
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding SEI evolution during the cycling test of anode-free lithium-metal batteries with LiDFOB salt†
Naufal Hanif Hawari ab,
Huiqing Xiea,
Achmad Prayogib,
Afriyanti Sumboja*b and
Ning Ding*a
ab,
Huiqing Xiea,
Achmad Prayogib,
Afriyanti Sumboja*b and
Ning Ding*a
aInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology, and Research), 138634, Singapore. E-mail: dingni@imre.a-star.edu.sg
bMaterial Science and Engineering Research Group, Faculty of Mechanical and Aerospace, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia. E-mail: sumboja@itb.ac.id
First published on 29th August 2023
Abstract
Anode-free lithium-metal batteries (AFLMBs) have the potential to double the energy density of Li-ion batteries, but face the challenges of mossy dendritic lithium plating and an unstable solid electrolyte interphase (SEI). Previous studies have shown that the AFLMBs with an electrolyte containing lithium difluoro(oxalato)borate (LiDFOB) salt outperform those with lithium hexafluorophosphate (LiPF6), but the mechanism behind this improvement is not fully understood. In this study, X-ray photoelectron spectroscopy (XPS) depth profile analysis and electrochemical impedance spectroscopy (EIS) were conducted to investigate the SEI on plated Li from the two conducting salts and their evolution in Cu‖NMC full cells during cycling. XPS results revealed that an inorganic-rich SEI layer is formed in the cell with LiDFOB-based electrolyte, with a low carbon/oxygen ratio of 0.56 compared to 1.42 in the LiPF6-based cell. With the inorganic-rich SEI, a dense electroplated Li with a shining surface on the Cu substrate can be retained after ten cycles. The inorganic-rich SEI enhances the reversibility of Li plating and stripping, with a high average CE of ∼98% and a stable charge/discharge voltage profile. The changes in SEI resistance and cathode electrolyte interphase resistance are more prominent compared to the changes in solution and charge transfer resistances, which further validate the role of the passivation films on Li deposits and NMC cathode surfaces in stabilizing AFLMB cycling performance.
Introduction
Lithium (Li) metal is hailed as the next holy grail of high-energy-density anodes, which promises the lowest electrochemical potential (−3.04 V vs. standard hydrogen electrode) and the highest theoretical specific capacity (3861 mA h g−1).1,2 However, Li metal is costly and handling Li metal requires ultra-low humidity, which increases the production costs.3 Furthermore, the use of excessive Li may pose safety concerns for the equipped battery.4 Thus, anode-free lithium-metal batteries (AFLMBs) with all the active lithium supplied from the cathode materials become the most promising choice as the next-generation rechargeable batteries, with energy densities of up to 423 W h kg−1 and 1514 W h L−1.5 It is also noteworthy that the production of AFLMBs is compatible with the existing manufacturing facilities for Li-ion batteries.Nonetheless, AFLMBs suffer fast capacity fading due to the limited Li supply.6–8 In particular, interfacial reactions that form the solid electrolyte interphase (SEI) layer consume the active Li and adversely affect the AFLMBs cell life.9,10 The undesirable heterogeneous SEI layer may form due to inherent electrolyte instability at low reduction potentials and inhomogeneous surface chemistry. This passivation layer produces unequal electric field distribution and Li concentration gradient, which may be the root cause of Li dendrites growth.11 The unstable Li dendrites could lose electrical contact with the Cu current collector during cycling, resulting in “dead” Li that is electrochemically irreversible.12 Thus, the formation and evolution of SEI influence Li plating and stripping during cell cycling, and affect the coulombic efficiency (CE) of AFLMBs.
Various strategies have been employed to suppress the dead Li formation in AFLMBs, focusing on modified current collectors,13,14 carbon host materials,15 artificial solid electrolyte interphase additives,16 and innovative electrolyte strategies.17 Modified current collectors, such as those coated with protective layers or porous structures, are reported to mitigate the dead Li formation by promoting more uniform lithium plating and stripping.18 Carbon-based hosts can serve as a physical barrier against dendrite growth and enhance lithium ion transport.15 However, Li host with high contact area may promote excessive SEI formation, leading to electrolyte depletion. An artificial SEI protective layer can be engineered through additives or coatings, enhancing electrolyte compatibility and preventing the continuous growth of detrimental Li dendrites.18 However, developing an artificial SEI remains challenging due to the trade-off between ionic conductivity and mechanical robustness.19 In the end, material choice of electrolyte (i.e., solvent, co-solvent, salt, and additives) still play a pivotal role in AFLMBs performance as it controls the Li+ ion flux, current density, and de-solvation mechanisms that can lead to homogenous and dense Li plating on the current collector.
The role of electrolyte solvents and salts in the formation and evolution of SEI film as well as Li electrodeposition, has been reported in the literature.20–23 Ether-based solvents, such as dimethyl ether (DME), are widely studied to achieve smooth lithium plating in Li‖Li or Li‖Cu symmetric cells.22,24 However, its low working voltage inhibits its compatibility with the high-voltage cathode. Using highly concentrated electrolytes can solve this problem, but the high production cost will be unavoidable.25 Notably, carbonate solvents can achieve AFLMBs with high cut-off voltage.26 Common combinations of carbonate solvents with a high dielectric constant (e.g., ethylene carbonate (EC)) and low viscosity solvent (e.g., diethylene carbonate (DEC)) can be employed in AFLMBs.27 However, the commercial electrolyte for Li-ion batteries with LiPF6 as the conducting salt results in poor Li deposits due to the autocatalytic reaction of LiPF6 with a trace amount of water.28 This reaction generates HF that damages SEI on the Li metal surface and causes poor cycling stability of AFLMBs. Besides, HF could also initiate transition metal dissolution on the cathode side, further deteriorating the battery performance.29
Therefore, choosing a compatible Li salt for carbonate electrolytes is crucial for enabling high-energy-density and stable AFLMBs. Lithium difluoro(oxalato)borate (LiDFOB) has emerged as a suitable choice to replace LiPF6 in prolonging the cycle life of AFLMBs with a carbonate-based electrolyte.30–32 In a LiFePO4 (LFP)‖Cu cells, the oxalate group in LiDFOB was reported to regulate the growth of LiF particles by serving as a capping agent, producing a uniform distribution of LiF particles on the LFP surface.33 Weber et al.32 employed different salt compositions in AFLMBs with LiNi0.5Ni0.3Co0.2O2 cathode. Cells with LiDFOB salt cycled between 3.6–4.5 V can reach 60 cycles with capacity retention above 80%, whereas cells with LiPF6 only lasted for 10 cycles. The performance improvement of AFLMBs with LiDFOB salt can be related to the SEI composition and morphology of the electrodeposited lithium.22,34 Reports using Li‖Li cells have shown that SEI influences the lithium morphology and CE, which can be demonstrated by measuring the changes in the internal resistance by electrochemical impedance spectroscopy (EIS).35 The cells that produce dense plated Li morphology are reported to have a stable solution resistance, an increase of charge transfer resistance, and an increase in interfacial film (i.e., SEI) resistance after the symmetric cell is cycled for 600 hours. However, these prior reports are limited to Li‖Li cells, where the Li supply is unlimited and is completely different from Cu‖NMC cell configuration.
