50
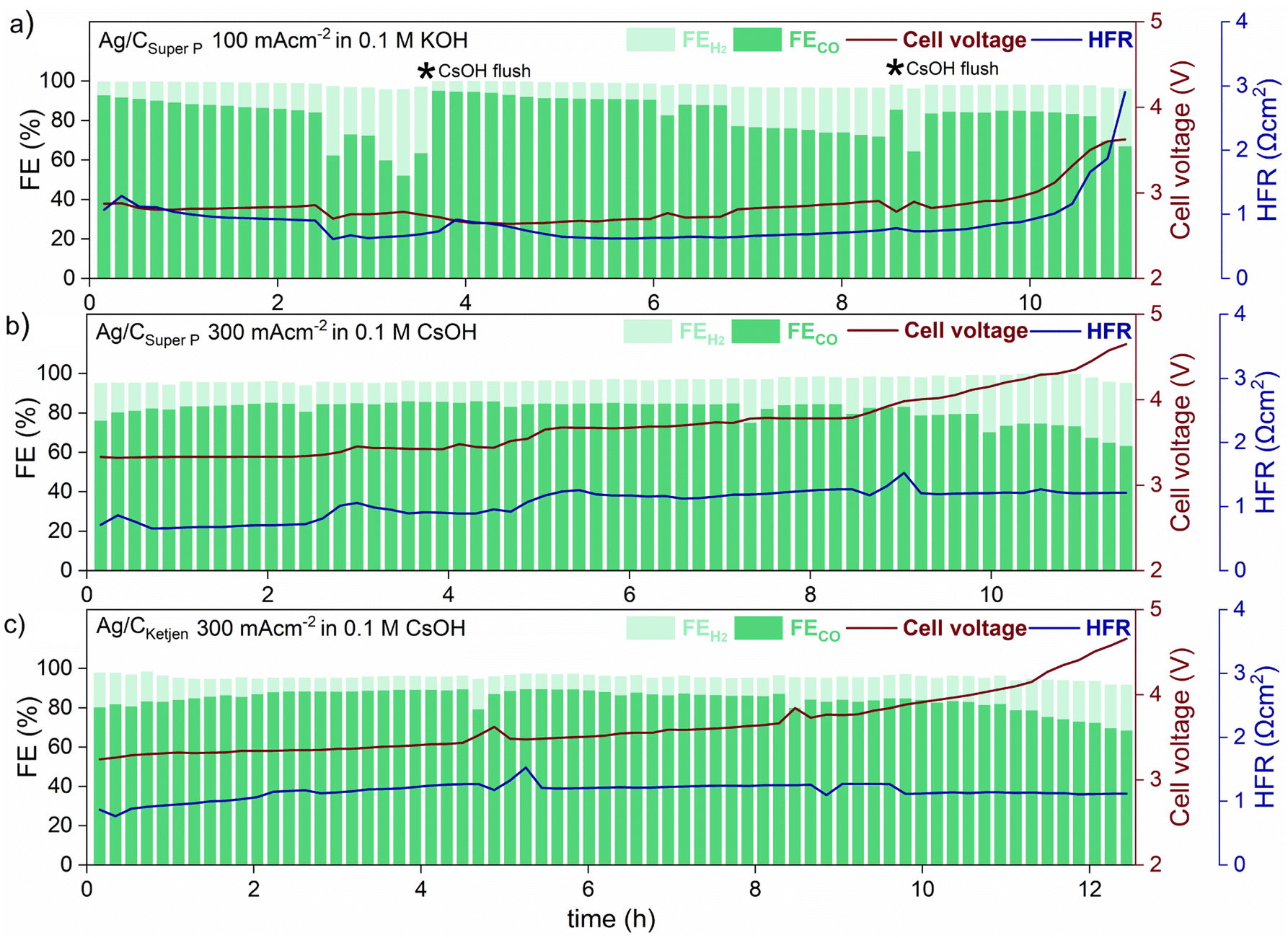 | ||
| Fig. 5 Long-term operation at ambient temperature of (a) Ag/CSuper P with a 0.05 mgAg cm−2 at 100 mA cm−2 in 0.1 M KOH, (b) Ag/CSuper P and (c) Ag/CKetjen with 0.05 mgAg cm−2 at 300 mA cm−2 in 0.1 M CsOH. A membrane coated with a IrO2 catalyst layer for the anode side was used for all Ag/C catalysts. The depicted * in (a) represents cathode flush by 50 μL 0.1 M CsOH to remove accumulated carbonates on the surface of the cathode. | ||
The formation of salt precipitation can be better controlled by changing the cation of the anolyte: Cofell et al. observed an increase in the performance caused by lower carbonate salt precipitation could be obtained by changing the electrolyte from KOH to CsOH.51 This effect can be ascribed to the larger Cs+ cations having a smaller hydration shell compared to K+ resulting in being more concentrated on the electrode surface. This is believed to stabilize the CO2 reduction intermediate species, contributing to improved kinetics.52,53 Moreover, cesium hydrogen carbonates are more soluble in water than potassium carbonates (CsHCO3 with 13.16 molkg−1 vs. KHCO3 with 3.64 molkg−1), indicating a reduced tendency for precipitation in Cs-based electrolytes.51,54 To make use of this, Ag/CSuper P and Ag/CKetjen with 0.05 mgAg cm−2 were tested in 0.1 M CsOH (Fig. 5b and c). As can be observed, switching the electrolyte from KOH to CsOH yields favorable long-term operation at 300 mA cm−2. The cell performance show that the FECO of the cell remained above 80% for a period of 9 h when using Ag/CSuper P, and over 11 h when using Ag/CKetjen. These findings were consistent with similar overall cell voltages and HFR values.
