DOI:
10.1039/D3RA03159D
(Paper)
RSC Adv., 2023,
13, 21746-21753
The investigation of the ultrafast excited state deactivation mechanisms for coumarin 307 in different solvents
Received
12th May 2023
, Accepted 11th July 2023
First published on 19th July 2023
Abstract
The intramolecular charge transfer (ICT) and twisted intramolecular charge transfer (TICT) processes of coumarin 307 (C307) in different solvents were investigated by performing steady-state/time-resolved transient absorption spectroscopic and steady-state photoluminescence spectroscopic characterizations in combination with time-dependent density functional theoretical calculation (TDDFT). The study unveiled the remarkable influence of solvent polarity and the strength of intermolecular hydrogen bonds formed between the solutes and solvents on the relaxation dynamics of the electronic excited state. Significantly, the emergence of the TICT state was observed in polar solvents, specifically dimethylformamide (DMF) and dimethyl sulfoxidemethanol (DMSO), owing to their inherent polarity as well as the enhanced intermolecular hydrogen bonding interactions. Interestingly, even in a weak polar solvent such as methanol (MeOH), the TICT state was also observed due to the intensified hydrogen bonding effects. Conversely, nonpolar solvents, exemplified by 1,4-dioxane (Diox), resulted in the stabilization of the ICT state due to the augmented N–H⋯O hydrogen bonding interactions. The experimental findings were corroborated by the computational calculations, thus ensuring the reliability of the conclusions drawn. Furthermore, schematic diagrams were presented to illustrate the mechanisms underlying the excited-state deactivation. Overall, this investigation contributes valuable mechanistic insights and provides a comprehensive understanding of the photochemical and photophysical properties exhibited by coumarin dyes.
1. Introduction
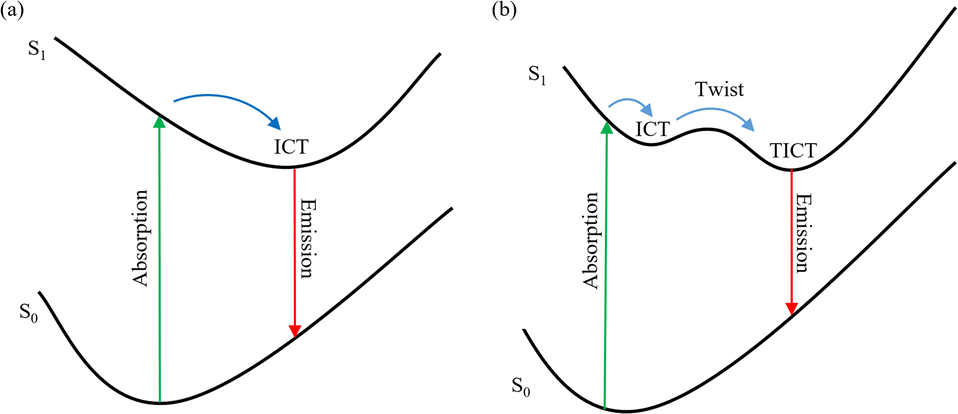
Twisted intramolecular charge transfer is a crucial process in dye molecules with significant implications for photochemical and photophysical applications, such as solar storage, molecular probes, and organic photovoltaics.1–5 As we know, the TICT process can be affected significantly by both intrinsic features (i.e. electron donating ability, electron accepting ability, connection mode, and spatial arrangement between the electron donor and electron acceptor) and the external factors.6–10 Intermolecular hydrogen bonds and the polarity of solvents as external factors play a vital role in the formation of the TICT state for dye molecules.11–14
The 7-aminocoumarin (7-NHEt) dyes, as a distinct subset within the coumarin dye family, serve as a valuable model system for investigating the dynamics of electronic excited states,9,15–17 which exhibit significant alterations in dipole moment between the ground state and electronic excited states, as well as substantial solvatochromic shifts in steady-state absorption and photoluminescence spectra, as reported in previous studies.18–21 And the solvatochromic shifts can be associated with the ICT process from the electron donor amino group to the electron acceptor carbonyl group.13,22,23 Dobek et al. conducted a study illustrating the crucial impact of the hydrogen bond between coumarin 102 and protic solvent on the deactivation process of the dye's excited state. Additionally, they highlighted that variations in the hydrogen bonding capacities among coumarins with similar structures could result in distinct photophysical properties across different states.14 Based on recent literature studies,24,25 there is compelling evidence supporting that coumarin dyes possess large conjugated systems and excellent electron-donating capabilities, thereby endowing them with significant potential in terms of non-linear optical (NLO) properties. Moreover, these dyes exhibit promising prospects as π-bridges in dye-sensitized solar cells, allowing for further enhancement of the cells' performance. These potential applications provide vast prospects for the utilization of coumarin dyes. And recently, Cong et al. investigated the excited state dynamics of C500 using steady-state and transient spectroscopy combing with time-dependent density functional theory calculations.26 The results from experimental data as well as theoretical analysis indicated that the change of hydrogen bond may facilitate the ICT process and induce the formation of a TICT state in MeOH. However, their discussion focused solely on the dynamics of the electronically excited state in solvents with different hydrogen bonding capacities, without addressing the influence of solvent polarity on the electronic excited state dynamics. Specifically, the impact of solvent polarity on the electronic excited state dynamics remains unclear due to the comparable polarities of Diox and MeOH. Thus, gaining a comprehensive insight into the excited state dynamics of 7-aminocoumarin dye molecules in distinct solvents with varying polarity scales is imperative.