In this work, we study the SEI evolutions governed by the two conducting salts (i.e., LiPF6 and LiDFOB) in a full-cell configuration during the cycling test. The effect of salt type on the performance and stability of AFLMBs is investigated. Cu‖NMC cell with high cathode mass-loading of 20 mg cm−2 is used, mimicking the industrial standard for cathode mass loading. X-ray photoelectron spectroscopy (XPS) depth profile analysis is used to examine the SEI composition formed by different salts after the formation cycle. The plated Li of different cycles is monitored with SEM to reveal its morphology. The resistance is checked by EIS to further reveal the evolution of SEI behaviour in cell operation. Our findings demonstrate that AFLMBs with LiDFOB-based electrolytes exhibit better performance and stability compared to those with LiPF6. Specifically, the inorganic-rich SEI formed with LiDFOB promotes stable SEI formation and better Li plating and stripping kinetics, resulting in ∼98% CE with a retained shining and dense plated Li. These mechanistic insights can further explain the behaviour of LiDFOB salts in SEI formation for prolonging the AFLMBs cycle life.
Experimental section
Materials preparation
A 9 μm thick electrodeposited copper foil (Jingliang Copper Co., Ltd) was used as a working electrode in AFLMBs. LiNi0.6Mn0.2Co0.2O2 (NMC622, ANR Technologies Pte., Ltd) powders were mixed with polyvinylidene fluoride (PVDF, Solef® 5130) and Super C65 conductive carbon (TIMCAL) with a ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5:0.5, respectively. The obtained slurry was coated on aluminum foil and punched into a disk with a diameter of 12 mm. The NMC622 active material mass loading is 20 ± 0.5 mg cm−2. The electrolyte is comprised of 1 M lithium difluoro(oxalate) borate (LiDFOB, water content < 500 ppm, Yacoo Science Co., Ltd) or 1 M lithium hexafluorophosphate (LiPF6, battery grade, ≥99.99%, Sigma Aldrich) dissolved in ethylene carbonate (EC, Sigma Aldrich):diethyl carbonate (DEC, Sigma Aldrich) (1:1, v/v), which has been dried with 4 Å molecular sieves for 48 hours prior to the mixing. The CR2032-cells were assembled inside an Ar-filled glove box (MBRAUN, Germany) with O2 and H2O concentrations maintained at less than 0.1 ppm. Each cell contains 60 μL of electrolyte with the Cellgard® 2400 separator (Φ = 16 mm).
:0.5:0.5, respectively. The obtained slurry was coated on aluminum foil and punched into a disk with a diameter of 12 mm. The NMC622 active material mass loading is 20 ± 0.5 mg cm−2. The electrolyte is comprised of 1 M lithium difluoro(oxalate) borate (LiDFOB, water content < 500 ppm, Yacoo Science Co., Ltd) or 1 M lithium hexafluorophosphate (LiPF6, battery grade, ≥99.99%, Sigma Aldrich) dissolved in ethylene carbonate (EC, Sigma Aldrich):diethyl carbonate (DEC, Sigma Aldrich) (1:1, v/v), which has been dried with 4 Å molecular sieves for 48 hours prior to the mixing. The CR2032-cells were assembled inside an Ar-filled glove box (MBRAUN, Germany) with O2 and H2O concentrations maintained at less than 0.1 ppm. Each cell contains 60 μL of electrolyte with the Cellgard® 2400 separator (Φ = 16 mm).
Electrochemical measurements
The formation cycle was done by applying a current rate of 5 mA g−1 to 3.8 V, then 25 mA g−1 to 4.5 V, followed by discharge at 50 mA g−1 to 2.8 V. The galvanostatic charge–discharge was conducted at room temperature with a battery analyzer (Neware Battery Testing System) within the potential range of 2.8 V to 4.5 V at 50 mA g−1. Linear scan voltammetry (LSV) was conducted with Autolab PGSTAT 302N (Metrohm AG) from the open circuit voltage (OCV) to −0.2 V. The electrochemical impedance spectroscopy measurements were measured at room temperature with Autolab PGSTAT 302N (Metrohm AG) in the frequency range of 1 MHz to 0.01 Hz, with an amplitude of 10 mV after fully charged to 4.5 V and rested for 24 hours. Subsequently, the EIS result was fitted with the NOVA Software. The electrolyte conductivity was measured with Pt microelectrode with TSC 1600 equipment (rhd Instruments) connected to Autolab M204 (Metrohm AG). Cell impedances with 1 M LiDFOB or 1 M LiPF6 in EC:DEC (1:1, v/v) were measured at the temperature range from −10 to 70 °C with 10 min for equilibration.
Surface characterizations
The cells were evaluated after 100% charge (SOC) in the 1st, 5th, and 10th cycles, respectively. All the cells were disassembled inside the glovebox and washed with anhydrous dimethyl carbonate (DMC, dried with 4 Å molecular sieves) to thoroughly remove the electrolyte's remnant. The lithium-plated copper foil was then dried under a vacuum at room temperature for one hour. The sample was transferred to the characterization chamber (e.g., SEM or XPS) using an airtight container. The samples were exposed to the air for a maximum of 10 seconds.JEOL JSM-7600F field emission scanning electron microscopy (FE-SEM) was used to observe the morphology of plated lithium. Characterization of X-ray photoelectron spectroscopy (XPS) was carried out at high vacuum (3.8 × 10−8 Torr) in the PHI Quantera SKM X-ray photoelectron spectrometer with Al kα source (1486.6 eV). The examined areas were 250 μm2. XPS depth profile analyses of the SEI were done with Ar+ sputter-etching at different stages: 0 min (surface), 6 min, and 12 min. The etching rate is 1.67 nm min−1 based on SiO2. The obtained XPS spectra were calibrated to a C–C bond with a binding energy of 285 eV and fitted using CasaXPS (version 2.3.17, Casa Software Ltd).
Results and discussion
In this study, the full-cell AFLMBs were assembled using electrolytes consisting of 1 M LiDFOB in EC:DEC (1:1, v/v) or 1 M LiPF6 in EC:DEC (1:1, v/v) and NMC622 cathode with high areal mass loading of 20 mg cm−2. The cells were cycled between 2.8 and 4.5 V in a full charge–discharge mode at a constant current density of 50 mA g−1 (or at a rate of C/4) after the formation cycle. The performance of AFLMBs depends on the salt choice in the electrolyte, suggesting that the salt participates in the SEI formation and influences the following Li plating process.36 The SEI composition can alter the distribution of electric current and Li+ flux across the current collector, directly impacting the shape and structure of electrodeposited Li metal.37 To assess the chemical composition of the SEI at different depths, X-ray photoelectron spectroscopy (XPS) depth profiling was employed on the deposited Li at Cu foil after the formation cycle. The depth profile of the SEI is determined through a sputter-etching process with accelerated Ar+ ions prior to the XPS measurement at different stages, namely the surface (i.e., 0 min), 6 min, and 12 min. The SEI surfaces were gradually removed through etching time, and then the distinctive photoelectron signature was collected.
