DOI:
10.1039/D3RA03187J
(Paper)
RSC Adv., 2023,
13, 22070-22078
A new strategy to exploit maximum rate performance for aqueous batteries through a judicious selection of MOF-type electrodes†
Received
13th May 2023
, Accepted 13th July 2023
First published on 21st July 2023
Abstract
A metal–organic framework (MOF) having a redox active 1,4,5,8-naphthalenetetracarboxdiimide (NDI) derivative in its organic linker shows excellent rate performance as an electrode material for aqueous batteries thanks to its large pores. Among aqueous electrolytes examined, K+-based ones exhibit the highest rate performance, which is caused by the highest mobility of the smallest hydrated K+ ion not only in the aqueous electrolyte but also in the electrode. Since the use of a counter electrode with insufficiently small pores for the full-cell configuration offsets this merit, our study may lead to a conclusion that the maximum rate performance for aqueous batteries will be accomplished only through further elaboration of both electrode materials with sufficiently large pores, in which hydrated ions can travel equally fast as those in the electrolyte.
Introduction
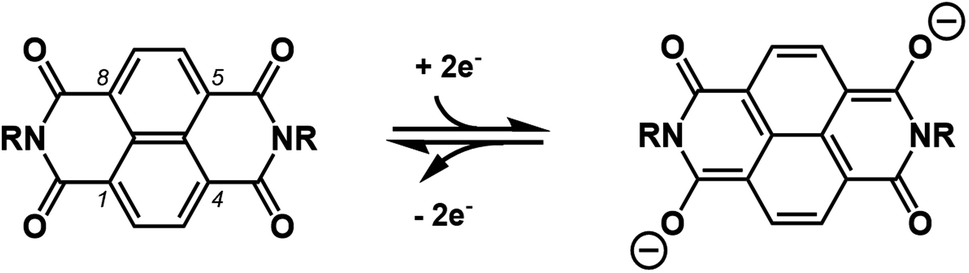
With the increasing demand for large-scale energy storage to meet the huge energy consumption, aqueous batteries have gained much attention as post lithium-ion batteries due to their attractive features such as safety, low cost, and high power.1–3 Recently, alkaline metal (Li, Na, K) ion batteries with non-conventional aqueous electrolytes showing electrochemical window beyond the theoretical value (1.23 V) have provided renewed opportunities for various active electrode materials.4 In contrast with inorganic electrode active materials, the redox active organic counterparts5–10 have a higher degree of flexibility in materials design and their molecular modification may not only finely tune their redox potential but also add properties that affect intermolecular forces to change their solubility, surface area (porosity), and so on. 1,4,5,8-naphthalenetetracarboxdiimide derivatives (NDIs), which are readily prepared by dehydrative condensation of naphthalene-1,4,5,8-tetracarboxylic dianhydride (NDA) with a variety of primary amines, have been one of the most promising organic anode materials for aqueous batteries.11–13 As illustrated in Scheme 1, the pair of two carbonyl groups either at 1 and 5 or at 4 and 8 positions undergo a reversible two electron redox reaction with concomitant change of resonance form in the naphthalene core. The parent 1,4,5,8-naphthalenetetracarboxdiimide (H2NDI) was an exceptionally poor anode material, possibly because its NH groups are susceptible to irreversible reduction to generate a dianion, which is not hydrophobic enough to persist in the electrode but dissolves into aqueous electrolyte prior to the reversible redox reaction.14 In contrast, a polymeric NDI derived from hydrazine was successfully employed as a durable anode for aqueous batteries.15,16 Unfortunately, however, its rate performance was only modest considering high ionic conductivity of aqueous electrolyte, which suggested that non-uniform surface of polymeric material could hinder smooth insertion of guest cation from aqueous electrolyte.17 In order to overcome above-mentioned shortcoming and to exploit reliable redox behaviors of NDIs, we focused on NDI-based metal–organic frameworks (MOFs)18 as active materials which enable the smooth insertion of guest cation. To the best of our knowledge, there have been no reports for rocking chair-type monovalent cation-conducting aqueous batteries using such MOFs, though Prussian-blue analogues (PBAs) may have been applied as conventional MOF-type electrodes.19 These situations may be caused mainly by the three reasons; (i) the MOF-type electrodes should have adequate water resistance, (ii) their redox potentials should locate within the stability window of aqueous electrolyte, (iii) they should not have unnecessarily large molecular weights because their redox-silent parts inevitably attenuate their theoretical capacity. On the other hand, Dincă and co-workers recently developed [Zn(dmpz)2NDI]n-MOF, which undergoes a two-electron reaction of NDI in aqueous electrolyte to serve as electrochromic materials.20,21 It has NDI units functionalized with two 3,5-dimethylpyrazole (dmpzH) ligands, which generate infinite [metal(μ-pyrazolate)2]n chains with tetrahedral Zn(II) in the orthogonal direction.22 Thus formed staggered parallelogram having (dmpz)2NDI molecule on each side provides a wide channel whose surface should be easily accessible by aqueous electrolyte. Most importantly, [Zn(μ-dmpz)2]n chains should be stable toward a range of aqueous solutions unless they are too acidic or basic.23–26 Therefore, we envisaged that [Zn(dmpz)2NDI]n-MOF should serve as a durable anode material in the aqueous battery, and it could show greater rate and cycle performances than conventional NDI-based anode materials.
