
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLasiodipline G and other diketopiperazine metabolites produced by Lasiodiplodia chiangraiensis†
Sarunpron Khruengsaiab,
Patcharee Pripdeevechabc,
Chutima Tanapichatsakulb,
Winnie Chemutai Suma,
Mahmoud A. A. Ibrahim de,
Marc Stadlera and
Sherif S. Ebada*af
de,
Marc Stadlera and
Sherif S. Ebada*af
aDepartment of Microbial Drugs, Helmholtz Centre for Infection Research GmbH (HZI), Inhoffenstraße 7, 38124 Braunschweig, Germany. E-mail: sherif.elsayed@helmholtz-hzi.de
bSchool of Science, Mae Fah Luang University, Chiang Rai, Thailand
cCenter of Chemical Innovation for Sustainability (CIS), Mae Fah Luang University, Chiang Rai, Thailand
dComputational Chemistry Laboratory, Chemistry Department, Faculty of Science, Minia University, Minia 61519, Egypt
eSchool of Health Sciences, University of KwaZulu-Natal, Westville, Durban 4000, South Africa
fDepartment of Pharmacognosy, Faculty of Pharmacy, Ain Shams University, Abbasia, 11566 Cairo, Egypt. E-mail: sherif_elsayed@pharma.asu.edu.eg
First published on 27th June 2023
Abstract
Lasiodiplodia fungi are known to colonize plants as both pathogens and/or endophytes; hence, they can be exploited for their beneficial roles. Many compound classes from the genus have exhibited their potential biotechnological applications. Herein, we report two new metabolites 1 and 2 together with three known cyclo-(D-Ala-D-Trp) (3), indole-3-carboxylic acid (4) and a cyclic pentapeptide clavatustide B (5), isolated from the submerged cultures of a recently described species L. chiangraiensis. Chemical structures of the isolated compounds were determined by extensive NMR spectroscopic analyses together with HRESIMS. The absolute configurations of the new compounds were established based on comparing experimental and calculated time-dependent density functional theory circular dichroism (TDDFT-ECD) spectra. Compound 1 exhibited significant cytotoxic activities against an array of cell lines with IC50 values of 2.9–12.6 μM, as well as moderate antibacterial effects.
1. Introduction
The genus Lasiodiplodia (Botryosphaeriaceae) comprises endophytic and/or saprobic fungi cosmopolitan in tropical and subtropical regions.1 The fungal genus consists currently of 87 species, typified by the widely studied L. theobromae.1 The fungi have been detected as significant pathogens on Acacia sp.,2 Eucalyptus3 and Ficus,4 among other hosts.1 Recently, a myriad of bioactive compounds have been isolated from L. theobromae, sparking interest in the further mining of metabolites from Lasiodiplodia metabolome. L. theobromae proved to be a proficient producer of indoles, jasmonates, phenols, lactones and diketopiperazines.5 Its preussomerins and cloropreussomerins had antitumor properties;6 lasiodiplodia (macrolides) were cytotoxic, antimicrobial and anti-inflammatory,7,8 whereas indoles, jasmonic acid and their derivatives were phytohormones.9 In addition, the antitumor drug Taxol was reported from L. theobromae endophytic strains.10Our ongoing research is directed towards exploring new fungal strains for fungal secondary metabolites with a special interest especially on those revealing potential cytotoxic and/or antimicrobial activities. In our recent studies, we investigated a strain of the genus Lasiodiplodia, L. chiangraiensis, designated as a new species by one of our partner departments in Mae Fah Luang University, Thailand.11
Based on the phylogenetic analyses, L. chiangraiensis was closely related to Lasiodiplodia iranensis but formed a distinct lineage. In addition, it exhibited one different base pair on the ITS region,11 compared to L. iranensis.12 Hitherto, the screening of L. chiangraiensis for secondary metabolites production remained unexplored. Thus, based on the earlier reported successes of bioactive compounds with innovative chemistry unearthed from genus Lasiodiplodia; we further investigated L. chiangraiensis for metabolite production that afforded two new diketopiperazine derivatives (1 and 2) along with three known compounds (3–5). Herein, we describe the isolation and structure elucidation of the new compounds together with their antimicrobial and cytotoxic activities.
2. Results and discussion
2.1. Structure elucidation of compounds
The fungal species L. chiangraiensis was cultivated using Q6 culture media whose extract featured the most significant antimicrobial activity rather than those obtained from other four culture media. Therefore, the fungus was cultivated on a larger scale using the same culturing conditions to explore its produced secondary metabolites exclusively in this media and it afforded two new (1 and 2) and one known (3) diketopiperazine derivatives together with two other known metabolites (4 and 5) that were all assessed for their antimicrobial and cytotoxic activities.Compound 1 (Fig. 1) was purified as an off-white solid powder. The HRESIMS of 1 established its molecular formula to be C15H15N3O3S3 by exhibiting pseudomolecular ion peaks at m/z 382.0347 [M + H]+ (calculated 382.0348) indicating its possession to ten degrees of unsaturation in its structure. The 1H NMR, 1H–1H COSY and HSQC spectra of 1 (Table 1, Fig. 2) revealed the presence of ortho-disubstituted aromatic ring via the noticed extending spin system over four different aromatic protons at δH 7.74 (dt, J = 8.0, 1.0 Hz, H-15; δc 120.1), δH 7.09 (ddd, J = 8.0, 7.0, 1.0 Hz, H-14; δc 120.7), δH 7.14 (ddd, J = 8.0, 7.0, 1.0 Hz, H-13; δc 123.2) and δH 7.37 (dt, J = 8.0, 1.0 Hz, H-12; δc 112.5) along with a singlet deshielded aromatic proton at δH 7.58 (H-9; δc 126.5) that appeared in DMSO-d6 as a doublet peak spin-coupled to an exchangeable pyrrole (NH) proton at δH 11.24 ppm with a coupling constant (J value) of 2.6 Hz that undoubtedly indicated the presence of an indole moiety in its chemical structure that accounts for six degrees of unsaturation. The 13C NMR data of 1 (Table 1) revealed the presence of two carbonyl carbons at δc 174.5 (C-4) and at δc 168.4 (C-1) that also account for two more degrees of unsaturation in the chemical structure of 1.