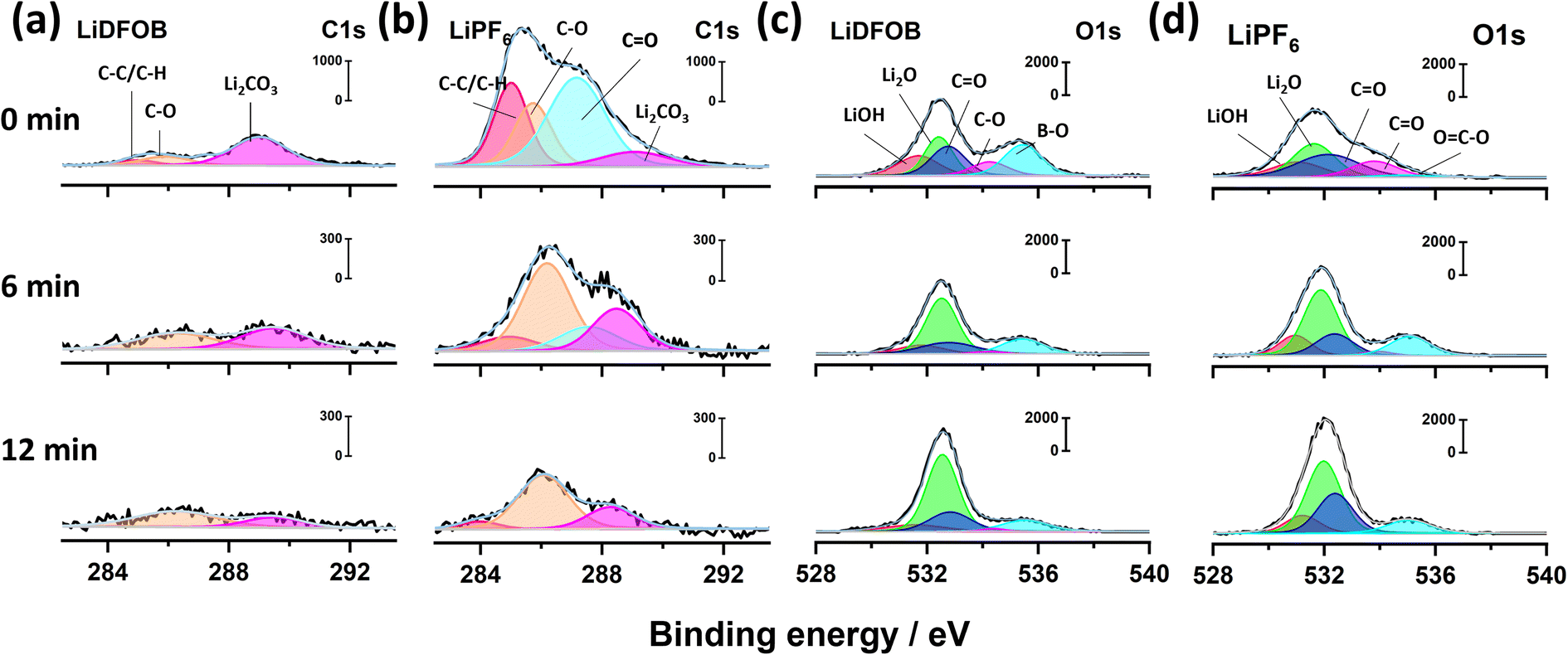
The XPS spectra of both samples at various etching times can indicate the presence of organic and inorganic components of the resultant SEI (Fig. 1).38 The C 1s spectra in Fig. 1a and b display the characteristic inorganic peak of Li2CO3 (289 eV) along with deconvoluted peaks of organic compounds, such as carbonyl group C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.2 eV), polyether carbon C–O (285.8 eV) and hydrocarbon C–C/C–H (285 eV).28,30,39 The C 1s XPS spectra of the cell with LiDFOB from 0 to 12 minutes of etching time are dominated by the inorganic compounds (Fig. 1a), as shown with the presence of Li2CO3 with less prominent C 1s spectra intensity compared to the cell with LiPF6 salt (Fig. 1b). The lower organic compound in the SEI may reflect less SEI breakage and formation during Li plating, resulting in a high CE during the charge–discharge cycle.40
O (287.2 eV), polyether carbon C–O (285.8 eV) and hydrocarbon C–C/C–H (285 eV).28,30,39 The C 1s XPS spectra of the cell with LiDFOB from 0 to 12 minutes of etching time are dominated by the inorganic compounds (Fig. 1a), as shown with the presence of Li2CO3 with less prominent C 1s spectra intensity compared to the cell with LiPF6 salt (Fig. 1b). The lower organic compound in the SEI may reflect less SEI breakage and formation during Li plating, resulting in a high CE during the charge–discharge cycle.40
 | ||
| Fig. 1 XPS depth profile analysis of electroplated Li after the formation cycle, with etching times of 0, 6, and 12 minutes. C 1s XPS spectra of plated Li with (a) LiDFOB and (b) LiPF6 electrolyte salt. O 1s XPS spectra of plated Li with (c) LiDFOB and (d) LiPF6 electrolyte salt. | ||
Compared to Li deposits from the cell with LiDFOB, the SEI formed in the cell with LiPF6 is enriched with organic compounds, evidenced by high intensity of carbonyl, polyether, and hydrocarbon peaks (Fig. 1b). The C 1s spectra intensity remains significant even at the inner SEI of LiPF6 cell (i.e., after 6 and 12 minutes of etching time), as shown with the polyether compound dominating the peak intensity. Therefore, the use of LiPF6-based electrolytes can lead to the formation of an organic-rich SEI layer. The organic-rich SEI strongly bonds with Li metal due to its low interfacial energy.41 Thus, it may experience the same volume change with Li during the plating and stripping, leading to SEI breakage during cycling. Moreover, its low interfacial energy also facilitates vertical and dendritic Li growth, which is detrimental to battery performance.12
The O 1s XPS spectra can further confirm the presence of inorganic compounds in the SEI.39 Fig. 1c and d show the O 1s spectra for the cells with LiDFOB and LiPF6 salt, respectively. The peaks associated with inorganic phases, such as LiOH (531.5 eV), Li2O (532.5 eV), and Li2CO3 (532.9 eV), along organic phases such as carbonyl (533.8 eV) and polyether (534.9 eV), are detected in both of O 1s spectra.39,42 The relative atomic concentrations of carbon with oxygen are calculated based on C 1s and O 1s peak area to quantify the organic or inorganic phases inside the SEI. The SEI of the cell with LiDFOB salt possesses significantly lower organic compounds (i.e., inorganic-rich), with a C/O ratio of 0.56 at the surface, compared to that with LiPF6 salt (C/O ratio of 1.42). Hence, the use of LiDFOB-based electrolytes can lead to the formation of an inorganic-rich SEI layer. The inorganic lithium compounds were reported to have weak bonding with high interfacial energy with Li metal.43,44 Thus, the produced inorganic-rich SEI can keep its integrity during Li plating and stripping.40 Besides, the inorganic-rich SEI also possesses a high Young's modulus that can suppress dendritic Li growth and penetration.45 The high mechanical stability of inorganic-rich SEI could also prevent the SEI from continuous breakage during the Li plating and stripping, retaining high CE and stable cycle life of the battery.40
Besides the SEI components, the energy required to dissociate the Li+ from the solvent component (i.e., de-solvation energy) electrolytes is critical for the kinetics performance because the solvent molecules around Li+ have to be completely stripped off before plating into the substrate.46 To show the de-solvation energy of LiPF6 and LiDFOB salt in the EC:DEC solvent, we have added the comparison of the Arrhenius plot between the 1 M LiPF6 and 1 M LiDFOB in EC:DEC, as shown in Fig. 2a. The activation energy (Ea) of 1 M LiDFOB is 0.168 eV, compared to 0.176 eV for 1 M LiPF6. The lower Ea value of LiDFOB salt suggests that the LiDFOB salt has a low de-solvation barrier for Li+ and promotes the facile de-solvation process of Li+ compared to LiPF6 salt in a carbonate-based solvent. Thus, Li+ could easily dissociate from EC:DEC solvent and easily plated to Li.