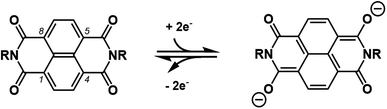 |
| | Scheme 1 Two electron reduction/oxidation reaction of NDIs. | |
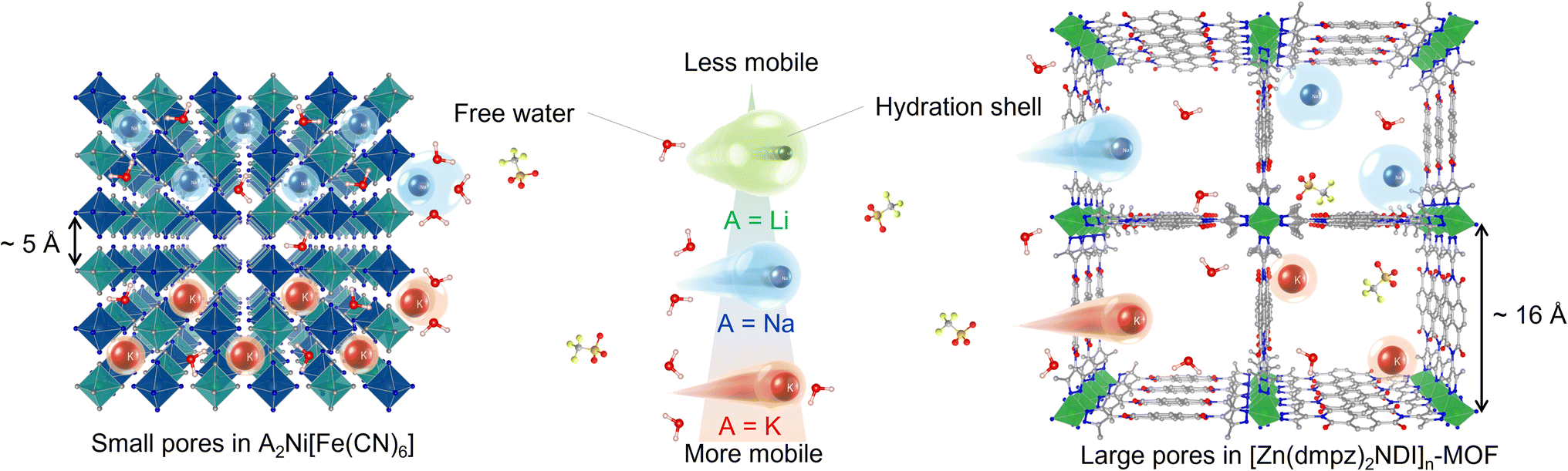
We have been engaged in the development of aqueous battery using PBAs as active material with non-conventional aqueous electrolytes.27,28 In these studies, we became interested in the generally accepted idea that K+ ion is less Lewis acidic compared with Li+ or Na+ ions, and its aqueous solutions display the lowest viscosity, and therefore K+-based aqueous electrolytes have an advantage in terms of rate performance.29–36 Significantly, however, this hypothesis may not be applicable unless these charge carriers may behave similarly within the electrode to those in the electrolyte. In fact, Cui and co-workers claimed37 that K+-based aqueous electrolytes have no kinetic advantage in the particular case of PBA electrodes.38–45 Their result suggested that the pore size around 5 Å provided by the cyano-bridged MOF structure of PBAs (Fig. 1) may restrict the mobility of K+ ions as compared to the aqueous solution state, thereby cancelling out its kinetic merit. On the other hand, the pore size around 16 Å provided by [Zn(dmpz)2NDI]n-MOF (Fig. 1) is large enough to allow accommodation of K+ ions in the similar environment to that in the aqueous solution, and has a greater chance of maximizing the rate performance of K+-based aqueous electrolytes. Therefore, we began our study by electrochemical characterization of [Zn(dmpz)2NDI]n-MOF as an anode material, initially in 17 m NaClO4 aqueous electrolyte to confirm the benefit provided by its open-framework structure. Next, aqueous electrolytes with several cations having identical anion and constant concentration (8 m AOTf, A = Li, Na, K, trifluoromethanesulfonate salts) were used with this anode to assess the rate performance of these cations. As a result, we obtained conclusive evidence that K+-based aqueous electrolytes may offer a non-negligible kinetic advantage, if they are combined with electrodes with large pore size and readily accessible uniform surface. Herein we describe these results.
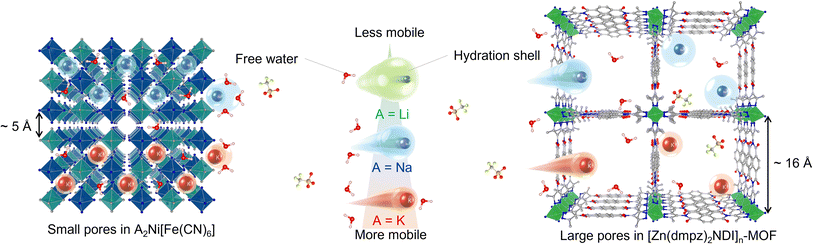 |
| | Fig. 1 Schematic images for illustration of relative size of hydrated ions, triflate anions, water molecules towards pores of electrode materials. | |
Results and discussion
Viability of MOF-type electrode
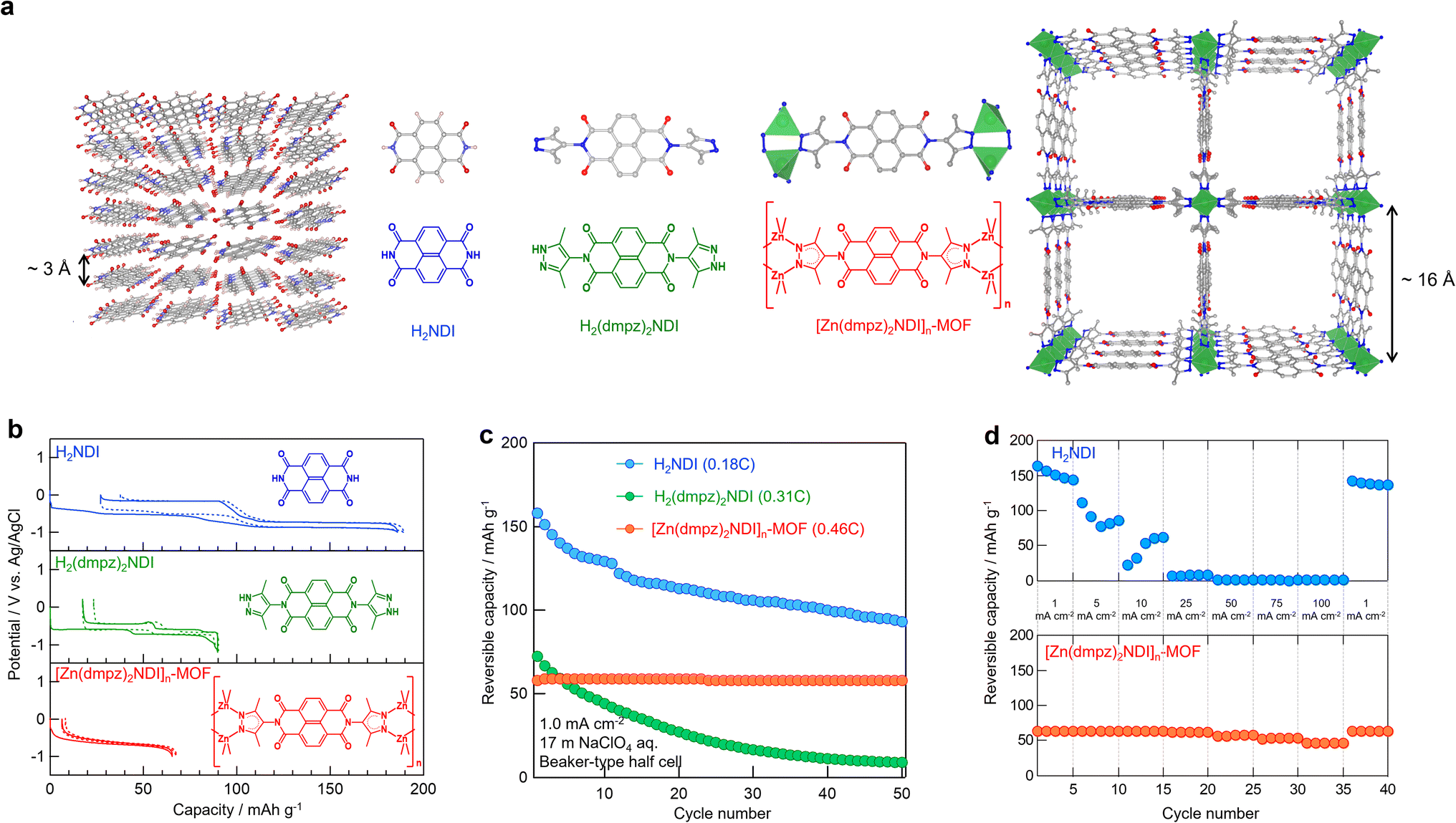
Our initial study focused on the electrochemical characterization of [Zn(dmpz)2NDI]n-MOF as an anode material in aqueous battery. [Zn(dmpz)2NDI]n-MOF was prepared according to the reported method,20 and its disc-shaped electrode composite was fabricated by mixing it with conductive carbon and binder to measure its electrochemical properties in 17 mol kg−1 (m) NaClO4 aqueous electrolyte.27,28,46–48 Control experiments were performed with H2NDI and H2(dmpz)2NDI, which lacks infinite [metal(μ-pyrazolate)2]n chains but still carries dmpzH end groups capable of intermolecular H-bonding, under otherwise