
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA method for the synthesis of unsymmetric bisphosphoric analogs of α-amino acids†
Anna Kuźnik *ab,
Dominika Kozickaab,
Agnieszka Październiok-Holewaab,
Alicja Dąbeka,
Karolina Juszczaka,
Gloria Sokołowskaa and
Karol Erfurtc
*ab,
Dominika Kozickaab,
Agnieszka Październiok-Holewaab,
Alicja Dąbeka,
Karolina Juszczaka,
Gloria Sokołowskaa and
Karol Erfurtc
aDepartment of Organic Chemistry, Bioorganic Chemistry and Biotechnology, Silesian University of Technology, B. Krzywoustego 4, 44-100 Gliwice, Poland. E-mail: anna.kuznik@polsl.pl
bBiotechnology Center of Silesian University of Technology, B. Krzywoustego 8, 44-100 Gliwice, Poland
cDepartment of Chemical Organic Technology and Petrochemistry, Silesian University of Technology, B. Krzywoustego 4, 44-100 Gliwice, Poland
First published on 23rd June 2023
Abstract
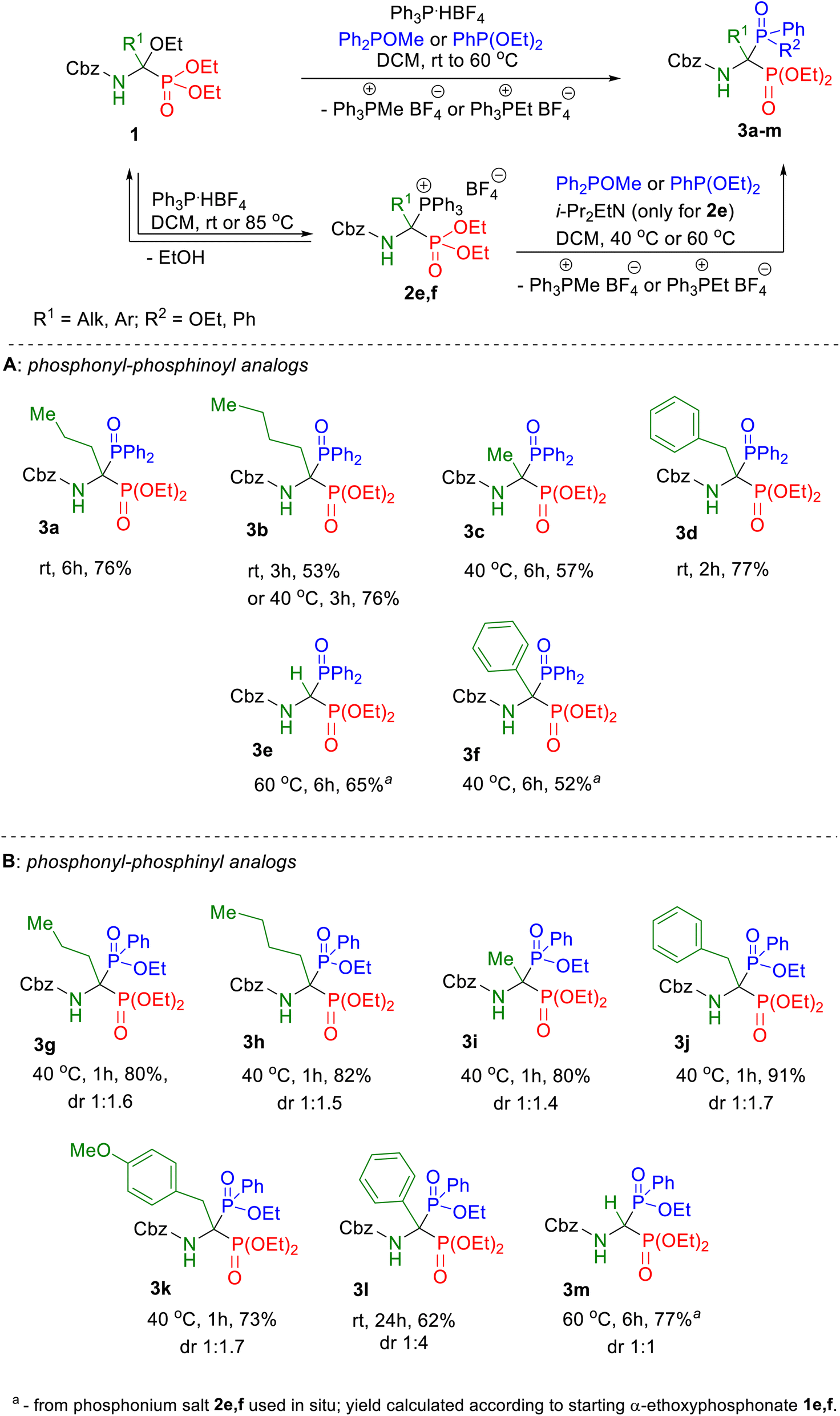
Herein, we describe the first universal strategy for the synthesis of unsymmetric phosphonyl–phosphinyl and phosphonyl–phosphinoyl analogs of N-protected 1-aminobisphosphonates. The proposed user-friendly procedure, based on a one-pot reaction of the α-ethoxy derivatives of phosphorus analogs of protein and non-protein α-amino acids with triphenylphosphonium tetrafluoroborate and an appropriate phosphorus nucleophile (diethyl phenylphosphonite or methyl diphenylphosphinite), provides good to very good yields of 53–91% under mild catalyst-free conditions (temperature: rt to 40 °C, time: 1 to 6 hours). The progress of the transformation, running through the corresponding phosphonium salt as a reactive intermediate, was monitored by 31P NMR spectroscopy, which is a convenient tool for the identification of the transient species formed here. In this paper, we present the full characteristics of the spectroscopic properties of all 13 synthesized models of structurally diverse N-protected unsymmetric bisphosphoric analogs of α-amino acids. Therefore, these results contribute to increasing the practical applicability of our recently reported synthesis protocol of symmetric models of α-aminobisphosphonates derivatives and thus justify its universality.
