
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of fluorous boronic acid catalysts integrated with sulfur for enhanced amidation efficiency†
Kevin Timothy Fridiantoa,
Ya-Ping Wenb,
Lee-Chiang Lo *b and
Yulin Lam*a
*b and
Yulin Lam*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmlamyl@nus.edu.sg
bDepartment of Chemistry, National Taiwan University, No. 1, Sec. 4 Roosevelt Road, Taipei 106, Taiwan. E-mail: lclo@ntu.edu.tw
First published on 9th June 2023
Abstract
A thermally stable, fluorous sulfur-containing boronic acid catalyst has been developed and was shown to efficiently promote dehydrative condensation between carboxylic acids and amines under environmentally friendly conditions. The methodology can be applied to aliphatic, aromatic and heteroaromatic acids as well as primary and secondary amines. N-Boc protected amino acids were also successfully coupled in good yields with very little racemization. The catalyst could be reused four times with no significant loss of activity.
Introduction
The amide moiety is the most fundamental functional group in biology and chemistry. Besides being the key constituent of proteins, it is found in a wide variety of natural products, pharmaceuticals, fine chemicals and materials. Amides also serve as reaction intermediates for accessing other compound classes like heterocycles and complex amines.1 Consequently the amide bond formation reaction is one of the most frequently performed reactions in organic chemistry research laboratories, and industrially it is commonplace in polymer, chemical, agrochemical and pharmaceutical production.2 In view of its relevance, a plethora of strategies to generate the amide bond from alkene, alkynes, carbonyl compounds and other functional groups have been reported.3 Despite these developments, the direct condensation of carboxylic acids and amines in the presence of stoichiometric quantities of coupling reagents remains the most frequently employed approach to amide bond formation.4 However this process leads to the generation of large quantities of waste making it nonoptimal from the perspective of atom economy and cost effectiveness. To mitigate this issue, novel approaches to access amide containing molecules via transition metal or metal-free catalysis were developed.5 Among these approaches, the catalytic dehydrative condensation between carboxylic acids and amines is one of the most attractive methods because the only by-product of the reaction is water.Using boronic acid as a catalyst for dehydrative condensation between carboxylic acids and amines have received much attention since the seminal study by Yamamoto and coworkers in 1996.5m Its popularity may be attributed to the catalyst's versatile reactivity, stability, low toxicity and its ultimate degradation to boric acid.6 Thus boronic acids are regarded as safe compounds and through the years, various arylboronic acid catalysts which promote the direct amidation reaction have been reported.7 However these reactions are almost universally carried out in toluene, o-xylene and chlorinated solvents which present safety and environmental issues and cost associated with their disposal. In addition, the water by-product is often scavenged by adding excess molecular sieves into the reaction mixture or by azeotropic reflux. The use of molecular sieves is common in the literature as it enables the catalytic amidation to occur at lower temperatures but they could possibly lead to increase solvent usage during the reaction and workup. Moreover molecular sieves are impractical in large-scale reactions due to the need to adequately dry them under high temperature and vacuum before use (large energy input).5b
The move towards the adoption of sustainable chemistry has stimulated renewed interest in new methodologies for boronic acid-catalyzed amide bond formation. The most prevalent strategy involves the development of reusable boronic acid such as solid-phase,8 mesocellular siliceous foam (MCF)-supported,9 fluorous10 and phase-transfer boronic acid catalysts.8b Despite the potential reusability of these boronic acids, the solvents employed in the amidation reactions remained unchanged. Since there is considerable merit in promoting the use of less harmful solvents11 which would also help to increase sustainability within the industry, we sought to develop an operationally simple catalytic protocol for the formation of amide bonds. We herein present a new recyclable boronic acid catalyst which is able to promote amide/peptide bond formation reaction under neat or environmentally friendlier solvent conditions and without the need for any additives or dehydrating agents.
Results and discussion
Inspired by earlier works on o-substituted phenylboronic acid11 catalyzed reaction between carboxylic acids and amines which suggested that the o-substituent (X) is capable of engaging in O–H⋯X hydrogen bonding to stabilize the rate-determining transition-state and thus facilitate hemiaminal dehydration, we designed two sulfur-containing catalysts 1a–b (Fig. 1) to appraise both the effects of the Lewis basicity of sulfur and the distance between the sulfur and boron on amide bond formation. A perfluoroalkyl chain (–C8F17) was tagged to each of the catalysts to enable easy purification and catalyst recovery. Catalysts 1a–b are classified as “light fluorous” compounds (fluorine content <60%)12 which have lower toxicity and persistence compared to “heavy fluorous” ones. They can be separated from the non-fluorous compounds using fluorous silica gel eluted with methanol and the fluorous silica gel can be reused multiple times after washing with acetone.13 Thus, compared to conventional column chromatography, this technique reduces the volume of solvent used and waste generated. | ||
| Fig. 1 Boronic acid catalysts. | ||
Initial assessment of the catalytic efficiencies of 1a–b (10 mol%) was conducted using phenylacetic acid A (1.1 equiv.) and benzylamine B (1 equiv.) in toluene under azeotropic reflux with removal of water via a Dean–Stark apparatus. The reaction was found to be completed after 17 h and provided N-benzyl-2-phenylacetamide 2a in 73 and 99% yields respectively (Table 1, entry 1). Similarly, lactamization of 6-aminohexanoic acid under the same reaction condition for 20 h with catalyst 1b also gave ε-caprolactam in higher yield than with catalyst 1a (Table 1, entry 2). In both reactions, the spent catalysts 1a–b were readily recovered in quantitative yields. Since 1b proved to be the more effective catalyst for this model reaction, it was used for subsequent reactions.
| Entry | Substrate | Yield (%) | |
|---|---|---|---|
| 1a | 1b | ||
| 1 |  |
73 | 99 |
| 2 |  |
70 | 90 |
To optimize the reaction conditions, we first varied the solvent (Table 2, entries 2–4) and found that the reaction proceeded most efficiently under neat condition. Encouraged by the result, we investigated if the reaction could be carried out in a sealed tube under microwave irradiation. A neat reaction mixture was thus microwave irradiated at 130 °C and the reaction was followed by TLC every 15 min. The reaction was completed after 1 h and provided 2a in 82% (Table 2, entry 5). Optimization of the reaction conditions by varying temperature, equivalence of A and B, and catalyst loading eventually gave 2a in 99% yield when the reaction was performed under neat condition with A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B = 1:1.3 equivalence and 5 mol% of catalyst 1b at 150 °C for 15 min (Table 2, entry 9). After the reaction, the excess base was removed by an aqueous acid wash and the product 2a and catalyst 1b were easily separated via fluorous solid-phase extraction (F-SPE) with fluorous silica and methanol–water as eluent. This process significantly reduced the reaction time and solvent requirements as there was no need for chromatographic purification.
:B = 1:1.3 equivalence and 5 mol% of catalyst 1b at 150 °C for 15 min (Table 2, entry 9). After the reaction, the excess base was removed by an aqueous acid wash and the product 2a and catalyst 1b were easily separated via fluorous solid-phase extraction (F-SPE) with fluorous silica and methanol–water as eluent. This process significantly reduced the reaction time and solvent requirements as there was no need for chromatographic purification.
| Entry | Solvent | Catalyst loading | A:B |
Temp, time | Yieldd (%) |
|---|---|---|---|---|---|
| a Azeotropic reflux.b Conventional heating with water removal via a Dean–Stark apparatus.c MW.d Isolated yields. | |||||
| 1a | Toluene | 10 mol% | 1.1:1 |
125 °C, 17 h | 99 |
| 2a | 2-MeTHF | 10 mol% | 1.1:1 |
100 °C, 17 h | 44 |
| 3a | Anisole | 10 mol% | 1.1:1 |
130 °C, 17 h | 71 |
| 4b | Neat | 10 mol% | 1.1:1 |
130 °C, 17 h | 82 |
| 5c | Neat | 10 mol% | 1.1:1 |
130 °C, 1 h | 82 |
| 6c | Neat | 10 mol% | 1:1.1 |
130 °C, 1 h | 87 |
| 7c | Neat | 10 mol% | 1:1.3 |
130 °C, 1 h | 91 |
| 8c | Neat | 10 mol% | 1:1.3 |
150 °C, 15 min | 94 |
| 9c | Neat | 5 mol% | 1:1.3 |
150 °C, 15 min | 99 |
| 10c | Neat | 1 mol% | 1:1.3 |
150 °C, 15 min | 93 |
To explore the generality of this process, we prepared a selection of amides using various primary amines and structurally different carboxylic acids. The reaction proceeded cleanly to give the desired secondary amides 2a–2g in excellent yields. Catalyst 1b was also useful for reactions between secondary amines and carboxylic acids to give tertiary amides (2k–2m, Fig. 2) in very good yields.
 | ||
| Fig. 2 Scope of 1b-catalyzed amide bond formation. | ||
To investigate if the methodology was applicable to peptide synthesis, we directed our efforts to the amidation of amino acids as they are important renewable raw materials for the synthesis of biological active targets. Initial assessment was conducted using the lactamization reaction of 5-aminovaleric acid. However under neat conditions, the isolated yield of the desired lactam was low with the side-products arising from intermolecular amidation reaction. To circumvent this problem, we diluted the reaction mixture with solvent such as anisole or benzotrifluoride14 and found that in the latter solvent, the side-product was absent and δ-valerolactam 2i was obtained in 95% yield. Similarly, 4-aminobutyric acid and 6-aminocaproic acid gave γ-butyrolactam 2h and ε-caprolactam 2j respectively in excellent yields. Encouraged by these results, we proceeded to explore other coupling partners for this reaction.
Amidation of N-Boc-protected amino acids with benzylamine under neat condition and microwave irradiation at 130 °C gave the corresponding amino amides (2n–2p, Fig. 2) in good yields. However when the same reaction condition was applied to dipeptide synthesis using Boc-glycine and phenylalanine methyl ester, the reaction was sluggish and did not proceed to completion. This was attributed to the heterogenous reaction mixture and thus solvent was added to keep the reaction mixture liquid. We explored different solvents (entries 2–6, Table 3) and found that the reaction proceeded most efficiently in benzotrifluoride providing the desired dipeptide 2q in 90% yield. However when the same reaction condition was applied to the dipeptide synthesis between Boc-leucine and phenylalanine, HPLC analysis of the product showed that the compound was a 60:40 mixture of two diastereomers. To address the problem of racemization, the reaction was carried out via azeotrope water removal (reverse Dean–Stark conditions) in benzotrifluoride (azeotropic temperature for 90% benzotrifluoride14b is 80 °C) at lower temperatures (entries 7–8, Table 3). Pleasingly, the reactions of phenylalanine with Boc-glycine and Boc-leucine both proceeded efficiently at 85 °C to provide compounds 2q and 2r in 87% and 73% yields respectively and there was also no detectable racemization in the reaction of Boc-leucine and phenylalanine. Amidation using other amino acids also gave the corresponding dipeptides in good yields.
| Entry | Solvent | Method, temp, time | Yield (%) |
|---|---|---|---|
| 1 | Neat | MW, 130 °C, 2 h | 30 |
| 2 | Diethyl carbonate | MW, 130 °C, 2 h | 60 |
| 3 | Ethylene carbonate | MW, 130 °C, 2 h | 40 |
| 4 | Propylene carbonate | MW, 130 °C, 2 h | 45 |
| 5 | Benzotrifluoride | MW, 130 °C, 2 h | 90 |
| 6 | Cyclopentyl methyl ether | MW, 130 °C, 2 h | 55 |
| 7 | Benzotrifluoride | Azeotropic reflux, 120 °C, 16 h | 65 |
| 8 | Benzotrifluoride | Azeotropic reflux, 85 °C, 36 h | 87 |
Next, we investigated the possibility of recycling and reusing catalyst 1b. The amidation reactions were performed under the optimized reaction conditions and the recycling experiments were carried out over 4 cycles (Table 4). Gratifyingly, the fluorous boronic acid catalyst 1b recovered from reaction under microwave irradiation at 150 °C or prolonged reflux conditions was indistinguishable from the original catalyst and showed minimal decline in catalytic activity even after multiple recovery steps. Comparison of the yields after each cycle after a fixed reaction time also showed that the recovery of catalyst 1b was very good.
 |
||||
| Cycle | 1 | 2 | 3 | 4 |
| Yield (%) | 99 | 99 | 99 | 92 |
| Recovered 1b (wt%) | 99 | 99 | 99 | 97 |
 |
||||
| Cycle | 1 | 2 | 3 | 4 |
| Yield (%) | 87 | 85 | 85 | 85 |
| Recovered 1b (wt%) | 99 | 99 | 99 | 99 |
Experimental
General
All chemicals purchased were used without further purification. Reactions were carried out under N2 with commercially obtained anhydrous solvents. Analytical thin-layer chromatography (TLC) was carried out on precoated F254 silica plates and visualized with UV light. F-SPF was performed with FluoroFlash® silica gel (40 micron). 1H and 13C NMR spectra were recorded at 298 K. Chemical shifts are expressed in terms of ppm relative to the internal standard tetramethylsilane (TMS). Mass spectra were performed under EI and ESI mode. Microwave reactions were performed on the Anton Paar Monowave 400 microwave synthesizer in quartz pressure tubes.Synthesis of fluorous boronic acid 1b
The synthesis of fluorous catalyst 1b is outlined in Scheme 1.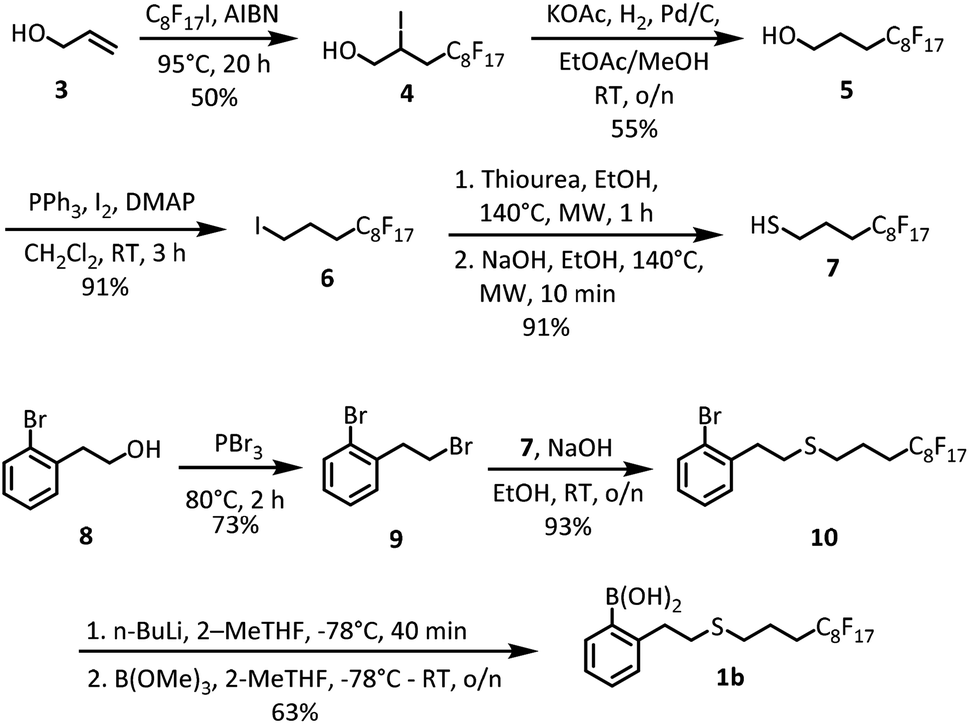 | ||
| Scheme 1 Synthesis of catalyst 1b. | ||
4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-Heptadecafluoro-2-iodo-1-undecanol (4)
A mixture of C8F17I (12.3 mL, 46.6 mmol) and AIBN (0.197 g, 1.2 mmol) was stirred at 95 °C for 15 min under N2. To the mixture, 3 (4.1 mL, 60.3 mmol) was added dropwise and the resulting solution was stirred for 20 h at 95 °C. After completion of reaction, the crude mixture was cooled to room temperature and the resulting solid dissolved in methanol. The crude mixture was evaporated under reduced pressure and recrystallized from hexane to give 4 as an off-white solid (13.9 g, 50%). 1H NMR (400 MHz, CDCl3): δ 4.47–4.41 (m, 1H), 3.86–3.77 (m, 2H), 3.09–2.94 (m, 1H), 2.85–2.69 (m, 1H), 1.81 (br s, 1H). 13C NMR (125 MHz, CDCl3): δ 67.9, 37.5 (t, J(C, F) = 20 Hz), 21.8. 19F NMR (377 MHz, CDCl3): δ −80.8 (t, J = 9.69 Hz, 3F), −112.2 to −114.5 (qt, J = 269.9, 13.8 Hz, 2F), −121.5 to −121.6 (m, 2F), −121.9 (m, 4F), −122.7 (m, 2F), −123.5 (m, 2F), −126.1 (m, 2F). HRMS (+EI) calcd. for C11H6F17OI: 603.9186; found: 603.9196.4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-Heptadecafluoro-1-undecanol (5)
To a solution of 4 (13.9 g, 23 mmol) in 2:1 EA:MeOH (90 mL), 10 wt% Pd/C (3 g) and KOAc (18.1 g, 0.184 mol) was added and stirred overnight under H2. The reaction mixture was then filtered through a pad of Celite and the solvent was removed under reduced pressure. The residue was taken up in CH2Cl2 and washed successively with H2O (3 × 50 mL) and brine (1 × 50 mL). The organic layer was dried over MgSO4 and dry-loaded onto silica. The crude product was purified through column chromatography using a gradient eluent system of hexane:EA (16:1 to 4:1) to give 5 as a colourless oil, which solidifies at room temperature to a white crystalline solid (6.03 g, 55%). 1H NMR (400 MHz, CDCl3): δ 3.74 (t, J = 6.12 Hz, 2H), 2.28–2.15 (m, 2H), 1.90–1.83 (m, 2H), 1.78 (br s, 1H). 13C NMR (100 MHz, CDCl3): δ 61.4, 27.6 (t, J = 22.1 Hz, C–F), 23.4. 19F NMR (377 MHz, CDCl3): δ −80.9 (t, J = 10.63 Hz, 3F), −114.4 (t, J = 13.3 Hz, 2F), −121.8 to −122.0 (m, 6F), −122.8 (m, 2F), −123.5 (m, 2F), −126.2 (m, 2F). HRMS (+EI) calcd. for C11H6F17O: 477.0142; found: 477.0146.
1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8-Heptadecafluoro-11-iodoundecane (6)
A mixture of 5 (6.03 g, 12.6 mmol), triphenylphosphine (3.97 g, 15.1 mmol) and 4,4-dimethylaminopyridine (2.31 g, 18.9 mmol) in CH2Cl2 (50 mL) was stirred at room temperature for 15 min. Iodine (4.8 g, 18.9 mmol) was added and the reaction was stirred for 3 h at room temperature. After completion of reaction, the mixture was quenched using 5% Na2S2O4 solution (15 mL) and washed successively with H2O (3 × 50 mL) and brine (1 × 50 mL). The organic layer was dried over MgSO4 and dry-loaded onto silica. The crude product was purified using column chromatography with pure hexane to give the desired product as a colourless oil, which solidifies at room temperature to a white, waxy solid (6.78 g, 91%). 1H NMR (400 MHz, CDCl3): δ 3.25 (t, J = 6.6 Hz, 2H), 2.29–2.10 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 31.9 (t, J = 22.2 Hz, C–F), 24.3, 3.9. 19F NMR (377 MHz, CDCl3): δ −80.8 (t, J = 11.31 Hz, 3F), −113.7 (quint, J = 16.1 Hz, 2F), −121.7 to −121.9 (m, 6F), −122.7 (m, 2F), −123.4 (m, 2F), −126.1 to −126.2 (m, 2F). HRMS (+EI) calcd. for C11H6F17I: 587.9237; found: 587.9241.4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-Heptadecafluoro-1-undecanethiol (7)
A solution of 6 (6.78 g, 11.5 mmol) and thiourea (1.05 g, 13.8 mmol) in deoxygenated EtOH (15 mL) was stirred at 140 °C under MW irradiation for 1 h. 4 M NaOH (3 mL) was then added into the clear solution and the mixture heated to 140 °C under MW irradiation for 10 min. The resulting light-yellow suspension was acidified using 4 M HCl (3 mL) and the cloudy suspension was extracted with diethyl ether (3 × 50 mL). The combined organic layer was washed with water (1 × 50 mL), brine (1 × 50 mL), dried over anhydrous MgSO4 and evaporated under reduced pressure. The crude mixture was reconstituted in 30 mL hexane and filtered through a short silica column to yield the desired product as a yellowish oil (5.19 g, 91%). 1H NMR (400 MHz, CDCl3): δ 2.63 (dt, J = 8.0, 7.0 Hz, 2H), 2.29–2.16 (m, 2H), 1.97–1.80 (m, 2H), 1.38 (t, J = 8.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 29.5 (t, J(C, F) = 22.3 Hz), 24.6 (t, J(C, F) = 3.6 Hz), 23.9. 19F NMR (376 MHz, CDCl3): −80.8 (t, J = 10.0 Hz, 3F), −113.9 to −114.03 (m, 2F), −121.7 to −121.9 (m, 6F), −122.7 (m, 2F), −123.5 (m, 2F), −126.1 to −126.2 (m, 2F).1-Bromo-2(2-bromoethyl)benzene (9)
The procedure reported by Blanckaert et al. was used.15 2-(2-Bromophenyl)ethanol (8) (1.48 g, 7.4 mmol) was added to a 25 mL vial, and the vial was cooled to 0 °C under N2. Phosphorus tribromide (2.4 g, 0.83 mL, 8.9 mmol) was added dropwise. The reaction mixture was heated at 80 °C for 2 h. The mixture was poured onto crushed ice, saturated NaHCO3 solution (20 mL) was added, and the mixture was stirred for 30 min. The mixture was extracted with chloroform (3 × 20 mL) and the combined extracts washed once with saturated NaHCO3 solution (20 mL) and once with brine (20 mL). The solution was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. A clear oil was obtained, which was sufficiently pure and was used directly without further purification (1.42 g, 73%). 1H NMR (400 MHz, CDCl3): δ 7.60–7.56 (m, 1H), 7.31–7.27 (m, 2H), 7.18–7.14 (m, 1H), 3.62 (t, J = 7.4 Hz, 2H), 3.32 (t, J = 7.68 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 138.1, 133.0, 131.1, 128.7, 127.5, 124.3, 39.5, 30.9. MS (+ESI) calcd. for C8H9Br2: 264.96; found: 264.79.(2-Bromophenethyl)(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)sulfane (10)
Compound 7 (5.19 g, 10.5 mmol) and NaOH (0.467 g, 11.7 mmol) were dissolved in degassed anhydrous EtOH (30 mL) and allowed to stir for 30 min at room temperature. A solution of 9 (3.06 g, 11.7 mmol) in 20 mL degassed anhydrous EtOH was added via syringe. The flask containing 9 was rinsed with 2 × 10 mL anhydrous EtOH and the combined reaction mixture was left to stir overnight at room temperature. Upon completion of the reaction, the colourless solution was evaporated to dryness before being redissolved in CH2Cl2 (50 mL), washed with water (25 mL), brine (1 × 25 mL) and dried over MgSO4. The evaporated crude sample was purified by column chromatography (0–5% ethyl acetate in hexane) to afford the desired product as a yellowish liquid (6.65 g, 93%). 1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 7.84 Hz, 1H), 7.26–7.24 (dd, J = 4.88, 0.72 Hz, 1H), 7.12–7.07 (m, 1H), 3.04–2.99 (m, 2H), 2.79–2.76 (m, 2H), 2.64 (t, J = 7.04 Hz, 2H), 2.27–2.14 (m, 2H), 1.93 (quint, J = 7.08 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 139.5, 132.9, 130.8, 128.3, 127.6, 124.2, 36.8, 31.4, 31.2, 29.8, 20.2. 19F NMR (377 MHz, CDCl3): δ −80.7 (t, J = 10.25 Hz, 3F), −114.0 (quint, J = 17.15 Hz, 2F), −121.7 to −121.9 (m, 7F), −122.7 (m, 2F), −123.4 (m, 2F), −126.0 to −126.1 (m, 2F). HRMS (APCI) calcd. for C19H14F17S: 597.0539; found: 597.0543.(2-(2-((4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-Heptadecafluoroundecyl)thio)ethyl)phenyl)boronic acid (1b)
The procedure by El Dine et al. was used.11e To a solution of 10 (6.65 g, 9.8 mmol) in 10 mL dry 2-methyltetrahydrofuran at −78 °C under N2 atmosphere was added dropwise n-BuLi solution (2 M in cyclohexane, 5.4 mL, 1.1 equiv.). The resulting mixture was stirred for 40 min, after which B(OMe)3 (10.4 mL, 93.3 mmol, 9.5 equiv.) was added at −78 °C. The mixture was allowed to warm slowly to room temperature and stirred overnight. After the addition of 10 mL distilled water, the mixture was acidified with 1 M HCl to pH 1 and extracted with 30 mL EtOAc. The combined organic layers were washed with brine, dried over MgSO4, and concentrated under vacuum. The resulting solid was recrystallized from hexane to give the desired product as a white solid (3.97 g, 63%). 1H NMR (400 MHz, acetone-d6): δ 7.62 (d, J = 7.32 Hz, 1H), 7.31–7.27 (td, J = 7.52, 1.52 Hz, 1H), 7.23–7.21 (m, 1H), 7.20 (s, 2H), 7.18–7.14 (td, J = 7.32, 1.36 Hz, 1H), 3.15–3.11 (m, 2H), 2.79–2.75 (m, 2H), 2.72 (t, J = 7.08 Hz, 2H), 2.44–2.31 (m, 2H), 1.96–1.89 (quint, J = 7.6 Hz, 2H). 13C NMR (125 MHz, acetone-d6): δ 135.0, 130.7, 130.3, 130.2, 126.4, 126.2, 37.7, 34.7, 31.3, 31.2, 21.3. 19F NMR (377 MHz, acetone-d6): δ −81.7 (t, J = 10.67 Hz, 3F), −114.4 (quint, J = 16.73 Hz, 2F), −122.3 to −122.5 (m, 6F), −123.3 (m, 2F), −123.9 (m, 2F), −126.7 to −126.8 (m, 2F). 11B NMR (128 MHz, acetone-d6): δ 30.2 (s, 1B). HRMS (+ESI) calcd. for C19H15[11B]F17O2S: 641.0609; found: 641.0617.General procedure for the synthesis of amides under azeotropic reflux
Into a two-neck round bottom flask equipped with a magnetic stirring bar was added the corresponding catalyst (0.05 mmol), phenylacetic acid (1 mmol), benzylamine (1.3 mmol), and solvent (5 mL). A Dean–Stark trap was fitted to the center neck, and a reflux condenser was fitted on top of the Dean–Stark trap. The reaction setup was wrapped with aluminium foil and heated in an oil bath for 17 h at the indicated temperature. The reaction mixture was then cooled down and transferred into a separatory funnel with ethyl acetate. The organic layer was washed with saturated Na2CO3 solution (3 mL), 5% citric acid solution (3 mL) and concentrated under reduced pressure. The resulting crude was reconstituted in 2-methyltetrahydrofuran (0.2 mL) and subjected to fluorous SPE. The non-fluorous and fluorous fractions were concentrated under reduced pressure to afford the amide and the recovered catalyst, respectively.General procedure for synthesis of amides under neat condition and microwave irradiation
Into a G10 Anton Paar microwave vial equipped with a magnetic stirring bar was added the corresponding catalyst (0.05 eq., 5 mol%), acid (1 eq.), and amine (1.3 eq.). The vial was capped and subjected to microwave irradiation for the indicated reaction time at 150 °C, maximum power. Upon complete consumption of acid as confirmed by TLC, the reaction mixture was reconstituted in 5 mL ethyl acetate. The organic layer was washed with saturated Na2CO3 solution (3 mL), 5% citric acid solution (3 mL) then concentrated under reduced pressure. The resulting crude was then reconstituted in 2-methyltetrahydrofuran (0.2 mL) and subjected to fluorous SPE. The non-fluorous and fluorous fractions were concentrated under reduced pressure to afford the amide and the recovered catalyst, respectively.General procedure for fluorous SPE
2 g of FluoroFlash silica was added into a 10 mL cartridge. The fluorous silica was conditioned with 80:20 MeOH/H2O (5 mL). The crude mixture was dissolved in 2-methyltetrahydrofuran (0.2 mL) and loaded onto the column using positive pressure, ensuring that all the solution is properly loaded. The flask containing the crude mixture was further rinsed with 3 × 1 mL 80:20 MeOH/H2O to transfer the remaining crude product. The column was then flushed with 20% H2O in MeOH (10 mL) to elute the non-fluorous fraction, and subsequently with MeOH (12 mL) to elute the fluorous fraction. Upon completion, the column was washed with acetone (5 mL). Prior to reuse, the column was reconditioned with 80:20 MeOH/H2O (5 mL).
General procedure for the synthesis of dipeptides under azeotropic reflux
Into a two-neck round bottom flask equipped with a magnetic stirring bar was added the corresponding catalyst (0.1 mmol), Boc-protected amino acid (1 mmol), amino acid methyl ester (1 mmol), and benzotrifluoride (5 mL). A reverse Dean–Stark trap was fitted to the center neck, and a reflux condenser was fitted on top of the reverse Dean–Stark trap. The reaction setup was wrapped with aluminium foil and heated in an oil bath for 18–36 h at 85 °C. The reaction mixture was then cooled down and transferred into a separatory funnel with ethyl acetate. The organic layer was washed with saturated Na2CO3 solution (3 mL), 5% citric acid solution (3 mL) and concentrated under reduced pressure. The resulting crude was reconstituted in 2-methyltetrahydrofuran (0.2 mL) and subjected to fluorous SPE. The non-fluorous and fluorous fractions were concentrated under reduced pressure to afford the amide and the recovered catalyst, respectively. In cases where the resulting dipeptide contained impurities, the dipeptide was further purified through a short pad of silica using ethyl acetate as eluent.Determination of diastereomeric purity through chiral HPLC
Diastereomeric purity was determined on an Agilent LC 1200 DAD HPLC system (Agilent Tech. Inc., Loveland, CO). Separation was carried out on a DAICEL Chiralpak ID (4.6 × 250 mm, 5 μm; Daicel Corporation, Japan), using an isocratic elution method (80% hexane/20% isopropanol, flow rate 1 mL min−1, detector channel 210 nm). The chromatogram was monitored for 20 min for the detection of the major peak of test compound, which was expressed as a percentage of total peaks detected during the run.Conclusions
Two versions of sulfur-containing fluorous boronic acid catalysts (1a and 1b) were developed. Catalyst 1b was shown to promote efficient dehydrative condensation between carboxylic acids and amines under environmentally friendly conditions. The methodology could be applied to the synthesis of secondary and tertiary amides, amino amides and dipeptides. Catalyst 1b could also be recovered and reused four times with minimal loss of catalytic activity.Author contributions
L.-C. L. and Y. L. conceptualized this work. Investigation was carried out by K. T. F. and Y.-P. W. and K. T. F., L.-C. L. and Y. L. contributed to the writing of the manuscript. Review and editing were performed by all authors. L.-C. L. and Y. L. contributed to funding acquisition. Supervision was performed by L.-C. L. and Y. L. Corresponding authors L.-C. L. and Y. L. contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Ministry of Education (MoE) Academic Research Fund A-0004133-00-00 to Y. L. and the Taiwan Ministry of Science and Technology (MOST) 110-2113-M-002-017 to L.-C. L.References
- (a) K. Marchildon, Macromol. React. Eng., 2011, 5, 22 CrossRef CAS; (b) A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Material Science, Wiley-VCH, New York, 2003 Search PubMed; (c) M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468 CrossRef CAS PubMed; (d) M. Funabashi, Z. Yang, K. Nonaka, M. Hosobuchi, Y. Fujita, T. Shibata, X. Chi and S. G. Van Lanen, Nat. Chem. Biol., 2010, 6, 581 CrossRef CAS PubMed; (e) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (f) E. Kovács, B. Rózsa, A. Csomos, I. G. Csizmadia and Z. Mucsi, Molecules, 2018, 23, 2859 CrossRef.
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC; (b) R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253 CrossRef CAS; (c) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443 CrossRef CAS; (d) J. Boström, D. G. Brown, R. J. Young and G. M. Keserű, Nat. Rev. Drug Discovery, 2018, 17, 922 CrossRef PubMed.
- (a) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790 CrossRef CAS PubMed; (b) H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796 CrossRef CAS PubMed; (c) W.-K. Chan, C.-M. Ho, M.-K. Wong and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 14796 CrossRef CAS PubMed; (d) E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641 CrossRef CAS.
- (a) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS; (b) K. Scheidt, Nature, 2010, 465, 1020 CrossRef CAS PubMed; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC; (d) J. S. Casey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 RSC.