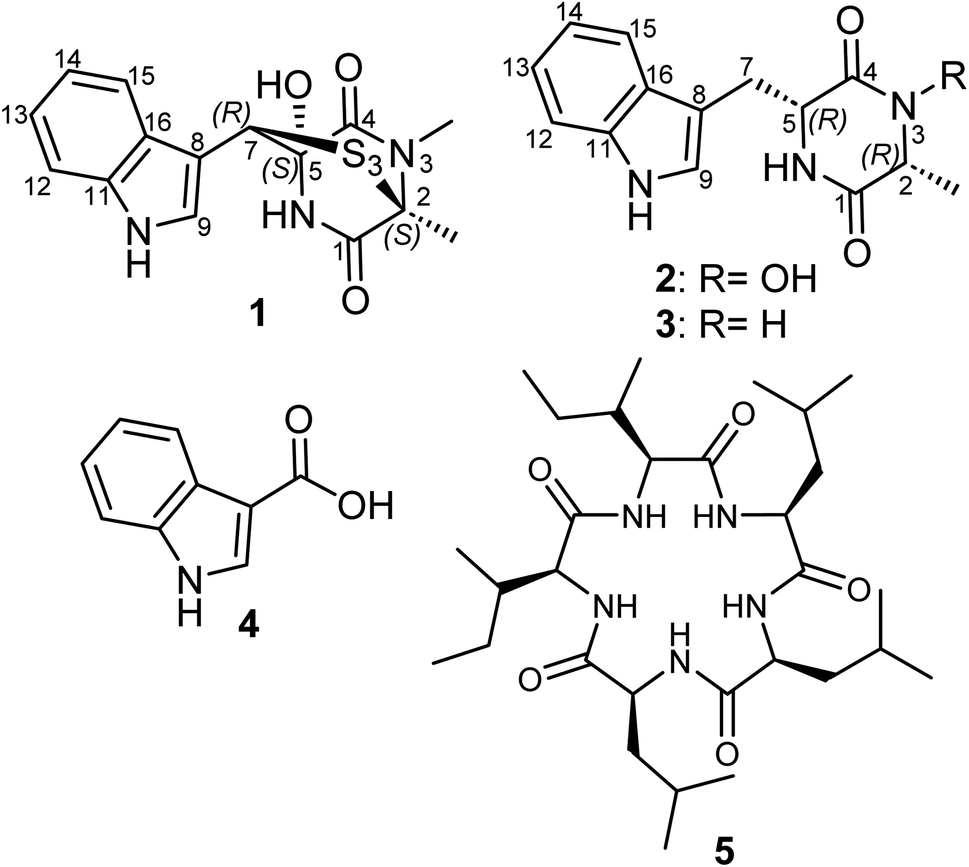 | ||
| Fig. 1 Chemical structures of 1–5. | ||
| Pos | 1 | 2 | |||||
|---|---|---|---|---|---|---|---|
| δC,a,c type | δHb (multi, J [Hz]) | δHd (multi, J [Hz]) | δC,a,c type | δHb (multi, J [Hz]) | δC,c,e type | δHd (multi, J [Hz]) | |
| a Measured in methanol-d4 at 175.b Measured in methanol-d4 at 700 MHz.c Assigned based on HMBC and HSQC spectra.d Measured in DMSO-d6 at 700.e Measured in DMSO-d6 at 175 MHz. | |||||||
| 1 | 168.4, CO | 169.2, CO | 166.3, CO | ||||
| 2 | 73.2, C | 60.0, CH | 3.90 (qd, 6.9, 1.0) | 58.4, CH | 3.77 (qd, 6.8, 1.0) | ||
| 3-NOH | 9.83 (br s) | ||||||
| 4 | 174.5, CO | 163.4, CO | 161.1, CO | ||||
| 5 | 84.6, C | 56.9, CH | 4.35 (td, 4.3, 1.0) | 55.1, CH | 4.21 (q, 3.7) | ||
| 6-NH | 8.82 (br s) | 8.28 (br d, 2.5) | |||||
| 7 | 64.1, CH | 5.99 (s) | 4.36 (s) | 30.4, CH2 | α 3.15 (dd, 14.7, 4.4) | 29.2, CH2 | α 3.03 (dd, 14.7, 4.4) |
| β 3.44 (dd, 14.8, 3.7) | β 3.24 (dd, 14.6, 3.9) | ||||||
| 8 | 107.1, C | 108.8, C | 108.1, C | ||||
| 9 | 126.5, CH | 7.58 (s) | 7.57 (d, 2.6) | 125.4, CH | 7.05 (s) | 124.5, CH | 7.01 (d, 2.4) |
| 10-NH | 11.24 (d, 1.4) | 10.91 (br s) | |||||
| 11 | 137.7, C | 137.5, C | 135.7, C | ||||
| 12 | 112.5, CH | 7.37 (dt, 8.0, 1.0) | 7.38 (dt, 8.0, 1.0) | 112.0, CH | 7.32 (dt, 8.1, 1.0) | 111.1, CH | 7.30 (dt, 8.1, 1.0) |
| 13 | 123.2, CH | 7.14 (ddd, 8.0, 7.0, 1.0) | 7.09 (ddd, 8.0, 7.0, 1.0) | 122.2, CH | 7.10 (td, 8.2, 1.2) | 120.8, CH | 7.03 (td, 8.2, 7.0, 1.3) |
| 14 | 120.7, CH | 7.09 (ddd, 8.0, 7.0, 1.0) | 7.00 (ddd, 8.0, 7.0, 1.0) | 119.9, CH | 7.01 (td, 8.1, 1.1) | 118.9, CH | 6.95 (ddd, 8.0, 6.9, 1.1) |
| 15 | 120.1, CH | 7.74 (dt, 8.0, 1.0) | 7.67 (d, 8.0) | 119.6, CH | 7.59 (dt, 8.0, 1.0) | 118.5, CH | 7.54 (dt, 7.8, 1.1) |
| 16 | 128.6, C | 128.9, C | 127.9, C | ||||
| 2-Me | 22.1, CH3 | 1.95 (s, 3H) | 1.82 (s, 3H) | 16.3, CH3 | 0.42 (d, 6.9, 3H) | 16.4, CH3 | 0.41 (d, 6.9, 3H) |
| 3-NMe | 29.5, CH3 | 3.17 (s, 3H) | 3.03 (s, 3H) | ||||