Introduction
1,1-Bisphosphoric derivatives, due to their confirmed biological activity, not only enjoy unwavering interest, but even gain more and more importance because of the huge potential of compounds with this type of structure. The essential feature that guarantees the biological activity of 1,1-bisphosphoric derivatives is the presence of the P–C–P skeleton, which is resistant to enzymatic hydrolysis and additionally provides them with an affinity for hydroxyapatite (HA) present in the bone matrix due to the ability to chelate metals such as magnesium and calcium by phosphonyl groups.1 One of the subclasses of bisphosphoric compounds are their derivatives that contain an amino group on the bridge carbon atom and thus are characterized by the presence of the P–C(N)–P moiety, of which the most known group are geminal 1-amino-1,1-bisphosphonates.2 These compounds can be considered phosphorus analogs of α-amino acids, functionalized with an additional phosphonyl group. The biological activity of 1,1-bisphosphonates is well known since they are an important class of drugs currently used to treat osteoporosis and other diseases such as Paget's disease, hypercalcemia, or bone metastases.3,4 The wide range of their potential applications is also associated with other properties including antiviral,5 antibacterial,6 antiparasitic,7,8 and herbicidal.9 Furthermore, they have also a significant potential to be used in targeted anticancer therapies as drug delivery systems to bone tissue10 or in the synthesis of new diagnostic agents for bone tissue imaging by MRI or PET.11 Another group of 1-amino-1,1-bisphosphoric compounds, namely their unsymmetric derivatives, including phosphonyl–phosphinyl and phosphonyl–phosphinoyl analogs (Fig. 1), may be equally interesting. However, such compounds of the P–C(N)–P skeleton have been much less explored so far; hence, their potential has not yet been fully discovered.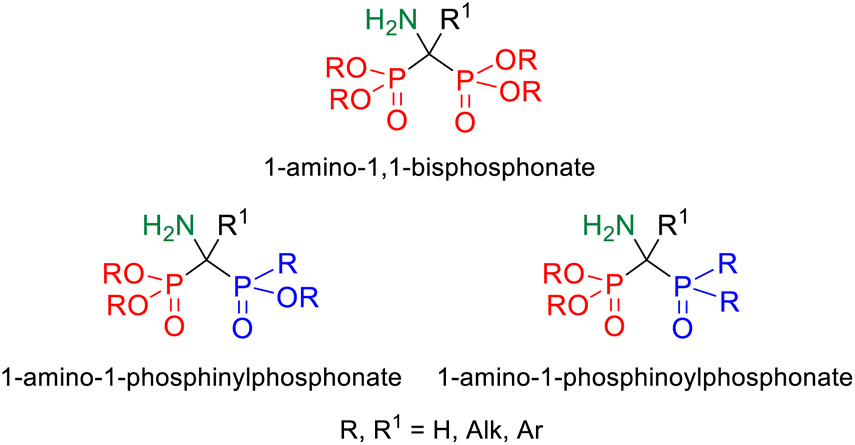 | ||
| Fig. 1 Comparison of the structures of 1-amino-1,1-bisphosphonate and its unsymmetric phosphonyl–phosphinyl and phosphonyl–phosphinoyl analogs. | ||
The only studies found in the literature that indicate the biological potential of these compounds were reported by Ebetino. In vitro studies on HA crystal growth inhibition and in vivo studies on antiresorptive activity of phosphonyl–phosphinyl derivatives of (N-pyridyl)-1-amino-1,1-bisphosphonates have shown that they have both the ability to interact with HA crystals and antiresorptive activity, but to a lesser extent compared to their symmetric bisphosphonate analogs.12 However, this feature may be desirable, especially when the high affinity for bone tissue and the strong chelating effects of symmetric bisphosphonates are the cause of their toxicity. This knowledge can then be used in the design of therapeutics with appropriate antiresorptive potency. The limited number of data on the biological activity of these compounds may be due to their much lower availability, the synthesis of which must be performed stepwise to allow the introduction into the molecule of two different phosphorus groups. This contrasts with the preparation of symmetric derivatives of 1-amino-1,1-bisphosphonates, where in most known synthetic routes both Cα–P bonds are formed simultaneously between a central carbon atom and two identical molecules of the phosphorus nucleophile.13–16 Therefore, the search for not only an effective but also a universal method to obtain this type of unsymmetric bisphosphoric derivatives remains a challenge that attracts the attention of scientists. It should be emphasized that the usefulness of most of the methods reported in the literature for the preparation of phosphonyl–phosphinoyl analogs of 1-aminobisphosphonates has been demonstrated for a limited number of synthesized models, often single (Scheme 1).