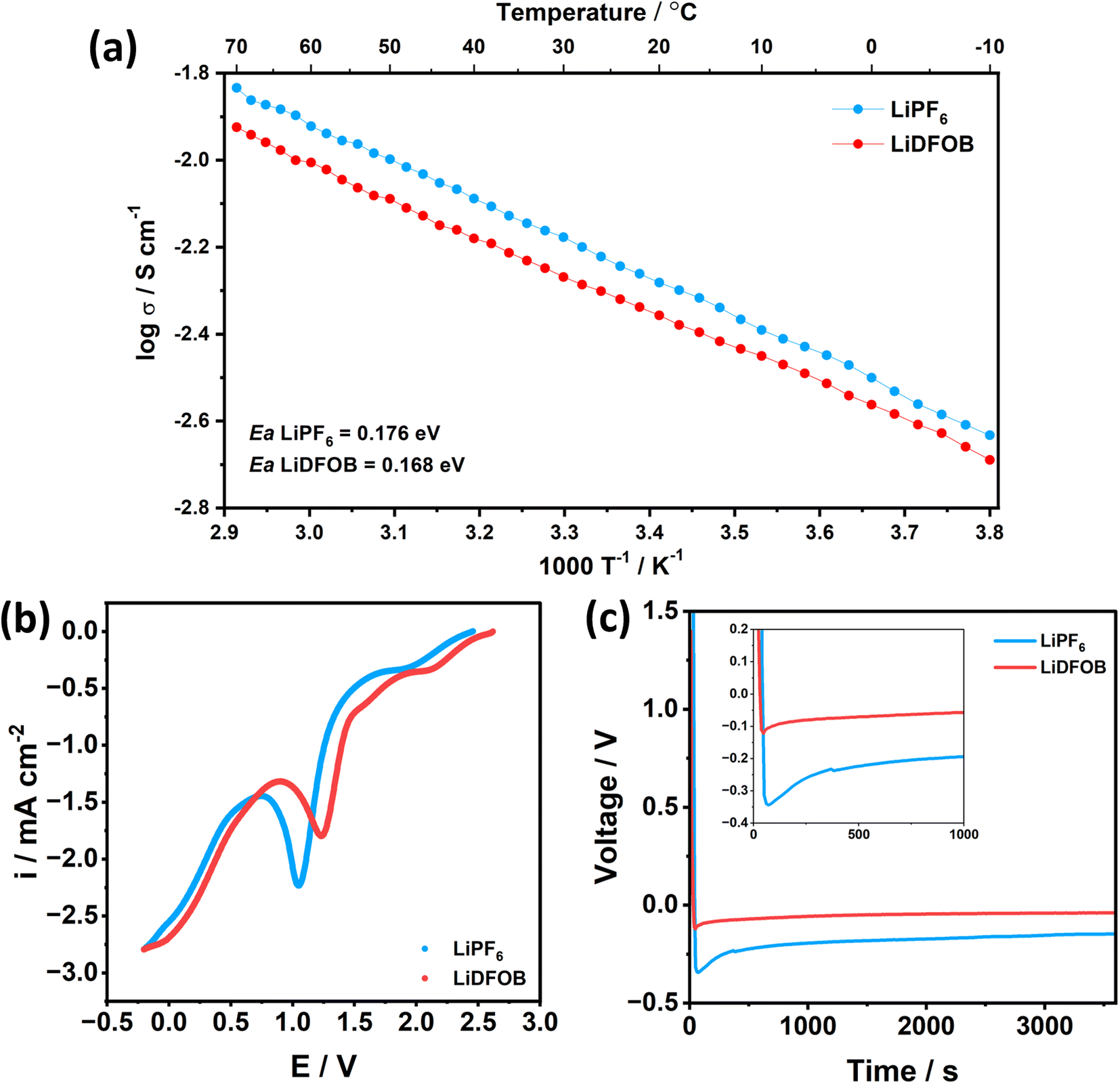 | ||
| Fig. 2 (a) Arrhenius plot of 1 M LiPF6 in EC:DEC (1:1, v/v) and 1 M LiDFOB in EC:DEC (1:1, v/v) electrolyte obtained with electrolyte conductivity measurement from 70 °C to −10 °C. The electrolyte conductivity was measured from −10 to 70 °C. (b) LSV scan of Li‖Cu half cell with 1 M LiPF6 and 1 M LiDFOB in EC:DEC (1:1, v/v) electrolyte with a scan rate of 0.1 V s−1. (c) Li plating overpotential at first discharge of Li‖Cu half cell with 1 M LiPF6 and 1 M LiDFOB in EC:DEC (1:1, v/v) electrolyte at a current density of 1 mA cm−2. | ||
LSV plot in Fig. 2b also supports this argument, where the electrolyte that contains LiDFOB salt reduced first at a potential of 1.24 V, compared to 1.05 V of Li‖Cu cell with LiPF6 salt. Faster de-solvation of LiDFOB leads to early SEI formation, suggested to be beneficial to protect the electrolyte from decomposition, as well as controlling the local current density across the current collector.47 With controlled local current density and synergistic effect of inorganic-rich SEI, the cell with LiDFOB salt achieves a low nucleation overpotential of 0.08 V, compared to 0.18 V of the cell with LiPF6 salt (Fig. 2c). Lower nucleation overpotential can be regarded as low energy to form Li nuclei.48 Thus, LiDFOB salt can be a beneficial contributor to achieving facile nucleation and homogenous spatial distribution of nuclei.