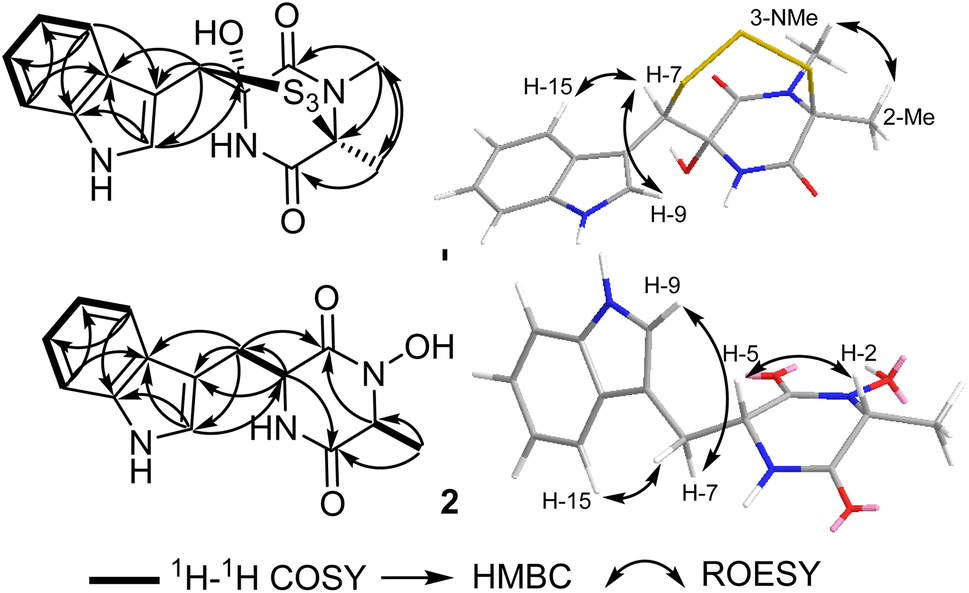 | ||
| Fig. 2 Key COSY, HMBC and ROESY correlations of 1 and 2. | ||
These results suggested that 1 includes in its structure two ring structures aside of the indole moiety. By searching the reported literature and comparing the obtained NMR data, compound 1 was found to closely resemble lasiodipline D, a dithiodiketopiperazine derivative featuring a characteristic α,β-dithio bridge between alanine and tryptophan amino acid residues, previously reported from the genus Lasiodiplodia13 and in outovirins A-C from an endophytic fungus Penicillium raciborskii.14 The obvious difference between 1 and lasiodipline D was in their molecular weights by 32 atomic units that could be interpreted by its inclusion of an additional sulphur atom. To further confirm the depicted structure of 1, the HMBC spectrum was measured (Table 1, Fig. 2) that displayed key HMBC correlations from a singlet proton resonating at δH 5.99 (δc 64.1) ascribed as H-7 to five carbon resonances at δc 174.5 (C-4), δc 128.6 (C-16), δc 126.5 (C-9), δc 107.1 (C-8) and δc 84.6 (C-5). In addition two singlet methyl groups at δH 1.95 (2-Me; δC 22.1) and δH 3.17 (s, 3-NMe; δC 29.5) disclosed key HMBC correlations to a carbon peak at δC 73.2 (C-2) confirming their suggested positions at C-2 and N-3, respectively.
The ROESY spectrum of 1 (Fig. 2) confirmed the depicted structure via key NOE correlations from H-7 to H-9 and H-15 together with the NOE correlations between two methyl groups at C-2 and N-3. The absolute configuration of 1 was determined by comparing the measured and calculated ECD spectra of all optional stereoisomers (Fig. 3) where an obvious accordance was noticed between the measured and the calculated ECD spectrum of (2S,5S,7R)-configuration over the whole range. These results unambiguously confirmed compound 1 as a new trithiodiketopiperazine derivative and was given a trivial name, lasiodipline G.