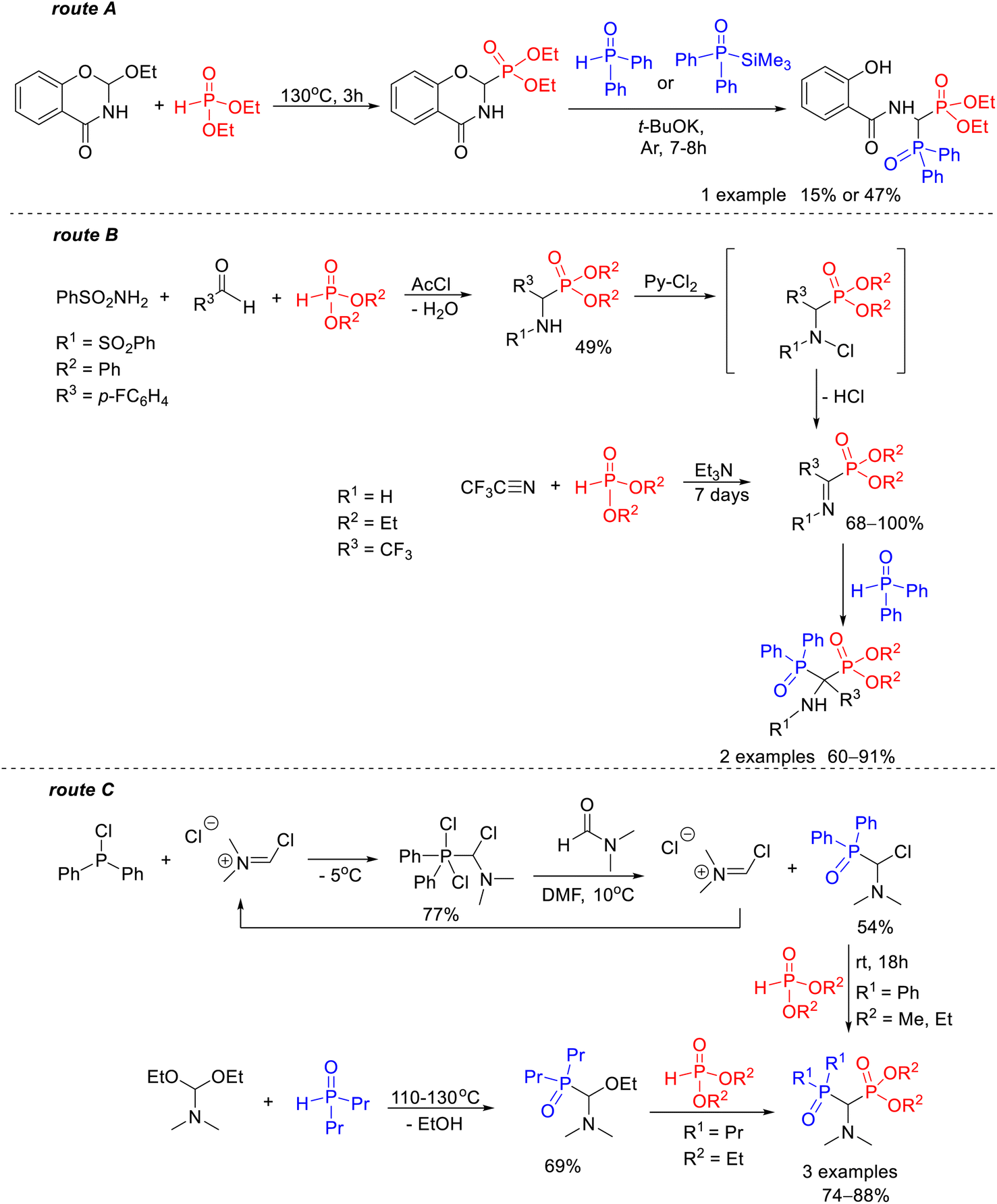 | ||
| Scheme 1 Known routes for the synthesis of 1-amino-1-phosphinoylalkylphosphonate derivatives: reaction of diethyl 2,3-dihydro-4H-1,3-benzoxazin-4-one-2-phosphonate with diphenylphosphine oxide17 or diphenyl(trimethylsilyl)phosphine oxide18 (route A); reaction of diphenylphosphine oxide with activated iminophosphonates (route B);19,20 reaction of dialkyl phosphite with N,N-dialkylamine derivatives of phosphine oxide functionalized at the α-position by chlorine atom21 or ethoxy group22 (route C). | ||
Such an example is the synthesis of diethyl 1-(N-salicyloylamino)-1-diphenylphosphinoylmethylphosphonate described by Kostka and Kotyński, one of the first methods for the preparation of this type of bisphosphoric derivative that was carried out with a yield of 15% or 47% depending on the type of nucleophile used (Scheme 1, route A).17,18 In addition to that, there are only two known routes for the preparation of 1-amino-1-phosphinoylalkylphosphonates. One of them consists of the reaction of diphenylphosphine oxide with iminophosphonate derivatives activated by the presence of trifluoromethyl19 or a p-fluorophenyl group20 at the α-carbon atom provided the desired product in yields of 60–91% (Scheme 1, route B). The other involves the reaction of dialkyl phosphite with N,N-dialkylamine derivatives of phosphine oxide functionalized at the α-position by a nucleofugal group such as chlorine21 or ethoxy22 (Scheme 1, route C).
Similarly, only a few mentions of synthetic methods have been found in the literature for phosphonyl–phosphinyl analogs of 1-amino-1,1-bisphosphonates. One of the first was the two-stage procedure, reported by Schrader and Steglich, in which diethyl 1-(N-2,2,2-trichloroethoxycarbonylamino)-1-(ethoxymethylphosphinyl)methylphosphonate was obtained via the corresponding 1-bromo-1-aminoethoxymethylphosphinic acid derivative (Scheme 2, route A).23 In 1997, Ebetino et al. described the synthesis of diethyl 1-[N-(3-methylpyridin-2-yl)amino]-1-(etoxymethylphosphinyl)methylphosphonate prepared in 32% yield in the reaction of diethyl dietoxymethylphosphonate with 2-amino-3-methylpyridine and ethyl methylphosphinate (Scheme 2, route B).12 In 2003, a four-step method was patented for the synthesis of two models of 1-aminophosphinylmethylphosphonic acids through azide derivatives, however, no yield data was provided (Scheme 2, route C).24
