
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceK2CO3-promoted synthesis of amides from 1-aryl-2,2,2-trifluoroethanones and amines under mild conditions†
Pinyong Zhonga,
Yu-Chao Wanga,
Jin-Biao Liu *a,
Linjun Zhang*b and
Nianhua Luo*c
*a,
Linjun Zhang*b and
Nianhua Luo*c
aJiangxi Provincial Key Laboratory of Functional Molecular Materials Chemistry, Jiangxi University of Science and Technology, Ganzhou 341000, China. E-mail: liujinbiao@jxust.edu.cn
bJiangxi Province Zhonggantou Survey and Design Co., Ltd., Nanchang 330029, China. E-mail: zhanglinjun2022@163.com
cSchool of Pharmaceutical Sciences, Gannan Medical University, Ganzhou 341000, China. E-mail: luoxiaoge102@163.com
First published on 15th June 2023
Abstract
A base-promoted amidation of 1-aryl-2,2,2-trifluoroethanones with amines via Haller–Bauer reaction has been developed. In this reaction, the direct transformation of 1-aryl-2,2,2-trifluoroethanones into amides via C(O)–C bond cleavage occurs without the use of any stoichiometric chemical oxidants or transition-metal catalysts. A series of primary and secondary amines are shown to be compatible with this transformation, and several pharmaceutical molecules were synthesized.
The introduction of amide bonds is one of the most important research topics in organic chemistry,1–5 as amides not only play an irreplaceable role in life, but also exist in a large number of drug molecules,6 materials7 and organic catalysts.8 In fact, ca. 25% of medicines are reported to contain amide bonds.9 The coupling of amines with pre-activated carboxylic acids or carboxylic acid derivatives (esters, aldehydes, acyl halides and acid anhydrides) is an effective method for constructing amide bonds.10–18 In recent years, alcohols,19–21 olefins,22–25 and alkynes26–28 have also been used as carboxylic acid substitutes to synthesize amides.
The carbon–carbon (C–C) bond is fundamental in organic compounds and has excellent stability.29–37 Developing new methods for constructing C–N bonds via C–C bond cleavage is attractive and challenging.38 Recently, several examples of constructing amides via copper catalyzed cleavage of C(O)–C bonds of ketones have been reported.39–43 In 2017, Liu et al.44 reported for the first time that the chemoselectivity C(α)–C(β) bond cleavage of saturated aryl ketones using copper catalyst led to α-ketoamides (Scheme 1a). Subsequently, Yang et al.45 also revealed the strategy of copper-catalyzed aerobic oxidation C–C-bond cleavage of simple ketones for the synthesized amides (Scheme 1b). However, the residue of transition-metal catalysts cannot be tolerated in some fields (such as pharmaceuticals). The method of constructing C–N bonds by activating C(O)–C bonds without metal catalysis has also been reported.46–50 In 2018, Xu and co-workers51 reported a TBHP/TBAl-mediated method for the direct oxidative of ketones and 2-aminopyridine to synthesize N-(pyridine-2-yl)amides (Scheme 1c). Subsequently, a method for selective synthesis of N-(pyridine-2-yl)amides from α-bromoketones and 2-aminopyridine was also developed (Scheme 1d).52 Although these methods are effective and well examined, there are also some limitations: amines are limited to the heterocyclic amines; using oxidants and additional additives.
 | ||
| Scheme 1 The formation of amide bond via C–C bond cleavage. (a) Using simple ketones and aliphatic amines. (b) Using simple ketones and amines. (c) Using heterocyclic amines. (d) Using α-bromoketones and 2-aminopyridines. (e) Using 1-aryl-2,2,2-trifluoroethanones and amines. | ||
Trifluoromethyl ketone, as an organofluorine compound, plays an important role in drug design and development.53 In 2022, Huang et al.54 conducted research on the reactivity of aryl perfluoroalkyl ketones, demonstrating that electron-deficient perfluoroalkyl groups possess the capability to function as formal leaving groups.