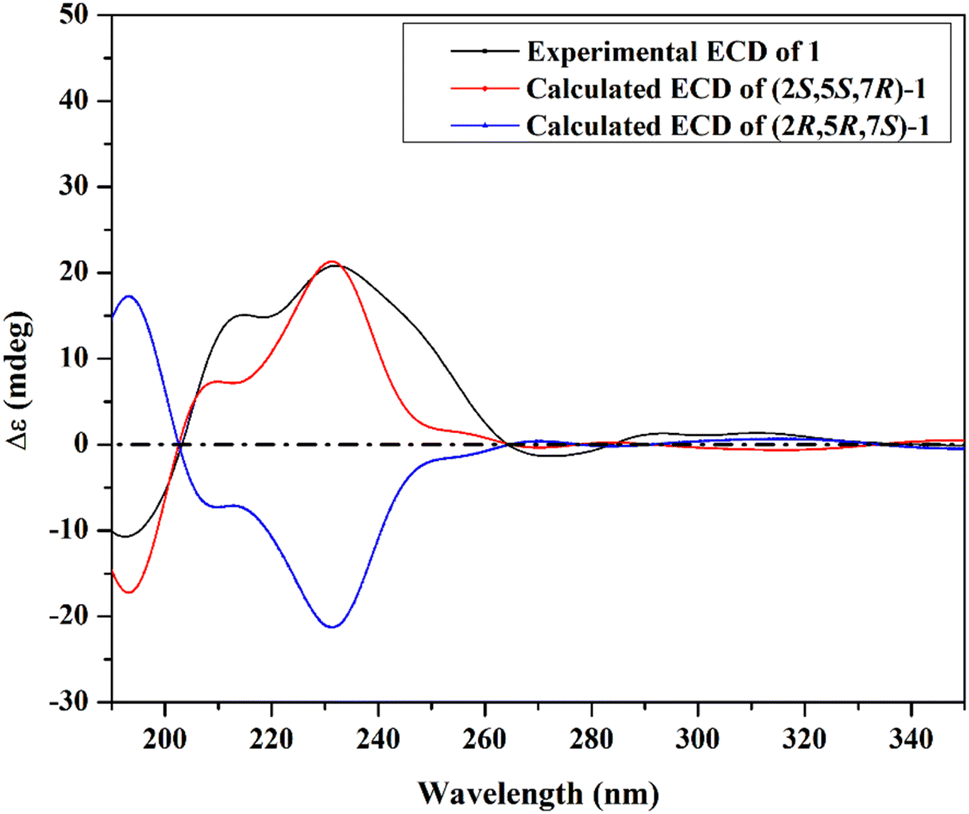 | ||
| Fig. 3 Experimental and calculated ECD spectra of lasiodipline G (1). | ||
Compound 2 was obtained as a white solid powder that revealed three absorption maxima (λmax) in its UV spectrum at 202, 227 and 280 nm. The molecular formula of 2 was determined to be C14H15N3O6 based on HRESIMS that showed three pseudomolecular ion peaks at m/z 274.1190 [M + H]+ (calculated 274.1186), m/z 296.1004 [M + Na]+ (calculated 296.1006) and m/z 547.2300 [2M + H]+ (calculated 547.2300) indicating the presence of nine degrees of unsaturation. The 1D (1H/13C) NMR and 2D (1H–1H COSY and HSQC) (Table 1, Fig. 2) suggested the existence of an indole moiety similar to that in 1. The molecular formula of 2 was deprived of any sulphur atoms that was supported by the presence of one methine at δH 3.77 (qd, J = 6.8, 1.0 Hz, H-2; δc 58.4) and one methylene at δH 3.03/δH 3.24 (H2-7) both directly correlated to a secondary carbon at δc 29.2 that were spin-coupled via 1H–1H COSY to a doublet methyl at δH 0.41 (d, J = 6.9 Hz, 2-Me; δc 16.4) and a methine proton at δH 4.21 (q, J = 3.7 Hz, H-5; δc 55.1), respectively. By comparing the obtained NMR data of 2 with the reported literature, it was found to be very similar to cyclo(D-Ala-D-Trp) (3) that was previously reported as a fungal metabolite produced by an endophytic fungus Chaetomium globosum from the fruits of Azadirachta indica.15 By comparing NMR data and molecular formulas of 2 and cyclo(D-Ala-D-Trp) (3), it was found that the former has an additional oxygen atom interpreting the difference of 16 atomic units in their molecular weights while their NMR data were closely similar except in the appearance of a deshielded exchangeable proton at δH 9.83 in 2 replacing one exchangeable amide NH proton in the diketopiperazine moiety indicating that one amide NH is converted to N–OH functionality. To unambiguously define the position of N–OH group in 2, 1H–1H COSY spectrum was measured in DMSO-d6 that revealed an obvious spin system from an exchangeable NH proton at δH 8.28 (br d, J = 2.5 Hz, 6-NH) to an aliphatic methine at δH 4.21 (q, J = 3.7 Hz, H-5) and extending to the methylene group at δH 3.03/δH 3.24 (H2-7). Hence, compound 2 was concluded to be 3-hydroxy derivative of cyclo-(D-Ala-D-Trp). The relative configuration of 2 was determined by ROESY spectrum (Fig. 2) that revealed key NOE correlations between H-2 and H-5 indicating that they are facing the same plane of the structure while another key NOE correlation was noticed between a doublet methyl at C-2 and the methylene group H2-7. By comparing the obtained 1D/2D NMR data and optical rotation of 2 to all possible stereoisomers of cyclo-(Ala-Trp) that have been previously described,15,16 compound 2 was unambiguously confirmed to feature (2R,2R) configuration and hence was trivially named as cyclo-(D-N-OH-Ala-D-Trp). To the best of our knowledge and by searching the reported literature, this is the first report of 2 as a natural product while it was formerly patented among other synthetic derivatives for the prevention and treatment of nephritis.17
Compounds (3–5) were identified as cyclo-(D-Ala-D-Trp),15,16 indole-3-carboxylic acid18 and a cyclic pentapeptide clavatustide B,19 respectively.