Both cells demonstrated gradual increase in cell voltage over time. This increase in cell voltage for Ag/CSuper P and Ag/CKetjen was found to be likely due to a change in pH value during electrolysis (Fig. S31†). It was observed that the pH values of all Ag/C catalysts undergo a shift from an initial range of ∼13–14 to a final range of ∼8–9. Erikson et al. reported a fast decrease in pH from 14 to 8 with AEMs, which resulted in a 1.2 V increase in cell voltage.55 The observed increase in cell voltage for Ag/C catalysts was also in a similar range, with Ag/CKetjen showing a 1.33 V increase (from 3.24 V to 4.57 V after 12.5 h) and Ag/CSuper P showing a 1.25 V increase (from 3.32 V to 4.57 V after 11.5 h). The pH value of the electrolyte plays a crucial role in OER kinetics, which impacts the overall cell voltage. After long-term tests, it was observed that Ni-felts degraded, since they are known to be highly unstable in a neutral medium causing the increase in cell voltage over time (Fig. S32†).45
After having clarified that the voltage increase does not stem from nanoparticle degradation, we conducted post-test TEM imaging of the Ag/C GDEs after long-term operation to show that the majority of the Ag NPs retain their size (Fig. S33†). Hence, in the presented measurements, no significant catalyst degradation was observed, as the performance degradation was superimposed by other degradation mechanisms.
This is in line with the fact that thiol-based linkers are widely used due to strong covalent bond with metal centers to prevent the degradation and aggregation of Ag NPs.56,57 As a result, the surface of the Ag NPs can be effectively passivated, which reduces the possibility of degradation and aggregation. Adding to this, citrate capped NPs can exhibit irreversible aggregation behavior due to the weak binding to the metal surface.57,58 These findings suggest thiol-based linkers as promising candidates for achieving long-term durability.