The morphologies of plated Li in the Cu‖NMC cells with 1 M LiDFOB or 1 M LiPF6 in EC:DEC (1:1, v/v) electrolyte after fully charged at the 1st, 5th, and 10th cycle are depicted in Fig. 3. Initial plating at the formation cycle shows the Li dendritic growth with LiDFOB salt (Fig. 3a). Due to the presence of surface cracks, pits, and subsurface impurities on Cu foil, the localized high electron density at these inhomogeneous sites may lead to the preferential gathering of Li-ions and electrons at the interface, thereby resulting in the formation of dendritic Li upon the initial cycle.6 As the cycle number increases, the plated Li with LiDFOB becomes dense and spherical-like, as shown in Fig. 3b and c. The inorganic-rich SEI layer is known to have a low Li+ diffusion barrier, which facilitates a fast and uniform Li-ion diffusion during the Li plating, as illustrated in Fig. 3d.49 Moreover, the dense plated lithium led to a good mechanical integrity of deposited lithium on the copper foil, proven by the retained shining Li deposits after 10 cycles (Fig. S1a–c†).
 | ||
| Fig. 3 (a–c) SEM images of plated Li on Cu foil after fully charged at different cycles with LiDFOB salt. (d) Illustration of dense Li plating with inorganic-rich SEI. (e–g) SEM image of plated Li on Cu foil after fully charged at different cycles with LiPF6 salt. (h) Illustration of dendritic Li plating with organic-rich SEI. All the cells are cycled from 2.8–4.5 V with a current density of 50 mA g−1. | ||
On the other hand, the Li morphology plated with LiPF6 salt tends to form a mossy structure, as shown in Fig. 3e–g. The organic-rich SEI has a high porosity, which promotes heterogeneous local current buildup, leading to uneven Li+ transfer inside SEI.34 Thus, the organic-rich SEI even can promote the growth of dendritic Li, as illustrated in Fig. 2h.49 This dendritic Li is easily detached from the current collector (Fig. S1d–f†).
The electrochemical performance of Cu‖NMC cells with LiDFOB and LiPF6 salt is depicted in Fig. 4. As the inorganic-rich SEI governed by LiDFOB promotes dense electroplated Li on Cu foil, the Li reversibility of the cell is positively affected and shows an overlapping voltage profile (Fig. 4a). On the other hand, organic-rich SEI in the cell with LiPF6 induces the mossy and dendritic Li growth on Cu foil. Thus, the Li loss caused by irreversible Li plating-stripping with LiPF6 salt is prominent, as illustrated by the cells' voltage degradation in Fig. 4b. The Cu‖NMC cell with LiDFOB salt demonstrates an impressive average coulombic efficiency of 98% and capacity retention of 52% even after 50 cycles (Fig. 4c). In contrast, cells utilizing LiPF6 salt exhibit a complete depletion of capacity after 40 cycles. This highlights the significant performance enhancement achieved by incorporating LiDFOB salt in anode-free lithium metal batteries for extended cycles. The relatively low stability of the cell with LiDFOB salt in this study can be justified by the use of a high cathode mass loading, a non-fluorinated solvent, and a wide operating voltage range, all of which collectively contribute to the observed stability levels.32
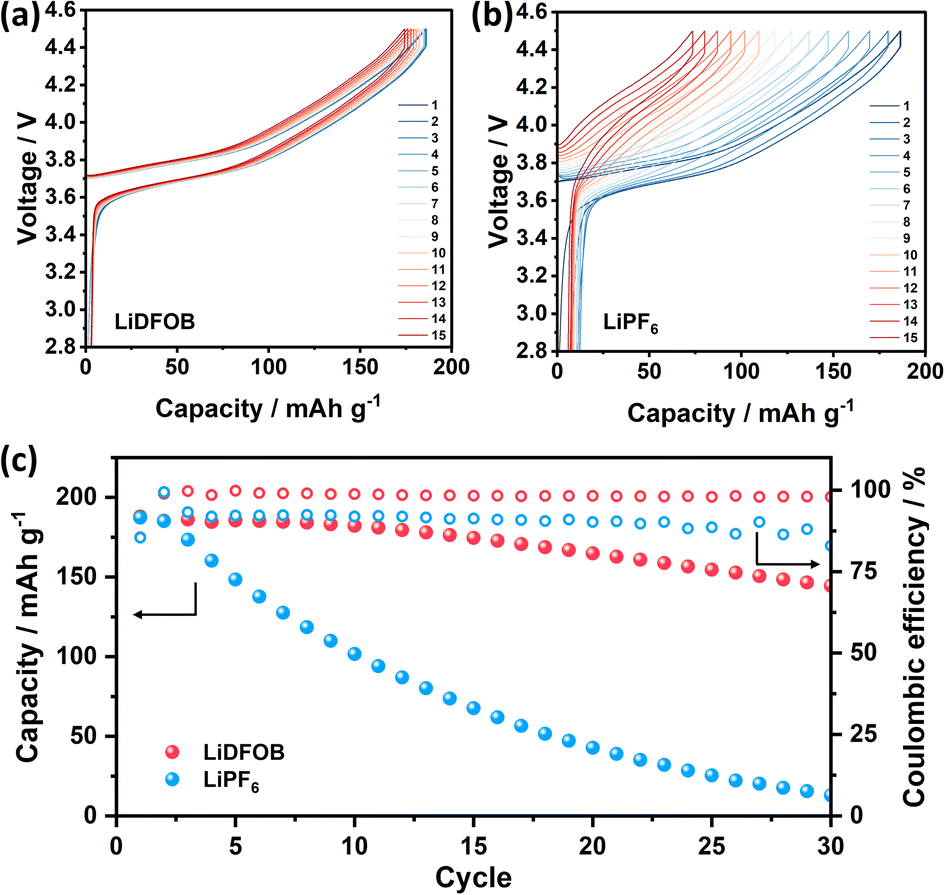 | ||
| Fig. 4 Galvanostatic charge–discharge voltage profiles of Cu‖NMC full cell with 1 M (a) LiDFOB and (b) LiPF6 in EC:DEC (1:1 v/v). (c) Cycling stability of Cu‖NMC full cell with 1 M LiDFOB and LiPF6 in EC:DEC (1:1 v/v). | ||
Besides shortening the battery cycle life, the SEI formation and evolution are important factors that govern the changes in the battery resistance.34,45 Therefore, the electrochemical impedance spectroscopy (EIS) of cells with LiDFOB and LiPF6 is measured at the 1st, 5th, 10th, and 15th cycle (at a fully charged state) to monitor the cell resistance change from electrolyte, cathode, and anode. The details of the fitting procedure and parameters are given in ESI S2 and S3,† respectively. The obtained Nyquist plot of both samples is composed of two semicircles with distinct characteristics. The first semicircle, which dominates the impedance at high frequency, corresponds to the electrode resistance. It can be further analyzed by deconvoluting it into two semicircles from the passivation layers on the cathode and anode, as depicted in Fig. S2a.† Meanwhile, the last semicircle at low frequency is associated with the charge transfer resistance (Rct).
The impedance of symmetric cells composed of Cu‖Cu (Fig. S2c†) and NMC‖NMC (Fig. S2d†) retrieved from the two fully charged Cu‖NMC cells was measured to define the contribution of the cathode and anode passivation layer. For the negative side, the symmetric Cu‖Cu cell (Fig. S2c†) exhibits smaller semicircles compared to NMC symmetric cell (Fig. S2d†). Comparing these to the full cell impedance spectra (Fig. S2a†), the negative lithium electrode contributes to high frequency (i.e., first semicircles), while the positive electrode contributes to the medium frequency (i.e., second semicircle). This result is in line with the study by Iurilli et al.,50 which explains that the high and medium frequencies observed in the first semicircle are attributed to the impedance of the passivation layer at the anode side (RSEI-anode) and the cathode side (RCEI-cathode), respectively. The fitted Nyquist plots and parameters are shown in Fig. S3 and Table S1,† respectively.
The changes in the resistance value of cells with LiDFOB and LiPF6 within the first 15 cycles are shown in Fig. 5a and b, respectively. The cell with LiDFOB shows a stable impedance over the cycles compared to the cell with LiPF6, which correlates well with the improved cycling stability and high CE. Furthermore, the changes in the RSEI-anode (Fig. 5c) and RCEI-cathode (Fig. 5d) are prominent. Thus, the battery's performance is related to the formation of the passivation film on Li and NMC622 cathode surfaces. In contrast, the changes in Rs (Fig. S4a†) and Rct (Fig. S4b†) are relatively small, indicating that the electrolyte conductivity and electrode kinetics are not major limiting factors in the electrochemical performance of the cells.
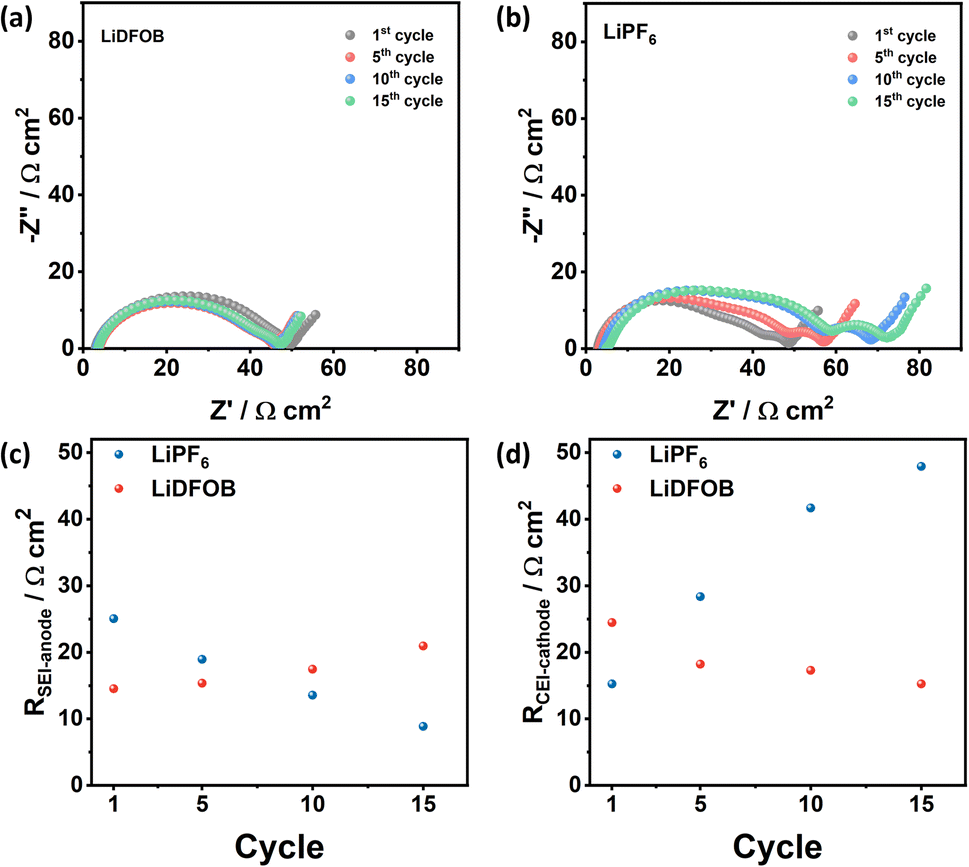 | ||
| Fig. 5 Nyquist plots of the fully charged Cu‖NMC cells with (a) LiDFOB and (b) LiPF6 salt at the 1st, 5th, 10th, and 15th cycle and the fitting data of (c) solid electrolyte interphase resistance at the anode (RSEI-anode) and (d) cathode electrolyte resistance (RCEI-cathode). The fitting parameters are listed in Table S1.† | ||
Fig. 5c shows the distinct behaviour of SEI resistance at the anode (RSEI-anode) for cells with LiDFOB and LiPF6. Changes in the SEI resistance may indicate the stability of SEI during charge and discharge cycles. The volume changes during Li plating can break the SEI, especially when Li is porous or dendritic.49 After the formation cycle, the cells with LiDFOB and LiPF6 exhibit RSEI-anode of 14.53 and 25.04 Ω cm2, respectively. As the cycle number increases, the RSEI-anode of the LiDFOB cell increases to 20.95 Ω cm2, while the LiPF6 cell decreases to 8.85 Ω cm2. The decrease of the RSEI-anode for the LiPF6 cell can be attributed to the formation of porous lithium structure and the continuous SEI breakage and formation in each cycle.49 Fresh cracks of SEI expose Li metal, and SEI formation occurs again.34 Thus, a thinner SEI may be formed, reflected in the decreased SEI resistance over the cycle number. In contrast, the increase in the RSEI-anode of the LiDFOB cell may reflect the stable SEI formed at Li metal surfaces. Previous research also reported that the increase in SEI resistance correlates with the dense lithium metal plating in the lithium metal battery.35 The presence of stable SEI and densely plated Li metal was also reflected in high CE and stable cycling stability of cells with LiDFOB.
The evolution of interphase at the cathode is observed in the evolution of the RCEI-cathode (Fig. 5d). The cathode electrolyte interphase (CEI) plays a role in mitigating the capacity decay due to transition metal dissolution at the cathode (i.e., the crosstalk effect).6,51 In this case, the transition metal with IV+ oxidation state in the NMC cathode is likely to escape during lithiation at a high cut-off voltage, which degrades the cathode and increases the resistance of the battery.52,53 The cell with LiPF6 salt exhibits an increased RCEI-cathode from 15.24 to 47.93 Ω cm2 after 15 cycles, while the cell with LiDFOB decreases its RCEI-cathode from 24.46 to 15.25 Ω cm2.
In particular, LiDFOB is known for stabilizing CEI by forming LiF-rich phases while the electrolyte decomposes at the cathode side, preventing continuous parasitic reactions at the cathode (i.e., transition metal dissolution and electrolyte decomposition).51 The presence of LiDFOB stabilizes the CEI layer at the cathode surface, proven by the decreased RCEI-cathode value following the cycle number. On the other hand, the increase of RCEI-cathode on the cell with LiPF6 salt can be related to the immature passivation layer formed at the NMC cathode surface. The accumulation of CEI produces from the interfacial reaction at the cathode side further increases the interfacial resistance of the cathode and consumes the electrolyte.54
Conclusions
The formation of mossy, dendritic lithium plating and unstable SEI has long been known to be significant challenges in the cycling performance of AFLMBs. Previous works showed that LiDFOB-based electrolytes had enhanced the life cycle of AFLMBs. Our study adds to this body of work by highlighting the role of LiDFOB salt in SEI formation. The XPS data indicates that the inorganic-rich SEI formed on the Li deposit in the cell with LiDFOB, which suppresses dendrite growth, promotes a dense Li deposition, thus delivering high CE. With the inorganic-rich SEI, a dense electroplated Li with a shining surface on the Cu substrate can be retained after 10 charge–discharge cycles of Cu‖NMC cell. The cell with LiDFOB shows a relatively stable impedance over the cycles compared to the cell with LiPF6, which correlates well with the improved cycling stability and high CE. The changes in RSEI-anode and RCEI-cathode in both cells are more prominent compared to the changes in Rs and Rct, indicating the improved cycling performance is related to the formation of the passivation film on Li and NMC622 cathode surfaces, rather than electrolyte conductivity and electrode kinetics. The use of LiDFOB is shown to provide advantages in the passivation of plated lithium and NMC cathodes, leading to enhanced cycling performance of AFLMBs. These findings demonstrate the fundamental behaviour of LiDFOB salts in governing stable inorganic-rich SEI and can provide insights for the design of future high-performance AFLMBs.Author contributions
N. H. H., A. S., and N. D. contributed to the conceptualization and methodology of the research. N. H. H., X. H., and N. D. conducted the investigation, while N. H. H. and N. D. curated and analyzed the data. A. S. and N. D. provided validation of the results. N. H. H., A. P., A. S., and N. D. were involved in writing and preparing the original draft, with N. H. H., A. S., and N. D. contributing to the reviewing and editing of the draft. N. H. H. and A. P. created the visualizations. A. S. and N. D. provided supervision for the project, oversaw the project administration, and secured the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. H. H. acknowledge the funding support from Singapore International Pre-Graduate Award (SIPGA) scheme under A*STAR Graduate Academy (A*GA). We acknowledge the funding from Institut Teknologi Bandung through the 2022 ITB research programme.References
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- S. S. Zhang, ACS Appl. Energy Mater., 2018, 1, 910–920 CrossRef CAS.
- C. Fang, X. Wang and Y. S. Meng, Trends Chem., 2019, 1, 152–158 CrossRef CAS.
- Q. Wang, B. Liu, Y. Shen, J. Wu, Z. Zhao, C. Zhong and W. Hu, Adv. Sci., 2021, 8, 2101111 CrossRef CAS PubMed.
- C. Heubner, S. Maletti, H. Auer, J. Hüttl, K. Voigt, O. Lohrberg, K. Nikolowski, M. Partsch and A. Michaelis, Adv. Funct. Mater., 2021, 31, 2106608 CrossRef CAS.
- C.-J. Huang, B. Thirumalraj, H.-C. Tao, K. N. Shitaw, H. Sutiono, T. T. Hagos, T. T. Beyene, L.-M. Kuo, C.-C. Wang, S.-H. Wu, W.-N. Su and B. J. Hwang, Nat. Commun., 2021, 12, 1452 CrossRef CAS PubMed.
- M. Genovese, A. J. Louli, R. Weber, S. Hames and J. R. Dahn, J. Electrochem. Soc., 2018, 165, A3321 CrossRef CAS.
- L. Su, H. Charalambous, Z. Cui and A. Manthiram, Energy Environ. Sci., 2022, 15, 843–854 RSC.
- S. Menkin, C. A. O'Keefe, A. B. Gunnarsdóttir, S. Dey, F. M. Pesci, Z. Shen, A. Aguadero and C. P. Grey, J. Phys. Chem. C, 2021, 125, 16719–16732 CrossRef CAS PubMed.
- J. Sun, S. Zhang, J. Li, B. Xie, J. Ma, S. Dong and G. Cui, Adv. Mater., 2022, 2209404 Search PubMed.
- B. Wu, J. Lochala, T. Taverne and J. Xiao, Nano Energy, 2017, 40, 34–41 CrossRef CAS.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- J. J. Hyebin Jeong and C. Jo, Chem. Eng. J., 2022, 446, 136860 CrossRef.
- A. B. Bingxin Zhou, I. Stosevski, B. Fang and D. P. Wilkinson, Prog. Mater. Sci., 2022, 130, 100996 CrossRef.
- Y. Y. Na Zhu, Y. Li, Y. Bai, J. Rong and C. Wu, Carbon Energy, 2023, e423 CrossRef.
- G. Z. Hang Yuan, D. Zhang and X. Yang, Energy Fuels, 2023, 37, 833–857 CrossRef.
- Y. C. Zhongzhe Li, X. Yun, P. Gao, C. Zheng and P. Xiao, Adv. Funct. Mater., 2023, 33, 2300502 CrossRef.
- C. C. Baolin Wu, L. H. J. Raijmakers, J. Liu, D. L. Danilov, R.-A. Eichel and P. H. L. Notten, Energy Storage Mater., 2023, 57, 508–539 CrossRef.
- J. Sun, S. Zhang, J. Li, B. Xie, J. Ma, S. Dong and G. Cui, Adv. Mater., 2023, 35, 2209404 CrossRef CAS PubMed.
- Y. Zhao, T. Zhou, T. Ashirov, M. E. Kazzi, C. Cancellieri, L. P. H. Jeurgens, J. W. Choi and A. Coskun, Nat. Commun., 2022, 13, 2575 CrossRef CAS PubMed.
- C.-C. Su, K. Amine, M. Cai and M. He, ACS Appl. Mater. Interfaces, 2023, 15, 2804–2811 CrossRef CAS PubMed.
- G. Hobold, K.-H. Kim and B. M. Gallant, Energy Environ. Sci., 2023, 16, 2247–2261 RSC.
- L. Li, G. Xu, S. Zhang, S. Dong, S. Wang, Z. Cui, X. Du, C. Wang, B. Xie, J. Du, X. Zhou and G. Cui, ACS Energy Lett., 2022, 7, 591–598 CrossRef CAS.
- H. Zhang, C. Shen, Y. Huang and Z. Liu, Appl. Surf. Sci., 2021, 537, 147983 CrossRef CAS.
- J. Liu, S. Ihuaenyi, R. Kuphal, J. Salinas, L. Xie, L. Yang, U. Janakiraman, M. E. Fortier and C. Fang, J. Electrochem. Soc., 2023, 170, 010535 CrossRef.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H.-M. Cheng, Adv. Mater., 2022, 2206009 Search PubMed.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- B. S. Parimalam, A. D. MacIntosh, R. Kadam and B. L. Lucht, J. Phys. Chem. C, 2017, 121, 22733–22738 CrossRef CAS.
- J. Zhang, J. Shi, L. W. Gordon, N. Shojarazavi, X. Wen, Y. Zhao, J. Chen, C.-C. Su, R. J. Messinger and J. Guo, ACS Appl. Mater. Interfaces, 2022, 14, 36679–36687 CrossRef CAS PubMed.
- S. Li, Q. Liu, W. Zhang, L. Fan, X. Wang, X. Wang, Z. Shen, X. Zang, Y. Zhao, F. Ma and Y. Lu, Adv. Sci., 2021, 8, 2003240 CrossRef CAS PubMed.
- J. Zhang, H. Zhang, L. Deng, Y. Yang, L. Tan, X. Niu, Y. Chen, L. Zeng, X. Fan and Y. Zhu, Energy Storage Mater., 2023, 54, 450–460 CrossRef.
- R. Weber, M. Genovese, A. J. Louli, S. Hames, C. Martin, I. G. Hill and J. R. Dahn, Nat. Energy, 2019, 4, 683–689 CrossRef CAS.
- S. Jurng, Z. L. Brown, J. Kim and B. L. Lucht, Energy Environ. Sci., 2018, 11, 2600–2608 RSC.
- D. T. Boyle, S. C. Kim, S. T. Oyakhire, R. A. Vilá, Z. Huang, P. Sayavong, J. Qin, Z. Bao and Y. Cui, J. Am. Chem. Soc., 2022, 144, 20717–20725 CrossRef CAS PubMed.
- R. Pathak, K. Chen, A. Gurung, K. M. Reza, B. Bahrami, J. Pokharel, A. Baniya, W. He, F. Wu, Y. Zhou, K. Xu and Q. Qiao, Nat. Commun., 2020, 11, 93 CrossRef CAS PubMed.
- A. J. Louli, A. Eldesoky, R. Weber, M. Genovese, M. Coon, J. deGooyer, Z. Deng, R. T. White, J. Lee, T. Rodgers, R. Petibon, S. Hy, S. J. H. Cheng and J. R. Dahn, Nat. Energy, 2020, 5, 693–702 CrossRef CAS.
- W. Deng, X. Yin, W. Bao, X. Zhou, Z. Hu, B. He, B. Qiu, Y. S. Meng and Z. Liu, Nat. Energy, 2022, 7, 1031–1041 CrossRef CAS.
- D. Kang, S. Sardar, R. Zhang, H. Noam, J. Chen, L. Ma, W. Liang, C. Shi and J. P. Lemmon, Energy Storage Mater., 2020, 27, 69–77 CrossRef.
- K. N. Wood and G. Teeter, ACS Appl. Energy Mater., 2018, 1, 4493–4504 CrossRef CAS.
- S. Liu, X. Ji, N. Piao, J. Chen, N. Eidson, J. Xu, P. Wang, L. Chen, J. Zhang, T. Deng, S. Hou, T. Jin, H. Wan, J. Li, J. Tu and C. Wang, Angew. Chem., Int. Ed., 2021, 60, 3661–3671 CrossRef CAS PubMed.
- A. A. Assegie, J.-H. Cheng, L.-M. Kuo, W.-N. Su and B.-J. Hwang, Nanoscale, 2018, 10, 6125–6138 RSC.
- Z. L. Brown and B. L. Lucht, J. Electrochem. Soc., 2019, 166, A5117 CrossRef CAS.
- J. Chen, Q. Li, T. P. Pollard, X. Fan, O. Borodin and C. Wang, Mater. Today, 2020, 39, 118–126 CrossRef.
- X. Fan, X. Ji, F. Han, J. Yue, J. Chen, L. Chen, T. Deng, J. Jiang and C. Wang, Sci. Adv., 2018, 4, eaau9245 CrossRef CAS PubMed.
- N. Ding, A. Sumboja, X. Yin, Y. Zheng, D. Wen Hui Fam and Y. Zong, J. Electrochem. Soc., 2023, 170, 010540 CrossRef.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- L. Xia, S. Lee, Y. Jiang, Y. Xia, G. Z. Chen and Z. Liu, ACS Omega, 2017, 2, 8741–8750 CrossRef CAS PubMed.
- B. Thirumalraj, T. T. Hagos, C.-J. Huang, M. A. Teshager, J.-H. Cheng, W.-N. Su and B.-J. Hwang, J. Am. Chem. Soc., 2019, 141, 18612–18623 CrossRef CAS PubMed.
- K. Dong, Y. Xu, J. Tan, M. Osenberg, F. Sun, Z. Kochovski, D. T. Pham, S. Mei, A. Hilger, E. Ryan, Y. Lu, J. Banhart and I. Manke, ACS Energy Lett., 2021, 6, 1719–1728 CrossRef CAS.
- P. Iurilli, C. Brivio and V. Wood, J. Power Sources, 2021, 505, 229860 CrossRef CAS.
- M. Mao, B. Huang, Q. Li, C. Wang, Y.-B. He and F. Kang, Nano Energy, 2020, 78, 105282 CrossRef CAS.
- Y. Tesfamhret, R. Younesi and E. J. Berg, J. Electrochem. Soc., 2022, 169, 010530 CrossRef CAS.
- J. Karunawan, P. N. Suryadi, L. Mahfudh, S. P. Santosa, A. Sumboja and F. Iskandar, Energy Fuels, 2023, 37, 754–762 CrossRef CAS.
- Y. Wu, X. Liu, L. Wang, X. Feng, D. Ren, Y. Li, X. Rui, Y. Wang, X. Han, G.-L. Xu, H. Wang, L. Lu, X. He, K. Amine and M. Ouyang, Energy Storage Mater., 2021, 37, 77–86 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03184e |
| This journal is © The Royal Society of Chemistry 2023 |