2.2. Bioassays
All isolated compounds were subjected to cytotoxicity and antimicrobial assays. Based on the obtained results (Table 2), lasiodipline G (1) proved to possess a significant pan-cytotoxic activity against the seven tested mammalian cell lines with IC50 values ranging between 2.9 and 12.6 μM. Nonetheless, its antimicrobial activity was moderate at a minimum inhibitory concentration (MIC) of 87.4 μM (33.3 μg mL−1), and only against Staphylococcus aureus rather than the other tested eleven microorganisms, revealing that activity can probably be attributed to its relatively potent cytotoxic activity. Intriguingly, compounds 2 and 3, deprived of the trithio bridge, exhibited neither significant cytotoxic nor antimicrobial activities illustrating the essential role of α,β-trithio bridge in inducing cytotoxic rather than antimicrobial activity.| Compound | IC50 | ||||||
|---|---|---|---|---|---|---|---|
| L929 | KB3.1 | PC-3 | MCF-7 | SKOV-3 | A431 | A549 | |
| Lasiodipline G (1) (μM) | 5.8 | 8.4 | 6.3 | 3.9 | 3.9 | 2.9 | 12.6 |
| Epothilon B (nM) | 0.65 | 0.17 | 0.09 | 0.07 | 0.09 | 0.06 | 0.05 |
3. Materials and methods
3.1. General experimental procedure
Ultraviolet-visible (UV/vis) spectra were obtained using a UV-vis spectrophotometer UV-2450 (Shimadzu, Kyoto, Japan). Optical rotations were determined using a MCP 150 polarimeter at 20 °C (Anton-Paar Opto Tec GmbH, Seelze, Germany). ECD spectra of new compounds were acquired on a J-815 spectropolarimeter (JASCO, Pfungstadt, Germany). High-resolution electrospray ionization mass spectra (HR-ESI-MS) were acquired with an Agilent 1200 Infinity Series HPLC-UV system (Agilent Technologies, Santa Clara, CA, USA) utilizing a C18 Acquity UPLC BEH column (2.1 × 50 mm, 1.7 μm: Waters, Milford, MA, USA), solvent A: H2O + 0.1% formic acid; solvent B: acetonitrile (MeCN) + 0.1% formic acid, gradient: 5% B for 0.5 min increasing to 100% B in 19.5 min, maintaining 100% B for 5 min, flow rate 0.6 mL min−1, UV/vis detection 190–600 nm connected to an time-of-flight mass spectrometer (ESI-TOF-MS, Maxis, Bruker, Billerica, MA, USA) (scan range 100–2500 m/z, rate 2 Hz, capillary voltage 4500 V, dry temperature 200 °C). NMR spectra were recorded with an Avance III 700 spectrometer (Bruker, Billerica, MA, USA, 1H-NMR: 700 MHz, and 13C-NMR: 175 MHz) dissolving compounds in deuterated methanol-d4 and deuterated DMSO-d6.3.2. Fermentation, extraction, and isolation
A decaying wood-derived strain of Lasiodiplodia chiangraiensis (MFLUCC21-0003) was collected in July and December 2019 from Mae Fah Luang University in Chiang Rai, Thailand. The samples were pre-sterilized by washing with sterile deionized water and further immersed in 70% ethanol for 60 s, 1% NaClO for 60 s prior washing with sterile deionized water for 60 s. The sterilized samples were cut into 0.5 cm2 size and then further placed on PDA plate mixed with chloramphenicol antibiotic to suppress the growth of bacteria. The dead wood samples were placed on PDA for 7 days until growth of the fungal hyphae was visible. All fungi that formed colonies were separated and cultured on new PDA plates and then incubated at room temperature for 7 days. The single spore and germinated spores were transferred to PDA plates. Cultures were deposited at Mae Fah Luang University Culture Collection (MFLUCC), the Institute of Excellence in Fungal Research, Mae Fah Luang University, Thailand. Its Genbank accession number was MW760853, MW815629, and MW815627 in ITS, tef, and tub2 genes, respectively.11 It was cultured in Yeast Malt medium (YMG) (10 g L−1 malt extract, 4 g L−1 yeast extract, 4 g L−1 D-glucose and 20 g L−1 agar in deionized water) plates with adjusted pH to 6.3 and incubated at 24 °C. After 7 days, five plugs of fungal colonies were obtained using a cork borer and further placed into 20 × 500 mL Erlenmeyer flasks, each containing 250 mL of cotton seed flour (Q6) medium broth (5 g L−1 cotton seed flour, 10 g L−1 glycerin, 2.5 g L−1 D-glucose and 20 g L−1 agar in deionized water and pH adjusted at 7.2). The cultures were incubated at 23 °C on a rotary shaker at 140 rpm for 7 days. After fermentation, filtration was conducted to separate mycelial and supernatant parts. The extraction method was done according to previous protocols.20 The ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ethyl acetate:supernatant) was used for extraction. After partition in a separatory funnel, the organic phase was rinsed through anhydrous sodium sulfate. The water phase was retained in separatory funnel for extraction three times. The mycelial part was soaked in 500 mL acetone and placed in an ultrasonic bath at 40 °C for 30 min. The supernatant phase was extracted with 500 mL of ethyl acetate twice after adjusting the volume to 500 mL using distilled water. The obtained solutions from ethyl acetate and acetone parts were concentrated using rotary evaporator and the yields of supernatant and mycelial crude extracts were 1.105 mg and 556 mg, respectively. The mycelial crude extract was dissolved in MeOH and subjected to preparative reversed phase HPLC (PLC 2020, Gilson, Middleton, WI, USA). As stationary phase, Gemini C18 column (250 × 21.2 mm, 10 μm, Phenomenex, Aschaffenburg, Germany) was used, while the mobile phase consisted of solvent A, deionized water with 0.1% formic acid and solvent B, MeCN with 0.1% formic acid. Purification of the crude extracts was performed by using a linear gradient elution of 45–100% solvent B at a flow rate of 30 mL min−1 for 65 min and eluting with isocratic elution at 100% solvent B for 10 min to afford compounds 1 (tR: 36.3–43.5 min, 1.58 mg), 5 (tR: 53.4–55.2 min, 0.94 mg), and other observed fractions (B1–B15). Compound 4 (tR: 26.4–28.6 min, 3.49 mg) was purified from fraction B13 (31.32 mg) by preparative reversed phase HPLC (PLC 2020, Gilson, Middleton, WI, USA). The Synergi C18 column (250 μm × 21.2 mm, 10 μm, Phenomenex, Aschaffenburg, Germany) was used as stationary phase, and the mobile phase consisted of solvent A, deionized water with 0.1% formic acid and solvent B, MeCN with 0.1% formic acid. The separation was performed with the elution gradient 40–100% solvent B for 55 min, followed by isocratic elution with 100% B for 10 min. Compounds 2 (tR: 9.30–10.2 min, 1.43 mg) and 3 (tR: 10.2–10.9 min, 2.65 mg) were purified from fraction B7 (12.62 mg) using semipreparative HPLC using Synergi C18 column (250 μm × 10 mm, 4 μm, Phenomenex, Aschaffenburg, Germany) implementing the following gradient elution 20–50% solvent B in 20 min, then from 50–100% solvent B in 10 min and holding 100% solvent B for 5 min.
:1 (ethyl acetate:supernatant) was used for extraction. After partition in a separatory funnel, the organic phase was rinsed through anhydrous sodium sulfate. The water phase was retained in separatory funnel for extraction three times. The mycelial part was soaked in 500 mL acetone and placed in an ultrasonic bath at 40 °C for 30 min. The supernatant phase was extracted with 500 mL of ethyl acetate twice after adjusting the volume to 500 mL using distilled water. The obtained solutions from ethyl acetate and acetone parts were concentrated using rotary evaporator and the yields of supernatant and mycelial crude extracts were 1.105 mg and 556 mg, respectively. The mycelial crude extract was dissolved in MeOH and subjected to preparative reversed phase HPLC (PLC 2020, Gilson, Middleton, WI, USA). As stationary phase, Gemini C18 column (250 × 21.2 mm, 10 μm, Phenomenex, Aschaffenburg, Germany) was used, while the mobile phase consisted of solvent A, deionized water with 0.1% formic acid and solvent B, MeCN with 0.1% formic acid. Purification of the crude extracts was performed by using a linear gradient elution of 45–100% solvent B at a flow rate of 30 mL min−1 for 65 min and eluting with isocratic elution at 100% solvent B for 10 min to afford compounds 1 (tR: 36.3–43.5 min, 1.58 mg), 5 (tR: 53.4–55.2 min, 0.94 mg), and other observed fractions (B1–B15). Compound 4 (tR: 26.4–28.6 min, 3.49 mg) was purified from fraction B13 (31.32 mg) by preparative reversed phase HPLC (PLC 2020, Gilson, Middleton, WI, USA). The Synergi C18 column (250 μm × 21.2 mm, 10 μm, Phenomenex, Aschaffenburg, Germany) was used as stationary phase, and the mobile phase consisted of solvent A, deionized water with 0.1% formic acid and solvent B, MeCN with 0.1% formic acid. The separation was performed with the elution gradient 40–100% solvent B for 55 min, followed by isocratic elution with 100% B for 10 min. Compounds 2 (tR: 9.30–10.2 min, 1.43 mg) and 3 (tR: 10.2–10.9 min, 2.65 mg) were purified from fraction B7 (12.62 mg) using semipreparative HPLC using Synergi C18 column (250 μm × 10 mm, 4 μm, Phenomenex, Aschaffenburg, Germany) implementing the following gradient elution 20–50% solvent B in 20 min, then from 50–100% solvent B in 10 min and holding 100% solvent B for 5 min.