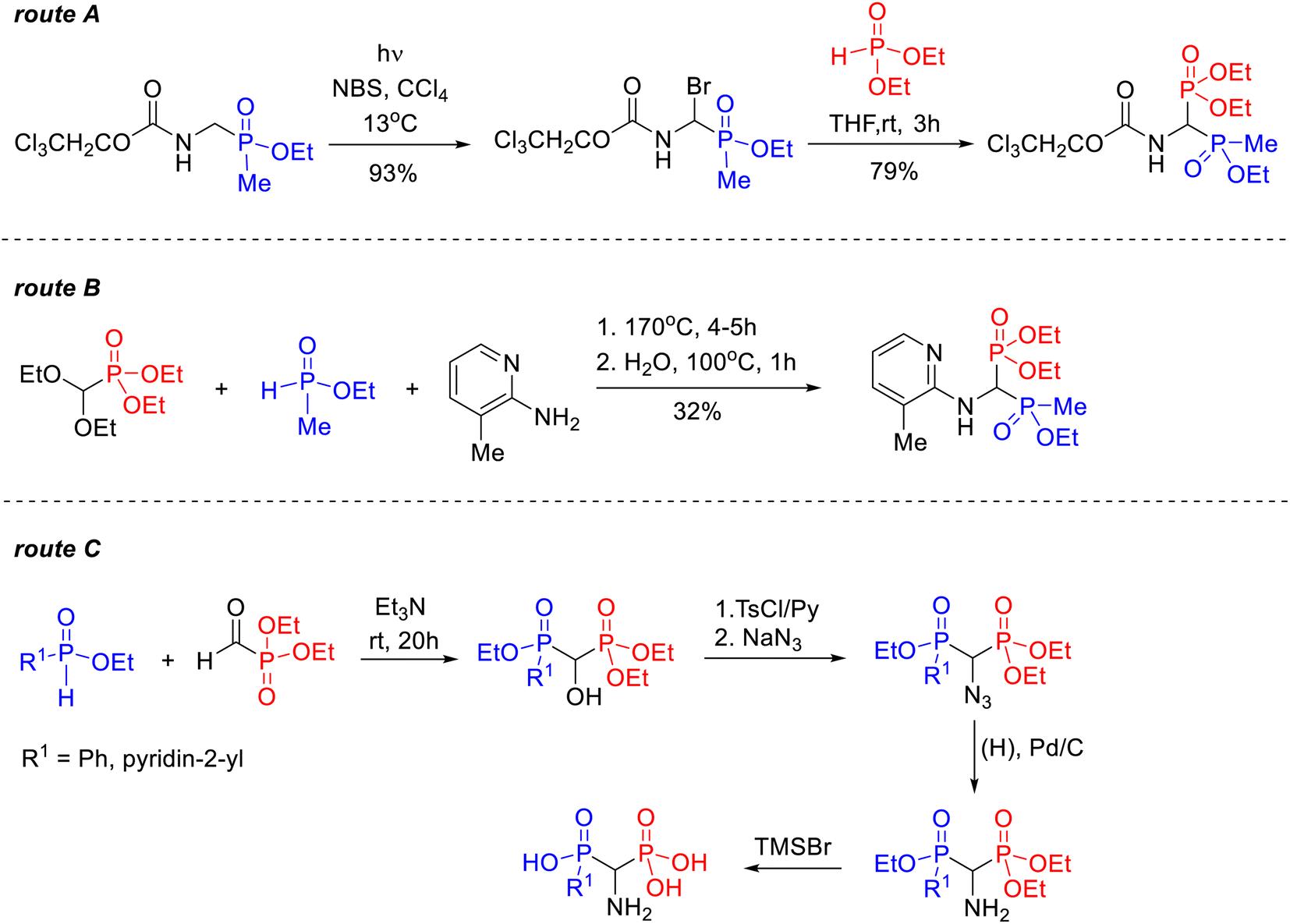 | ||
| Scheme 2 Known routes for the synthesis of 1-amino-1-phosphinylalkylphosphonate derivatives: reaction of 1-bromo-1-aminoethoxymethylphosphinic acid derivative with diethyl phosphonate (route A);23 reaction of diethyl diethoxymethylphosphonate with 2-amino-3-methylpyridine and ethyl methylphosphinate (route B);12 synthesis of 1-aminophosphinylmethylphosphonic acids through reduction of azide derivatives (route C).24 | ||
In recent years, our research group has developed effective procedures for the transformation of α-alkoxyphosphonates into target 1-amino-1,1-bisphosphoric derivatives in the Michaelis–Arbuzov-type reaction.25–28 These routes proceed through 1-(diethoxyphosphoryl)phosphonium salts as reactive intermediates, and they have been tested in the synthesis of both symmetric and unsymmetric bisphosphoric derivatives. The starting α-alkoxy derivatives of the phosphorus analogs of α-amino acids were synthesized by electrochemical oxidation of diethyl 1-(N-acylamino)alkylphosphonates25 or by adding diethyl phosphite to ethyl N-acylimidates.26 Recently, we published a simplified variant of this procedure, performed using the one-pot method in a catalyst-free system, whose usefulness has been tested so far in the synthesis of a number of symmetric 1-amino-1,1-bisphosphonates.27 Since this procedure has great potential to become a general route in the synthesis of 1-amino-1,1-bisphosphoric derivatives, it seemed justified to explore its applicability in the synthesis of unsymmetric phosphonyl–phosphinyl and phosphonyl–phosphinoyl derivatives.
In this paper, we present the synthesis and full spectroscopic characteristics of selected analogs of 1-aminophosphinyl and 1-aminophosphinoylphosphonate derivatives (Scheme 3, pathway C) that can be obtained according to our one-pot strategy.