identical conditions. These results are listed in Fig. 2b–d along with the schematic images of molecular and crystal structures for H2NDI, H2(dmpz)2NDI, and [Zn(dmpz)2NDI]n-MOF (Fig. 2a). The theoretical capacity for 2e− redox reaction (Table 1) for H2NDI, H2(dmpz)2NDI, and [Zn(dmpz)2NDI]n-MOF calculated by their molecular weights is 202, 118, 80 mA h g−1, respectively, and their initial practical reversible capacity proved to be approximately equal as shown in the Fig. 2b. These results indicated that the electrochemical behavior for the three materials commonly originates from the redox reaction of their NDI moiety and that the [Zn(dmpz)2]n chains in [Zn(dmpz)2NDI]n-MOF may remain unchanged in 17 m NaClO4 aqueous electrolyte. Also of note is that the overpotential within the initial discharge/charge cycles proved to be the smallest for [Zn(dmpz)2NDI]n-MOF and increased in the order of [Zn(dmpz)2NDI]n-MOF, H2(dmpz)2NDI, and H2NDI. We believe that these results reflect how easily the charge carrier Na+ ion (guest) can insert into and extract from the three NDIs (host). As shown in Fig. 2a, [Zn(dmpz)2NDI]n-MOF has a porous structure to easily accommodate the guests from all directions. In contrast, H2NDI stacks in a dense manner because of its flat molecular structure and, therefore, the accommodation of the guests should require some structural change in the solid state. H2(dmpz)2NDI may form a less dense solid-state structure compared with H2NDI, since its dmpzH groups are non-coplanar to the NDI group and its molecular structure is no longer as flat as H2NDI. Nevertheless, H2(dmpz)2NDI could not accommodate the guests as easily as [Zn(dmpz)2NDI]n-MOF. These speculations may be consistent with the cyclability shown in Fig. 2c. The reversible capacity for H2NDI as well as H2(dmpz)2NDI gradually decreased from the initial values (158 and 72 mA h g−1, and 78% and 61% compared with their theoretical capacities respectively) along with repeated cycles, while the reversible capacity for [Zn(dmpz)2NDI]n-MOF never attenuated throughout examined cycles (58 mA h g−1 for 1–50 cycles, 73% of capacity utilization compared with it theoretical capacity). Therefore, the repeated insertion and extraction of Na+ ion into and out of these materials should deteriorate the pristine solid-state structures of H2NDI and H2(dmpz)2NDI having only weak intermolecular forces but should not affect the original porous structure of [Zn(dmpz)2NDI]n-MOF thanks to its robust [Zn(dmpz)2]n framework. Fig. 2d illustrates the rate performance for H2NDI and [Zn(dmpz)2NDI]n-MOF at different current densities. While the reversible capacity for H2NDI decreased with increasing current density and it became unobservable above 25 mA cm−2 (12C), [Zn(dmpz)2NDI]n-MOF showed almost constant reversible capacity around 60 mA h g−1 within the range of 1–100 mA cm−2 (0.46–46C). These results clearly suggested that the porous and robust structure of electrode materials should be beneficial for high-rate and long-life battery.