4. Conclusion
In this study, we synthesized Ag catalysts supported on three carbon blacks, Ketjenblack, Vulcan and Super P carbon, for electrochemical CO2 reduction to CO. The synthesized Ag nanoparticles were all evenly dispersed on the three carbon supports with a loading of ∼30 wt% Ag on carbon. The obtained average diameter of about 5 nm is believed to be the ideal particle size towards electrochemical CO2 reduction to CO. A slight trend for the surface area of the carbon supports was observed with increasing surface area leading to smaller Ag particles. As a consequence the electrochemical active surface area as determined by Pb under potential deposition is also higher for the catalyst with higher surface area carbons. TEM tomography revealed that the mesoporous structure of the carbon support has a major effect on the final position of the deposited particles, as known from carbon supported Pt catalysts. Ag particles were found to be more outside for the low surface carbon, while the majority was also found inside for the mesoporous high surface carbon Ketjenblack.However, in electrochemical characterization in a gas diffusion electrode of a zero-gap electrolyzer cell, all three carbon supported Ag catalysts showed similar performance. This is a strong indicator that the peak performance and the mass activity are not limited by the catalyst kinetics but by mass transport limitations similarly affecting all three supported catalysts. This further means that the catalyst support surface area does not play a role in the investigated systems, which used PTFE as an electrode binder. This is in strong contrast to fuel cell electrodes, where the surface area of the carbon support and the resulting uptake of ion-conductive binder has a strong effect on performance and durability.33 With this, one could assume that when using an anion conductive electrode binder, which could be beneficial especially with increasing water consumption at high current densities (e.g., > 200 mA cm−2), the surface area of the carbon could play an important role.
Compared to literature the here synthesized catalysts show outstanding mass activities for AEM-based CO2 reduction under real operating conditions, with a maximum Ag mass activity up to 3920 mA mg−1 at 3.2 V in 0.1 M KOH and 5400 mA mg−1 at 3.3 V with 0.1 M CsOH, outperforming reported Ag and Au supported catalysts. The results of our study indicate that the catalyst is not a limiting factor in cell measurements. Specifically, we observed stable FECO greater than 80% for over 11 h at 300 mA cm−2 in 0.1 M CsOH, with no significant degradation of Ag/C catalysts. These findings suggest thiol-based linkers as promising candidates for achieving long-term durability. To further increase the activity, the wettability and transport properties of the electrode need to be controlled via tailoring hydrophobicity and particle sizes of the additive (binder, ionomer, linker and catalyst support). Furthermore, increasing the Ag/C loading or the Ag loading on carbon may result in higher current densities. Such optimization will provide further insight into the distinction between the three Ag/C catalysts.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge funding from the Vector Foundation (Project “CO2-to-X”) and would like to thank Ralf and Yi Thomann for their help with electron microscopy and Ines Ketterer for TGA measurements.References
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87 CrossRef CAS PubMed.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451 CrossRef CAS.
- Q. Lu and F. Jiao, Nano Energy, 2016, 29, 439 CrossRef CAS.
- S. Liang, N. Altaf, L. Huang, Y. Gao and Q. Wang, J. CO2 Util., 2020, 35, 90 CrossRef CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631 RSC.
- S. Park, D. T. Wijaya, J. Na and C. W. Lee, Catalysts, 2021, 11, 253 CrossRef CAS.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571 CAS.
- C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Modern Aspects of Electrochemistry, Springer New York, New York, NY 2008 Search PubMed.
- K. C. Poon, W. Y. Wan, H. Su and H. Sato, RSC advances, 2022, 12, 22703 RSC.
- H.-K. Lim, H. Shin, W. A. Goddard, Y. J. Hwang, B. K. Min and H. Kim, J. Am. Chem. Soc., 2014, 136, 11355 CrossRef CAS PubMed.
- C. Kim, H. S. Jeon, T. Eom, M. S. Jee, H. Kim, C. M. Friend, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2015, 137, 13844 CrossRef CAS PubMed.
- X. Deng, D. Alfonso, T.-D. Nguyen-Phan and D. R. Kauffman, ACS Catal., 2022, 34, 5921 CrossRef.
- S. Liu, C. Sun, J. Xiao and J.-L. Luo, ACS Catal., 2020, 10, 3158 CrossRef CAS.
- A. Gawel, T. Jaster, D. Siegmund, J. Holzmann, H. Lohmann, E. Klemm and U.-P. Apfel, iScience, 2022, 25, 104011 CrossRef CAS PubMed.
- S. Malkhandi and B. S. Yeo, Curr. Opin. Chem. Eng., 2019, 26, 112 CrossRef.
- J. Disch, L. Bohn, S. Koch, M. Schulz, Y. Han, A. Tengattini, L. Helfen, M. Breitwieser and S. Vierrath, Nat. Commun., 2022, 13, 6099 CrossRef CAS PubMed.