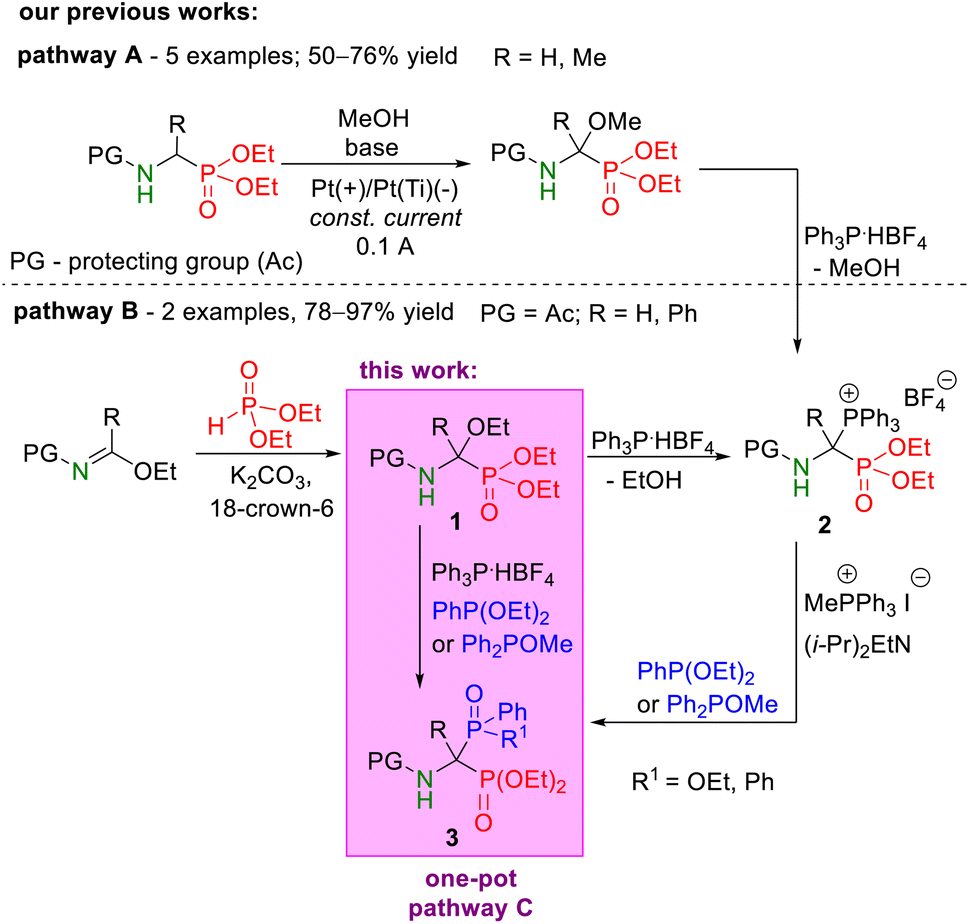 | ||
| Scheme 3 Synthetic pathways for the preparation of phosphonyl–phosphinyl and phosphonyl–phosphinoyl analogs of 1-aminobisphosphonates from α-functionalized derivatives of phosphorus analogs of α-amino acids, such as diethyl 1-(N-acetylamino)-1-triphenylphosphoniumalkylphosphonate tetrafluoroborates 2 obtained from α-methoxyphosphonates (pathway A)25 or α-ethoxyphosphonates (pathway B).26,28 | ||
Results and discussion
By analogy to the recently published synthesis of 1-amino-1,1-bisphosphonates, here also the models of unsymmetric bisphosphonate derivatives, which we defined as the target compounds, possessed at the α position a side chain identical to those characteristic for natural α-amino acids, both protein and non-protein, since we assumed that its type may have an impact on the biomedical profile of the compound. In all synthesized products, the benzyloxycarbonyl group was used as the protection of the amino group because of the ease of its deprotection.The starting diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxyalkylphosphonates 1 were synthesized according to a recently described two-step protocol27 consisting of acylation of imidate hydrochlorides with benzyl chloroformate followed by the Michaelis–Becker-like addition of diethyl phosphite to ethyl N-acylimidates. Our research on the transformation of diethyl 1-(N-acylamino)-1-ethoxyalkylphosphonates into unsymmetric phosphonyl–phosphinoyl derivatives of 1-amino-1,1-bisphosphonates has begun from a series of exploratory experiments with the use of selected models of α-ethoxy derivatives 1 according to the newly developed one-pot procedure, using methyl diphenylphosphinite as nucleophile (Scheme 4). After performing initial optimization experiments for all models of the intended phosphonyl–phosphinoyl analogs of 1-aminobisphosphonates, the syntheses were repeated on a larger scale according to optimized procedures to isolate and characterize the spectroscopic properties of compounds 3 (Scheme 4A). Therefore, for the 1a–d substrate models, a one-pot procedure was successfully applied, and the Michaelis–Arbuzov-type reaction proceeded smoothly in dichloromethane, at room temperature or 40 °C in 2–6 hours (Scheme 4A). It was observed that in the case of product 3b, despite the satisfactory course of its synthesis at room temperature (53% yield), it turned out to be advantageous to increase the reaction temperature to 40 °C, resulting in a significantly higher yield of 76%. However, the synthesis of compound 3d carried out for 1 hour at elevated temperature provided a slightly lower yield of the expected product (64%) compared to that obtained at room temperature (77%). Only, syntheses from the phosphorus analog of glycine 1e and the phosphorus analog of phenylglycine 1f did not lead to the expected products 3e or 3f, even at elevated temperature. It was not surprising at all in the case of the bisphosphorus analog of glycine, because the same result was observed for the reaction of this model with triethyl phosphite, which we wrote in our previous work (see ref. 27). As we found, this is probably due to the lack of a substituent at the α position of the substrate model 1e that stabilizes the iminophosphonate derivative, formed here as an intermediate. For substrate model 1f, the main obstacle in the effective performance of the intended reaction may be the steric hindrance associated with the accumulation of aromatic rings (present both in the substrate structure and in the nucleophile) at the reaction center. Furthermore, according to the plausible mechanism for the formation of the intermediate diethyl 1-(N-acylamino)-1-triphenylphosphoniumalkylphosphonate tetrafluoroborate 2 proposed in our previous article,27 the low conversion of α-ethoxyphosphonate 1 during the one-pot transformation can also be explained by the unfavorable equilibrium state associated with the reversible nature of this reaction, which in the presence of the released ethanol is shifted toward substrate 1 (Scheme 4). To overcome this problem, it was decided to divide the procedure into two steps, consisting of ethanol removal after phosphonium salt preparation and subsequent use of the synthesized salts 2e,f in situ in the Michaelis–Arbuzov-type reaction with methyl diphenylphosphinite. Therefore, phosphonium salts 2e,f were obtained by dissolving the α-ethoxyphosphonate 1e,f and phosphonium tetrafluoroborate in dichloromethane at room temperature, then after 40 minutes, the solvent from the reaction mixture was evaporated and the residue was dried under reduced pressure for 1 or 5 hours at room or elevated temperature (Table 1).