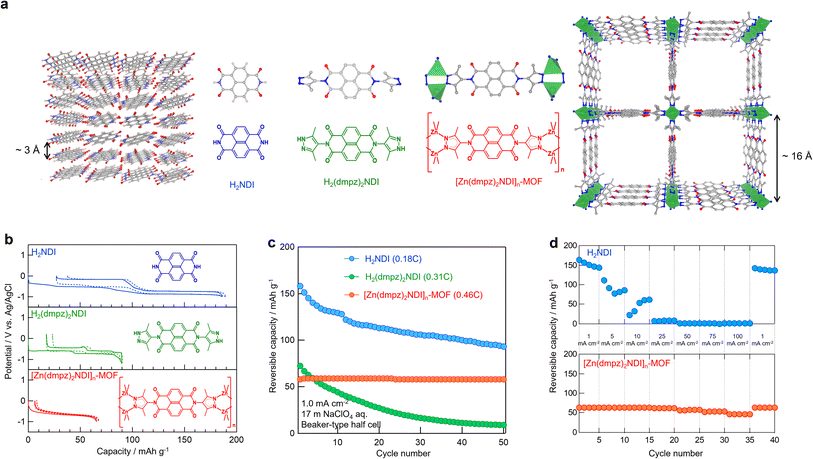 |
| | Fig. 2 (a) Schematic images of crystal and molecule structures. (b) 1st and 2nd discharge/charge profiles and (c) the cyclability of half cell in 17 m NaClO4 aq electrolyte, for H2NDI (blue), H2(dmpz)2NDI (green), and [Zn(dmpz)2NDI]n-MOF (red), respectively. (d) Rate performance of H2NDI (upper) and [Zn(dmpz)2NDI]n-MOF (lower) of half cell in 17 m NaClO4 aq electrolyte. | |
Table 1 Molecular weight and theoretical capacity of active materials
| |
MW/g mol−1 |
2e− theoretical capacity/mA h g−1 |
| H2NDI |
266 |
202 |
| H2(dmpz)2NDI |
454 |
118 |
| [Zn(dmpz)2NDI]n-MOF·2DMF |
666 |
80 |
Redox and Na+-ion storage mechanisms of [Zn(dmpz)2NDI]n-MOF
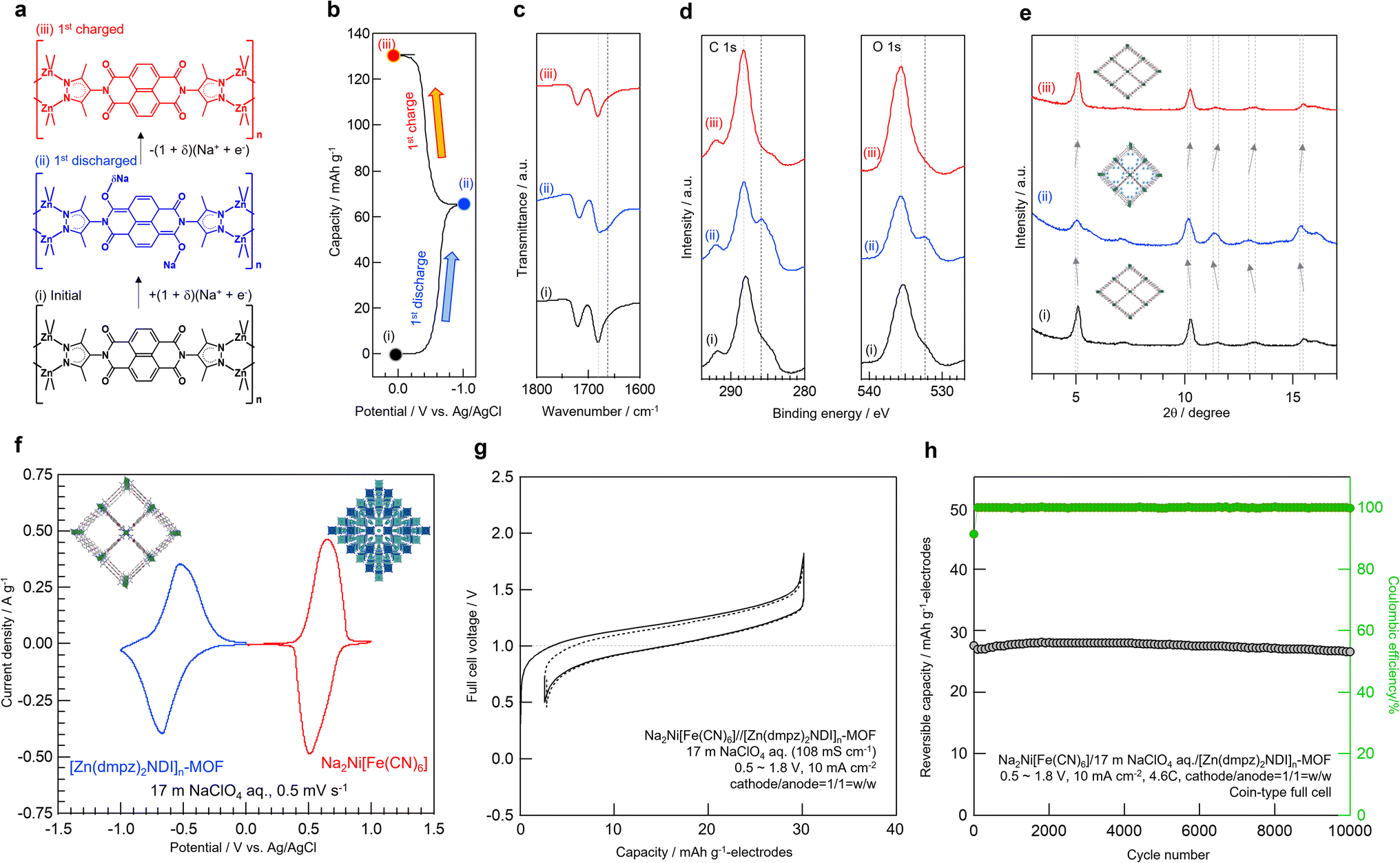
To gain deeper insight on the electrochemical behavior of [Zn(dmpz)2NDI]n-MOF in 17 m NaClO4 aqueous electrolyte along with possible structural change (Fig. 3a), the disc pellets were disassembled from the electrochemical cell either after the first discharge or the first charge (Fig. 3b) to compare these used electrodes with the unused one by FT-IR (Fig. 3c), XPS (Fig. 3d), and XRD (Fig. 3e). In this ex situ FT-IR study, the unused sample (i) showed two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peaks at 1681 and 1720 cm−1, while the new shoulder peak at 1665 cm−1 appeared in the discharged sample (ii) and it disappeared in the charged sample (iii). This new peak may be assignable to the elongated C–O bonds13 in the reduced form of the NDI group depicted in Scheme 1. Similar change during the discharge/charge sequence was also observed in ex situ XPS (Fig. 3d). Only in the case of the sample (ii), noticeable new signals appeared at 286 eV for C1s and 532 eV for O1s, respectively. These signals were not observed in the samples (i) or (iii), both of which displayed signals at 288 eV for C1s and 535 eV for O1s, respectively. We believe that the new signals are typical for C–O− structure and the original signals are for CO structure, though overlaps by the signal of inseparable conductive carbon made these interpretation inconclusive.49 Furthermore, ex situ XRD showed shift in peaks towards lower angles along with discharge from (i) to (ii), whereas that towards higher angles with charge from (ii) to (iii). These results may represent the reversible accommodation of Na+ ion into [Zn(dmpz)2NDI]n-MOF from 17 m NaClO4 aqueous electrolyte with concurrent redox reaction of the NDI moiety.
O stretching peaks at 1681 and 1720 cm−1, while the new shoulder peak at 1665 cm−1 appeared in the discharged sample (ii) and it disappeared in the charged sample (iii). This new peak may be assignable to the elongated C–O bonds13 in the reduced form of the NDI group depicted in Scheme 1. Similar change during the discharge/charge sequence was also observed in ex situ XPS (Fig. 3d). Only in the case of the sample (ii), noticeable new signals appeared at 286 eV for C1s and 532 eV for O1s, respectively. These signals were not observed in the samples (i) or (iii), both of which displayed signals at 288 eV for C1s and 535 eV for O1s, respectively. We believe that the new signals are typical for C–O− structure and the original signals are for CO structure, though overlaps by the signal of inseparable conductive carbon made these interpretation inconclusive.49 Furthermore, ex situ XRD showed shift in peaks towards lower angles along with discharge from (i) to (ii), whereas that towards higher angles with charge from (ii) to (iii). These results may represent the reversible accommodation of Na+ ion into [Zn(dmpz)2NDI]n-MOF from 17 m NaClO4 aqueous electrolyte with concurrent redox reaction of the NDI moiety.
