
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper borate (CuB4O7)-promoted multi-component green synthesis of 2,4,5-triarylimidazole derivatives and evidence of in situ conversion of copper borate (CuB4O7) into Cu(OAc)2 in the presence of NH4OAc†
Sailesh Chettria,
Sumiran Tamanga,
Kiran Pradhanb,
Biswajit Sinha b and
Dhiraj Brahman*a
b and
Dhiraj Brahman*a
aDepartment of Chemistry, St. Joseph's College, Darjeeling, 734104, India. E-mail: dhirajslg2@gmail.com
bDepartment of Chemistry, University of North Bengal, Darjeeling, 734013, India
First published on 4th July 2023
Abstract
A green, efficient, and straightforward methodology for the three-component synthesis of 2,4,5-triarylimidazole has been developed under solvent-free conditions using unconventional CuB4O7 as a promoter. This green approach encouragingly provides access to a library of 2,4,5-tri-arylimidazole. Also, we have been able to isolate the compound (5) and (6) in situ, which provides an insight into the direct conversion of CuB4O7 into copper acetate in the presence of NH4OAc under solvent-free condition. The main advantage of this protocol includes an easy reaction procedure, short reaction time, and easy work up of the product without using any tedious separation method.
Introduction
Of late, the design and development of reaction methodology in an environmentally benign, greener, and sustainable way to reduce the use of hazardous chemicals and to explore the alternative reaction conditions or reaction media to bring about the desired chemical transformations by minimizing the byproducts or waste and the exclusion of the conventional organic solvents has gained considerable interest in the field of organic transformation reactions.1–10 Fortunately, multi-component reactions (MCRs) serve as good example to meet the requirements for the green chemical processes and it may be viewed as an art of performing efficient chemical transformation by coupling three or more reactants in a single pot in single operation, avoiding toxic reagents, solvents, and costly purification techniques. MCR also has an advantage in the development and formulation of novel and cost-effective approaches in synthetic chemistry research, leading to environmental protection and have emerged as powerful tools for drug discovery.11–19 The MCR technique has gained special attention and drawn considerable interest in modern synthetic chemistry and pharmaceutical chemistry owing to its advantages, such as high atom-economy, high selectivity, operational simplicity, structural diversity, short reaction time, low cost, low energy consumption, high yield, and easy purification process.20–24 The imidazole scaffold belongs to an important heterocyclic compound due to their richness in various natural products and their widespread use in medicinal fields.25,26 Imidazole ring system possesses a variety of pharmaceutical properties and plays a fundamental role in numerous biochemical processes.27 Over the past few years, highly substituted imidazoles have found a special place owing to their biological relevance in many natural products, extensive applications in the area of material science,28 and potential use as herbicide,29 fungicide,30 inhibitors of P38 MAP kinase,31 B-Raf kinase,32 antiinflammatory,33 antitumor,34 and antithrombotic35 agents. In addition, some of the highly substituted imidazole compounds serve as a photosensitive material in photography.36 Clemizole (antihistaminic agent),37 nocodazole (antinematodal),38 azathioprine (antirheumatoid arthritis),39 and dacarbazine (anticancer)40,41 are commercially available drugs that contain the imidazole scaffold and have been used for the treatment of different diseases (Fig. 1).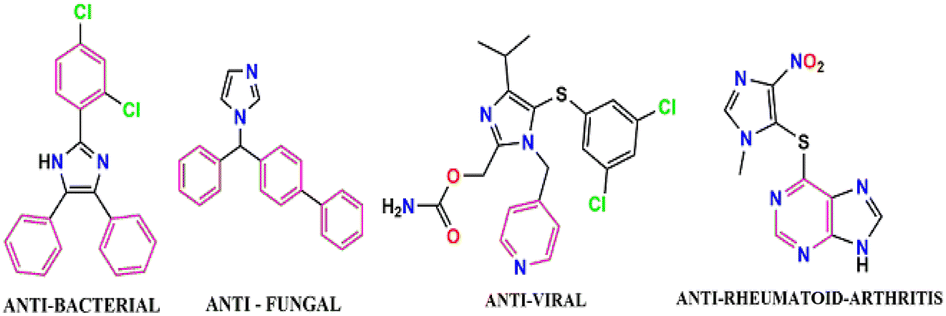 | ||
| Fig. 1 Representative example of some commercially available drugs containing the imidazole core. | ||
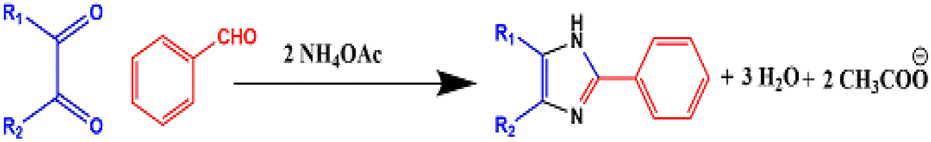
Japp and Radziszewski independently reported the first synthesis of 2,4,5-triarylimidazole in 1882 by the cyclocondensation of 1,2-dicarbonyl compound with an aldehyde in the presence of ammonia as a nitrogen source in acidic medium42,43 (Scheme 1a and b).
 | ||
| Scheme 1 (a) Japp imidazole synthesis and (b) Radziszewski imidazole synthesis. | ||
After the successful synthesis of triarylimidazoles by Radziszewski and Japp, various synthetic modifications have been made in this approach. The most common modification that has been made is the use of ammonium acetate in place of ammonia as a nitrogen source44 (Scheme 2).
 | ||
| Scheme 2 Synthesis of 2,4,5-triarylimidazole using NH4OAc as nitrogen source. | ||
In view of the varied properties and applications of imidazole core, numerous synthetic methodologies have been formulated using various catalytic systems under different reaction conditions for the efficient and rapid synthesis of substituted imidazole derivatives by the three-component cyclocondensation of a 1,2-diketone, α-hydroxyketone, or α-ketomonoxime with an aldehyde and ammonium acetate45–57 (Scheme 3a). In addition to the use of conventional 1,2-diketone, α-hydroxyketone or α-ketomonoxime, α-methylene ketone has also been used for the synthesis of substituted imidazoles58,59 (Scheme 3b).
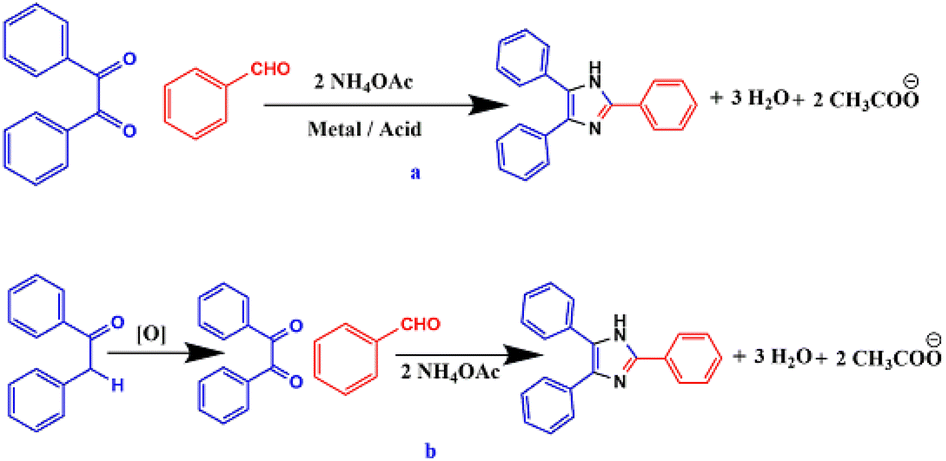 | ||
| Scheme 3 (a) Triarylimidazole synthesis from cyclocondensation of 1,2-diketone, aldehyde and ammonium acetate and (b) synthesis of tri-substituted imidazole using α-hydroxyketone, aldehyde, and ammonium acetate. | ||
Although these documented procedures offer several beneficial features, yet most of them have serious drawbacks such as the use of expensive catalyst, lower yield, use of toxic solvent, harsh reaction condition, longer reaction time, and tedious work-up procedures.60–63 Interestingly, there are several reports on the synthesis of 2,4,5-tri-aryl imidazole using different copper salts as catalyst,64–69 but there are no reports on the study of catalytic activities of copper borate salt so far.
All these facts have encouraged us to find an ecofriendly, environmentally benign, greener, and sustainable method for the synthesis of substituted imidazole and stimulated us to employ copper(II) borate (CuB4O7) for the one-pot three-component synthesis of 2,4,5 triarylimidazole derivatives by the condensation of benzil, ammonium acetate, and aromatic/heteroaromatic aldehydes in solvent-free condition.
Results and discussion
Synthesis of 2,4,5 triarylimidazole derivatives
To ascertain the optimized reaction conditions for the designed protocol, benzil (1), 2-nitrobenzaldehyde (2), and NH4OAc (3) were chosen as the model reactants (Scheme 4).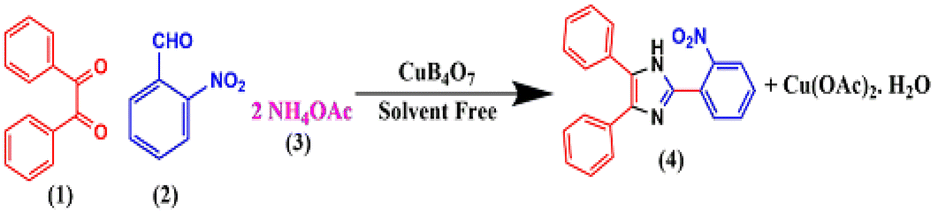 | ||
| Scheme 4 Synthetic route for the preparation of 2,4,5-triarylimidazoles. | ||
We investigated the model reaction with various Cu(II) salts including copper borate, and the results are listed in Table 1. We found that product 4 is formed in 60% yield after 120 min in ethanol at 80 °C using CuSO4 is as a catalyst (Table 1, entry 1). Furthermore, we extended the model reaction with other copper salts such as CuO, CuCl2, CuBr2, Cu(OAc)2, and CuB4O7 at 80 °C in ethanol medium and obtained 64%, 70%, 71%, 68%, and 86% yields of product 4, respectively (Table 1, entries 2–6). Thus, from the optimization reaction, we obtained the maximum yield of product 4 in the presence of copper borate.
We further investigated the amount of catalyst loading for the designed reaction and set up the model reaction employing different mol% (0, 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0) of copper borate (Table 2). Interestingly, we observed that the reaction goes to completion when the amount of copper borate is 2 mol% at 80 °C (92%, Table 2, entry 5). Also, we carried out the same reaction in the absence of copper borate and observed that only a minimum amount (12%) of product 4 was formed (Table 2 entry 1).
We screened the catalytic efficiency of the catalyst with the model reactants in different solvents, and the result is summarized in Table 3. We found that in more polar solvents such as alcohol and DMF, the yield of the products were good (Table 3, entries 1–3) but the reaction needed more time (2 h) to achieve completion and required a tedious technique for the recovery of the product. Again, by considering the green chemistry principles, we were interested in employing water as a green solvent for the designed reaction, but in this case, we obtained only 32% yield of the product in the stipulated time (Table 3, entry 5). Due to this drawback, we focused our study on solid phase synthesis using silica gel as a reaction medium, and 91% yield of the product 4 was obtained; however, in this case also, we encountered problems during the work-up of the desired product 4 (Table 3, entry 6).
| Entry | Catalysta | Solventb | Yieldc (%) |
|---|---|---|---|
| a The reaction was performed with benzil 1 (1 mmol), 2-nitrobenzaldehyde 2 (1 mmol), and NH4OAc 3 in the presence of different copper(II) salts in different solvent/neat conditions at 80 °C for 2 h except for neat condition.b Under neat condition, the reaction completed in 10 min.c Isolated yield. | |||
| 1 | CuB4O7 | Methanol | 92 |
| 2 | Ethanol | 88 | |
| 3 | DMF | 86 | |
| 4 | DMSO | 82 | |
| 5 | Water | 32 | |
| 6 | Silica gel | 91 | |
| 7 | Neat | 98 | |
Therefore, these drawbacks prompted us to carry out the desired reaction in neat conditions without using any organic solvents. Encouragingly, the desired product 2,4,5-triarylimidazole (4) was obtained in excellent yield in less time (8–10 min) at 80 °C under solvent-free condition (Table 3, entry 7). The green chemistry metric E-factor for the designed reaction has been included in the ESI.†
Also, we observed that the yield of the product is maximum when the amount of NH4OAc is 2.5 mmol with respect to other substrate, and with the decrease or increase in the amount of NH4OAc, the yield of product 4 did not changed significantly.
Moreover, having recognized the optimized reaction conditions, we subsequently examined the catalytic efficiency and applicability of this protocol by extending this protocol to other aromatic/heteroaromatic aldehydes to prepare 2,4,5-triarylimidazole derivatives (4a–4z) (Fig. 2). We observed that all the reactions proceeded smoothly in a very short reaction time to afford the desired 2,4,5-triarylimidazoles in good to excellent yields (88–98%).
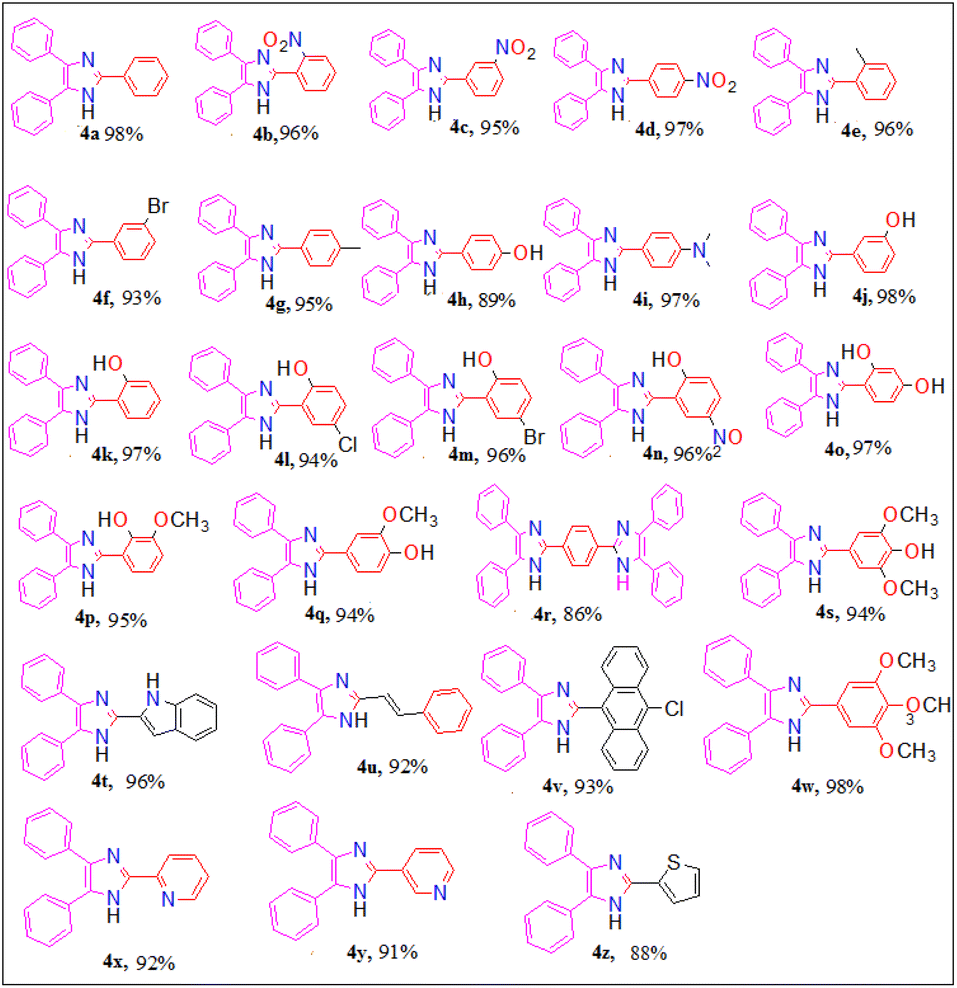 | ||
| Fig. 2 Substrate scope for the synthesis of 2,4,5-triarylimidazole (4a–z). | ||
It was observed that para-substituted benzaldehyde with electron withdrawing group successfully afforded the corresponding 2,4,5-triarylimidazoles in excellent yield under optimized reaction condition compared to the para-substituted benzaldehyde with electron-releasing group. Other aromatic/heteroaromatic aldehydes also afforded the corresponding imidazoles in good yield but they required more time for completion (15–20) min. All the synthesized compounds (4a–4z) were recrystallized from ethanol (10 mL), characterized by their analytical (melting point) and spectroscopic data (FT-IR and 1H NMR), and compared with the literature value. The analytical results (yield and melting point) and the spectroscopic details (FT-IR and 1H NMR) data of the synthesized compounds are given in the Experimental section, and the corresponding spectra are provided in ESI 1.†
Comparisons of efficacy of CuB4O7 with those of the reported catalysts70–91 for the synthesis of 2,4,5-trisubstituted imidazole have shown that the present system involving CuB4O7 has proven to be more efficient in terms of the short reaction time, milder reaction condition, and product yields (Table 4).
| Entry | Catalyst | Solvent | Temperature | Time | Yield (%) |
|---|---|---|---|---|---|
| 1 | L-Proline | MeOH | 60 °C | 9 h | 90 [lit. 70] |
| 2 | Zeolite ZSM-11 | Neat | Reflux | 60 min | 80 [lit. 71] |
| 3 | Zr(acac)4 | EtOH | Reflux | 2.5 h | 90 [lit. 72] |
| 4 | Yb(OTf)3 | OHAc | 70 °C | 2 h | 92 [lit. 73] |
| 5 | ZnO nanorod | H2O | Reflux | 1.45 h | 83 [lit. 74] |
| 6 | I2 | Neat | RT | 15 min | 90 [lit. 75] |
| 7 | Fe3O4 nanoparticles | Neat | 80 °C | 20 min | 96 [lit. 76] |
| 8 | Nano TiCl4·SiO2 | Neat | 110 °C | 1 h | 90 [lit. 77] |
| 9 | Nano MgO | Neat | 100 °C | 0.6 h | 94 [lit. 78] |
| 10 | InF3 | Neat | 60 °C | 0.3 h | 90 [lit. 79] |
| 11 | CoFe2O4 NPs | EtOH | 40 °C | 20 min | 95 [lit. 80] |
| 12 | Y(TFA)3 | Neat | 100 °C | 3 h | 97 [lit. 81] |
| 13 | Boric acid | H2O | Ultrasound | 30 min | 98 [lit. 82] |
| 14 | ZSM-5–SO3H | Neat | 110 °C | 90 min | 90 [lit. 83] |
| 15 | Trichloromelamine | Neat | 110 °C | 1 h | 92 [lit. 84] |
| 16 | TiO2 | Neat | 120 °C | 2.5 h | 92 [lit. 85] |
| 17 | H2SO4·SiO2 | Neat | 110 °C | 60 min | 94 [lit. 86] |
| 18 | Copper nanoparticles | Neat | RT | 60 min | 94 [lit. 87] |
| 19 | HMDS | Neat | 120 °C | 3 h | 96 [lit. 88] |
| 20 | KMnO4/CuSO4 | EtOH | Reflux | 70 min | 85 [lit. 89] |
| 21 | SiO2–NaHSO4 | Neat | 120 °C | 30 min | 95 [lit. 90] |
| 22 | Zeolite HY/silica gel | Neat | MW | 6 min | 89 [lit. 91] |
| 23 | CuB4O7 | Neat | 80 °C | 10 min | 98 [this work] |
Since copper borate was insoluble in most of the solvents and we carried out the reaction under neat condition, therefore, we were hopeful of recycling copper borate in fair amount. Surprisingly, we found that during the addition of copper borate to the reaction mixture, the reaction mixture turned blue and after the work-up of the product, unfortunately, the desired copper borate could not be recovered. Although it efficiently afforded the desired product 4 in good to excellent yield, yet still there were some doubts due to the nonrecyclability of copper borate.
Fortunately, when 2-OH-benzaldehyde derivatives were employed as an aldehyde substrate (2), we found the formation of some blue colored products in the reaction mixture along with the desired product 4. We carefully recrystallized the blue colored compound in N,N-dimethylformamide (DMF) solvent (2 mL) and after 5 days, a blue colored crystal suitable for X-ray single crystal diffraction was isolated from the reaction, where 2-OH-5-NO2-benzaldehyde was used as an aldehyde substrate. After successful X-ray single crystal diffraction study, it was revealed that the desired 2,4,5-triarylimidazole product was formed during the reaction but the product itself acted as a bidentate chelating ligand for the copper(II) ion in the reaction mixture, thereby forming bis[2-(4,5-diphenyl-1H-imizol-2-yl)-4-nitro-phenolato]copper(II) dihydrate complex with molecular formula [Cu(C21H14N3O3)2]·2H2O (5) (CCDC no: 1958158)92 (Fig. 3a and b).
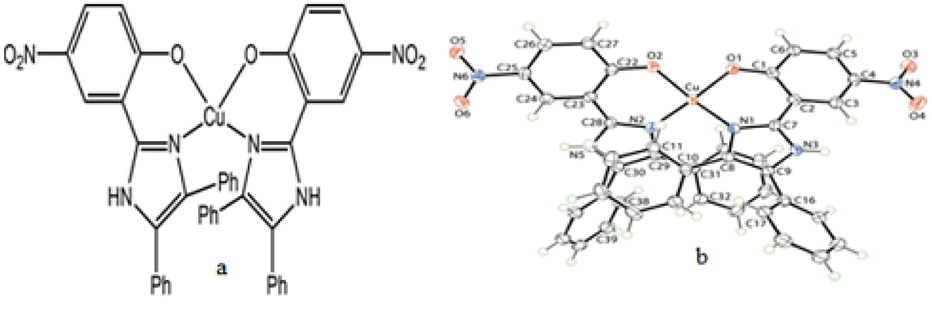 | ||
| Fig. 3 (a) Molecular structure of compound 5 and (b) crystal structure of compound 5 with displacement ellipsoid drawn at 70% probability level. | ||
Therefore, we carried out the detailed study of transformation of copper borate into other intermediate in the reaction mixture under the given conditions separately.
Firstly, we synthesized 2-(4,5-diphenyl-1H-imidazol-2-yl)-4-nitrophenol (4m) ligand and subsequently reacted with CuB4O7 in neat condition at 100 °C for 8–10 h; however, we failed to get the desired product (5) (Scheme 5a). But when we added stoichiometric amount of NH4OAc (3) to the same reaction, the reaction mixture instantaneously turned blue and after reacting for 4 h, the compound was extracted with DMF (5 mL) and the filtrate was kept for 5–8 days for slow evaporation at room temperature. Surprisingly, we got the same blue colored copper(II) complex (5) in excellent yield (60%) (Scheme 5b).
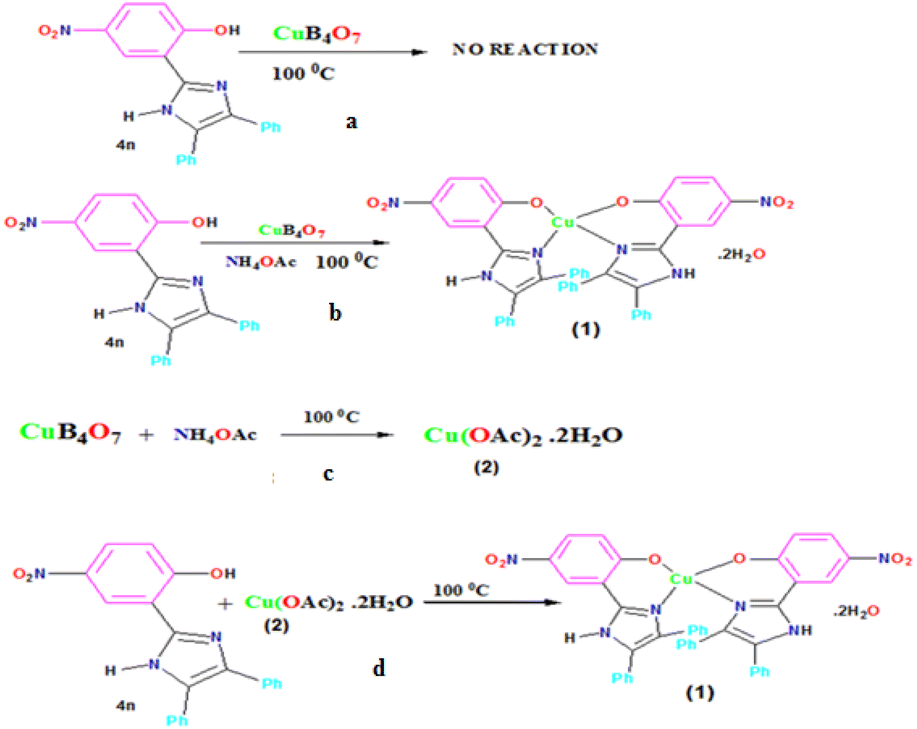 | ||
| Scheme 5 (a) Reaction between ligand 4n and CuB4O7, (b) reaction between ligand 4n and CuB4O7 in the presence of NH4OAc for the synthesis of 5, (c) reaction between CuB4O7 and NH4OAc to form compound 6, and (d) reaction between ligand 4n and compound 6 to form 5. | ||
Secondly, to understand the role of NH4OAc, a separate reaction was set up in which 1 mmol CuB4O7 was directly added to 50 mL water containing 3 mmol NH4OAc in a 100 mL round bottomed flask (Scheme 5c), and the reaction mixture was allowed to stir at room temperature for 8 h. It was seen that CuB4O7 slowly discharged into the NH4OAc solution and with the passage of time, the entire CuB4O7 dissolved in the reaction mixture. The reaction mixture was allowed to evaporate at room temperature and finally after 14 days, a blue colored single crystal suitable for X-ray single crystal diffraction study appeared in the solution. After successful X-ray single crystal diffraction of the compound, it was found that the blue colored complex was copper(II) acetate monohydrate (6) (molecular formula Cu2C8H16O10) with monoclinic crystal system and (C2/c) space group having paddle wheel structure (Fig. 4a–c).
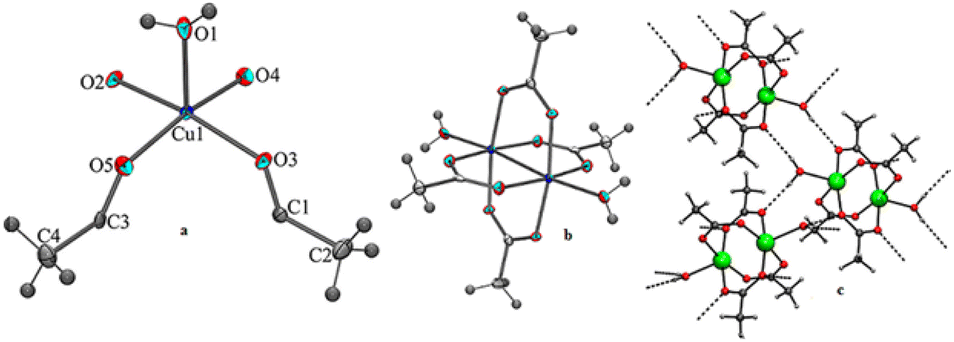 | ||
| Fig. 4 (a) Asymmetric unit of compound 6, (b) ortep diagram of 6 with 50% probability and (c) H-bonding pattern of compound 6. | ||
The crystallographic description, selected bond lengths bond distances, and hydrogen bonded geometry of the compound (6) are given in the ESI 1.†
Thus, it IS evident from the above reaction that CuB4O7 in the presence of NH4OAc is converted to intermediate Cu(OAc)2 monohydrate (6) in the reaction mixture and undergoes metallation with ligand (4n) to give the complex 5 under the given reaction conditions. We again set up a reaction directly with compound 4n employing Cu(OAc)2 monohydrate 6 under the same reaction conditions, and we obtained the same compound 5 (Scheme 5d).
This finding encouraged us to understand the reaction of NH4OAc with other copper salts, and we set up a reaction between different copper(II) salts such as CuSO4, CuCl2, and CuO to check the conversion of these salts into Cu(OAc)2 monohydrate under the same reaction. Fascinatingly, it was found that these copper salts are readily converted to Cu(OAc)2 monohydrate 6, as evident by the comparisons of the FT-IR spectra of the respective reaction with commercially available Cu(OAc)2 monohydrate (Fig. 5).
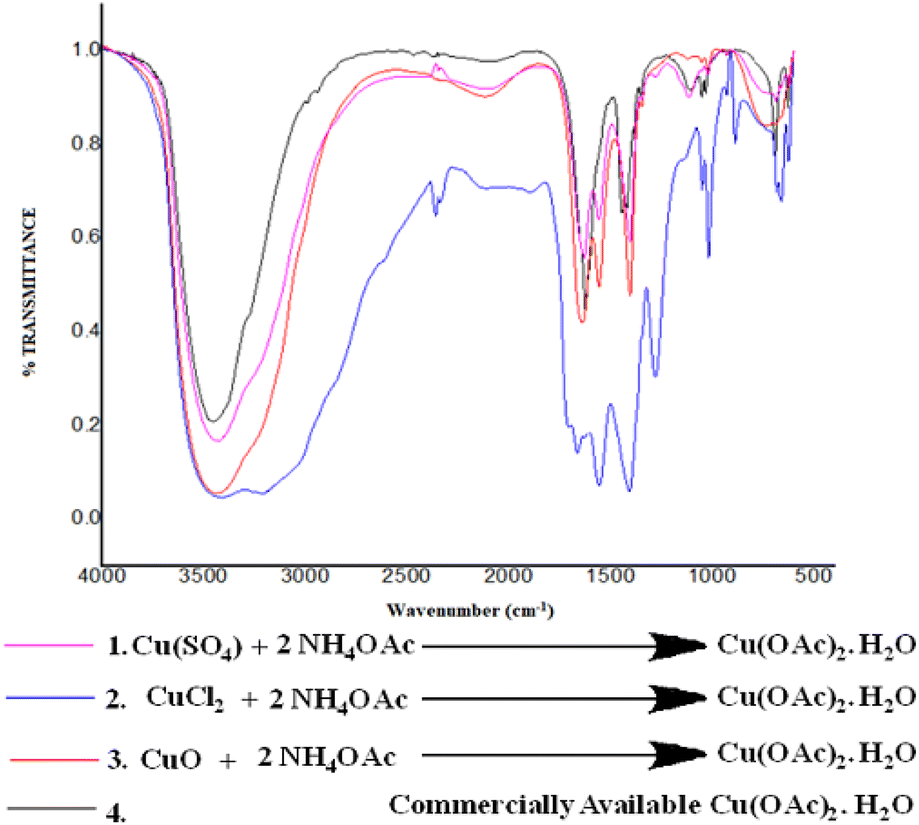 | ||
| Fig. 5 (1) FT-IR spectrum of the reaction mixture of CuSO4 and NH4OAc, (2) FT-IR spectrum of the reaction mixture of CuCl2 and NH4OAc, (3) FT-IR spectrum of the reaction mixture of CuO and NH4OAc, and (4) FT-IR spectrum of commercially available Cu(OAc)2·H2O. | ||
Furthermore, we also investigated the catalytic activity of Cu(OAc)2 monohydrate for the designed reaction under solvent-free condition, but it was observed that the desired product formed in less amount (72%), and the reaction takes a longer time to reach completion.58 Thus, we can infer that copper borate helps to ionize NH4OAc more efficiently, which acts as a nitrogen source for the schemed reaction to give the desired product (Table 5, entry 2).
| Entry | Catalyst | Solventa | Temperature | Time (min) | Yieldb (%) |
|---|---|---|---|---|---|
| a The reaction was performed with, benzil 1 (1 mmol), 2-nitrobenzaldehyde 2 (1 mmol), and NH4OAc (3) in the presence of CuB4O7 and Cu(OAc)2. H2O under solvent-free condition.b Isolated yield. | |||||
| 1 | CuB4O7 | Neat | 80 °C | 10 | 98 |
| 2 | Cu(OAc)2·H2O | Neat | 80 °C | 120 | 72 |
Moreover, a plausible mechanism for the formation of 2,4,5-triarylimidazole 4 by the condensation reaction between benzil 1, aldehyde 2 and ammonium acetate 3 catalyzed by copper borate (CuB4O7) is described in Scheme 4. Different pathways have been reported in the literature for the synthesis of 2,4,5-triarylimidazole using benzil as the starting material.44,93 In our proposed mechanism, it was hypothesized that copper borate reacts with ammonium acetate to generate ammonia and copper acetate 5 in situ at the initial stage of the reaction. Furthermore, the generated ammonia reacts with 2-nitrobenzaldehyde to produce intermediate I and, subsequently, intermediate I adds a second molecule of ammonia to give intermediate (2-nitrophenyl)methanediamine (II). Intermediate II reacts with benzil to afford intermediate III, and the loss of water molecule from intermediate III affords intermediate IV. At the final stage of the reaction, intermediate IV undergoes 1,5-H shift to form the desired product 4b (Scheme 6).
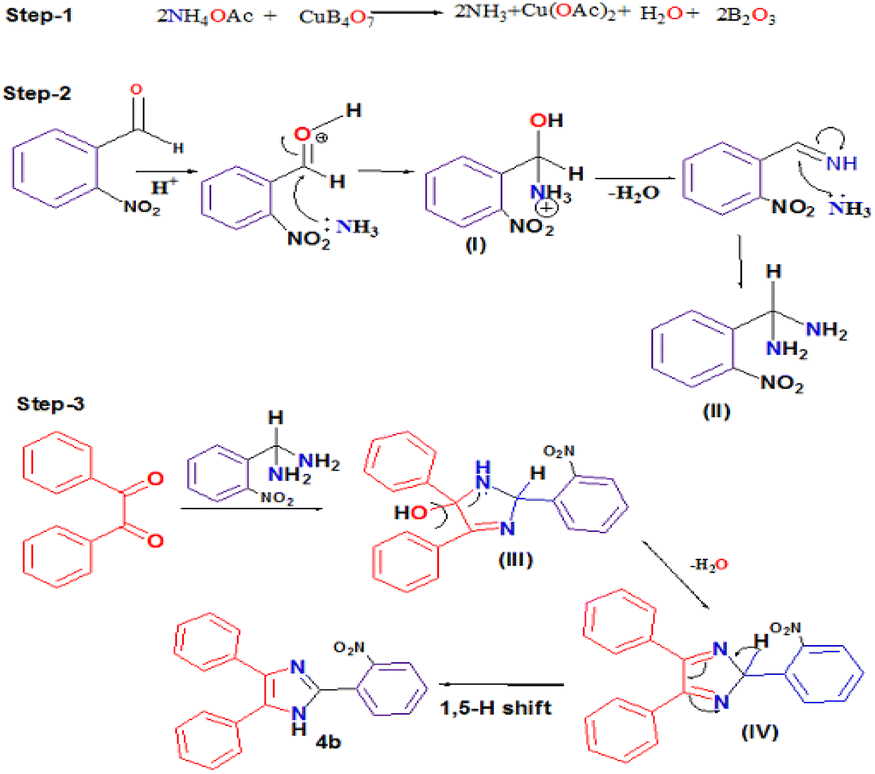 | ||
| Scheme 6 Proposed mechanism for the synthesis of 2,4,5-triarylimidazole using CuB4O7. | ||
Moreover, CuB4O7 under the schemed protocol could not be recovered after the reaction since it gets transformed into Cu(CH3COO)2·H2O. Thus, we repeated the same reaction with fresh CuB4O7 to ensure the repeatability of the desired product, and it was observed that the product can be reproduced under the given reaction protocol.
Conclusion
Herein, we have successfully developed a simple, environmentally benign, and efficient green methodology for the synthesis of 2,4,5-triarylimidazole derivatives using unconventional CuB4O7 utilizing one-pot three-component reaction. This environmentally green approach provides access to 2,4,5-tri-arylimidazole derivatives in good to excellent yield using an unconventional and inexpensive CuB4O7. The developed procedure was found to be operative for a wide range of aldehyde (aromatic/heteroaromatic) substrate, and we have been able to report the in situ formation of copper acetate in the reaction mixture from the reaction between different copper salts in the presence of ammonium acetate.Experimental section
Materials and methods
All starting materials of high purity for the aforementioned synthesis were purchased commercially and were used as received. The FT-IR spectra of the prepared compound were recorded using a Bruker Alpha III spectrophotometer operating in the wavenumber region from 4000 to 400 cm−1 in dry KBr. Single crystal X-ray diffraction study was performed using an Xcalibur Sapphire 3 (manufactured by Oxford Diffraction, Poland) equipped with CCD camera. The melting point of the synthesized compounds was determined by the open capillary method. The 1H-NMR spectra of the synthesized tri-aryl imidazole derivatives were recorded at room temperature on an FT-NMR (Bruker Avance-II 400 MHz) spectrometer using DMSO-d6 as solvent, and chemical shifts were quoted in ppm downfield of internal standard tetramethylsilane (TMS).General procedure for the synthesis of 2,4,5-triarylimidazole
In a typical procedure, a mixture of benzil (1) (1.0 mmol), substituted benzaldehyde (2) (1.0 mmol), ammonium acetate (3) (2.5 mmol), and copper borate (2 mol%) were thoroughly ground and mixed in a motor and pestle to make a homogeneous mixture. The mixture was then transferred to a test tube. The reaction was heated at 80 °C for 10 min. The progress of the reaction was monitored by TLC using hexane/ethyl acetate (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20) solvent (1 mL). After the completion of the reaction, the reaction mixture was dissolved in methanol (10 mL) and filtered. The filtrate was evaporated under vacuum and subsequently dried to afford the desired product (4a–z). Finally, the products were recrystallized from dry ethanol (10 mL) to afford the pure product.
:20) solvent (1 mL). After the completion of the reaction, the reaction mixture was dissolved in methanol (10 mL) and filtered. The filtrate was evaporated under vacuum and subsequently dried to afford the desired product (4a–z). Finally, the products were recrystallized from dry ethanol (10 mL) to afford the pure product.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1503 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.69 (s, 1H, NH), 7.22–7.55 (m, Ar H, 13H), 8.09 (d, 2H, J = 7.2 Hz) ppm; 13C NMR (100 MHz, DMSO-d6): δ 145.4, 138.2, 136.1, 131.5, 133.4, 130.1, 129.4, 128.7, 128.2, 127.8, 126.2 ppm [lit. 94].C), 1602 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.98 (s, 1H, NH), 7.21–7.97 (m, Ar H, 12H), 8.01 (d, 2H, J = 8.0 Hz) ppm; 13C NMR (100 MHz, DMSO d6): δ 148.7, 141.4, 138.2, 135.2, 132.6, 131.2, 130.5, 130.2, 129.6, 129.4, 128.6, 128.5, 127.5, 127.2, 124.3, 123.7 ppm [lit. 95].C), 1598 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.56 (s, 1H, NH), 7.61–8.37 (m, 12H, Ar H), 8.62–8.68 (d, 2H, Ar H); 13C NMR (100 MHz, DMSO d6): δ 148.7, 143.6, 138.1, 135.6, 132.4, 131.8, 131.3, 131.1, 197.7, 129.4, 128.9, 128.8, 128.1, 127.8 ppm [lit. 96].C), 1600 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.60 (s, 1H, NH), 7.24–8.45 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6): δ 147.3, 143.9, 138.8, 136.7, 135.3, 130.5, 129.6, 129.3, 128.9, 127.5, 127.7, 126.4, 124.6 ppm [lit. 96].C), 1602 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.66 (s, 1H, NH), 7.21–7.93 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6): δ 143.8, 137.6, 135.6, 132.3, 132.3, 131.4, 130.8, 130.5, 129.3, 128.8, 128.6, 128.4, 127.9, 127.6, 127.3 ppm [lit. 96].C), 1601 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.83 (s, 1H, NH), 8.30 (s, Ar H, 1H), 7.24–8.10 (m, Ar H, 13H) ppm; 13C NMR (100 MHz, DMSO d6): δ 144.4, 138.8, 135.4, 134.9, 133.5, 131.3, 130.2, 129.6, 129.4, 128.7, 127.7, 127.5, 126.3, 119.7, 110.8 ppm [lit. 97].C), 1610 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.03 (s, 1H, NH), 7.23–7.96 (m, Ar H, 12H), 8.26 (d, 2H, J = 8.2 Hz) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 144.4, 138.8, 135.4, 134.9, 133.5, 131.3, 130.2, 129.6, 129.4, 128.7, 127.7, 127.5, 126.3, 119.7, 110.8 ppm [lit. 96].C), 1612 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.41 (s, 1H, NH), 9.73 (s, 1H, OH), 6.86–8.31 (m, Ar H, 14H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ: 159.4, 144.9, 126.8, 126.3, 125.5, 125.3, 120.1, 113.9 ppm [lit. 96].C), 1552 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.03 (s, 1H, NH), 8.26 (d, 2H), 7.96 (d, 2H), 7.25–7.56 (m, Ar H, 10H) ppm; 13C NMR (DMSO d6, 100 MHz): δ 150.3, 146.6, 136.7, 135.4, 131.5, 128.8, 128.5, 128.2, 127.8, 127.2, 126.4, 118.5, 112.1, 39.6 ppm [lit. 96].N), 1H NMR (400 MHz, DMSO-d6): δ 13.51 (s, 1H, NH), 9.75 (s, 1H, OH), 6.87 (d, 2H, Ar–H), 7.30–7.93 (m, Ar H, 12H) ppm; 13C NMR (100 MHz, DMSO-d6): δ 157.4, 145.2, 137.1, 135.0, 131.4, 131.3, 129.5, 128.3, 128.4, 128.1, 128.0, 127.6, 127.2, 126.4, 116.2, 115.5, 112.3 ppm [lit. 98].C), 1603 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.97 (s, 1H, NH), 12.7 (s, 1H, OH), 6.95–8.13 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6) δ: 147.8, 133.2, 130.4, 129.6, 128.3, 122, 118.6, 116.5 ppm [lit. 99].C), 1595 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H, NH), 10.27 (s, 1H, OH), 6.95–8.23 (m, Ar H, 13H) ppm; 13C NMR (100 MHz, DMSO d6): δ 145.1, 137.6, 135.6, 133.4, 131.6, 129.8, 129.4, 129.3, 129.2, 129.1, 128.7, 128.5, 127.7, 127.6, 127.3 ppm [lit. 99].C), 1610 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H, NH), 13.09 (s, 1H, OH), 6.91–8.31 (m, Ar H, 13H), 13C NMR (100 MHz, DMSO-d6, 100 MHz): δ 144.7, 137.6, 135.2, 132.6, 132.4, 130.1, 129.9, 129.2, 129.1, 128.4, 127.8, 128.1, 127.8, 126.6, 121.5 ppm [lit. 99].C), (CN), 1H NMR (400 MHz, DMSO-d6): δ = 13.3 (s, 1H, NH), 8.6 (s, 1H, OH), 7.20–7.50 (m, Ar H, 13H) ppm; 13C NMR (100 MHz, DMSO-d6): δ 148.2, 147.4, 145.8, 137.8, 134.9, 131.7, 131.4, 130.5, 129.8, 128.5, 126.8, 126.6, 124.3 ppm [lit. 99].C), 1606 (CN), 1H NMR (400 MHz, DMSO-d6): δ = 12.943 (s, 1H, NH), 9.73 (s, 1H, OH), 6.18–8.11 (m, Ar H, 13H), 4.049 (s, 1H, OH) ppm.C), 1604 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.97 (s, 1H, NH), 6.86–7.67 (m, Ar H, 13H), 4.28 (s, 1H, OH), 3.87 (s, 3H, OCH3) ppm; 13C NMR (100 MHz, DMSO-d6): δ 155.6, 147.3, 146.1, 145.1, 129.8, 127.4, 127.3, 126.6, 126.3, 117.1, 112.1, 110.9, 54.3 ppm [lit. 100].C), 1606 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.41 (s, 1H, NH), 6.83–7.67 (m, Ar H, 13H), 4.03 (s, 1H, OH), 3.90 (s, 3H, OCH3) ppm; 13C NMR (100 MHz, DMSO d6): δ 146.8, 146.3, 132.3, 127.5, 127.3, 126.2, 121.1, 118.1, 114.6, 108.5, 55.1 ppm [lit. 96].C), 1605, 1531 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.72 (s, 1H, NH), 8.21 (d, 2H), 7.25–7.55 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6): δ 148.2, 137.7, 133.3, 130.4, 128.9, 128.6, 127.1, 126.9, 124.2 ppm [lit. 101].C), 1543 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.97 (s, 1H, NH), 9.15 (s, 1H, OH), 7.19–8.18 (m, Ar H, 12H), 3.36 (s, OCH3, 6H) ppm [lit. 102].C), 1598 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.31 (s, 1H, NH), 11.40 (s, 1H, NH), 7.13–8.50 (m, Ar H, 15H), 13C NMR (100 MHz, DMSO d6): δ 143.5, 135.8, 135.4, 135.1, 131.4, 128.6, 127.8, 127.3, 126.4, 125.9, 125.5, 124.8, 123.5, 121.5, 121.4, 119.1, 111.4, 106.6 ppm [lit. 96].C), 1600 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.6 (s, 1H, NH), 7.58–7.92 (m, Ar H, 15H), 6.52 (d, 2H, CHCH), 13C NMR (100 MHz, DMSO d6): δ 146.2, 138.1, 134.3, 130.1, 128.6, 128.6, 128.2, 128.0, 127.7, 125.8, 123.5, 110.8 ppm [lit. 103].C), 1605 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.07 (s, 1H, NH), 8.56 (d, 2H Ar–H J = 8.0 Hz), 7.29–8.51 (m, Ar H, 16H) ppm.C), 1590 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.3 (s, 1H, NH), 7.20–7.62 (m, Ar H, 12H), 3.89 (s, 9H, OCH3) ppm.C), 1486 (CN), 1H NMR (400 MHz, DMSO-d6): δ 9.46 (s, 1H, NH), 7.42–7.69 (m, Ar H, 5H), 8.11–8.13 (m, 4H, J = 7.2 Hz) ppm; 13C NMR (100 MHz, DMSO d6): δ 150.1, 149.6, 139.2, 137.6, 135.8, 131.5, 130.1, 129.3, 129.1, 128.5, 128.1, 127.8, 127.4, 123.6, 120.5 [lit. 96].C), 1503 (CN), 1H NMR (400 MHz, DMSO-d6): δ = 12.93 (s, 1H, NH), 7.25–7.94 (m, Ar H, 5H), 8.08–8.51 (m, 4H) ppm [lit. 104].C), 1501 (CN), 1H NMR (400 MHz, DMSO-d6): δ = 12.81 (s, 1H, NH), 7.31–7.68 (m, Ar H, 5H), 8.10–8.13 (m, 4H, J = 7.2 Hz) ppm; 13C NMR (100 MHz, DMSO d6): δ 142.4, 137.6, 135.4, 134.6, 131.3, 130.1, 128.6, 128.3, 128.1, 127.7, 127.3, 126.9, 124.7 ppm [lit. 96].
C), 1503 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.69 (s, 1H, NH), 7.22–7.55 (m, Ar H, 13H), 8.09 (d, 2H, J = 7.2 Hz) ppm; 13C NMR (100 MHz, DMSO-d6): δ 145.4, 138.2, 136.1, 131.5, 133.4, 130.1, 129.4, 128.7, 128.2, 127.8, 126.2 ppm [lit. 94].C), 1602 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.98 (s, 1H, NH), 7.21–7.97 (m, Ar H, 12H), 8.01 (d, 2H, J = 8.0 Hz) ppm; 13C NMR (100 MHz, DMSO d6): δ 148.7, 141.4, 138.2, 135.2, 132.6, 131.2, 130.5, 130.2, 129.6, 129.4, 128.6, 128.5, 127.5, 127.2, 124.3, 123.7 ppm [lit. 95].C), 1598 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.56 (s, 1H, NH), 7.61–8.37 (m, 12H, Ar H), 8.62–8.68 (d, 2H, Ar H); 13C NMR (100 MHz, DMSO d6): δ 148.7, 143.6, 138.1, 135.6, 132.4, 131.8, 131.3, 131.1, 197.7, 129.4, 128.9, 128.8, 128.1, 127.8 ppm [lit. 96].C), 1600 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.60 (s, 1H, NH), 7.24–8.45 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6): δ 147.3, 143.9, 138.8, 136.7, 135.3, 130.5, 129.6, 129.3, 128.9, 127.5, 127.7, 126.4, 124.6 ppm [lit. 96].C), 1602 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.66 (s, 1H, NH), 7.21–7.93 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6): δ 143.8, 137.6, 135.6, 132.3, 132.3, 131.4, 130.8, 130.5, 129.3, 128.8, 128.6, 128.4, 127.9, 127.6, 127.3 ppm [lit. 96].C), 1601 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.83 (s, 1H, NH), 8.30 (s, Ar H, 1H), 7.24–8.10 (m, Ar H, 13H) ppm; 13C NMR (100 MHz, DMSO d6): δ 144.4, 138.8, 135.4, 134.9, 133.5, 131.3, 130.2, 129.6, 129.4, 128.7, 127.7, 127.5, 126.3, 119.7, 110.8 ppm [lit. 97].C), 1610 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.03 (s, 1H, NH), 7.23–7.96 (m, Ar H, 12H), 8.26 (d, 2H, J = 8.2 Hz) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 144.4, 138.8, 135.4, 134.9, 133.5, 131.3, 130.2, 129.6, 129.4, 128.7, 127.7, 127.5, 126.3, 119.7, 110.8 ppm [lit. 96].C), 1612 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.41 (s, 1H, NH), 9.73 (s, 1H, OH), 6.86–8.31 (m, Ar H, 14H) ppm; 13C NMR (DMSO-d6, 100 MHz) δ: 159.4, 144.9, 126.8, 126.3, 125.5, 125.3, 120.1, 113.9 ppm [lit. 96].C), 1552 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.03 (s, 1H, NH), 8.26 (d, 2H), 7.96 (d, 2H), 7.25–7.56 (m, Ar H, 10H) ppm; 13C NMR (DMSO d6, 100 MHz): δ 150.3, 146.6, 136.7, 135.4, 131.5, 128.8, 128.5, 128.2, 127.8, 127.2, 126.4, 118.5, 112.1, 39.6 ppm [lit. 96].N), 1H NMR (400 MHz, DMSO-d6): δ 13.51 (s, 1H, NH), 9.75 (s, 1H, OH), 6.87 (d, 2H, Ar–H), 7.30–7.93 (m, Ar H, 12H) ppm; 13C NMR (100 MHz, DMSO-d6): δ 157.4, 145.2, 137.1, 135.0, 131.4, 131.3, 129.5, 128.3, 128.4, 128.1, 128.0, 127.6, 127.2, 126.4, 116.2, 115.5, 112.3 ppm [lit. 98].C), 1603 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.97 (s, 1H, NH), 12.7 (s, 1H, OH), 6.95–8.13 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6) δ: 147.8, 133.2, 130.4, 129.6, 128.3, 122, 118.6, 116.5 ppm [lit. 99].C), 1595 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.13 (s, 1H, NH), 10.27 (s, 1H, OH), 6.95–8.23 (m, Ar H, 13H) ppm; 13C NMR (100 MHz, DMSO d6): δ 145.1, 137.6, 135.6, 133.4, 131.6, 129.8, 129.4, 129.3, 129.2, 129.1, 128.7, 128.5, 127.7, 127.6, 127.3 ppm [lit. 99].C), 1610 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.15 (s, 1H, NH), 13.09 (s, 1H, OH), 6.91–8.31 (m, Ar H, 13H), 13C NMR (100 MHz, DMSO-d6, 100 MHz): δ 144.7, 137.6, 135.2, 132.6, 132.4, 130.1, 129.9, 129.2, 129.1, 128.4, 127.8, 128.1, 127.8, 126.6, 121.5 ppm [lit. 99].C), (CN), 1H NMR (400 MHz, DMSO-d6): δ = 13.3 (s, 1H, NH), 8.6 (s, 1H, OH), 7.20–7.50 (m, Ar H, 13H) ppm; 13C NMR (100 MHz, DMSO-d6): δ 148.2, 147.4, 145.8, 137.8, 134.9, 131.7, 131.4, 130.5, 129.8, 128.5, 126.8, 126.6, 124.3 ppm [lit. 99].C), 1606 (CN), 1H NMR (400 MHz, DMSO-d6): δ = 12.943 (s, 1H, NH), 9.73 (s, 1H, OH), 6.18–8.11 (m, Ar H, 13H), 4.049 (s, 1H, OH) ppm.C), 1604 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.97 (s, 1H, NH), 6.86–7.67 (m, Ar H, 13H), 4.28 (s, 1H, OH), 3.87 (s, 3H, OCH3) ppm; 13C NMR (100 MHz, DMSO-d6): δ 155.6, 147.3, 146.1, 145.1, 129.8, 127.4, 127.3, 126.6, 126.3, 117.1, 112.1, 110.9, 54.3 ppm [lit. 100].C), 1606 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.41 (s, 1H, NH), 6.83–7.67 (m, Ar H, 13H), 4.03 (s, 1H, OH), 3.90 (s, 3H, OCH3) ppm; 13C NMR (100 MHz, DMSO d6): δ 146.8, 146.3, 132.3, 127.5, 127.3, 126.2, 121.1, 118.1, 114.6, 108.5, 55.1 ppm [lit. 96].C), 1605, 1531 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.72 (s, 1H, NH), 8.21 (d, 2H), 7.25–7.55 (m, Ar H, 14H) ppm; 13C NMR (100 MHz, DMSO d6): δ 148.2, 137.7, 133.3, 130.4, 128.9, 128.6, 127.1, 126.9, 124.2 ppm [lit. 101].C), 1543 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.97 (s, 1H, NH), 9.15 (s, 1H, OH), 7.19–8.18 (m, Ar H, 12H), 3.36 (s, OCH3, 6H) ppm [lit. 102].C), 1598 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.31 (s, 1H, NH), 11.40 (s, 1H, NH), 7.13–8.50 (m, Ar H, 15H), 13C NMR (100 MHz, DMSO d6): δ 143.5, 135.8, 135.4, 135.1, 131.4, 128.6, 127.8, 127.3, 126.4, 125.9, 125.5, 124.8, 123.5, 121.5, 121.4, 119.1, 111.4, 106.6 ppm [lit. 96].C), 1600 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.6 (s, 1H, NH), 7.58–7.92 (m, Ar H, 15H), 6.52 (d, 2H, CHCH), 13C NMR (100 MHz, DMSO d6): δ 146.2, 138.1, 134.3, 130.1, 128.6, 128.6, 128.2, 128.0, 127.7, 125.8, 123.5, 110.8 ppm [lit. 103].C), 1605 (CN), 1H NMR (400 MHz, DMSO-d6): δ 13.07 (s, 1H, NH), 8.56 (d, 2H Ar–H J = 8.0 Hz), 7.29–8.51 (m, Ar H, 16H) ppm.C), 1590 (CN), 1H NMR (400 MHz, DMSO-d6): δ 12.3 (s, 1H, NH), 7.20–7.62 (m, Ar H, 12H), 3.89 (s, 9H, OCH3) ppm.C), 1486 (CN), 1H NMR (400 MHz, DMSO-d6): δ 9.46 (s, 1H, NH), 7.42–7.69 (m, Ar H, 5H), 8.11–8.13 (m, 4H, J = 7.2 Hz) ppm; 13C NMR (100 MHz, DMSO d6): δ 150.1, 149.6, 139.2, 137.6, 135.8, 131.5, 130.1, 129.3, 129.1, 128.5, 128.1, 127.8, 127.4, 123.6, 120.5 [lit. 96].C), 1503 (CN), 1H NMR (400 MHz, DMSO-d6): δ = 12.93 (s, 1H, NH), 7.25–7.94 (m, Ar H, 5H), 8.08–8.51 (m, 4H) ppm [lit. 104].C), 1501 (CN), 1H NMR (400 MHz, DMSO-d6): δ = 12.81 (s, 1H, NH), 7.31–7.68 (m, Ar H, 5H), 8.10–8.13 (m, 4H, J = 7.2 Hz) ppm; 13C NMR (100 MHz, DMSO d6): δ 142.4, 137.6, 135.4, 134.6, 131.3, 130.1, 128.6, 128.3, 128.1, 127.7, 127.3, 126.9, 124.7 ppm [lit. 96].Author contributions
Sailesh Chettri and Dhiraj Brahman: conceptualization, experimental and data curation; Biswajit Sinha, Sumiran Tamang, Dhiraj Brahman and Sailesh Chettri: formal analysis, writing original draft and software; Biswajit Sinha, Kiran Pradhan and Dhiraj Brahman: supervision, review, editing and visualization.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Authors are grateful to the research facilities provided by Incubation Centre of St. Joseph's College, Darjeeling, India and University of North Bengal, Darjeeling, India. All authors greatly acknowledge the analytical facilities provided by SAIF NEHU Shillong, India.Notes and references
- P. T. Anastas and T. C. Williamson, Green Chemistry: Frontiers in Benign Chemical Syntheses and Processes, Oxford Science Publications, New York, 1998 Search PubMed.
- M. Poliakoff and P. T. Anastas, Nature, 2001, 413, 257 CrossRef CAS PubMed.
- J. M. DeSimone, Science, 2002, 297(5582), 799–803 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2016, 18, 3180–3183 RSC.
- P. Gupta and A. Mahajan, RSC Adv., 2015, 5, 26686–26705 RSC.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- T. Welton, Green Chem., 2011, 13, 225 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2003, 77(4), 287–297 CrossRef CAS.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 22, 3–13 CrossRef.
- A. Domling and I. Ugi, Angew. Chem., 2000, 39(18), 3168–3210 CrossRef CAS.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14(3), 371–382 CrossRef CAS PubMed.
- B. Jiang, T. Rajale, W. Wever, S. J. Tu and G. Li, Chem.–Asian J., 2010, 5, 2318–2335 CrossRef CAS PubMed.
- S. Brauch, S. S. V Berkela and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
- C. Graaff, E. Ruijter and R. V. A. Orru, Chem. Soc. Rev., 2012, 41, 3969–4009 RSC.
- M. Zhang, Y.-H. Liu, Z.-R. Shang, H.-C. Hu and Z.-H. Zhang, Catal. Commun., 2017, 88, 39–44 CrossRef CAS.
- M.-N. Chen, L.-P. Mo, Z.-S. Cui and Z.-H. Zhang, Curr. Opin. Green Sustainable Chem., 2019, 15, 27–37 CrossRef.
- H. Liu, Z.-J. Cai and S.-J. Ji, Chin. Chem. Lett., 2022, 33, 4303–4305 CrossRef CAS.
- N. Li, S. Xub, X. Wang, L. Xua, J. Qiaoa, Z. Liang and X. Xu, Chin. Chem. Lett., 2021, 32(12), 3993–3997 CrossRef CAS.
- M. C. Pirrung and K. D. Sarma, Tetrahedron, 2005, 61, 11456–11472 CrossRef CAS.
- C. G. Neochoritis, T. Z. Tzitzikas, K. KHodgetts and A. Dömling, J. Chem. Educ., 2020, 97, 3739–3745 CrossRef CAS PubMed.
- S. Garbarino, D. Ravelli, S. Protti and A. Basso, Angew. Chem., 2016, 55, 15476–15484 CrossRef PubMed.
- G. Heijden, E. Ruijter and R. V. A. Orru, Synlett, 2013, 24, 666–685 CrossRef.
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Molecules, 2020, 25, 505 CrossRef PubMed.
- J. G. Lambardino and E. H. Wiseman, J. Med. Chem., 1974, 17, 1182–1188 CrossRef PubMed.
- A. Chawla, A. Sharma and A. K. Sharma, Der Pharma Chem., 2012, 4, 116 CAS.
- R. Breslow, Acc. Chem. Res., 1995, 28, 146–153 CrossRef CAS.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- L. Streit, M. Moreau, J. Gaudin, E. Ebert and H. V. Bossche, Pestic. Biochem. Physiol., 1991, 40(2), 162–168 CrossRef CAS.
- A. Husain, S. Drabu, N. Kumar, M. M. Alam and S. Bawa, J. Pharm. BioAllied Sci., 2013, 5(2), 154–161 CrossRef PubMed.
- T. Scior, D. M. Domeyer, K. Cuanalo-Contreras and S. A. Laufer, Curr. Med. Chem., 2011, 18(10), 1526–1539 CrossRef CAS PubMed.
- A. K. Takle, M. J. B. Brown, S. Davies, D. K. Dean, G. Francis, A. Gaiba, A. W. Hird, F. D. K. King, P. J. Lovell, A. Naylor, A. D. Reith, J. G. Steadman and D. M. Wilson, Bioorg. Med. Chem. Lett., 2006, 16, 378–381 CrossRef CAS PubMed.
- J. G. Lombardino and E. H. Wiseman, J. Med. Chem., 1974, 17(11), 1182–1188 CrossRef CAS PubMed.
- L. Wang, K. W. Woods, Q. Li, K. J. Barr, R. W. McCroskey, S. M. Hannick, L. Gherke, R. B. Credo, Y.-H. Hui, K. Marsh, R. Warner, J. Y. Lee, N. Zielinski-Mozng, D. Frost, S. H. Rosenberg and H. L. Sham, J. Med. Chem., 2002, 45(8), 1697–1711 CrossRef CAS PubMed.
- A. R. Phillips, H. L. White and S. Rosen, Eur. Pat. Appl., EP 58 890, Sep. 1 1982Chem. Abstr., 98, 1983, 53894z Search PubMed.
- R. A. Turner, C. F. Huebner and C. R. Scholz, J. Am. Chem. Soc., 1949, 71(8), 2801–2803 CrossRef CAS.
- J. Miller, Ann. Allergy, 1963, 21, 692–697 CAS.
- A. Goyal, J. Singh and D. P. Pathak, J. Pharm. Technol. Res. Manag., 2013, 1, 69–79 CrossRef.
- K. Gaffney and D. G. I. Scott, Br. J. Rheumatol., 1998, 37, 824–836 CrossRef CAS PubMed.
- J. Baharara, E. Amini, N. Nikdel and F. Salek-Abdollahi, Avicenna J. Med. Biotechnol., 2016, 8(3), 112–119 Search PubMed.
- J. B. Tagne, S. Kakumanu and R. J. Nicolosi, Mol. Pharm., 2008, 5(6), 1055–1063 CrossRef CAS PubMed.
- F. R. Japp and H. H. Robinson, Chem. Ber., 1882, 15, 1268 CrossRef.
- B. Radziszewski, Chem. Ber., 1882, 15, 1493 CrossRef.
- D. Davidson, M. Weiss and M. Jelling, J. Org. Chem., 1937, 2, 328–334 CrossRef CAS.
- R. S. Joshi, P. G. Mandhane, M. U. Shaikh, R. S. Kale and C. H. Gill, Chin. Chem. Lett., 2010, 21, 429 CrossRef CAS.
- C. M. Yu, M. Lei, W. K. Su and Y. Y. Xie, Synth. Commun., 2007, 37, 3301–3309 CrossRef CAS.
- S. D. Sharma, P. Hazarika and D. Konwar, Tetrahedron Lett., 2008, 49, 2216–2220 CrossRef.
- J. Sangshetty, N. D. Kokare, S. A. Kotharkara and D. B. Shinde, J. Chem. Sci., 2008, 120, 463–467 CrossRef.
- S. N. Murthy, B. Madhav and Y. V. D. Nageswar, Tetrahedron Lett., 2010, 51, 5252–5257 CrossRef CAS.
- S. Subhasis, C. N. Ganesh, S. Pallavi and M. S. Singh, Tetrahedron, 2009, 65, 10155–10161 CrossRef.
- R. Khosropour, Ultrason. Sonochem., 2008, 15, 659–664 CrossRef PubMed.
- R. K. Sharma and C. Sharma, Catal. Commun., 2011, 12, 327–331 CrossRef CAS.
- K. Mazaahir, M. Poonam, B. Vikas, K. S. Rishi, S. E. Abdul, D. Sharmistha and P. S. Tej, J. Mol. Catal. A: Chem., 2007, 265, 177–182 CrossRef.
- G. H. Mahdavinia, A. M. Amani and H. Sepehrian, Chin. J. Chem., 2012, 30, 703–708 CrossRef CAS.
- H. Zang, Q. Su, Y. Mo, B. W. Cheng and S. Jun, Ultrason. Sonochem., 2010, 17, 749–751 CrossRef CAS PubMed.
- M. Xia and Y. D. Lu, J. Mol. Catal. A: Chem., 2007, 265, 205–208 CrossRef CAS.
- M. V. Reddy and Y. T. Jeong, J. Fluorine Chem., 2012, 142, 45–51 CrossRef.
- J. Jayram and V. Jeena, Green Chem., 2017, 19, 5841–5845 RSC.
- J. Jayram and V. Jeena, RSC Adv., 2018, 18, 37557–37563 RSC.
- K. Nikoofar, M. Haghighi, M. Lashanizadegan and Z. Ahmadvand, J. Taibah Univ. Sci., 2015, 9, 570–578 CrossRef.
- M. Kalhor and Z. Zarnegar, RSC Adv., 2019, 9, 19333–19346 RSC.
- M. Kalhor and N. Khodaparast, Res. Chem. Intermed., 2015, 41, 3235–3242 CrossRef CAS.
- A. Bamoniri, B. B. F. Mirjalili and S. Saleh, RSC Adv., 2018, 8, 6178–6182 RSC.
- A. Khorramabadi-zad, M. Azadmanesh and S. Mohammadi, S. Afr. J. Chem., 2013, 66, 244–247 CAS.
- N. V. Gandhare, R. G. Chaudhary, V. P. Meshram, J. A. Tanna, S. Ladee, M. P. Gharpure and H. D. Juneja, J. Chin. Adv. Mater. Soc., 2015, 1–10 Search PubMed.
- Z. Varzi, M. S. Esmaeili, R. T. Ledari and A. Maleki, Inorg. Chem. Commun., 2021, 125, 108465 CrossRef CAS.
- D. Kumar, D. N. Kommi, N. Bollineni, A. R. Patel and A. K. Chakraborti, Green Chem., 2012, 14, 2038–2049 RSC.
- K. Sivakumar, A. Kathirvel and A. Lalitha, Tetrahedron Lett., 2010, 51, 3018–3021 CrossRef CAS.
- V. D. Kadu, G. A. Mali, S. P. Khadul and G. J. Kothe, RSC Adv., 2021, 11, 21955–21963 RSC.
- K. F. Shelke, S. S. Sonar, S. A. Sadaphal, B. B. Shingate and M. S. Shingare, Bull. Korean Chem. Soc., 2009, 30(9), 1963–1966 CrossRef.
- S. S. Dipake, M. K. Lande, A. S. Rajbhoj and S. T. Gaikwad, Res. Chem. Intermed., 2021, 47, 2245–2261 CrossRef CAS.
- A. R. Khosropour, Ultrason. Sonochem., 2008, 15, 659–664 CrossRef CAS PubMed.
- L.-M. Wang, Y.-H. Wang, W. Wu, Y.-F. Yao, J.-H. Shao and B. Liu, J. Fluorine Chem., 2006, 127, 1570–1573 CrossRef CAS.
- K. Nikoofar, M. Haghigi, M. Lashanizadegan and Z. Ahmadvand, J. Taibah Univ. Sci., 2015, 9, 570–578 CrossRef.
- M. Kidwai, P. Mothsra, V. Bansal and R. Goyal, Monatsh. Chem., 2006, 137, 1189–1194 CrossRef CAS.
- M. Banazadeh, S. Amirnejat and S. Javanshir, Front. Chem., 2020, 8, 1–15 CrossRef PubMed.
- J. Safari, S. Khalili and S. H. Banitaba, Synth. Commun., 2011, 41, 2359–2373 CrossRef CAS.
- J. Safari, S. D. Khalili, M. Rezaei, S. H. Banitaba and F. Meshkani, Monatsh. Chem., 2010, 141(12), 1339–1345 CrossRef CAS.
- M. V. Reddy and Y. T. Jeong, J. Fluorine Chem., 2013, 142(2), 45–51 Search PubMed.
- E. Eidi, M. Z. Kassaee and Z. Nasresfahani, Appl. Organomet. Chem., 2016, 30, 561–565 CrossRef CAS.
- R. Wang, C. Liu and G. Luo, Green Chem. Lett. Rev., 2010, 3(2), 101–104 CrossRef CAS.
- K. F. Shelke, S. Sapkal, S. Sonal, B. Madje, B. B. Shingate and M. S. Shingare, Bull. Korean Chem. Soc., 2009, 30(5), 1057–1060 CrossRef CAS.
- M. Vosoughi, F. Mohebali, A. P. S. Bonakdar, H. A. Lordegani and A. R. Massah, Bulg. Chem. Commun., 2015, 47(2), 607–612 Search PubMed.
- F. Mirjalili, A. Bamoniri and N. Mohaghegh, Curr. Chem. Lett., 2013, 2(1), 35–42 CrossRef.
- G. Brahmachari and S. Das, Indian J. Chem., 2013, 52B, 387–393 CAS.
- B. Maleki, H. K. Shirvan, F. Taimazi and E. Akbarzadeh, Int. J. Org. Chem., 2012, 2, 93–99 CrossRef CAS.
- R. G. Chaudhary, N. V. Gandhare, V. P. Meshram, J. Tanna, M. P. Gharpure, S. Lade and H. S. Juneja, J. Chin. Adv. Mater. Soc., 2015, 1–10 Search PubMed.
- A. Sedrpoushan, Z. Joshani and L. Fatollahi, Lett. Org. Chem., 2014, 11(4), 287–292 CrossRef CAS.
- A. Khorramabadi-zad, M. Azadmanesh and S. Mohammadi, S. Afr. J. Chem., 2013, 66, 244–247 CAS.
- F. Hatamjafari and H. Khojastehkouhi, Orient. J. Chem., 2014, 30(1), 329–331 CrossRef CAS.
- S. Balalaie, A. Arabanian and M. Hashtroudi, Monatsh. Chem., 2000, 131, 945–948 CrossRef CAS.
- S. Chettri, D. Brahman, B. Sinha, M. M. Jotani and E. R. Tiekink, Acta Crystallogr., 2019, E75, 1664–1671 CrossRef PubMed.
- D. Davidson, M. Weiss and M. Jelling, J. Org. Chem., 1937, 2, 319–327 CrossRef CAS.
- A. A. Marzouk, V. M. Abbasov, A. H. Talybov and S. K. Mohamed, World J. Org. Chem., 2013, 1(1), 6–10 Search PubMed.
- A. Puratchikody and M. Doble, Chem. Biol. Drug Des., 2009, 74, 173–182 CrossRef CAS PubMed.
- V. D. Kadu, G. A. Mali, S. P. Khadul and G. J. Kothe, RSC Adv., 2021, 11, 21955–21963 RSC.
- N. Thimmaraju and S. Z. M. Shamshuddin, RSC Adv., 2016, 6, 60231–60243 RSC.
- H. Sharghi, S. F. Razavi and M. Aberi, Catal. Lett., 2021, 152, 1769–1783 CrossRef.
- J. Peng, H.-J. Yang, Z. Weia and C.-Y. Guo, RSC Adv., 2015, 5(65), 53063–53072 RSC.
- M. Yar, M. Bajda, S. Shahzad, N. Ullah, M. A. Gilani, M. Ashraf, A. Rauf and A. Shaukat, Bioinorg. Chem., 2015, 58, 65–71 CrossRef CAS PubMed.
- D. B. Chugunov, E. V. Okina, A. S. Timonina, L. A. Klimaeva and Y. M. Selivanova, Russ. J. Org. Chem., 2020, 56(7), 1222–1227 CrossRef CAS.
- C. G. L. Veale, J. Jayram, S. Naidoo, D. Laming, T. Swart, T. Olivier, M. P. Akerman, K. A. de Villiers, H. C. Hoppe and V. Jeena, RSC Med. Chem., 2020, 11, 85–91 RSC.
- J. Banothu, R. Gali, R. Velpula and R. Bavantula, Arabian J. Chem., 2017, 10(2), S2754–S2761 CrossRef CAS.
- E. Rafiee, M. Joshaghani and P. Ghaderi-Shekhi Abadi, RSC Adv., 2015, 5, 74091–74101 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1958158. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra03183g |
| This journal is © The Royal Society of Chemistry 2023 |