 | ||
| Scheme 4 Scope of the Michaelis–Arbuzov-type reaction in the synthesis of phosphonyl–phosphinoyl and phosphonyl–phosphinyl analogs of 1-aminobisphosphonates. Conditions: substrate 1 (1.0 mmol), Ph3P·HBF4 (1.1 mmol, 1.1 equiv.), Ph2POMe or PhP(OEt)2 (1.5 mmol, 1.5 equiv.), DCM (5.0 mL). Isolated yield. | ||
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substrate model | R1 | Drying temperature of salt 2 [°C] | Drying time [h] | Solvent | Reaction temperature [°C] | Hünig's base | Substrate 1 conversionb [%] |
| a Conditions: substrate 1e,f (0.1 mmol), Ph3P·HBF4 (0.11 mmol, 1.1 equiv.), DCM (0.5 mL), next, after drying crude salt 2e,f, Ph2POMe (0.15 mmol, 1.5 equiv.), i-Pr2EtN (0.03 mmol, 0.3 equiv.), DCM (0.5 mL). +Denotes the addition of Hünig's base as a catalyst. −Denotes that the experiment was carried out without the addition of Hünig's base.b The conversion was estimated from the 31P NMR spectrum after 6 h of reaction. | ||||||||
| 1 | 1e | H | 70 | 1 | DCM | 60 | + | 52 |
| 2 | 1e | H | 70 | 1 | MeCN | 70 | + | 42 |
| 3 | 1e | H | 85 | 5 | DCM | 60 | + | 92 |
| 4 | 1f | Ph | rt | 1 | DCM | 25 | − | 60 |
| 5 | 1f | Ph | rt | 1 | MeCN | 40 | − | 75 |
| 6 | 1f | Ph | rt | 1 | DCM | 40 | − | 89 |

Subsequently, the crude phosphonium salts 2e,f were dissolved in situ in the selected solvent and a phosphorus nucleophile was added, performing the further reaction at room temperature or increased temperature. A catalytic amount of Hünig's base was additionally introduced into the reaction mixture in the case of the phosphonium salt model 2e. To monitor the progress of the reaction, samples were taken from the reaction system for spectroscopic analysis, usually after 3, 6 and 24 hours of reaction (see Table 1).
This modification of the one-pot procedure brought surprising results. In the case of model 3e, slight progress of the reaction course was observed (Table 1, entries 1–3), with the 31P NMR spectra showing the intense signal of substrate 1e, still dominant in the reaction mixture in relation to the low signals of the expected product 3e. As for model 3f, a significant improvement was found in the progress of the explored reaction. In each of the variants tested, the 3f product signals were observed in the 31P NMR spectra of the reaction mixtures. In the case of the reaction performed at room temperature in the second step, the conversion of substrate 1f was around 60% (Table 1, entry 4). Comparing the recorded spectra of reaction mixtures obtained in syntheses carried out in different solvents, it was again found that for the reaction in dichloromethane, the resulting spectrum was of the higher purity of the baseline and a higher degree of substrate conversion (Table 1, entries 5, 6).
Finally, unsymmetric products 3e,f were successfully synthesized in a stepwise procedure by the primary formation of the intermediate phosphonium salt 2e,f and its further use in situ in the Michaelis−Arbuzov-type reaction. The difference in the synthesis of both models was that compound 3f was obtained by heating the reaction mixture in the second step at 40 °C for 6 h, after the previous synthesis of the phosphonium salt 2f carried out at room temperature for 40 minutes, while model 3e was synthesized similarly to our recently published procedure,27 that is, first, the phosphonium salt 2e was obtained by heating the homogeneous residue after evaporation of the solvent from a solution, prepared by combining α-ethoxyphosphonate 1e and triphenylphosphonium tetrafluoroborate, at 85 °C under reduced pressure for 5 hours. Next, the crude phosphonium salt 2e, after dissolving in DCM and treatment with methyl diphenylphosphinite in the presence of a catalytic amount of Hünig's base, was subjected to the Michaelis–Arbuzov-type reaction carried out at 60 °C for 6 hours, resulting in a good yield of the target product 3e (65%, Scheme 4A).
The one-pot protocol was further explored for the synthesis of 3g–m derivatives of 1-amino-1-phosphinylalkylphosphonates (Scheme 4B). It was found that selected N-protected α-ethoxyaminophosphonates 1 subjected to the Michaelis–Arbuzov-type reaction with triphenylphosphonium tetrafluoroborate and diethyl phenylphosphonite at 40 °C for 1 hour resulted in the desired products 3g–k with high yields (73–91%). We showed that the phosphonyl–phosphinyl derivative of phenylglycine 3l can be synthesized in the one-pot protocol with a good yield of 62%, while this result was obtained in the reaction carried out at room temperature for 24 h unlike the standard reaction conditions (40 °C, 1 h) used in the synthesis of most models of bisphosphoric phosphonyl–phosphinyl analogs. As expected, the one-pot method failed to give the glycine derivative product 3m. The desired product 3m was successfully synthesized in a stepwise procedure identical to that used in the synthesis of the unsymmetric product 3e. Similarly to the synthesis of the phosphonyl–phosphinoyl analogs 3a–f, also in that case for the synthesis of the phosphonyl–phosphinyl analogs 3g–m, the progress of the reaction was controlled by 31P NMR spectroscopy to evaluate the conversion of α-ethoxyaminophosphonate 1 (Fig. 2) The phosphonyl–phosphinyl analogs of 1-aminobisphosphonates 3g–m were obtained as mixtures of diastereomers (dr 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–1:4) with very good yields (62–91%). Only product 3l (dr 1:4) was separated and isolated as single isomers by gravity column chromatography. Unfortunately, attempts to separate other diastereomeric mixtures 3g–k,m, both by gravity column chromatography and by HPLC, failed (see ESI†).