- (a) B. Mahjour, Y. Shen, W. Liu and T. Cernak, Nature, 2020, 580, 71 CrossRef CAS PubMed; (b) M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10 CrossRef CAS; (c) D. C. Lenstra, F. P. Rutjes and J. Mecinovic, Chem. Commun., 2014, 50, 5763 RSC; (d) R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512 CrossRef CAS PubMed; (e) H. Lundberg, F. Tinnis and H. Adolfsson, Chem.–Eur. J., 2012, 18, 3822 CrossRef CAS PubMed; (f) R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 2013, 7453 CrossRef CAS; (g) N. A. Stephenson, J. Zhu, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 10003 CrossRef CAS PubMed; (h) F. Tinnis, H. Lundberg and H. Adolfsson, Adv. Synth. Catal., 2012, 354, 2531 CrossRef CAS; (i) C. L. Allen, A. R. Chhatwal and J. M. Williams, Chem. Commun., 2012, 48, 666 RSC; (j) H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271 CrossRef CAS; (k) M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef; (l) C. W. Cheung, M. L. Ploeger and X. Hu, Nat. Commun., 2017, 8, 14878 CrossRef PubMed; (m) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem, 1993, 61, 4196 CrossRef PubMed; (n) G. M. Torres, Y. Liu and B. A. Arndtsen, Science, 2020, 368, 318 CrossRef CAS PubMed; (o) L. Bering, E. J. Craven, S. A. Sowerby Thomas, A. A. Shepherd and J. Micklefield, Nat. Commun., 2022, 13, 380 CrossRef CAS PubMed; (p) R. Yamashita, A. Sakakura and K. Ishihara, Org. Lett., 2013, 15, 3654 CrossRef CAS PubMed; (q) E. Dimitrijević and M. S. Taylor, ACS Catal., 2013, 3, 945 CrossRef; (r) E. Dimitrijević and S. Taylor, ACS Catal, 2013, 3, 945 CrossRef.
- (a) S. J. Baker, C. Z. Ding, Y. K. Zhang, V. Hernandez and Y. Xie, Future Med. Chem., 2009, 1, 1275 CrossRef CAS PubMed; (b) M. P. Silva, L. Saraiva, M. Pinto and M. E Sousa, Molecules, 2020, 25, 4323 CrossRef CAS PubMed.
- (a) D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475 RSC; (b) B. Pan, D.-M. Huang, H.-T. Sun, S.-N. Song and X.-B. Su, J. Org. Chem., 2023, 88, 2832 CrossRef CAS PubMed; (c) N. Shimada, M. Hirata, M. Koshizuka, N. Ohse, R. Kaito and K. Makino, Org. Lett., 2019, 21, 4303 CrossRef CAS PubMed; (d) R. M. Al-zoubi, W. K. Al-Jammal and R. McDonald, New J. Chem., 2020, 44, 3612 RSC.
- (a) R. Latta, G. Springsteen and B. Wang, Synthesis, 2001, 11, 1611 CrossRef; (b) T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2005, 7, 5043 CrossRef CAS PubMed; (c) Y. Lu, K. Wang and K. Ishihara, Asian J. Org. Chem., 2017, 6, 1191 CrossRef CAS.
- L. Gu, J. Lim, J. L. Cheong and S. S. Lee, Chem. Commun., 2014, 50, 7017 RSC.
- K. Ishihara, S. Kondo and H. Yamamoto, Synlett, 2001, 9, 1371 CrossRef.
- (a) T. Marcelli, Angew. Chem., Int. Ed., 2010, 49, 6840 CrossRef CAS PubMed; (b) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Green Chem., 2008, 10, 124 RSC; (c) R. M. Al-Zoubi, O. Marion and D. G. Hall, Angew. Chem., Int. Ed., 2008, 47, 2876 CrossRef CAS PubMed; (d) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8486 CrossRef PubMed; (e) T. M. El Dine, W. Erb, Y. Berhault, J. Rouden and J. Blanchet, J. Org. Chem., 2015, 80, 4532 CrossRef PubMed; (f) H. Charville, D. A. Jackson, G. Hodges, A. Whiting and M. R. Wilson, Eur. J. Org. Chem., 2011, 5981 CrossRef CAS.
- W. Zhang, Tetrahedron, 2003, 59, 4475 CrossRef CAS.
- W. Zhang and D. P. Curran, Tetrahedron, 2006, 62, 11837 CrossRef CAS PubMed.
- (a) N. Winterton, Clean Technol. Environ. Policy, 2021, 23, 2499 CrossRef PubMed; (b) J. J. Maul, P. J. Ostrowski, G. A. Ublacker, B. Linclau and D. P. Curran, Top. Curr. Chem., 1999, 206, 79 CrossRef.
- P. Blanckaert, M. Vandecapelle, L. Staelens, I. Burvenich, R. A. Dierckx and G. Slegers, J. Labelled Compd. Radiopharm., 2004, 47, 591 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03300g |
| This journal is © The Royal Society of Chemistry 2023 |