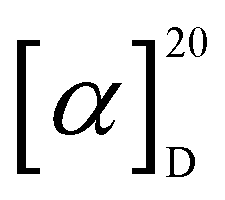 [α]D20 + 140 (c 0.1, acetonitrile); UV (MeOH) λmax 281, 224 and 200 nm; NMR data (1H NMR: 700 MHz, 13C NMR: 175 MHz, methanol-d4 or DMSO-d6) see Table 1; in LRESIMS m/z 382.05 [M + H]+, m/z 785.04 [2M + Na]+, m/z 379.87 [M − H]− and m/z 760.91 [2M−H]−; HRESIMS m/z 364.0241 [M–H2O + H]+ (calcd. 364.0243 for C15H14N3O2S3+), 382.0347 [M + H]+ (calcd. 382.0348 for C15H16N3O3S3+), 404.0168 [M + Na]+ (calcd. 404.0168 for C15H15N3NaO3S3+); tR = 7.98 min (HR-LC-ESIMS). C15H15N3O3S3 (381.03 g mol−1) cyclo-(D-N-OH-Ala, D-Trp) (2): white solid powder;
[α]D20 + 140 (c 0.1, acetonitrile); UV (MeOH) λmax 281, 224 and 200 nm; NMR data (1H NMR: 700 MHz, 13C NMR: 175 MHz, methanol-d4 or DMSO-d6) see Table 1; in LRESIMS m/z 382.05 [M + H]+, m/z 785.04 [2M + Na]+, m/z 379.87 [M − H]− and m/z 760.91 [2M−H]−; HRESIMS m/z 364.0241 [M–H2O + H]+ (calcd. 364.0243 for C15H14N3O2S3+), 382.0347 [M + H]+ (calcd. 382.0348 for C15H16N3O3S3+), 404.0168 [M + Na]+ (calcd. 404.0168 for C15H15N3NaO3S3+); tR = 7.98 min (HR-LC-ESIMS). C15H15N3O3S3 (381.03 g mol−1) cyclo-(D-N-OH-Ala, D-Trp) (2): white solid powder; 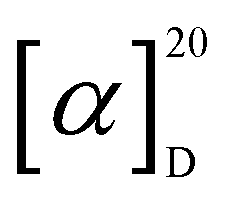 [α]D20 − 8.4 (c 0.1, MeOH); UV (MeOH) λmax 280, 227, and 202 nm; NMR data (1H NMR: 700 MHz, 13C NMR: 175 MHz, methanol-d4 or DMSO-d6) see Table 1; in LRESIMS m/z 274.08 [M + H]+, m/z 547.23 [2M + H]+, m/z 271.89 [M − H]− and m/z 545.07 [2M − H]−; HRESIMS m/z 274.1190 [M + H]+ (calcd. 274.1186 for C14H16N3O3+), 296.1004 [M + Na]+ (calcd. 296.1006 for C14H15N3NaO3+), 547.2300 [2M + H]+ (calcd. 547.2300 for C28H31N6O6+); tR = 2.55 min (HR-LC-ESIMS). C14H15N3O3 (273.05 g mol−1).
[α]D20 − 8.4 (c 0.1, MeOH); UV (MeOH) λmax 280, 227, and 202 nm; NMR data (1H NMR: 700 MHz, 13C NMR: 175 MHz, methanol-d4 or DMSO-d6) see Table 1; in LRESIMS m/z 274.08 [M + H]+, m/z 547.23 [2M + H]+, m/z 271.89 [M − H]− and m/z 545.07 [2M − H]−; HRESIMS m/z 274.1190 [M + H]+ (calcd. 274.1186 for C14H16N3O3+), 296.1004 [M + Na]+ (calcd. 296.1006 for C14H15N3NaO3+), 547.2300 [2M + H]+ (calcd. 547.2300 for C28H31N6O6+); tR = 2.55 min (HR-LC-ESIMS). C14H15N3O3 (273.05 g mol−1).3.3. Biological assays
All isolated compounds with sufficient amounts were assessed for their antimicrobial activity according to the previously reported protocol,22 over a concentration range from 66.6 to 0.1 μg mL−1 to determine their minimum inhibitory concentrations (MIC) against five pathogenic fungi including Candida albicans, Mucor hiemails, Pichia anomala, Rhodotorula glutinis and Schizosaccharomyces pombe, three Gram-positive bacteria including Bacillus subtilis, Mycobacterium smegmatis, and Staphylococcus aureus, and four Gram-negative bacteria including Chromobacterium violaceum, Acinetobacter baumanii, Escherichia coli, and Pseudomonas aeruginosa. Nystatin was used as an antifungal positive control whereas oxytetracycline, ciprofloxacin, gentamycin, kanamycin were used as positive controls against Gram-positive and Gram-negative bacteria. All compounds were also tested their cytotoxicity (IC50) using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) test following the standard protocols.21 These compounds were tested against seven mammalian cell lines including human endocervical adenocarcinoma KB 3.1, breast cancer MCF-7, lung cancer A549, ovary cancer SK-OV-3, prostate cancer PC-3, squamous cancer A431, and mouse fibroblasts L929. Epothilone B was used as the positive control.3.4. Computational functional theory calculations
Conformational analysis was performed using Omega2 software23 to obtain the possible conformers for compound 1 within an energy window value of 10 kcal mol−1. All resulting conformers were optimized at B3LYP/6-31G* level of theory using Gaussian09 software.24 Frequency calculations were then performed on the optimized structures to ensure the nature of the local minima as well as to estimate the Gibbs free energies. Time-dependent density functional theory (TDDFT) calculations incorporating a polarizable continuum model (PCM) using methanol as a solvent were carried out at the B3LYP/6-31G* level of theory to calculate the first fifty excitation states. Electronic circular dichroism (ECD) spectra were finally generated using SpecDis 1.71 (SpecDis 2017 (ref. 25)) by applying Gaussian band shapes with sigma = 0.20–30 eV. The generated ECD spectra were Boltzmann-averaged.4. Conclusions
Two new compounds, 1 and 2 were isolated from the submerged cultures of the recently isolated fungus L. chiangraiensis alongside three known molecules. This forms the first report of compounds from the fungal species, and clearly depicts that various species belonging to the genus Lasiodiplodia, can synthesize a vast array of secondary metabolites. Compound 1 was significantly cytotoxic against various cancer cell lines, thus sparking a significant discussion concerning the potential application of its derivatives as starting points in the discovery of potent antitumor agents. It will be of particular interest to perform further structure activity-relationship studies to establish the role of α,β-trithio bridge in inducing cytotoxicity.Author contributions
S. K. and C. T.: cultivation of the fungus. S. K. and P. P.: methodology, purification and isolation of pure compounds. S. K. and S. S. E. wrote draft manuscript. W. C. S. performed spectroscopic experiments and data curation. S. S. E.: structure elucidation. M. A. A. I.: CFT calculations. S. S. E. and M. S.: supervision and editing manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We appreciate Centre of Excellence in Fungal Research; Mae Fah Luang University for providing the fungus. This work was also financially supported by Mae Fah Luang University. S. S. E. acknowledges Alexander von Humboldt (AvH) Foundation for granting him Georg-Forster Fellowship for Experienced Researchers (ref. 3.4-1222288-EGY-GF-E). S. K. and P. P. acknowledge the Mae Fah Luang University grant and the Royal Golden Jubilee (RGJ) Ph. D. Program for the financial support of this work through grant no. PHD/0193/2560. The authors also thank the German Academic Exchange Service (DAAD) project number 57521414, (Programm Projektbezogener Personenaustausch, PPP), Thailand 2020.References
- N. I. de Silva, A. J. Phillips, J. K. Liu, S. Lumyong and K. D. Hyde, Sci. Rep., 2019, 9, 1–11 CrossRef CAS PubMed.
- M. Tarigan, M. J. Wingfield, M. van Wyk, B. Tjahjono and J. Roux, South. For., 2011, 73, 187–191 CrossRef.
- G. Li, R. J. Arnold, F. Liu, J. Li and S. Chen, J. Phytopathol., 2015, 163, 956–967 CrossRef CAS.
- S. R. Mohali, F. Castro-Medina, J. R. Úrbez-Torres and W. D. Gubler, Forensic Pathol., 2017, 47, e12355 Search PubMed.
- M. M. Salvatore, A. Andolfi and R. Nicoletti, Agriculture, 2020, 10, 488 CrossRef CAS.
- S. Chen, D. Chen, R. Cai, H. Cui, Y. Long, Y. Lu, C. Li and Z. She, J. Nat. Prod., 2016, 79, 2397–2402 CrossRef CAS PubMed.
- J. Mathew Valayil, G. C. Kuriakose and C. Jayabaskaran, Anticancer Agents Med. Chem., 2016, 16, 865–874 CrossRef PubMed.
- S. Chen, Z. Liu, H. Liu, Y. Long, D. Chen, Y. Lu and Z. She, Org. Biomol. Chem., 2017, 15, 6338–6341 RSC.
- M. M. Salvatore, A. Alves and A. Andolfi, Toxins, 2020, 12, 457 CrossRef CAS PubMed.
- B. Sah, K. Subban and J. Chelliah, FEMS Microbiol. Lett., 2017, 364, fnx253 CrossRef.
- N. Wu, A. J. Dissanayake, K. T. Chethana, K. D. Hyde and J. K. Liu, Phytotaxa, 2021, 508, 142–154 CrossRef.
- J. Abdollahzadeh, A. Javadi, E. M. Goltapeh, R. Zare and A. J. L. Phillips, Pers.: Mol. Phylogeny Evol., 2010, 25, 1–10 CAS.
- W. Wei, N. Jiang, Y. N. Mei, Y. L. Chu, H. M. Ge, Y. C. Song, S. W. Ng and R. X. Tan, Phytochemistry, 2014, 100, 103–109 CrossRef CAS PubMed.
- M. Kajula, J. M. Ward, A. Turpeinen, M. V. Tejesvi, J. Hokkanen, A. Tolonen, H. Häkkänen, P. Picart, J. Ihalainen, H.-G. Sahl, A. M. Pirttilä and S. Mattila, J. Nat. Prod., 2016, 79, 685–690 CrossRef CAS PubMed.
- M. A. M. Mondol, J. Farthouse, M. T. Islam, A. Schüffler and H. Laatsch, Nat. Prod. Commun., 2016, 11, 1865–1868 CrossRef PubMed.
- E. Caballero, C. Avendaño and J. C. Menéndez, Tetrahedron: Asymmetry, 1998, 9, 967–981 CrossRef CAS.
- H. Tone, S. Sato, H. Sato, K. Tamura and S. Tamada, World Intellectual Property Organization, WO9009380A1, 1990-08-23.
- S. Bano, V. U. Ahmad, S. Perveen, N. Bano, S. Shafiuddin and M. Shameel, Planta Med., 1987, 53, 117–118 CrossRef CAS.
- P. Ye, L. Shen, W. Jiang, Y. Ye, C.-T. A. Chen, X. Wu, K. Wang and B. Wu, Mar. Drugs, 2014, 12, 3203–3217 CrossRef PubMed.
- C. Chepkirui, T. Cheng, J. Matasyoh, C. Decock and M. Stadler, Phytochem. Lett., 2018, 25, 141–146 CrossRef CAS.
- R. B. Teponno, S. R. Noumeur, S. E. Helaly, S. Hüttel, D. Harzallah and M. Stadler, Molecules, 2017, 22, 1674 CrossRef PubMed.
- F. Surup, B. Thongbai, E. Kuhnert, E. Sudarman, K. D. Hyde and M. Stadler, J. Nat. Prod., 2015, 78, 934–938 CrossRef CAS PubMed.
- OMEGA, 2.5.1.4; OpenEye Scientific Software, Santa Fe, NM, USA, 2013 Search PubMed.
- J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- T. Bruhn, A. Schaumloffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03242f |
| This journal is © The Royal Society of Chemistry 2023 |