:1–1:4) with very good yields (62–91%). Only product 3l (dr 1:4) was separated and isolated as single isomers by gravity column chromatography. Unfortunately, attempts to separate other diastereomeric mixtures 3g–k,m, both by gravity column chromatography and by HPLC, failed (see ESI†).
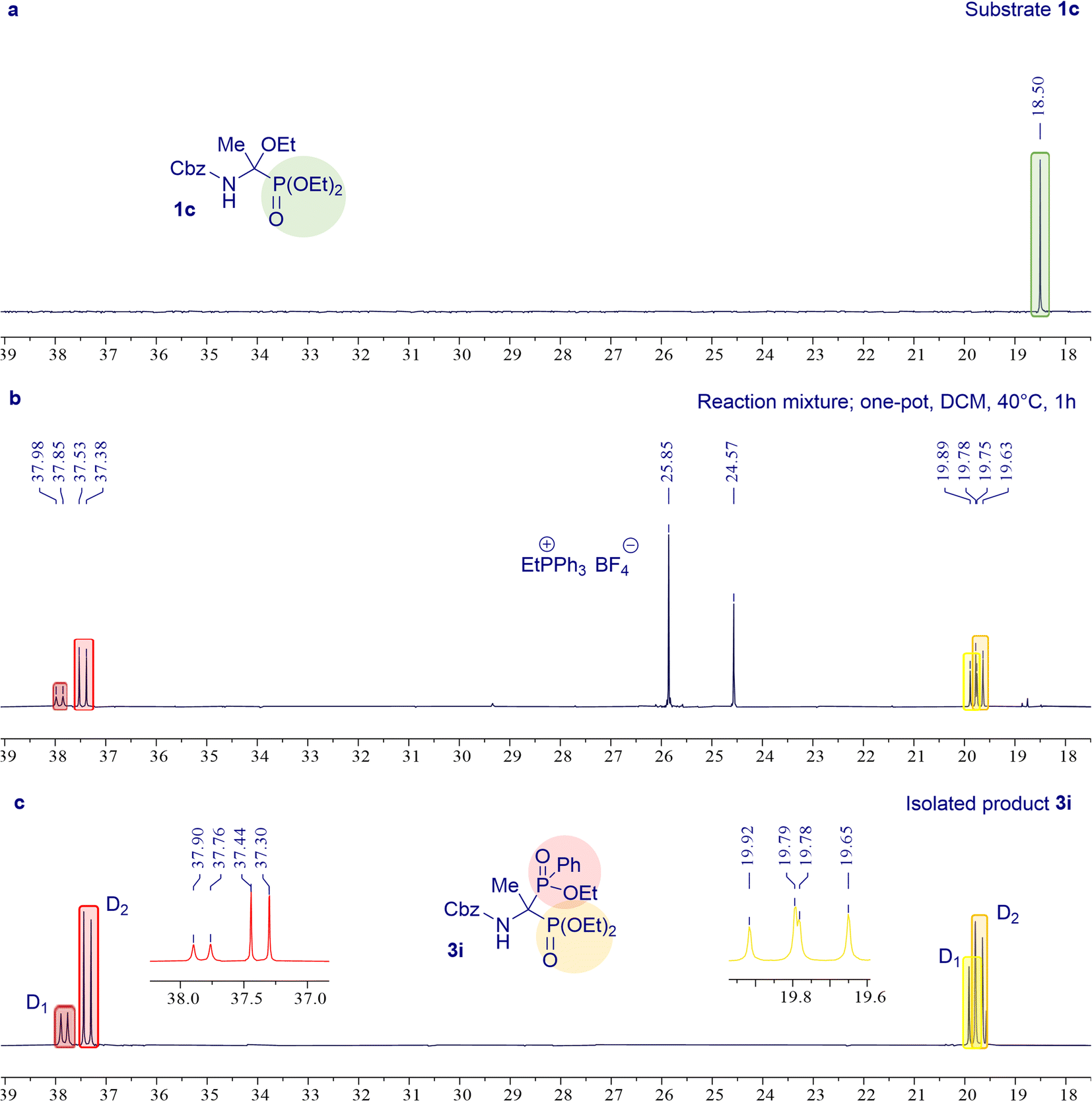 | ||
| Fig. 2 (a) 31P NMR spectrum of α-ethoxyphosphonate 1c. (b) 31P NMR spectrum of the reaction mixture obtained in the synthesis of 3i carried out at 40 °C for 1 h. (c) 31P NMR spectrum of isolated product 3i. | ||
The purification process for all of the synthesized esters of 1-(N-benzyloxycarbonylamino)alkylenebisphosphoric acids 3a–m, regardless of the procedure used for their synthesis, consisted of extraction with toluene from the reaction mixture and further purification of the extracts by twice column chromatography. For all reactions performed, yields of 52–91% for isolated products 3a–m were reported (Scheme 4A and B).
Considering the prospects for the further use of synthesized models of unsymmetric analogs of 1-aminobisphosphonates 3, it is planned to subject them to a hydrolysis reaction and then to biological tests to evaluate their cytotoxicity in selected tumor cell lines. Another research direction involves the acylation of obtained bisphosphoric derivatives with the use of appropriate chloroacyl chlorides to produce the building blocks that are useful in the synthesis of ligands, which after complexing with paramagnetic ions can be further used as potential contrast agents for imaging of bone tissue by MRI.