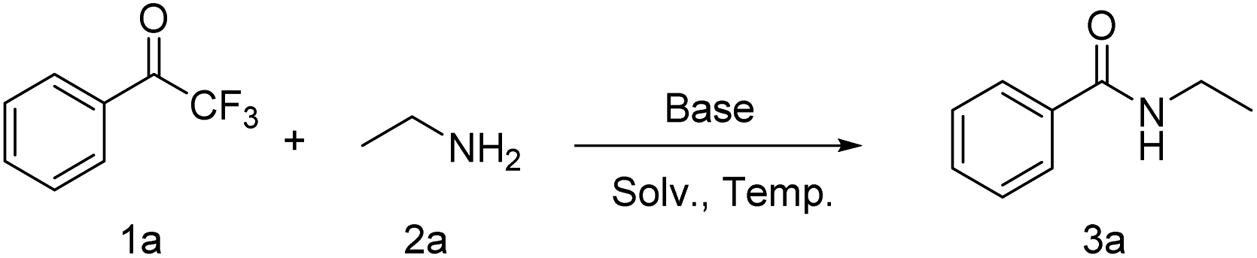
The base-induced cleavage of non-enolizable ketones leading to a carboxylic acid derivative and a neutral fragment in which the carbonyl group is replaced by a hydrogen is referred to as the Haller–Bauer (HB) reaction.55 However, to the best of our knowledge, there have been no reports on the preparation of amides using 1-aryl-2,2,2-trifluoroethanones under mild conditions. Based on our group's research on amide bonds,56–59 we report the activation of the C(O)–C bond of 1-aryl-2,2,2-trifluoroethanones, using amine as the N source to construct amides (Scheme 1e). This method does not require the use of transition-metal catalysts and additional oxidants, providing a convenient approach to C–N bond construction.
Initially, 2,2,2-trifluoro-1-phenylethanone (1a) and ethylamine (2a) were selected as model substrates, and to our delight, when they were treated in a reaction system containing KOH in acetonitrile at 40 °C, N-ethylbenzamide (3a) was obtained in 13% yield (Table 1, entry 1). Subsequently, several solvents were screened and the results showed that DMSO was the best solvent, with a yield of 90% for 3a (Table 1, entry 6). It is worth noting that increasing the temperature does not improve the reaction efficiency, but reducing the temperature from 40 °C to room temperature only provides 20% of the product 3a (Table 1, entry 7–11). Subsequently, various bases including K2CO3, KHCO3, NaHCO3, KOAc, NaH2PO4, NaH, DMAP, and Et3N, were screened (Table 1, entries 12–19), and it was found that K2CO3 was the most suitable base with a maximum yield of 93% (Table 1, entry 12). Furthermore, products 3a could not be obtained without the addition of base (Table 1, entry 20). Based on the reaction parameters above, the optimal reaction conditions were obtained: 1a (0.2 mmol), 2a (0.24 mmol), K2CO3 (0.4 mmol) in DMSO (2 mL) at 40 °C for 2 h.
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Temp./°C | Yield of 3ab |
| a Reaction conditions: 1a (0.2 mmol), 2a (70% in water, 0.24 mmol), base (2.0 eq.), solvent (2.0 mL), air, 2 h.b Isolated yield based on 1a. | ||||
| 1 | KOH | CH3CN | 40 | 13% |
| 2 | KOH | DMF | 40 | 82% |
| 3 | KOH | THF | 40 | 0% |
| 4 | KOH | EtOH | 40 | 0% |
| 5 | KOH | H2O | 40 | 0% |
| 6 | KOH | DMSO | 40 | 90% |
| 7 | KOH | DMSO | R.T | 20% |
| 8 | KOH | DMSO | 60 | 85% |
| 9 | KOH | DMSO | 80 | 84% |
| 10 | KOH | DMSO | 100 | 85% |
| 11 | KOH | DMSO | 120 | 86% |
| 12 | K2CO3 | DMSO | 40 | 93% |
| 13 | KHCO3 | DMSO | 40 | 41% |
| 14 | NaHCO3 | DMSO | 40 | 39% |
| 15 | KOAc | DMSO | 40 | 34% |
| 16 | NaH2PO4 | DMSO | 40 | 0% |
| 17 | NaH | DMSO | 40 | 60% |
| 18 | DMAP | DMSO | 40 | 0% |
| 19 | Et3N | DMSO | 40 | 0% |
| 20 | — | DMSO | 40 | 0% |
With the optimized conditions in hand, we investigated the substrate scope of this transformation using a series of trifluoroacetophenones and amines (Table 2). We first explored various aryl trifluoroacetophenones with different substituents. Excitingly, regardless of whether the aryl ring of the aryl trifluoroacetophenone contained electron-donating substituents (–Me, –OMe) or electron-withdrawing substituents (–F, –Br), it could be converted into the desired products with satisfactory yields (3b–3f). Additionally, disubstituted aryl trifluoroacetophenone was also tolerated in this reaction (product 3g). Subsequently, various amines were utilized to further explore the scope of reactions. Ethylamine containing halogen substituents was compatible with this reaction (product 3h). To our delight, ethylenediamine can produce the desired product 3i in moderate yield. Anilines were also tolerated in this reaction. Aniline derivatives with strong electron-withdrawing substituent (such as –NO2) or electron-donating substituent (such as –OMe) were capable of producing both the desired products (3k and 3l) in moderate yields. Benzylamine derivatives with either electron-donating or electron-withdrawing substituents could