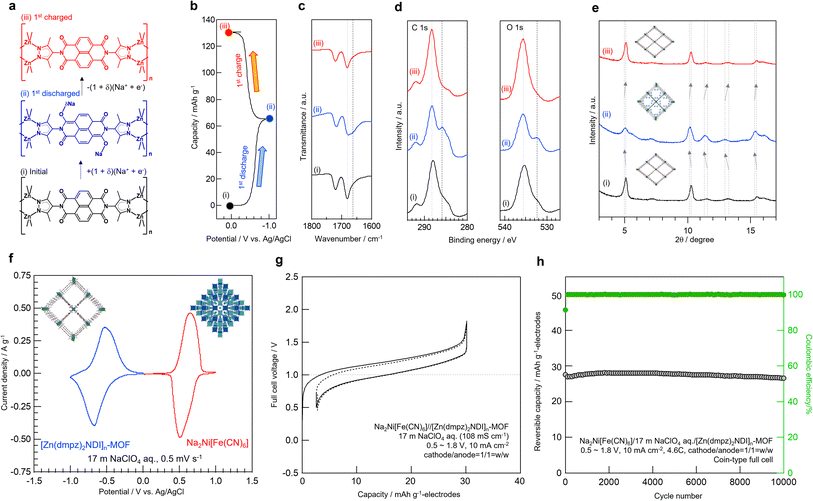 |
| | Fig. 3 (a) Possible structural change of [Zn(dmpz)2NDI]n-MOF upon electrochemical reaction in 17 m NaClO4 aq. (b) Origin of the samples; (i) unused sample, (ii) used sample after discharging down to −1.0 V, and (iii) used sample after charging up to 0.0 V vs. Ag/AgCl. (c) Ex situ FT-IR. (d) Ex situ XPS. (e) Ex situ XRD. (f) Cyclic voltammetry of [Zn(dmpz)2NDI]n-MOF anode (blue) and Na2Ni[Fe(CN)6] cathode (red) in 17 m NaClO4 aq along with the schematic image of their initial structures. (g) 1st and 2nd charge/discharge profiles for the full-cell of Na2Ni[Fe(CN)6]//[Zn(dmpz)2NDI]n-MOF in 17 m NaClO4 aq electrolyte. (h) Cyclability for the full-cell. | |
Encouraged by the stable electrochemical performance of [Zn(dmpz)2NDI]n-MOF in 17 m NaClO4 aqueous electrolyte, we next assembled a Na-ion battery comprised of [Zn(dmpz)2NDI]n-MOF and Na2Ni[Fe(CN)6] PBA as electrodes and 17 m NaClO4 as aqueous electrolyte. As shown in Fig. 3g, this rocking chair-type aqueous battery displayed cell voltage of ca. 1 V, which was in good agreement with cyclic voltammogram of separate electrodes shown in Fig. 3f; −0.5 V for [Zn(dmpz)2NDI]n-MOF and +0.5 V for Na2Ni[Fe(CN)6] PBA (vs. Ag/AgCl) in 17 m NaClO4 aqueous electrolyte. Notably, this battery showed virtually no capacity decay over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (Fig. 3h).
000 cycles (Fig. 3h).
Better compatibility of hydrated K+-ion electrolyte with [Zn(dmpz)2NDI]n-MOF
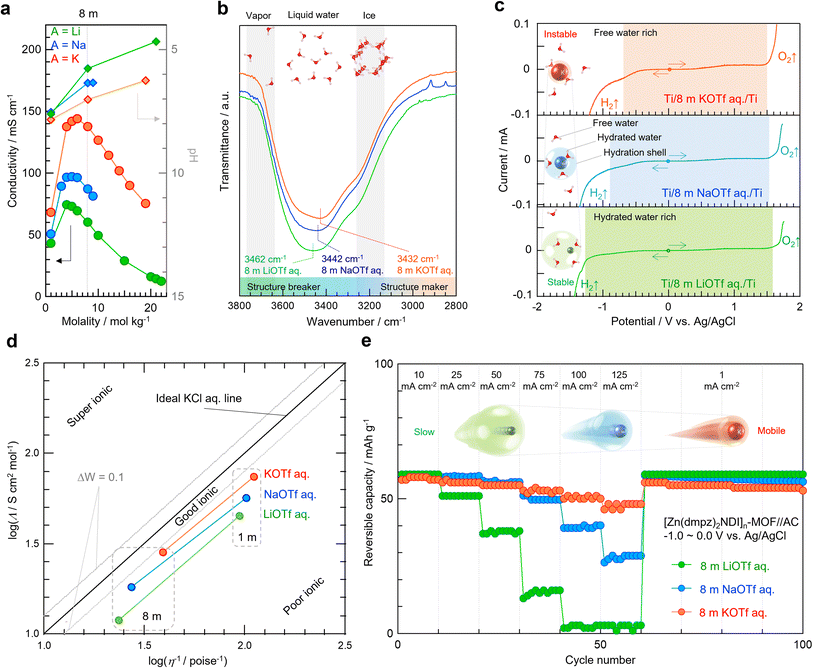
To identify which alkaline ion would perfectly suit the present battery of [Zn(dmpz)2NDI]n-MOF anode and Na2Ni[Fe(CN)6] PBA cathode, we next turned our attention on aqueous electrolytes having alkaline triflates (SO3CF3) because they show commonly high solubility in water irrespective of alkaline ion, which is a marked contrast to those having alkaline ClO4 (Table 2). We first studied pH (◆) and ionic conductivity (●) of aqueous AOTf solutions (A = Li, Na, K) at varying concentration (Fig. 4a). The pH of AOTf solution ranked in the order of K+ > Na+ > Li+ at the same concentration, but their individual values linearly decreased along with the increasing concentration. These results were not surprising because the Lewis acidity of the solute may give a proportional effect on the Brønsted acidity of its aqueous solution. The ionic conductivity (●) also ranked in the order of K+ > Na+ > Li+ at the same concentration. However, their individual values became maximum at different concentrations around 5–7 m and decreased either at lower or higher concentrations. Since all AOTf solutions showed acceptably good ionic conductivity at the concentration of 8 m (molar ratio of solute and water is ca. 1:7), we next studied the structure of these particular AOTf solutions by FT-IR and roughly estimated their electrochemical stability window by linear sweep voltammetry (LSV). Fig. 4b focuses on the broad peaks centered at 3462, 3442, and 3432 cm−1, which were observed in the FT-IR measurement for 8 m LiOTf, NaOTf, and KOTf solutions, respectively. It has been well documented that the O–H stretching in water is blue shifted upon destruction of its ice-like structure, which is caused by the hydration of Lewis acidic metal ion.35,36,46,47,50 Predictably, the more Lewis acidic Li+ ion should bind a greater number of Lewis basic water molecules to form a larger hydrated ion in 8 m LiOTf solution, whereas the less Lewis acidic K+ ion may form a smaller hydrated ion in 8 m KOTf solution. The highlighted areas in Fig. 4c denote the electrochemical stability windows, which were derived from oxidation/reduction waves obtained by LSV measurements using titanium mesh. The window became wider in the order of K+ < Na+ < Li+, which may be reasonable if Lewis acidic metal ion decreases the number of free water molecules, which are more readily subjected to electrolysis than coordinated water molecules. Also noteworthy is that the three potentials for the oxidation of 8 m AOTf solutions were almost identical, while those for the reduction varied significantly. This may be attributable to the different solubility of passivation layers on the electrode, whose large part should be composed of alkaline fluorides that are generated upon the reductive decomposition of OTf.51–53 Specifically, sparingly soluble LiF can form a robust passivation layer which can lower the reduction boundary effectively, whereas readily soluble KF cannot play a similar role.