Conclusions
In conclusion, our efforts to extend the procedure previously developed for the synthesis of N-protected bisphosphonic analogs of α-amino acids, to their unsymmetric diethyl phosphonyl–ethoxyphenylphosphinyl and diethyl phosphonyl–diphenylphosphinoyl analogs resulted in success. In most cases, the one-pot strategy performed under mild conditions in an autocatalytic system worked very well here. This easy-to-follow procedure consisted only of mixing the starting 1-(N-acylamino)-1-ethoxyphosphonates with triphenylphosphonium tetrafluoroborate and the appropriate phosphorus nucleophile (diethyl phenylphosphonite or methyl diphenylphosphinite) in dichloromethane and then leaving the reaction mixture at room temperature or slightly elevated temperature to 40 °C for 1 to 6 hours. Further extraction of the reaction mixture followed by purification by column chromatography furnished the expected structurally diverse both protein and non-protein N-protected 1-aminobisphosphoric derivatives with good to very good yields (52–91%). The one-pot procedure was not applicable only in the case of both unsymmetric bisphosphoric analogs of glycine and for the phosphonyl–phosphinoyl derivative of phenylglycine; therefore, a stepwise method of reaching the final compounds turned out to be necessary. The results presented in this article in the form of 13 new compounds, combined with our achievements published in the previous article,27 constitute the universality of the protocol developed for the transformation of ethoxyphosphonate into a 1-amino-1,1-bisphosphoric derivative through the corresponding phosphonium salt as a reactive intermediate. The wide range of various bisphosphoric compounds with the P–C(N)–P skeleton obtainable by this procedure emphasizes its synthetic importance.Conflicts of interest
There are no conflicts to declare.References
- Aminophosphonic and aminophosphinic acids: chemistry and biological activity, ed. V. P. Kukhar', Wiley, Chichester; New York, 2000 Search PubMed.
- V. D. Romanenko and V. P. Kukhar, Arkivoc, 2012, 2012, 127–166 Search PubMed.
- T. Hiraga, S. Tanaka, M. Yamamoto, T. Nakajima and H. Ozawa, Bone, 1996, 18, 1–7 CrossRef CAS PubMed.
- S. Zhang, G. Gangal and H. Uludağ, Chem. Soc. Rev., 2007, 36, 507–531 RSC.
- E. Chmielewska and P. Kafarski, Open Pharm. Sci. J., 2016, 3, 56–78 CrossRef.
- L. Wang, A. Kamath, H. Das, L. Li and J. F. Bukowski, J. Clin. Invest., 2001, 108, 1349–1357 CrossRef CAS PubMed.
- S. H. Szajnman, E. L. Ravaschino, R. Docampo and J. B. Rodriguez, Bioorg. Med. Chem. Lett., 2005, 15, 4685–4690 CrossRef CAS PubMed.
- S. Yajima, K. Hara, J. M. Sanders, F. Yin, K. Ohsawa, J. Wiesner, H. Jomaa and E. Oldfield, J. Am. Chem. Soc., 2004, 126, 10824–10825 CrossRef CAS PubMed.
- A. Occhipinti, Ł. Berlicki, S. Giberti, G. Dziedzioła, P. Kafarski and G. Forlani, Pest Manage. Sci., 2010, 66, 51–58 CrossRef CAS PubMed.
- K. B. Farrell, A. Karpeisky, D. H. Thamm and S. Zinnen, Bone Rep., 2018, 9, 47–60 CrossRef PubMed.
- A. Kuźnik, A. Październiok-Holewa, P. Jewula and N. Kuźnik, Eur. J. Pharmacol., 2020, 866, 172773 CrossRef PubMed.
- F. H. Ebetino and L. A. Jamieson, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 51, 23–26 Search PubMed.
- B. Kaboudin and S. Alipour, Tetrahedron Lett., 2009, 50, 4243–4245 CrossRef CAS.
- A.-E. Wang, Z. Chang, W.-T. Sun and P.-Q. Huang, Org. Lett., 2015, 17, 732–735 CrossRef CAS PubMed.
- R. E. Islas and J. J. García, ChemCatChem, 2019, 11, 1337–1345 CrossRef CAS.
- B. Kaboudin, H. Esfandiari, A. Moradi, F. Kazemi and H. Aoyama, J. Org. Chem., 2019, 84, 14943–14948 CrossRef CAS PubMed.
- K. Kosta and A. Kotyński, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 47, 261–265 CrossRef CAS.
- K. Kostka and A. Kotyński, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 83, 215–221 CrossRef CAS.
- Y. Rassukana, M. Kolotylo, O. Sinitsa, V. Pirozhenko and P. Onys'ko, Synthesis, 2007, 2007, 2627–2630 CrossRef.
- Y. V. Rassukana, A. A. Sinitsa and P. P. Onys'ko, Russ. Chem. Bull., 2005, 54, 2652–2655 CrossRef CAS.
- V. P. Morgalyuk, T. V. Strelkova and E. E. Nifant'ev, Bull. Chem. Soc. Jpn., 2012, 85, 93–100 CrossRef CAS.
- A. A. Prishchenko, M. V. Livantsov, O. P. Novikova, L. I. Livantsova and V. S. Petrosyan, Heteroat. Chem., 2009, 20, 319–324 CrossRef CAS.
- T. Schrader, W. Steglich and R. Kober, Synthesis, 1986, 5, 372–375 CrossRef.
- C. A. III Metcalf, W. C. Shakespeare, T. K. Sawyer, Y. Wang and R. Bohacek, WO 03/000270 A1, 2003.
- A. Kuźnik, R. Mazurkiewicz, M. Grymel, K. Zielińska, J. Adamek, E. Chmielewska, M. Bochno and S. Kubica, Beilstein J. Org. Chem., 2015, 11, 1418–1424 CrossRef PubMed.
- A. Kuźnik, R. Mazurkiewicz, M. Zięba and K. Erfurt, Tetrahedron Lett., 2018, 59, 3307–3310 CrossRef.
- A. Kuźnik, D. Kozicka, W. Hawranek, K. Socha and K. Erfurt, Molecules, 2022, 27, 3571 CrossRef PubMed.
- A. Kuźnik, D. Kozicka and J. Adamek, Molbank, 2022, 2022, M1470 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, NMR spectra. See DOI: https://doi.org/10.1039/d3ra02981f |
| This journal is © The Royal Society of Chemistry 2023 |