- J. J. Carroll, J. D. Slupsky and A. E. Mather, J. Phys. Chem. Ref. Data, 1991, 20, 1201 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C.-T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, e1807166 CrossRef PubMed.
- R. Küngas, J. Electrochem. Soc., 2020, 167, 44508 CrossRef.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS PubMed.
- Y.-M. Zhu, Int. J. Biomed. Imaging, 2018, 5, 44005 Search PubMed.
- I. Arganda-Carreras, V. Kaynig, C. Rueden, K. W. Eliceiri, J. Schindelin, A. Cardona and H. Sebastian Seung, Bioinformatics, 2017, 33, 2424 CrossRef CAS PubMed.
- B. D. A. Levin, Y. Jiang, E. Padgett, S. Waldon, C. Quammen, C. Harris, U. Ayachit, M. Hanwell, P. Ercius, D. A. Muller and R. Hovden, Microsc. Today, 2018, 26, 12 CrossRef.
- M. van Truong, M.-K. Yang and H. Yang, Int. J. Precis. Eng. Manuf. - Green Technol., 2019, 6, 711 CrossRef.
- Y.-C. Hsieh, L. E. Betancourt, S. D. Senanayake, E. Hu, Y. Zhang, W. Xu and D. E. Polyansky, ACS Appl. Energy Mater., 2019, 2, 102 CrossRef CAS.
- B. A. W. Mowbray, D. J. Dvorak, N. Taherimakhsousi and C. P. Berlinguette, Energy Fuels, 2021, 35, 19178 CrossRef CAS.
- V. Palomares, A. Goñi, I. G. d. Muro, I. de Meatza, M. Bengoechea, I. Cantero and T. Rojo, J. Power Sources, 2010, 195, 7661 CrossRef CAS.
- B. T. Sneed, D. A. Cullen, K. S. Reeves, O. E. Dyck, D. A. Langlois, R. Mukundan, R. L. Borup and K. L. More, ACS Appl. Mater. Interfaces, 2017, 9, 29839 CrossRef CAS PubMed.
- M. Bevilacqua, C. Bianchini, A. Marchionni, J. Filippi, A. Lavacchi, H. Miller, W. Oberhauser, F. Vizza, G. Granozzi, L. Artiglia, S. P. Annen, F. Krumeich and H. Grützmacher, Energy Environ. Sci., 2012, 5, 8608 RSC.
- J. Speder, A. Zana, I. Spanos, J. J. K. Kirkensgaard, K. Mortensen and M. Arenz, Electrochem. Commun., 2013, 34, 153 CrossRef CAS.
- L.-L. Zhang, Z.-L. Wang, D. Xu, X.-B. Zhang and L.-M. Wang, Int. J. Smart Nano Mater., 2013, 4, 27 CrossRef CAS.
- V. Yarlagadda, M. K. Carpenter, T. E. Moylan, R. S. Kukreja, R. Koestner, W. Gu, L. Thompson and A. Kongkanand, ACS Energy Lett., 2018, 3, 618 CrossRef CAS.
- E. Padgett, N. Andrejevic, Z. Liu, A. Kongkanand, W. Gu, K. Moriyama, Y. Jiang, S. Kumaraguru, T. E. Moylan, R. Kukreja and D. A. Muller, J. Electrochem. Soc., 2018, 165, F173–F180 CrossRef CAS.
- E. Padgett, N. Andrejevic, Z. Liu, A. Kongkanand, W. Gu, K. Moriyama, Y. Jiang, S. Kumaraguru, T. E. Moylan and R. Kukreja, J. Electrochem. Soc., 2018, 165, F173 CrossRef CAS.
- G. S. Harzer, A. Orfanidi, H. El-Sayed, P. Madkikar and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F770–F779 CrossRef CAS.
- G. Ding, Y. Liu, R. Zhang and H. L. Xin, Sci. Rep., 2019, 9, 12803 CrossRef PubMed.
- S. I. Sadovnikov, E. G. Vovkotrub and A. A. Rempel, Dokl. Phys. Chem., 2018, 480, 81 CrossRef CAS.