also give the desired product with high yields (3m–3q). Secondary amines were also compatible with this scheme (product 3r). As privileged scaffolds in drugs, the derivatization of nitrogen heterocycles has received widespread research attention. Interestingly, tetrahydropyrrole, piperidine, and morpholine were also tolerated in this scheme, obtaining the required amides with yields of 81–90% (3s–3u). More encouragingly, amino acid derivatives could also survive the process after the reaction temperature was raised to 70 °C and deliver the desired products with yields 51% (3v) and 81% (3x), respectively. These results indicate that this strategy of C–N bond construction has potential for late-stage functionalization of bioactive molecules. Nevertheless, both amide and trifluoromethyl alkyl ketone were not suitable for this reaction.
| a Reaction conditions: 1 (0.2 mmol), 2 (0.24 mmol), K2CO3 (2.0 eq.) DMSO (2.0 mL), air, 40 °C, 2 h.b Ethylamine (70% in water).c Reaction conditions: 1 (0.2 mmol), 2 (0.24 mmol), K2CO3 (2.0 eq.) DMSO (2.0 mL), air, 40 °C, 4 h.d Dimethylamine (40% in water).e Reaction conditions: 1 (0.2 mmol), 2 (0.24 mmol), K2CO3 (3.0 eq.) DMSO (2.0 mL), air, 70 °C, 4 h. |
|---|
 |
To demonstrate the utility of this strategy, we attempted to apply it to the synthesis of two pharmaceutical molecules (Scheme 2). Procainamide and moclobemide were obtained in yields of 72% and 89%, respectively.
 | ||
| Scheme 2 Synthetic of pharmaceutical molecules. | ||
To shed light on the mechanism of the reaction, we conducted several control experiments. When aniline was used as the substrate, benzoic acid was detected (Scheme 3a). However, under standard conditions, benzoic acid could not produce N-phenylbenzamide (Scheme 3b), indicating that it is a byproduct of amide formation. This result also suggests that this reaction may have undergone the HB reaction process. We then attempted to use benzaldehyde and acetophenone instead of aniline as substrates to react under standard conditions, but did not obtain the desired product 3j (Scheme 3c and d). These results indicate that neither benzaldehyde nor acetophenone are potential intermediates in this transformation.
 | ||
| Scheme 3 Control experiments.. (a) Identification of byproducts. (b) Using 6a instead of 1a. (c) Using 7a instead of 1a. (d) Using 8 instead of 1a. | ||
Based on the experimental results described above and previous literature,60 a possible reaction mechanism for this transformation has been proposed in Scheme 4. Initially, the 1-aryl-2,2,2-trifluoroethanones (1) was attacked by the amine (2) under the promotion of base, leading to the formation of the intermediate A. Then the amide 3 was obtained via the electron transfer and C–C bond cleavage of intermediate A.
 | ||
| Scheme 4 A possible mechanism. | ||
In summary, we have developed a mild and efficient methodology for the cleavage of C(O)–C bonds for the synthesis of amides. This recation can tolerate multiple primary and secondary amines, with yields ranging from moderate to good. This strategy enriches the substrate scope for constructing amides through C–C bond cleavage. The mild reaction conditions and simple operation process do not require the use of transition-metal catalysts or additional oxidants, making this protocol highly suitable for broad synthetic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Natural Science Foundation of China (21961014), the Jiangxi Provincial Key Laboratory of Functional Molecular Materials Chemistry (20212BCD42018), the Postdoctoral Merit Program of Jiangxi Province (2021KY21), and the Jinggang Scholars Program in Jiangxi Province.Notes and references
- R. M. de Figueiredo, J. Suppo and J. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 4, 345–3415 Search PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- L. Jiang-Sheng, X. Xie, S. Jiang, Y. Pan-Pan, L. Zhi-Wei, L. Cui-Hong and L. Wei-Dong, Org. Chem. Front., 2021, 8, 697–701 RSC.
- X. C. Wang, L. Li, Z. J. Quan, H. P. Gong, H. L. Ye and X. F. Cao, Chin. Chem. Lett., 2009, 20, 651–655 CrossRef CAS.
- F. Hamilton and A. MacGowan, Nat. Microbiol., 2019, 4, 1604–1605 CrossRef CAS PubMed.
- S. Han and J. Wu, Biomacromolecules, 2022, 23, 1892–1919 CrossRef CAS PubMed.
- F. Bourgeois, J. A. Medlock, W. Bonrath and C. Sparr, Org. Lett., 2020, 22, 110–115 CrossRef CAS PubMed.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- Z. Fu, X. Wang, S. Tao, Q. Bu, D. Wei and N. Liu, J. Org. Chem., 2021, 86, 2339–2358 CrossRef CAS PubMed.
- G. N. Papadopoulos and C. G. Kokotos, J. Org. Chem., 2016, 81, 7023–7028 CrossRef CAS PubMed.
- T. W. Bousfield, K. P. R. Pearce, S. B. Nyamini, A. Angelis-Dimakis and J. E. Camp, Green Chem., 2019, 21, 3675–3681 RSC.
- Y. Li, L. Ma, F. Jia and Z. Li, J. Org. Chem., 2013, 78, 5638–5646 CrossRef CAS PubMed.
- S. Jamalifard, J. Mokhtari and Z. Mirjafary, RSC Adv., 2019, 9, 22749–22754 RSC.
- S. A. Rzhevskiy, A. A. Ageshina, G. A. Chesnokov, P. S. Gribanov, M. A. Topchiy, M. S. Nechaev and A. F. Asachenko, RSC Adv., 2019, 9, 1536–1540 RSC.
- X. Xu, A. Amuti and A. Wusiman, Adv. Synth. Catal., 2020, 362, 5002–5008 CrossRef CAS.
- N. Iqbal and E. J. Cho, J. Org. Chem., 2016, 81, 1905–1911 CrossRef CAS PubMed.
- B. K. Zambroń, S. R. Dubbaka, D. Marković, E. Moreno-Clavijo and P. Vogel, Org. Lett., 2013, 15, 2550–2553 CrossRef PubMed.
- L. Bao, B. Zhang, Z. Wang and X. Chen, Org. Chem. Front., 2023, 10, 1375–1379 RSC.
- L. U. Nordstrom, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef CAS PubMed.
- Y. Zheng, X. Nie, Y. Long, L. Ji, H. Fu, X. Zheng, H. Chen and R. Li, Chem. Commun., 2019, 55, 12384–12387 RSC.
- Y. Yu, Y. Yuan and K. Ye, Chem. Commun., 2023, 59, 422–425 RSC.
- M. Maraswami, J. Goh and T. Loh, Org. Lett., 2020, 22, 9724–9728 CrossRef CAS PubMed.
- S. Liu and M. Klussmann, Chem. Commun., 2020, 56, 1557–1560 RSC.
- Y. Guan, X. Min, G. He, D. Ji, S. Guo, Y. Hu and Q. Chen, Iscience, 2021, 24, 102969 CrossRef CAS PubMed.
- A. Álvarez-Pérez, M. A. Esteruelas, S. Izquierdo, J. A. Varela and C. Saá, Org. Lett., 2019, 21, 5346–5350 CrossRef.
- S. Mahato, S. Santra, G. V. Zyryanov and A. Majee, J. Org. Chem., 2019, 84, 3176–3183 CrossRef CAS PubMed.
- X. Ji, B. Gao, X. Zhou, Z. Liu and H. Huang, J. Org. Chem., 2018, 83, 10134–10141 CrossRef CAS PubMed.
- M. Subaramanian, P. M. Ramar, J. Rana, V. K. Gupta and E. Balaraman, Chem. Commun., 2020, 56, 8143–8146 RSC.
- C. Jun, Chem. Soc. Rev., 2004, 33, 610–618 RSC.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
- A. Masarwa and I. Marek, Chem. - Eur. J., 2010, 16, 9712–9721 CrossRef CAS PubMed.