Table 2 Saturated concentration of alkaline salts with different anions in water
| Concentration/mol kg−1 |
Li+ |
Na+ |
K+ |
| F− |
0.05 |
1 |
18 |
| SO3CF3− |
21 |
9 |
19 |
| N(SO2CF3)2− |
21 |
9 |
1 |
| ClO4− |
6 |
17 |
0.1 |
 |
| | Fig. 4 (a) pH and ionic conductivity of AOTf aq. (b) FT-IR spectra. (c) LSVs on Ti current collector of 8 m AOTf aq (the hatch sections show estimated potential windows). (d) Walden plot of 1 m and 8 m AOTf aq. (e) Rate performance of [Zn(dmpz)2NDI]n-MOF in 8 m AOTf aq (A = Li, Na, K). | |
Fig. 4d summarizes the Walden plot54 for the three AOTf solutions at the concentration of 1 m as well as 8 m. These data were collected by the calculation using parameters listed in Table 3. Under both diluted and concentrated conditions, all data were close to “good ionic” area and KOTf solutions were slightly better ionic and less viscous than NaOTf or LiOTf. Fig. 4e illustrates the rate performance for the half-cell of [Zn(dmpz)2NDI]n-MOF electrode in 8 m AOTf aqueous electrolytes at different C-rates. The difference of reversible capacity between three electrolytes became apparent when the current density was gradually increased. The initial reversible capacity at 10 mA cm−2 (4.6C) was almost halved or lost at 125 mA cm−2 (58C) in the case of NaOTf or LiOTf, respectively, whereas it was hardly attenuated in the case of KOTf albeit with slight loss under the influence of its narrower stability window. When the current density was finally lowered from 125 to 1 mA cm−2 at the 61st cycle and thereafter, all half-cells recovered the capacity nearly equal or slightly inferior to their initial value. These results may indicate that there was virtually no deterioration of [Zn(dmpz)2NDI]n-MOF electrode after repeated cycles at a wide range of current density. The excellent rate performance observed with the KOTf electrolyte especially at higher current density was largely caused by its better ionic conductivity and lower viscosity discussed in Fig. 4a, b, and d. Furthermore, we believe that the smaller hydrated K+ ion could travel at higher speed even within the pore of [Zn(dmpz)2NDI]n-MOF electrode, in marked contrast to the larger Li+ and Na+ ions at lower speeds, though the detailed study on the ion diffusivity such as EIS and GITT in the near future may reinforce these qualitative discussions.
Table 3 Parameters of alkaline metal triflate aqueous electrolytes
| |
LiOTf aq |
NaOTf aq |
KOTf aq |
| Concentration |
1 m |
8 m |
1 m |
8 m |
1 m |
8 m |
| Conductivity/mS cm−1 |
42 |
59 |
52 |
88 |
68 |
133 |
| Density/g cm−3 |
1.07 |
1.40 |
1.09 |
1.45 |
1.09 |
1.47 |
| Viscosity/mPa s |
1.14 |
5.91 |
1.07 |
5.33 |
0.98 |
3.77 |
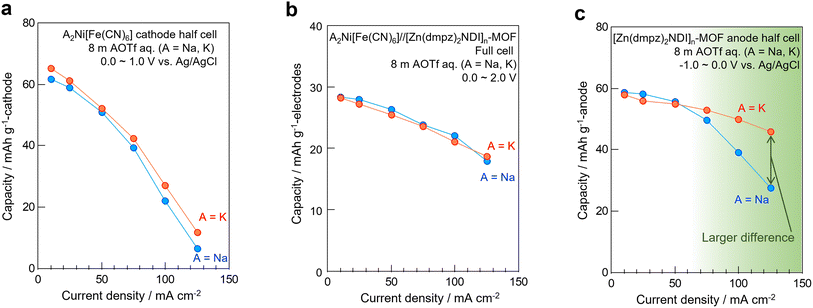
Unlike the half-cell of [Zn(dmpz)2NDI]n-MOF electrode (Fig. 5c), the half-cell of A2Ni[Fe(CN)6] PBA electrode in 8 m AOTf aqueous electrolytes (A = Na, K) showed only little difference in the rate performance even at higher current density above 100 mA cm−2 as shown in Fig. 5a. These results may suggest that the pore size provided by the PBA electrode was not large enough for hydrated Na+ and K+ ions. Therefore, both could go into the pore only after dehydration and the difference in the total travel time became negligible. Furthermore, the full-cell combining these two electrodes no longer showed kinetic difference between Na+ and K+-based aqueous electrolytes at a broad range of current density as shown in Fig. 5b. Therefore, we may conclude that K+-based aqueous electrolyte is fundamentally beneficial in terms of rate performance, but we can only appreciate the merit when we employ appropriate electrode materials that have sufficiently large pores to accommodate its hydrated ion without difficulty. To emphasize this conclusion, we separately illustrated the Ragone plot for the full-cells separately in Fig S3.† However, we have to be careful not to confuse the kinetic information with the potential difference of cathodes.
 |
| | Fig. 5 Rate capability of (a) A2Ni[Fe(CN)6], (c) [Zn(dmpz)2NDI]n-MOF, and (b) their full-cell in 8 m AOTf aq (A = Na, K), respectively. These were rebuilt based on the raw data shown in Fig. S3.† | |
Experimental section/methods
Preparation of electrode materials
H2(dmpz)2NDI and [Zn(dmpz)2NDI]n-MOF were prepared according to the reported procedure20 as shown in Scheme S1.† H2(dmpz)2NDI was characterized by JEOL JNM-ECA 600 spectrometer (1H NMR at 600 MHz, 13C{1H} NMR at 150 MHz) using (CD3)2SO, to check its identity and purity by comparison with the reported spectra. [Zn(dmpz)2NDI]n-MOF was characterized by powder X-ray diffraction (XRD) using Rigaku Miniflex 600 X-ray diffractometer using Cu-Kα radiation, to confirm its crystal structure20 (Fig. S1a†). To determine the remaining content of DMF in thus obtained [Zn(dmpz)2NDI]n-MOF, thermogravimetric analysis (TGA) was performed on a Rigaku Thermo Plus TG8110 under a nitrogen gas flow (Fig. S1b†). The local structures depicted in Fig. 1a as well as Fig. S2† were graphically rebuilt based on the reported data. Na2Ni[Fe(CN)6] and K2Ni[Fe(CN)6] were prepared by co-precipitation methods according to the reported methods.55,56 The chemical compositions of Na2Ni[Fe(CN)6] and K2Ni[Fe(CN)6] were estimated by EDS (JEOL JCM-7000) as Na1.0Ni[Fe(CN)6]0.8·2.8H2O (theoretical capacity is approximately 75 mA h g−1) and K1.5Ni[Fe(CN)6]0.9·1.8H2O (theoretical capacity is approximately 71 mA h g−1), respectively.
Preparation of aqueous electrolytes
Aqueous electrolytes were prepared by dissolving the appropriate amounts of NaClO4 (98%; Kishida), LiOTf (98.0%; TCI), NaOTf (98.0%; TCI), or KOTf (98.0%; TCI) in ultra-pure water (Merck KGaA, Direct-Q UV 3). These electrolytes were characterized by ATR-FT-IR (Thermo Fisher Scientific, Nicolet iS5), conductivity meter (Mettler Toledo, SevenExcellence S479), pH meter (HORIBA, LAQUAact D73-T), viscosity meter (A&D, SV-1A), and density meter (Anton Paar, DMA 35).
Electrochemical measurements
To improve the electronic conductivity, acetylene black (AB, DENKA) was mixed with electrode materials at a weight ratio of 25:70. The composites were mixed with polytetrafluoroethylene binder (PTFE) (Polyflon F-104; Daikin Industries) at a weight ratio of 95:5 to prepare the electrode pellets. They were shaped into disc of 3 mm diameter, which were sandwiched between sheets of titanium mesh (Thank-Metal) for electrochemical measurements. Three-electrode electrochemical beaker-type half cells with aqueous electrolytes were used in cyclic voltammetry (CV) and galvanostatic charge/discharge tests. Ag/AgCl electrode with saturated KCl (BAS Inc., RE-1CP) and activated carbon (AC) electrode were used as the reference and counter electrodes, respectively. For coin-type full cell, the cathode/anode weight balance was adjusted to 1/1 to fix cathode/anode capacity balance as approximately 1/1 (Fig. S4†). Linear sweep voltammetry (LSV) and CV were performed at a scan rate of 0.5 mV s−1 (AMETEK, VersaSTAT 3), and galvanostatic charge/discharge tests were carried out using a cycler (NAGANO & Co.) at constant current density.
Ex situ IR, XPS and XRD
Ex situ IR (JASCO, FT-IR 680 plus), X-ray photoelectron spectroscopy (XPS) (JEOL, JPS-9010 MC/IV; Mg-Kα) and ex situ XRD were carried out to study the electrochemical change of [Zn(dmpz)2NDI]n-MOF after the first discharge and the first charge in 17 m NaClO4 aqueous electrolyte as follows; The used cell was disassembled in an Ar-filled glove box and the retrieved anode material was immersed in distilled water for 30 min and then vacuum dried for 3 h to prepare samples, which were transferred under Ar atmosphere to the IR, XPS and XRD instruments.
Conclusion
A new strategy to exploit maximum rate performance in aqueous battery was demonstrated by use of [Zn(dmpz)2NDI]n-MOF electrode, which showed excellent electrochemical properties in aqueous electrolytes. This study showed that a rational design of electrode material that allows hydrated ions travel freely even in its pore may contribute significantly to the improvement of rate performance. Such electrode materials with sufficiently large pores allow hydrated charge carriers to undergo co-insertion directly from the electrolyte. Along this line, exploration of suitable cathode materials is now underway to complete the configuration of high-rate aqueous K+-ion battery.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was financially supported by Grant-in-Aid for Scientific Research program (Grant Number JP19K15679 and JP23K04910), supported in part by the Elements Strategy Initiative for Catalysts and Batteries (ESICB) project (Grant Number JPMXP0112101003), Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT), and supported in part by “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” from the MEXT.
References
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- N. Alias and A. A. Mohamad, J. Power Sources, 2015, 274, 237–251 CrossRef CAS.
- J. Liu, C. Xu, Z. Chen, S. Ni and Z. X. Shen, Green Energy Environ., 2018, 3, 20–41 CrossRef.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- Q. Zhao, Y. Lu and J. Chen, Adv. Energy Mater., 2017, 7, 1601792 CrossRef.
- X. Yin, S. Sarkar, S. Shi, Q.-A. Huang, H. Zhao, L. Yan, Y. Zhao and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908445 CrossRef CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS PubMed.
- W. Zhang, W. Huang and Q. Zhang, Chem.–Eur. J., 2021, 27, 6131–6144 CrossRef PubMed.
- K. Holguin, M. Mohammadiroudbari, K. Qin and C. Luo, J. Mater. Chem. A, 2021, 9, 19083–19115 RSC.
- D. J. Kim, Y. H. Jung, K. K. Bharathi, S. H. Je, D. K. Kim, A. Coskun and J. W. Choi, Adv. Energy Mater., 2014, 4, 1400133 CrossRef.
- A. E. Lakraychi, K. Fahsi, L. Aymard, P. Poizot, F. Dolhem and J. P. Bonnet, Electrochem. Commun., 2017, 76, 47–50 CrossRef CAS.
- Y. Shi, H. Tang, S. Jiang, L. V. Kayser, M. Li, F. Liu, F. Ji, D. J. Lipomi, S. P. Ong and Z. Chen, Chem. Mater., 2018, 30, 3508–3517 CrossRef CAS.
- M. Na, Y. Oh and H. R. Byon, Chem. Mater., 2020, 32, 6990–6997 CrossRef CAS.
- H. Qin, Z. P. Song, H. Zhan and Y. H. Zhou, J. Power Sources, 2014, 249, 367–372 CrossRef CAS.
- S. Qiu, Y. Xu, X. Li, S. K. Sandstrom, X. Wu and X. Ji, Electrochem. Commun., 2021, 122, 106880 CrossRef CAS.