- A. Zana, J. Speder, N. E. A. Reeler, T. Vosch and M. Arenz, Electrochim. Acta, 2013, 114, 455 CrossRef CAS.
- Y. Tzeng, C.-Y. Jhan, Y.-C. Wu, G.-Y. Chen, K.-M. Chiu and S. Y.-E. Guu, Nanomaterials, 2022, 12 Search PubMed.
- D. Tashima, K. Kurosawatsu, M. Taniguchi, M. Uota and M. Otsubo, Electr. Eng. Jpn., 2008, 165, 1 CrossRef.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748 CrossRef CAS PubMed.
- Á. Vass, A. Kormányos, Z. Kószó, B. Endrődi and C. Janáky, ACS Catal., 2022, 12, 1037 CrossRef PubMed.
- R. Wang, H. Haspel, A. Pustovarenko, A. Dikhtiarenko, A. Russkikh, G. Shterk, D. Osadchii, S. Ould-Chikh, M. Ma, W. A. Smith, K. Takanabe, F. Kapteijn and J. Gascon, ACS Energy Lett., 2019, 4, 2024 CrossRef CAS.
- S. Ma, Y. Lan, G. M. J. Perez, S. Moniri and P. J. A. Kenis, ChemSusChem, 2014, 7, 866 CrossRef CAS PubMed.
- S. Ma, R. Luo, J. I. Gold, A. Z. Yu, B. Kim and P. J. A. Kenis, J. Mater. Chem. A, 2016, 4, 8573 RSC.
- C. E. Tornow, M. R. Thorson, S. Ma, A. A. Gewirth and P. J. A. Kenis, J. Am. Chem. Soc., 2012, 134, 19520 CrossRef CAS PubMed.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Accounts of chemical research, 2022, 55, 1900 CrossRef CAS PubMed.
- E. R. Cofell, U. O. Nwabara, S. S. Bhargava, D. E. Henckel and P. J. A. Kenis, ACS Appl. Mater. Interfaces, 2021, 13, 15132 CrossRef CAS PubMed.
- M. R. Thorson, K. I. Siil and P. J. A. Kenis, J. Electrochem. Soc., 2012, 160, F69–F74 CrossRef.
- S. S. Bhargava, F. Proietto, D. Azmoodeh, E. R. Cofell, D. A. Henckel, S. Verma, C. J. Brooks, A. A. Gewirth and P. J. A. Kenis, ChemElectroChem, 2020, 7, 2001 CrossRef CAS.
- D. Pabsch, P. Figiel, G. Sadowski and C. Held, J. Chem. Eng. Data, 2022, 67, 2706 CrossRef CAS.
- B. Eriksson, T. Asset, F. Spanu, F. Lecoeur, M. Dupont, F. A. Garcés-Pineda, J. R. Galán-Mascarós, S. Cavaliere, J. Rozière and F. Jaouen, J. Electrochem. Soc., 2022, 169, 34508 CrossRef CAS.
- G. Pacchioni, Nat. Rev. Mater., 2019, 4, 226 CrossRef.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819 CrossRef CAS PubMed.
- L. Jiang, J. Guan, L. Zhao, J. Li and W. Yang, Colloids Surf., A, 2009, 346, 216 CrossRef CAS.
- D. R. Kauffman, J. Thakkar, R. Siva, C. Matranga, P. R. Ohodnicki, C. Zeng and R. Jin, ACS Appl. Mater. Interfaces, 2015, 7, 15626 CrossRef CAS PubMed.
- V. K. Kulkarni, B. N. Khiarak, S. Takano, S. Malola, E. L. Albright, T. I. Levchenko, M. D. Aloisio, C.-T. Dinh, T. Tsukuda, H. Häkkinen and C. M. Crudden, J. Am. Chem. Soc., 2022, 144, 9000 CrossRef CAS PubMed.
- Z. Yin, H. Peng, X. Wei, H. Zhou, J. Gong, M. Huai, L. Xiao, G. Wang, J. Lu and L. Zhuang, Energy Environ. Sci., 2019, 12, 2455 RSC.
- Y. Gu, J. Wei, L. Wang and X. Wu, J. Electrochem. Soc., 2022, 169, 54513 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03424k |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |