
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChalcogen-substituted carbenes: a density functional study of structure, stability, and donor ability†
Jamie S. Ritch
Department of Chemistry, The University of Winnipeg, 515 Portage Avenue, Winnipeg, MB R3B 2E9, Canada. E-mail: j.ritch@uwinnipeg.ca; Fax: +1-204-774-2401; Tel: +1-204-786-9730
First published on 5th June 2023
Abstract
Chalcogen-substituted carbenes are examined computationally using density functional theory. Several approaches are used to assess the stability and reactivity of chalcogenazol-2-ylidene carbenes (NEHCs; E = O, S, Se, Te). The known unsaturated species 1,3-dimethylimidazol-2-ylidene is studied at the same level of theory as the NEHC molecules, as a reference. Electronic structures, stability towards dimerization, and ligand properties are discussed. The results highlight the NEHCs as potentially valuable ancillary ligands for stabilizing low-valent metals or paramagnetic main group molecules. A simple, effective computational method for evaluating σ donor ability and π acidity of carbenes is presented.
Introduction
N-heterocyclic carbenes (NHCs) are popular spectator ligands due to their modularity, strong σ-donor strength, and in some cases π-acidity.1 Carbenes featuring heteroatoms other than nitrogen have been of interest since the early days of stable carbene research, including the report of a persistent (phosphino)(silyl)carbene in 1989.2 In 1997, a benzoxazolyl metal complex was reported,3 and a thiazol-2-ylidene and its dimer was disclosed.4 More recently, mesoionic carbenes (MICs, 2001)5–7 and cyclic (alkyl)(amino) carbenes (CAACs, 2005)8–10 have yielded examples with unsaturated and saturated carbon centres replacing a nitrogen atom, respectively. These variations on the NHC framework provide ligands with differing substitution patterns and electronic properties (Scheme 1).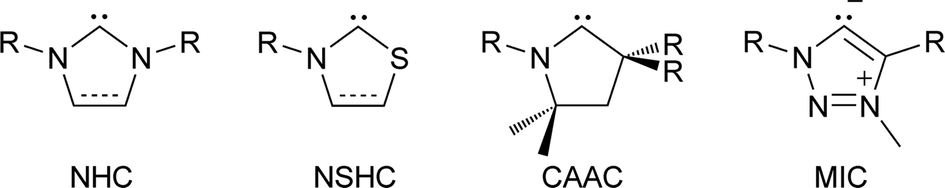 | ||
| Scheme 1 Structures of common carbenes. | ||
For chalcogen-substituted carbenes in particular, thioazol-2-ylidenes (NSHCs) can be prepared using numerous routes,11–13 and have seen significant use as organocatalysts for transformations such as 1,2 and 1,4 conjugate addition reactions,14,15 though their coordination chemistry is underexplored.16 By contrast, NOHCs have only been made using post-coordination ligand modifications of isocyanides,3 while NSeHC and NTeHCs remain unreported. Given the drastically different electronic properties of the heavier group 16 elements selenium and tellurium compared to lighter congeners, these are potentially valuable synthetic targets with unique chemical properties. Furthermore, NEHC dimerization to give electron-rich alkenes is of interest, as dimer molecules of this type are possible electron donors in conductive organic materials or precursors to stable radical cations. A previous computational study evaluated NEHCs (E = O, S, Se) and proposed NOHC and NSeHC derivatives may show reasonable thermodynamic stability,17 though there has been no comprehensive evaluation of NEHC (E = O, S, Se, Te) stability, both kinetic and thermodynamic, and their properties for thus far.
There have been many experimental and theoretical studies aimed at quantifying salient properties and finding useful descriptors of NHCs, including donor ability by Tolman electronic parameters (TEPs)18,19 and 77Se NMR of selenourea derivatives,20,21 steric encumbrance via percent buried volume,22 and stability and reactivity by calculation of singlet-triplet gaps23,24 and dimerization energy.25 The carbene dimerization mechanism has also been modeled computationally,26–28 and energies of isodesmic reactions between carbenes and methane have been correlated to the carbene dimerization energies.29
In this contribution the properties of N-methylchalcogenazol-2-ylidene (NEHC; E = O, S, Se, Te) ligands are examined computationally, highlighting their expected properties and possible applications. The electronic structures, stability towards dimerization, and ligand properties of the NEHCs are probed using density functional theory. This is done in comparison to the experimentally known and structurally similar NHC analogue N,N′-dimethylimidazol-2-ylidene (Me2im). Me2im is stable in THF solution for several days and thus provides a suitable reference for molecules which may be experimentally accessible.
Results and discussion
Ligand geometry and electronic properties
Optimized gas-phase geometries of the five carbenes (ωB97X-D/def2-TZVP) are depicted in Fig. 1. As the MeN group is replaced with increasingly larger chalcogen atoms the obvious effect is distortion of the ring as the carbon–chalcogen bond distances elongate and the C–E–C bond angle contracts. The MeN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C distance also shortens from 1.357 Å in the chalcogenated structures, consistent with increased π donation from the nitrogen lone pair to the divalent carbon atom outcompeting π donation from the chalcogen atom.
C distance also shortens from 1.357 Å in the chalcogenated structures, consistent with increased π donation from the nitrogen lone pair to the divalent carbon atom outcompeting π donation from the chalcogen atom.
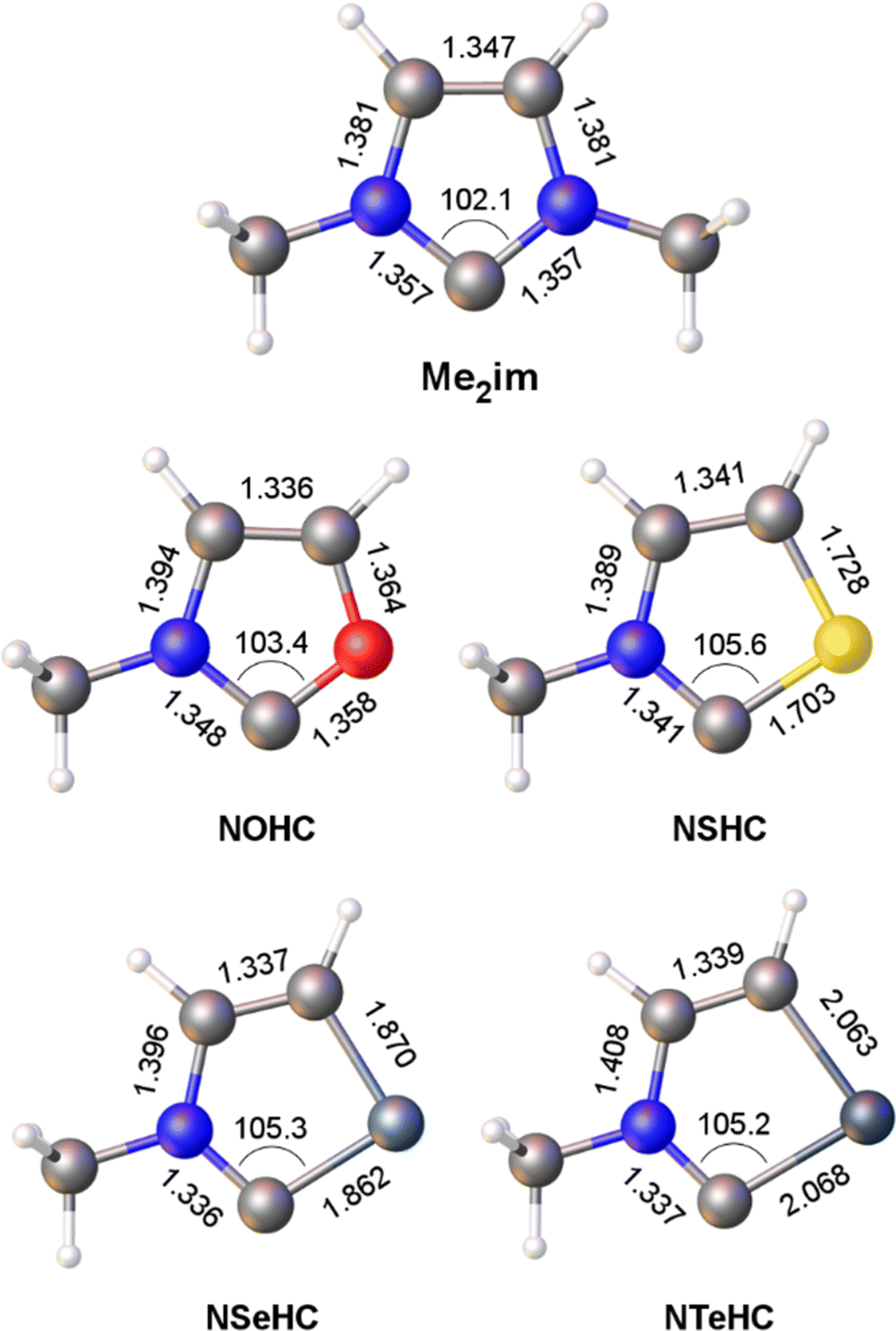 | ||
| Fig. 1 Calculated bond lengths (Å) and angles (°) for the heterocyclic ligands Me2im and NEHC (E = O, S, Se, Te). | ||
The contribution of resonance structures A and B was estimated by Natural Bond Order (NBO) analysis and used to assess the π donor capability of the various groups adjacent to the divalent carbon atom in these structures (Fig. 2), according to the amount of contribution from structure B. This yielded the trend N > S > O > Se > Te. That is, the larger chalcogen atoms are generally poorer at π donation, as would be expected. The greater π donor ability of sulfur versus oxygen reflects the very high electronegativity of oxygen. The qualitative trend in π donation was verified by examining the intramolecular donor–acceptor interaction strengths for n(E)→π*(CN) (kJ mol−1): 376 (N); 278 (S); 260 (O); 217 (Se); 167 (Te). The differing electron distribution of the NEHCs is also reflected in the natural atomic charges of the N–C–E fragment (Table 1). The carbene C atom in Me2im is roughly neutral in charge, with flanking negatively charged N atoms. For the NEHCs, the single nitrogen atom retains about the same negative charge as in Me2im, while the chalcogen atom and carbon atom change significantly. In the NOHC the carbon becomes positively charged with a negative O atom, while for S, Se, and Te the charges are reversed with a negative C atom and positively charged chalcogen centres.
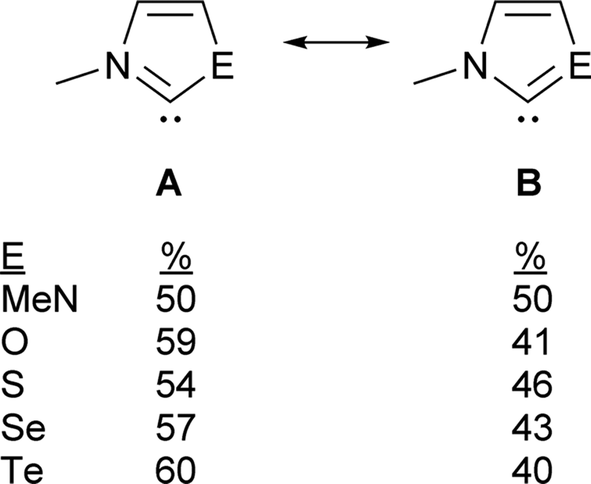 | ||
| Fig. 2 Resonance contributors of Me2im (E = NMe) and NEHC (E = O, S, Se, Te). | ||
| Carbene | C | N | E |
|---|---|---|---|
| Me2im | +0.07 | −0.38 | −0.38 |
| NOHC | +0.22 | −0.41 | −0.42 |
| NSHC | −0.22 | −0.38 | +0.41 |
| NSeHC | −0.21 | −0.40 | +0.46 |
| NTeHC | −0.26 | −0.42 | +0.60 |
The frontier orbitals of the optimized ligands are depicted in Fig. 3. As the NMe group is replaced with progressively less electronegative chalcogen atoms, the LUMO energy drops steadily. The LUMOs of the chalcogenated rings also have significant contributions from the divalent carbon π-symmetric p orbital, which is absent in Me2im. The HOMOs of the chalcogenated rings still have the σ-symmetric p orbital on the divalent carbon atom as the main contributor, but increased contributions from the chalcogen centre are seen moving from E = O to E = Te, raising the HOMO energies.
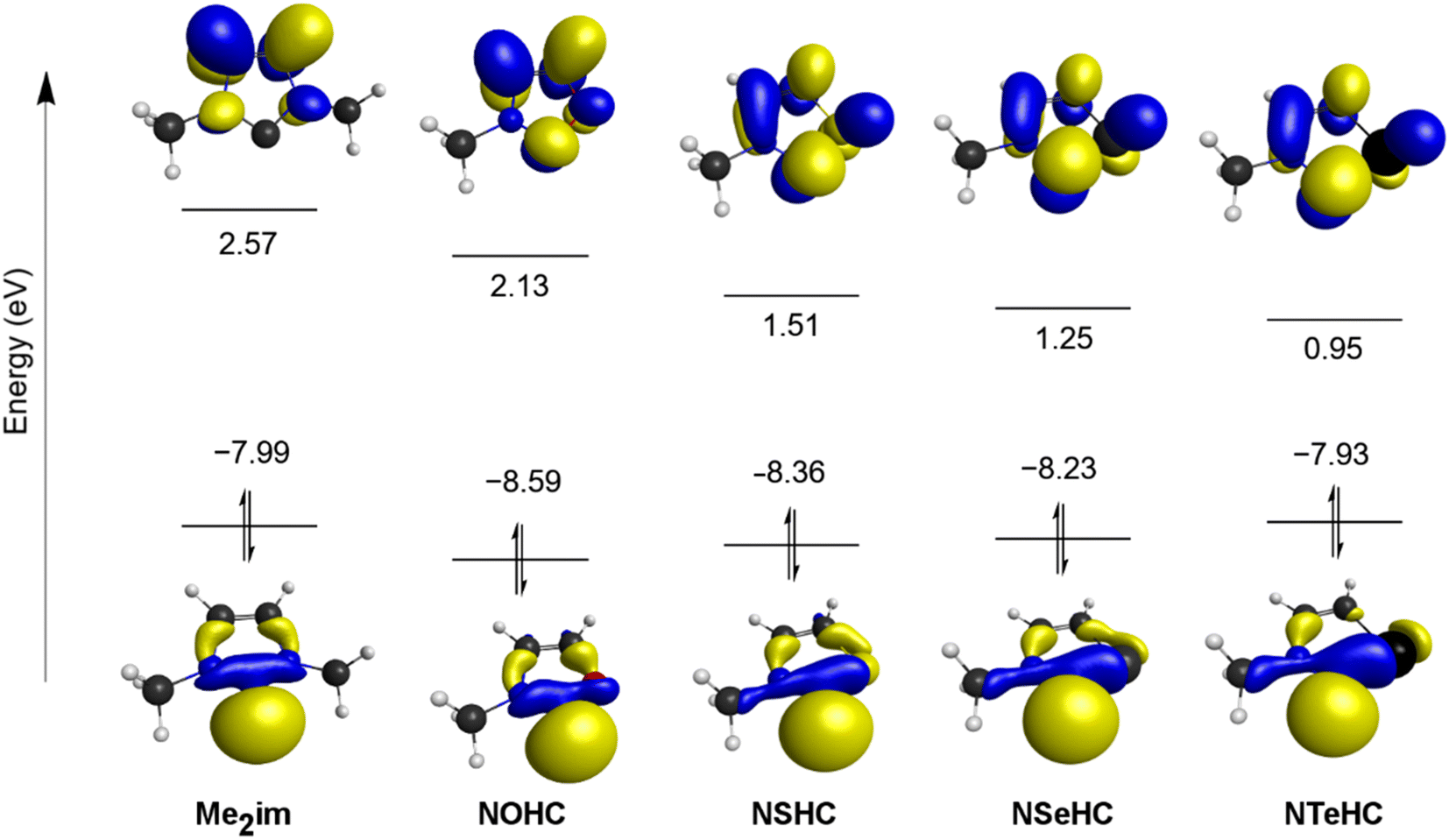 | ||
| Fig. 3 HOMO and LUMO orbitals for Me2im and the NEHCs (orbital isosurfaces at 0.05 contour level). | ||
Energies of excitation are listed in Table 2. HOMO–LUMO gaps and vertical singlet–triplet gaps were determined from the transition energy to the first singlet and triplet excited states, respectively, using time-dependent DFT (gap energies calculated using LUMO eigenvalues, i.e. values shown in Fig. 3, are normally prone to large errors for hybrid meta GGA functionals).30 Both vertical (EST(v)) and adiabatic (EST(a)) singlet–triplet gaps are presented. Values for the reference molecule 1,3-dimethylimidazol-2-ylidene (Me2im)31 corresponds closely to previously-reported values using B3LYP/aug-cc-PVTZ (the HOMO–LUMO gap, EST(v) and EST(a) values differ by <5% from the values in this study),32 though quite different from results using PBE/TZ2P.33 The trend in excitation energies predicts the NEHCs should become progressively less thermodynamically stable towards dimerization (vide infra), as the singlet-triplet gap of carbenes has shown to be inversely correlated with the dimer CC bond strength.23
| Compound | HOMO–LUMO gap | EST(v) | EST(a) | SD(C) | SD(N) | SD(E) |
|---|---|---|---|---|---|---|
| Me2im | 5.86 | 4.30 | 3.75 | 82.8 | 5.5 | 5.5 |
| NOHC | 5.61 | 3.84 | 3.47 | 83.9 | 5.8 | 5.3 |
| NSHC | 4.62 | 3.34 | 2.92 | 79.4 | 5.5 | 9.0 |
| NSeHC | 4.28 | 3.07 | 2.66 | 78.4 | 5.4 | 10.1 |
| NTeHC | 3.73 | 2.67 | 2.27 | 75.4 | 4.7 | 13.0 |
Spin density (SD) analysis for the optimized triplet states of the compounds revealed >93% of the spin resides on three atoms: the carbene carbon, and its two neighbours N and E. The spin density on the chalcogen atom is inversely proportional to the electronegativity for the atoms, peaking at 13% of the total for E = Te. These results raise the possibility that heavy chalcogen containing NEHC ligands are more able to bear unpaired electron density when attached to a paramagnetic Lewis acid, compared to the nitrogen atoms of NHCs (vide infra).
Donor/acceptor properties of NEHCs
Various descriptors of the electronic properties of NHCs have been proposed. Their σ donor ability has been correlated with computed HOMO eigenvalues and N–C–N bond angles.32 Spectroscopic measures of NHC derivatives including the νCO(A1) mode of [Ni(CO)3L] (Tolman Electronic Parameter, TEP),34 thought to reflect both σ-donor and π-acceptor properties, and the δ(13Ccarbene) of 1,3-diisopropylbenzimidazolin-2-ylidene (iPr2-bimy) in trans-[PdBr2L(iPr2bimy)] (Huynh Electronic Parameter, HEP) which measures primarily σ-donor behaviour.35 The 1J(13C–1H) value of protonated carbene precursors has been correlated to σ donor ability of the carbene.20,36 Bertrand et al. have disclosed the use of δ(31P) of NHC-phosphinidene adducts as a gauge of π-acidity, while Ganter et al. reported using the δ(77Se) of easily-prepared NHC = Se derivatives to ascertain π-acidity of the parent carbene,21 and the 1J(77Se–13C) to measure σ-donor ability. Computational work later showed that isotropic shielding of the Se atoms calculated at the BP86/TZ2P level correlated well with the experimental δSe values.37In the present study, a series of calculations was performed to determine if both the 77Se chemical shift values and 1J(77Se–13C) coupling constants of selenoureas could be replicated using rapid and readily-available electronic structure methods. In this way a predictive model could be developed and applied to the NEHCs under study, and provided for use by other researchers. A set of nine reported selenoureas were modeled, as the experimental 77Se chemical shifts (in acetone-d6) and coupling constants (in CDCl3) for each were determined under the same experimental conditions.20
While the range-separated hybrid functional ωB97X-D is used in this study for reaction energetics, the computationally less expensive pure GGA functional PBE was investigated for NMR property calculations. The nine gas-phase reference structures were optimized at the PBE/def2-TZVP level and the isotropic shielding at Se as well as the (C,Se) spin–spin coupling constants were calculated for these structures. The zeroth-order relativistic approximation (ZORA) was used along with the appropriate relativistically-contracted basis set for the property calculations. Plotting the shielding values (σiso) versus the measured chemical shifts (δSe) and performing linear regression yielded excellent agreement (R2 = 0.997) with the equation δSe = −0.744σiso + 1220.6 (Fig. 4a). The values predicted from this equation give a mean absolute error (MAE) of 15.6 ppm, and yielded the correct ordering of compounds from low to high predicted chemical shift. This method thus is recommended for 77Se shift calculations of selenoureas as it has a low computational cost and high accuracy. It is also convenient to evaluate both σ donor ability and π acidity of a carbene from NMR calculations of a single optimized structure. Given the large structural variety of the nine reference carbenes the model is very promising, though additional carbene experimental data would further verify its utility.
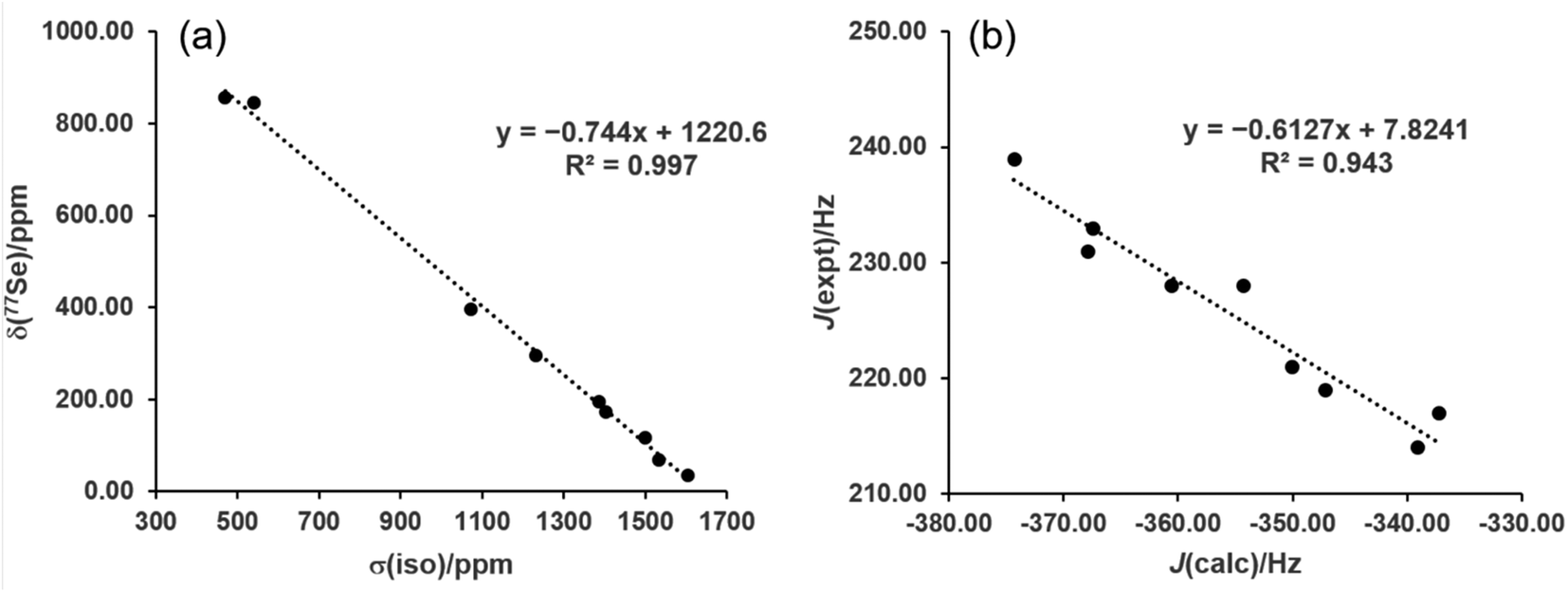 | ||
| Fig. 4 (a) Experimental 77Se chemical shifts versus computed isotropic shielding, and (b) experimental versus computed 77Se–13C spin–spin coupling constants. | ||
The same level of theory, when used to calculate the 1J(77Se–13C) spin–spin coupling constants, gave very good linear fitting (R2 = 0.943, MAE = 1.7 Hz) to experimental values with the equation JSe,C = −0.6127Jcalc + 7.8241 (Fig. 4b). The overall trend in values is reproduced, though due to the relatively small spread between the nine experimental J values the predicted ordering was not always correct for values within a few Hz of each other.
Applying these new models to the NEHCs and Me2im yielded predicted 77Se parameters as shown in Table 3. The predicted ordering of σ donor ability is thus: Me2im > NTeHC > NSHC > NSeHC ≫ NOHC; the π-acidity trend is NTeHC > NSeHC > NSHC > NOHC > Me2im. Fig. 5 and 6 display the nine experimental δSe and JSeC values along with the predicted values. The NEHCs under study are thus predicted to be similar to IMes in terms of σ donor ability, though slightly weaker (except for the NOHC which is significantly weaker), while being stronger π-acceptors. Such carbenes are valued for stabilizing low-valent metal complexes and open-shell species.38 The reported 77Se resonance for a CAAC selone derivative with aromatic N-substitution (+492 ppm)39 indicates a similar π acidity to the NSHC and NSeHC ligands. Since CAACs have found use in preparing low valent d-block complexes40 and main group radicals,41 this bodes well for the future use of NEHCs in this manner.
| Carbene | 77Se (ppm) | 1JSe,C (Hz) | ||
|---|---|---|---|---|
| Me | tBu | Me | tBu | |
| Me2im | 68.39 | — | 233.83 | — |
| NOHC | 141.85 | 191.67 | 253.15 | 253.17 |
| NSHC | 408.06 | 445.10 | 234.94 | 235.95 |
| NSeHC | 506.83 | 543.62 | 238.46 | 240.69 |
| NTeHC | 715.76 | 752.00 | 234.62 | 236.64 |
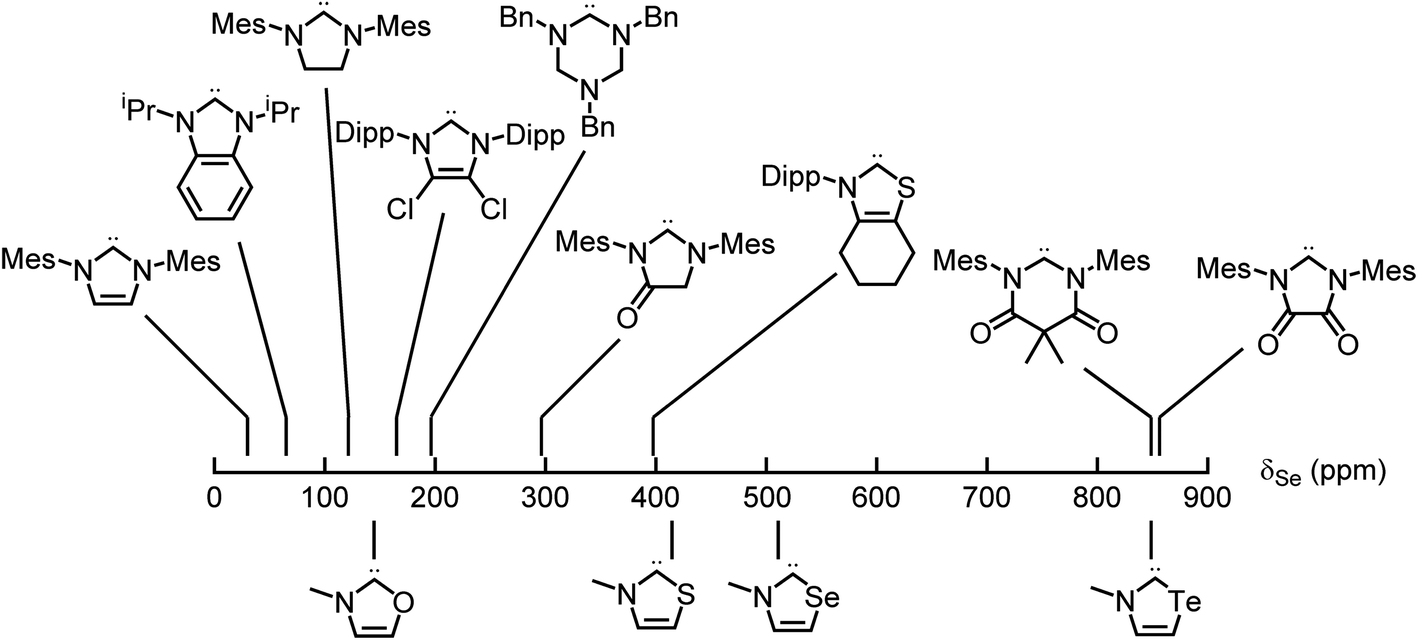 | ||
| Fig. 5 Experimental (top) and predicted (bottom) 77Se chemical shifts of the selone derivatives of carbenes – π acidity increases to the right. | ||
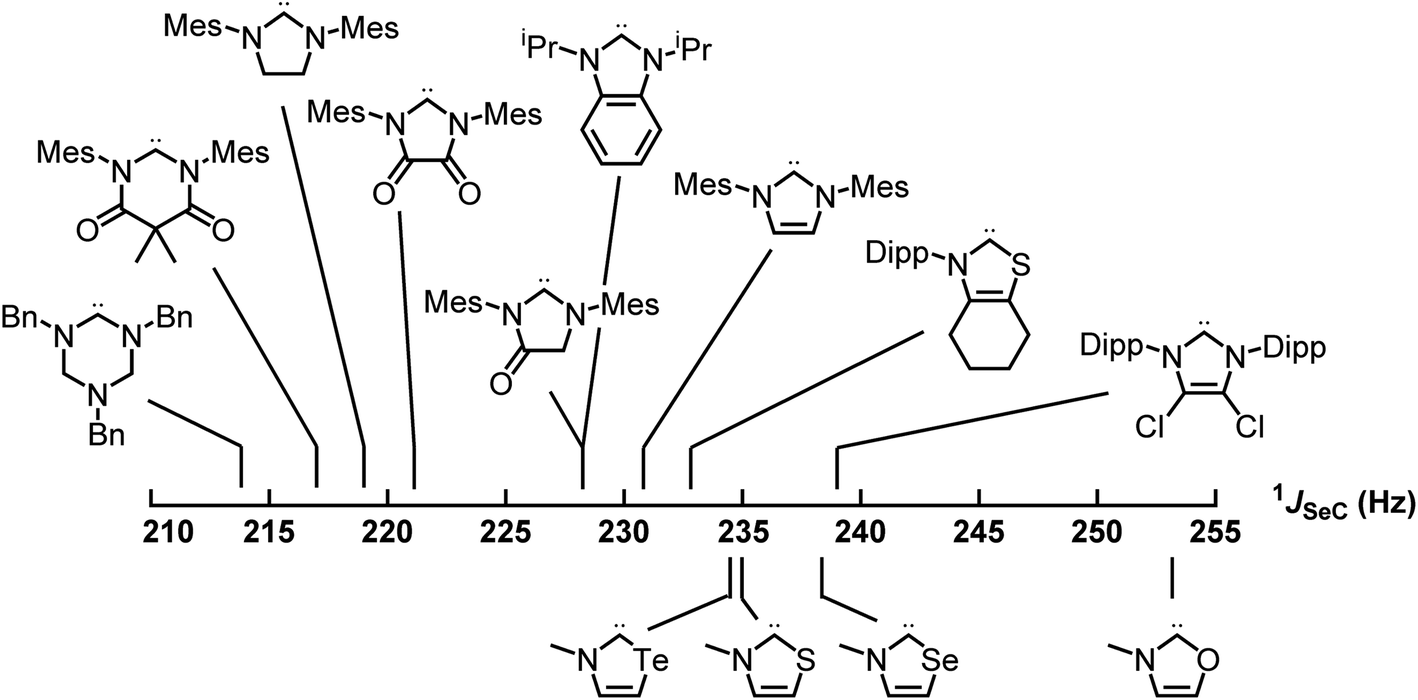 | ||
| Fig. 6 Experimental (top) and predicted (bottom) 77Se–13C spin–spin coupling constants of the selone derivatives of carbenes – σ donor strength increases to the left. | ||
To probe steric effects, the NMR properties of tert-butyl substituted NEHC selenoureas have been determined using the new model (Table 3). The predicted 77Se chemical shift values are higher by 50 ppm for the oxygen derivative and ca. 37 ppm for the heavier derivatives. The 1JSe–C values are nearly identical for oxygen but higher by 1–2 Hz for the heavier derivatives. Interestingly, this indicates the tert-butyl substituted carbenes are, compared to R = Me, slightly more π-acidic and slightly less σ-donating. The seemingly unintuitive result of a less donating when a more electron-releasing alkyl group is present has precedent – Ganter and coworkers20 revealed that the introduction of one or two carbonyl groups in the backbone of N-mesityl-substituted carbenes increased their σ donor ability relative to IMes.
Dimerization
Bulky N-substituents generally disfavour the dimerization of NHCs to give electron-rich olefins. In the case of imidazole-2-ylidenes, aromaticity of the five-membered ring also disfavours this process. For instance, the relatively unhindered carbene Me2im is not known to dimerize but rather decomposes slowly via other pathways upon storage at −30 °C.31 However, some imidazoline-2-ylidenes and benzimidazole-2-ylidenes with smaller substituents and/or less aromatic stabilization, are known to dimerize. The interconversion of NHCs and other cyclic carbenes and their dimer tetraaminoethylene derivatives has been studied experimentally and theoretically. It is generally accepted that a proton-catalyzed pathway operates in most instances as opposed to a direct one-step dimerization, however the latter is possible in some instances.27,42The smaller the singlet-triplet gap, the stronger the CC bond in the resulting carbene dimer, and hence the more thermodynamically favoured dimerization will be.23,24 In the context of the chalcogen-substituted NEHC carbenes, given their small singlet–triplet gaps, the direct uncatalyzed dimerization process is possible. Arduengo and coworkers reported a stable thiazolylidene (featuring methylation on the backbone and a Dipp substituent on nitrogen) which formed an anti-configuration dimer when catalyzed by the protonated thiazolium salt.4 In a previous report,17 dimerization energies for NEHCs (E = O, S, Se) were estimated using a correlation between Δ(E) of an isodesmic reaction of the carbene with methane and the dimerization energy, predicting the dimers are thermodynamically favoured.29 This approach accounts for only electronic factors, and estimates thermodynamic stability. There have been no previous computational dimerization studies on the entire series of group 16 NEHCs, nor any including both kinetic and thermodynamic aspects of dimerization.
Direct dimerization of NEHCs and Me2im was examined using DFT, and a set of t-butyl-substituted NEHC ligands was also examined to probe the effect of sterics at the nitrogen centres. These tert-butyl analogues had computed HOMO–LUMO gaps and vertical triplet excitation energies only slightly lower (by ca. 0.5–1.5%) compared to the methyl-substituted carbenes. Thus, their electronic structures are very similar, and significant differences in reaction energetics will be reflective of the effect of increased steric bulk at nitrogen. Both E/Z stereochemical outcomes were studied for each carbene (Scheme 2). One transition state was located for each reaction, and their structures indicated a non-least-motion pathway for each process. Using the intrinsic reaction coordinate method, dimer structures were located from the transition states and optimized, revealing that all (Z)-dimers had anti-orientations of the five-membered rings (oriented towards different faces of the alkene), while the (E)-dimers were syn (oriented towards the same face of the alkene). One of the studied NEHC dimers has been previously experimentally observed: the sulfur (E)-dimer with R = Me was crystallographically characterized by Arduengo and coworkers.4 Compared to the experimental atomic coordinates, the computed structure was very similar (RMSD of 0.126 Å for atomic coordinates of non-H atoms) indicating the suitability of the computational methodology for structures of this type.
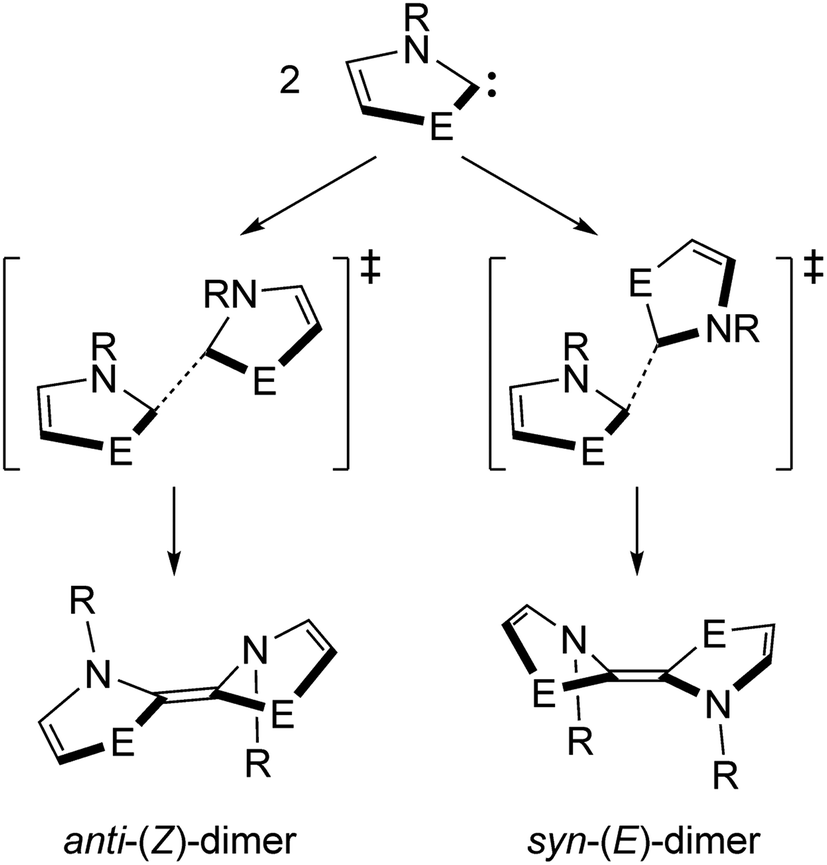 | ||
| Scheme 2 The E/Z dimerization of NEHCs. | ||
The reaction energetics for the dimerization processes are shown in Fig. 7. For Me2im, the dimerization is both kinetically and thermodynamically disfavoured (ΔG‡ and ΔG° of +132 and +25.7 kJ mol−1, respectively), consistent with the experimental observation that this carbene does not form a dimer. For NEHCs, the E dimerizations had, on average, lower activation barriers by 11 kJ mol−1 and more negative reaction Gibbs energies by 6 kJ mol−1. The two exceptions to this were for E = S and R = Me, where the Z dimerization had a roughly equivalent barrier to the (E) process (1.5 kJ mol−1 lower), and for E = O and R = tBu, where the (Z)-dimer was ca. 10 kJ mol−1 more stable (vide infra). Energetic differences between E and Z processes were more pronounced for tert-butyl-substituted NEHCS and, for a given NEHC, changing from methyl to tert-butyl substituents increased the reaction barrier by an average of 30 kJ mol−1 and decreased stability of the dimer relative to the monomers by 6 kJ mol−1.
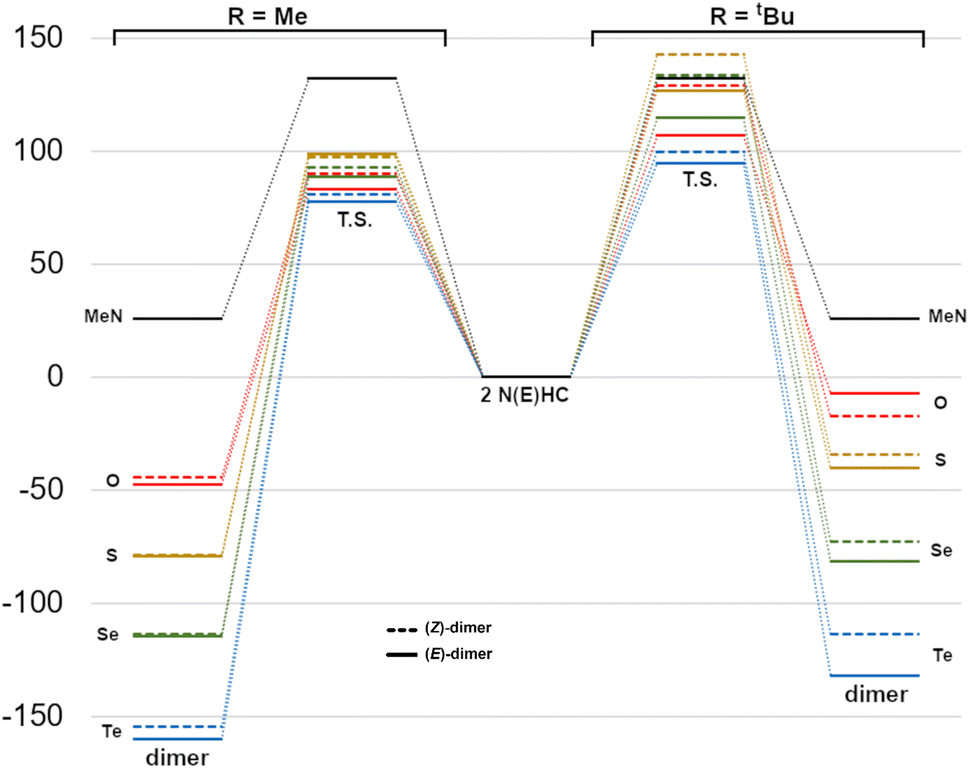 | ||
| Fig. 7 Reaction Gibbs energy diagram for dimerization (kJ mol−1) of Me2im and the NEHCs (centre) to give methyl- (left) or tert-butyl-substituted dimers (right). | ||
The reaction barriers can be attributed to the energy required to cause electronic reorganization of the carbenes, i.e. disruption of π bonding and loss of aromaticity. This is evidenced by pyramidalization of the nitrogen centres and lengthening of the carbene C–N and C–E bonds in the transition states (Table 4). The tert-butyl-substituted derivatives require more energy to pyramidalize the nitrogen centres and feature more intra- and intermolecular steric repulsions in the transition state geometries, increasing their activation energies. Whereas for the NEHC dimer products, the interplanar angles between the two five-membered rings are in the narrow range of ca. 0°–13° (and 17.4° for the Me2im dimer), the transition state values vary from ca. 0–89°, with the Z geometries generally featuring higher angles than E.
| NEHC | θTS | θdimer | d(CC)TS |
d(CC)dimer |
Conformation | ||||
|---|---|---|---|---|---|---|---|---|---|
| Me | tBu | Me | tBu | Me | tBu | Me | tBu | ||
| MeN | 49.2 | 17.4 | 1.79 | 1.35 | anti | ||||
| (E)-O | 2.8 | 0 | 0.2 | 0 | 1.94 | 1.91 | 1.33 | 1.34 | anti |
| (Z)-O | 30 | 50.7 | 7.9 | 13.1 | 1.92 | 1.93 | 1.33 | 1.34 | anti |
| (E)-S | 30.2 | 49.1 | 8.7 | 10.9 | 2.01 | 1.94 | 1.34 | 1.34 | syn |
| (Z)-S | 63.5 | 54.4 | 8.7 | 9.1 | 1.96 | 1.96 | 1.34 | 1.35 | anti |
| (E)-Se | 38.1 | 51.7 | 9.3 | 11.3 | 2.06 | 2 | 1.34 | 1.33 | syn |
| (Z)-Se | 69.9 | 58.1 | 9.6 | 8.8 | 2 | 1.99 | 1.34 | 1.34 | anti |
| (E)-Te | 89.3 | 53.7 | 10.1 | 11.9 | 2.14 | 2.08 | 1.34 | 1.33 | syn |
| (Z)-Te | 74.3 | 82.9 | 10.4 | 9.2 | 2.08 | 1.96 | 1.34 | 1.34 | anti |
The CC bond length of all the NEHC dimers were in the narrow range of 1.33–1.34 Å, whereas the C⋯C distance in the transition states varied from 1.91–2.08 Å (and was 1.79 Å for Me2im dimerization). The methyl-substituted transition states had longer distances by, on average, 0.04 Å. The bending of the five-membered rings out of the plane of the alkene to either syn or anti configurations was also more pronounced for the bulkier tert-butyl derivatives. For the E = O, R = Me dimers the deviation from planarity was only slight, making the (E)-dimer higher in energy than (Z) due to oxygen⋯tert-butyl group repulsive interactions between the two rings, which are avoided in the other NEHCS due to more substantial ring bending.
Overall, the thermodynamics of dimerization reveal that all methyl-substituted NEHCs have a ΔG‡ ranging from ∼75–100 kJ mol−1 and would consequently dimerize at room temperature to give an E/Z mixture, favouring the (E)-dimer in most cases. Dimerization would be rapid, except for the sulfur derivative which has barriers near 100 kJ mol−1. Substitution with tert-butyl groups has a stabilizing effect on the NEHCs and is predicted to yield kinetically stable carbenes in all cases (barriers > ∼95 kJ mol−1). Lower energy proton-catalyzed pathways are likely, and so experimentally prepared NEHCs should be handled carefully under aprotic conditions. The dimers are thermodynamically favoured in all cases, and none of the NEHC dimers studied have thermally accessible monomers at reasonable temperature via the studied pathway. These NEHC dimers may in and of themselves be intriguing synthetic targets, due to their possible use as organic electron donors akin to the tetrathiafulvalenes.43
Stabilization of main group radicals
Given the predicted high π-accepting properties of NEHCs, they may have utility to stabilize paramagnetic species via spin delocalization onto the carbene ligand. To probe this possibility, the ability of NEHCs to stabilize a main-group radical system was also investigated. The species [NHC–BH2]˙ are known to be generated by photolysis in the presence of a radical initiator and are observable by EPR spectroscopy,44 and find utility in promoting radical reactions and polymerization processes.45 While organosubstituted NHC–BR2 analogues can be rendered persistent,46 the BH2 analogues rapidly decompose in the absence of steric protection and/or increased delocalization. While NEHCs have only one nitrogen substituent for steric protection versus two in NHCs, the chalcogen atom is expected to have a significant effect on electronic stabilization of radicals. To model the influence of chalcogen substitution on this class of compound, energetics were computed using Me-substituted NEHCs and Me2im for boryl radical formation, and dimerization of the boryl radical to a diborane, which has been observed experimentally for monosubstituted systems NHC–B(R)H,44 and during the reduction of NHC–BBr3.47 The former process was modelled as the hydrogen atom abstraction from the borane adducts of the ligands by tert-butoxide radical (eqn (1)), while the latter was modelled as the dimerization of two NEHC–BH2˙ radicals (eqn (2)).| NEHC–BH3 + tBuO˙ → [NEHC–BH2]˙ + tBuOH | (1) |
| 2[NEHC–BH2]˙ → [(NEHC)BH2–BH2(NEHC)] | (2) |
The Gibbs energy changes (Table 5) for eqn (1) show that hydrogen atom abstraction is more favourable for the chalcogenated NEHC ligands versus Me2im, to an increasing degree as the chalcogen size increases. Natural population analysis was performed to analyze the spin density of the radical products (Table 6). In all cases the spin density on N–C–E atoms and the boron centre comprised >96% of the total spin. As chalcogen size increases, spin density on boron decreases with a concomitant increase at the chalcogen atom, while the sum of the spin densities on the carbene C and N atoms remains essentially the same at ca. 38–40%. In the case of Me2im, over 55% of the spin density resides on the boron atom, whereas for the NEHCs less than half of the spin density is found at the B atom. The tendency of the radicals to dimerize also decreases with increasing chalcogen size, with ΔG° values for the NEHCs ranging from ca. −88 to −34 kJ mol−1, cf. −122 for Me2im. The NEHCs are thus feasible candidates for the stabilization of paramagnetic species such as BH2, particularly for the heavier Se and Te analogues and with a large substituent at nitrogen for additional steric protection.
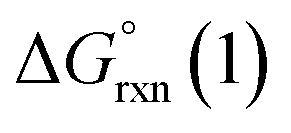
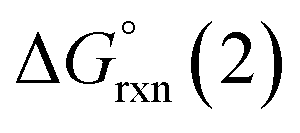
| Ligand | Me2im | NOHC | NSHC | NSeHC | NTeHC |
|---|---|---|---|---|---|
| SD(B) | 55.9 | 48.0 | 38.8 | 36.3 | 33.2 |
| SD(C) | 10.9 | 20.7 | 20.4 | 21.8 | 22.8 |
| SD(N) | 15.4 | 17.3 | 17.8 | 17.6 | 17.4 |
| SD(E) | 15.4 | 12.0 | 20.5 | 21.6 | 23.0 |
Conclusions
The first computational study of a full series of chalcogenazol-2-ylidene (NEHC) carbenes indicates they are attractive synthetic targets with a number of potentially useful ligand properties. While more likely to undergo dimerization than NHCs, the species should be useable in situ, and stabilization by additional steric protection, or the use of a silver complex as a transfer agent may increase their utility. The simple and rapid computational model for carbene donor properties presented is promising, and warrants validation using experimental data from an expanded library of carbenes. The predicted weaker σ donor and stronger π acceptor properties of NEHCs compared to commonly-used NHCs make them suitable for coordination to electron-rich metals, or for stabilizing paramagnetic main group species. The NEHC dimers are also valuable targets as possible sources of organic electron donors, organocatalysts, and precursors to stable organic radicals. Experimental studies of possible synthetic routes to NEHCs and their dimers are currently underway.Experimental section
Computational details
Geometry optimizations were performed with the GAMESS48 package without symmetry constraints utilizing the ωB97X-D49 density functional, from starting coordinates calculated at the Hartree–Fock level of theory. This hybrid meta-GGA functional was used as it a generally strong performer for main group thermochemistry.50 The triple-ζ basis set def2-TZVP51 was used for all atoms. For tellurium, the core electrons were modelled using an effective core potential (ECP). Closed shell species were modeled using the spin-restricted formalism, while open shell species were modeled with the spin-unrestricted formalism. Hessian calculations were performed to confirm the nature of all stationary points as either local minima or maxima on the potential energy surface, and to obtain thermochemical corrections at standard conditions. Gibbs energies are calculated at 1 atm and 298.15 K. For reaction studies, transition states were evaluated using the intrinsic reaction coordinate (IRC) method to ensure they were connected to the proposed reactants and products. Geometric analyses were performed on transition state geometries using the Olex2 software package.52 Interplanar angles (θ) are the minimum angles between the normals of the least-squares planes for the five ring atoms on each NEHC fragment, when viewed down the CC bond.
Natural bond orbital (NBO) calculations, including natural atomic charges and natural atomic spin densities by Natural Population Analysis (NPA), and donor–acceptor interactions using second-order perturbation theory analysis of the Fock matrix, were performed using the program NBO7,53 as interfaced with Gaussian16,54 at the same level of theory as the geometry optimizations (ωB97X-D/def2-TZVP).
NMR properties were evaluated with the ORCA55 package (version 5.0.3) by first optimizing geometries of the selenoureas at the PBE/def2-TZVP level of theory, and then performing NMR shielding and spin–spin coupling calculations including the zeroth-order regular approximation (ZORA)56 using the PBE57 functional and relativistically-contracted all-electron basis sets (SARC58 for tellurium, and ZORA-def2-TZVP as implemented in ORCA for all other elements). The Resolution of Identity approximation for Coulomb integrals (RI-J) was used for ORCA calculations.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from NSERC (Canada) and The University of Winnipeg, as well as computational resources from Digital Research Alliance of Canada, are gratefully acknowledged. Dr Joshua Hollett is thanked for useful discussions.References
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1989, 28, 621–622 CrossRef.
- F. E. Hahn, M. Tamm and T. Lügger, Angew. Chem., Int. Ed. Engl., 1994, 33, 1356–1359 CrossRef.
- A. J. Arduengo, J. R. Goerlich and W. J. Marshall, Liebigs Ann., 1997, 1997, 365–374 CrossRef.
- S. Gründemann, A. Kovacevic, M. Albrecht, J. W. F. Robert and H. Crabtree, Chem. Commun., 2001, 2274–2275 RSC.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- R. Lebeuf, K. Hirano and F. Glorius, Org. Lett., 2008, 10, 4243–4246 CrossRef CAS PubMed.
- I. Piel, M. D. Pawelczyk, K. Hirano, R. Fröhlich and F. Glorius, Eur. J. Org. Chem., 2011, 2011, 5475–5484 CrossRef CAS.
- J. F. Binder, A. M. Corrente and C. L. B. Macdonald, Dalton Trans., 2016, 45, 2138–2147 RSC.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- J. Zhang, T. Li, X. Li, A. Lv, X. Li, Z. Wang, R. Wang, Y. Ma, R. Fang, R. Szostak and M. Szostak, Commun. Chem., 2022, 5, 1–11 CrossRef CAS PubMed.
- Z. Kelemen, O. Hollóczki, J. Oláh and L. Nyulászi, RSC Adv., 2013, 3, 7970–7978 RSC.
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS.
- G. Ciancaleoni, N. Scafuri, G. Bistoni, A. Macchioni, F. Tarantelli, D. Zuccaccia and L. Belpassi, Inorg. Chem., 2014, 53, 9907–9916 CrossRef CAS PubMed.
- K. Verlinden, H. Buhl, W. Frank and C. Ganter, Eur. J. Inorg. Chem., 2015, 2416–2425 CrossRef CAS.
- A. Liske, K. Verlinden, H. Buhl, K. Schaper and C. Ganter, Organometallics, 2013, 32, 5269–5272 CrossRef CAS.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- E. A. Carter and W. A. Goddard III, J. Phys. Chem., 1986, 90, 998–1001 CrossRef CAS.
- M. Vasiliu, K. C. Edwards, D. Tapu, C. E. Castillo, T. H. Stein, R. Craciun, A. J. I. Arduengo and D. A. Dixon, J. Phys. Chem. A, 2022, 126, 2658–2669 CrossRef CAS PubMed.
- A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679–2681 CrossRef CAS.
- M.-J. Cheng and C.-H. Hu, Chem. Phys. Lett., 2001, 349, 477–482 CrossRef CAS.
- R. W. Alder, M. E. Blake, L. Chaker, J. N. Harvey, F. Paolini and J. Schütz, Angew. Chem., Int. Ed., 2004, 43, 5896–5911 CrossRef CAS PubMed.
- D. C. Graham, K. J. Cavell and B. F. Yates, J. Phys. Org. Chem., 2005, 18, 298–309 CrossRef CAS.
- L. Nyulászi, T. Veszprémi and A. Forró, Phys. Chem. Chem. Phys., 2000, 2, 3127–3129 RSC.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554–1561 CrossRef CAS PubMed.
- A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530–5534 CrossRef CAS.
- J. C. Bernhammer, G. Frison and H. V. Huynh, Chem.–Eur. J., 2013, 19, 12892–12905 CrossRef CAS PubMed.
- A. A. Tukov, A. T. Normand and M. S. Nechaev, Dalton Trans., 2009, 7015–7028 RSC.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS.
- G. Meng, L. Kakalis, S. P. Nolan and M. Szostak, Tetrahedron Lett., 2019, 60, 378–381 CrossRef CAS.
- S. V. C. Vummaleti, D. J. Nelson, A. Poater, A. Gómez-Suárez, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and L. Cavallo, Chem. Sci., 2015, 6, 1895–1904 RSC.
- D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- M. Tretiakov, Y. G. Shermolovich, A. P. Singh, P. P. Samuel, H. W. Roesky, B. Niepötter, A. Visscher and D. Stalke, Dalton Trans., 2013, 42, 12940–12946 RSC.
- S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed.
- S. Kundu, S. Sinhababu, V. Chandrasekhar and H. W. Roesky, Chem. Sci., 2019, 10, 4727–4741 RSC.
- V. P. W. Böhm and W. A. Herrmann, Angew. Chem., Int. Ed., 2000, 39, 4036–4038 CrossRef.
- J. A. Murphy, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed.
- J. C. Walton, M. M. Brahmi, J. Monot, L. Fensterbank, M. Malacria, D. P. Curran and E. Lacôte, J. Am. Chem. Soc., 2011, 133, 10312–10321 CrossRef CAS PubMed.
- T. Taniguchi, Chem. Soc. Rev., 2021, 50, 8995–9021 RSC.
- R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, Wisconsin, USA, 2018 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, M. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzweski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (version Revision C.01), Gaussian, Inc., Wallingford, CT, USA, 2019 Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. D. Rolfes, F. Neese and D. A. Pantazis, J. Comput. Chem., 2020, 41, 1842–1849 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic coordinates and energies of all optimized structures (.xyz format). See DOI: https://doi.org/10.1039/d3ra03324d |
| This journal is © The Royal Society of Chemistry 2023 |