- S. Jhulki, C. H. Feriante, R. Mysyk, A. M. Evans, A. Magasinski, A. S. Raman, K. Turcheniuk, S. Barlow, W. R. Dichtel, G. Yushin and S. R. Marder, ACS Appl. Energy Mater., 2020, 4, 350–356 CrossRef.
- Y. Zhou and L. Han, Coord. Chem. Rev., 2021, 430, 213665 CrossRef CAS.
- L. Kong, M. Cheng, H. Huang, J. Pang, S. Liu, Y. Xu and X. Bu, EnergyChem, 2022, 4, 100090 CrossRef CAS.
- C. R. Wade, T. Corrales-Sanchez, T. C. Narayan and M. Dincă, Energy Environ. Sci., 2013, 6, 2172–2177 RSC.
- C. R. Wade, M. Li and M. Dincǎ, Angew. Chem. Int., Ed., 2013, 52, 13377–13381 CrossRef CAS PubMed.
- N. Masciocchi, S. Galli and A. Sironi, Comments Inorg. Chem., 2005, 26, 1–37 CrossRef CAS.
- C. Wang, X. Liu, N. Keser Demir, J. P. Chen and K. Li, Chem. Soc. Rev., 2016, 45, 5107–5134 RSC.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC.
- H. J. Choi, M. Dincǎ, A. Dailly and J. R. Long, Energy Environ. Sci., 2010, 3, 117–123 RSC.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef.
- K. Nakamoto, R. Sakamoto, M. Ito, A. Kitajou and S. Okada, Electrochemistry, 2017, 85, 179–185 CrossRef CAS.
- K. Nakamoto, R. Sakamoto, Y. Sawada, M. Ito and S. Okada, Small Methods, 2019, 3, 1800220 CrossRef CAS.
- S. Dhir, S. Wheeler, I. Capone and M. Pasta, Chem, 2020, 6, 2442–2460 CAS.
- X. Zhang, T. Xiong, B. He, S. Feng, X. Wang, L. Wei and L. Mai, Energy Environ. Sci., 2022, 15, 3750–3774 RSC.
- T. Hosaka, A. Noda, K. Kubota, K. Chiguchi, Y. Matsuda, K. Ida, S. Yasuno and S. Komaba, ACS Appl. Mater. Interfaces, 2022, 14, 23507–23517 CrossRef CAS PubMed.
- D. Su, A. McDonagh, S. Qiao and G. Wang, Adv. Mater., 2017, 29, 1604007 CrossRef.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y. S. Hu, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- K. Miyazaki, N. Takenaka, E. Watanabe, Y. Yamada, Y. Tateyama and A. Yamada, ACS Appl. Mater. Interfaces, 2020, 12, 42734–42738 CrossRef CAS.
- S. Ko, Y. Yamada and A. Yamada, Electrochem. Commun., 2020, 116, 106764 CrossRef CAS.
- Q. Zheng, S. Miura, K. Miyazaki, S. Ko, E. Watanabe, M. Okoshi, C. Chou, Y. Nishimura, H. Nakai, T. Kamiya, T. Honda, J. Akikusa, Y. Yamada and A. Yamada, Angew. Chem. Int., Ed., 2019, 58, 14202–14207 CrossRef CAS PubMed.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef CAS.
- K. Hurlbutt, S. Wheeler, I. Capone and M. Pasta, Joule, 2018, 2, 1950–1960 CrossRef CAS.
- B. Wang, Y. Han, X. Wang, N. Bahlawane, H. Pan, M. Yan and Y. Jiang, iScience, 2018, 3, 110–133 CrossRef CAS PubMed.
- S. Qiu, Y. Xu, X. Wu and X. Ji, Electrochem. Energy Rev., 2022, 5, 242–262 CrossRef CAS.
- M. Ishizaki, H. Ando, N. Yamada, K. Tsumoto, K. Ono, H. Sutoh, T. Nakamura, Y. Nakao and M. Kurihara, J. Mater. Chem. A, 2019, 7, 4777–4787 RSC.
- M. Asai, A. Takahashi, K. Tajima, H. Tanaka, M. Ishizaki, M. Kurihara and T. Kawamoto, RSC Adv., 2018, 8, 37356–37364 RSC.
- S. Yagi, M. Fukuda, T. Ichitsubo, K. Nitta, M. Mizumaki and E. Matsubara, J. Electrochem. Soc., 2015, 162, A2356–A2361 CrossRef CAS.
- S. Phadke, R. Mysyk and M. Anouti, J. Energy Chem., 2020, 40, 31 CrossRef.
- S. Saeed, S. Boyd, W.-Y. Tsai, R. Wang, N. Balke and V. Augustyn, Chem. Commun., 2021, 57, 6744–6747 RSC.
- R. Sakamoto, M. Yamashita, K. Nakamoto, Y. Zhou, N. Yoshimoto, K. Fujii, T. Yamaguchi, A. Kitajou and S. Okada, Phys. Chem. Chem. Phys., 2020, 22, 26452–26458 RSC.
- K. Nakamoto, R. Sakamoto, Y. Nishimura, J. Xia, M. Ito and S. Okada, Electrochemistry, 2021, 89, 415–419 CrossRef CAS.
- M. H. Lee, S. J. Kim, D. Chang, J. Kim, S. Moon, K. Oh, K.-Y. Park, W. M. Seong, H. Park, G. Kwon, B. Lee and K. Kang, Mater. Today, 2019, 29, 26–36 CrossRef CAS.
- Z. Meng, C. G. Jones, S. Farid, I. U. Khan, H. M. Nelson and K. A. Mirica, Angew. Chem. Int., Ed., 2021, 61, e202113569 CrossRef PubMed.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Nat. Energy, 2016, 1, 16129 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem. Int., Ed., 2016, 55, 7136–7141 CrossRef CAS.
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y.-S. Hu, K. Xu and C. Wang, Adv. Energy Mater., 2017, 7, 1701189 CrossRef.
- W. Xu, E. I. Cooper and C. A. Angell, J. Phys. Chem. B, 2003, 107, 6170–6178 CrossRef CAS.
- X. Wu, C. Wu, C. Wei, L. Hu, J. Qian, Y. Cao, X. Ai, J. Wang and H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 5393–5399 CrossRef CAS PubMed.
- J. Peng, J. Wang, H. Yi, W. Hu, Y. Yu, J. Yin, Y. Shen, Y. Liu, J. Luo, Y. Xu, P. Wei, Y. Li, Y. Jin, Y. Ding, L. Miao, J. Jiang, J. Han and Y. Huang, Adv. Energy Mater., 2018, 8, 1702856 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Junwen Baib,
Minyan Zhaob,
Ryo Sakamoto
*a,
Junwen Baib,
Minyan Zhaob,
Ryo Sakamoto