- P. Sivaguru, Z. Wang, G. Zanoni and X. Bi, Chem. Soc. Rev., 2019, 48, 2615–2656 RSC.
- M. Murakami and N. Ishida, Chem. Rev., 2021, 121, 264–299 CrossRef CAS PubMed.
- L. Deng and G. Dong, Trends Chem., 2020, 2, 183–198 CrossRef CAS.
- M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed.
- T. Zou, Y. He, R. Liu, Y. Zhang, S. Wei, J. Lu, J. Wang, L. Wang, Q. Fu and D. Yi, Chin. Chem. Lett., 2023, 34, 107822 CrossRef CAS.
- B. Das, P. R. Reddy, C. Sudhakar and M. Lingaiah, Tetrahedron Lett., 2011, 52, 3521–3522 CrossRef CAS.
- W. Fan, Y. Yang, J. Lei, Q. Jiang and W. Zhou, J. Org. Chem., 2015, 80, 8782–8789 CrossRef CAS PubMed.
- K. Wu, Z. Huang, Y. Ma and A. Lei, RSC Adv., 2016, 6, 24349–24352 RSC.
- N. Vodnala, R. Gujjarappa, C. K. Hazra, D. Kaldhi, A. K. Kabi, U. Beifuss and C. C. Malakar, Adv. Synth. Catal., 2019, 361, 135–145 CrossRef CAS.
- P. Subramanian, S. Indu and K. P. Kaliappan, Org. Lett., 2014, 16, 6212–6215 CrossRef CAS.
- W. Ding and Q. Song, Org. Chem. Front., 2015, 2, 765–770 RSC.
- C. Liu, Z. Yang, Y. Zeng, Z. Fang and K. Guo, Org. Chem. Front., 2017, 4, 2375–2379 RSC.
- G. Yang, K. Li, W. Liu, K. Zeng and Y. Liu, Org. Biomol. Chem., 2020, 18, 6958–6964 RSC.
- R. Ballini, G. Bosica and D. Fiorini, Tetrahedron, 2003, 59, 1143–1145 CrossRef CAS.
- P. Biallas, A. P. Häring and S. F. Kirsch, Org. Biomol. Chem., 2017, 15, 3184–3187 RSC.
- S. N. Rao, D. C. Mohan and S. Adimurthy, Tetrahedron, 2016, 72, 4889–4894 CrossRef CAS.
- R. Guo, C. Zhu, Z. Sheng, Y. Li, W. Yin and C. Chu, Tetrahedron Lett., 2015, 56, 6223–6226 CrossRef CAS.
- X. Sun, M. Wang, P. Li, X. Zhang and L. Wang, Green Chem., 2013, 15, 3289 RSC.
- Y. Liu, H. Sun, Z. Huang, C. Ma, A. Lin, H. Yao, J. Xu and S. Xu, J. Org. Chem., 2018, 83, 14307–14313 CrossRef CAS PubMed.
- Y. Liu, L. Lu, H. Zhou, F. Xu, C. Ma, Z. Huang, J. Xu and S. Xu, RSC Adv., 2019, 9, 34671–34676 RSC.
- X. Liu, L. Liu, T. Huang, J. Zhang, Z. Tang, C. Li and T. Chen, Org. Lett., 2021, 23, 4930–4934 CrossRef CAS PubMed.
- J. Huang, X. Yan and Y. Xia, Angew. Chem., Int. Ed., 2022, 134, e202211080 Search PubMed.
- G. Mehta and R. V. Venkateswaran, Tetrahedron, 2000, 56, 1399–1422 CrossRef CAS.
- J. Li, Y. Wang, H. Xie, S. Ren, J. Liu, N. Luo and G. Qiu, Mol. Catal., 2021, 516, 111993 CrossRef CAS.
- P. Zhong, J. Wu, J. Wu, K. Liu, C. Wan and J. Liu, Tetrahedron Lett., 2022, 107, 154099 CrossRef CAS.
- Y. Wang, H. Xie, K. Liu, J. Li and J. Liu, Catalysts, 2022, 12, 1278 CrossRef.
- J. B. Liu, M. Ren, X. Lai and G. Qiu, Chem. Commun., 2021, 57, 4259–4262 RSC.
- K. Ishihara and T. Yano, Org. Lett., 2004, 6, 1983–1986 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03329e |
| This journal is © The Royal Society of Chemistry 2023 |