Herein, we investigate the effect of solvent polarity and intermolecular hydrogen bonds on the excited state dynamics of C307, a typical 7-aminocoumarin dye. Our study involves a series of solvents, including Diox, MeOH, DMF, and DMSO, and utilizes steady-state/transient spectroscopy and theoretical calculations to analyze the system.27–32 The results obtained from steady-state/ultrafast transient absorption spectroscopy and steady-state photoluminescence spectroscopy reveal the significant impact of strengthened hydrogen bonds and solvent polarity on the excited state dynamics. A stable ICT state can be formed in nonpolar solvents (i.e. Diox), while a TICT state can be formed stably in weak polar solvents (i.e. MeOH) because of strengthened hydrogen bonds at the position B and A and weakened hydrogen bond at the position C, as well as in polar solvents (i.e. DMF, and DMSO) because of the large dipole moment of solvents. The lifetimes of the conversion from ICT to TICT state are presented according to transient absorption spectroscopy. Moreover, the frontier molecular orbitals of the C307 monomer and hydrogen bonded complexes are performed to evaluate the ICT process. Also, the potential energy surfaces (PESs) of C307 complexes are scanned to verify the formation of the TICT state which aligns with the results of experiments. The in-depth mechanistic understanding of excited state dynamics can assist in photophysical and photochemical applications for coumarin dyes.
2. Experiments
2.1. Experimental methods
C307 was purchased from Exciton Inc. (USA) and used without further purification. The Diox, MeOH, DMF, and DMSO solvents were spectrum pure quality reagents. The concentrations of C307 in Diox, MeOH, DMF, and DMSO were 80 μM. The samples were stored in a quartz cuvette with a light path length of 1 mm.
The steady-state absorption and photoluminescence (PL) spectra were acquired using the FluoroMax+ fluorescence spectrometer from Horiba, employing the principle of single photon counting. The excitation wavelength for the PL spectrum was determined by identifying the position of the strongest absorption peak. The excitation wavelengths used were based on the maximum absorption peak positions: 379 nm in Diox, 394 nm in MeOH, 396 nm in DMF, and 405 nm in DMSO.
The femtosecond time-resolved transient spectroscopy measurements were performed by a Transpec-FS pump–probe system (Intelligent Scientific System) in combination with an amplified Ti:sapphire laser system (Astrella, Coherent). The pump pulse centered at 400 nm, which was generated according to an optical parametric amplifier (TOPAS-Prime, Coherent) by 800 nm laser pump with a pulse duration of 35 fs from the Coherent Ti:sapphire regenerative amplifier. The white-light continuum probe pulses (400–650 nm) were generated by focusing the 800 nm beam onto a CaF2 crystal. The time delay between the pump and probe pulses was controlled by a motorized optical delay line extending to 4 ns. The temporal and spectral profiles of pump-induced absorbance change of the white-light continuum probe pulses were registered by a calibrated spectrometer (AvaSpec-ULS2048CL-EVO, Avantes). All the TA measurements were performed under an atmospheric environment.
2.2. Computational methods
The geometric optimizations of the C307 monomer and hydrogen-bonded complexes in the ground state were performed using DFT.33,34 The geometric optimizations, vertical excitation energies, fluorescence emission energies, oscillator strengths, and the PESs of the first electronically excited-state for C307 monomer and hydrogen-bonded complexes were investigated using TDDFT.35,36 Frequency checks were performed for all optimized geometries. The Becke's three-parameter and Lee–Yang–Parr hybrid functional (B3LYP) and the TZVP basis sets were employed for all calculations unless otherwise noted. The integral equation formalism version of the polarizable continuum model (IEFPCM) was used to consider the solvent effects.37,38 Based on the optimized molecular structure of the ground state, the orbital energy values and transition contributions of HOMO and the LUMO were calculated using the Mulliken population analysis. In addition, the PESs of the S1 state were constructed to search for the TICT state. Since CAM-B3LYP functional can accurately describe the overall PESs in systems involving different amounts of charge transfer, the range separated hybrid (RSH) DFT function CAM-B3LYP was employed to calculate the PESs.39 All calculations were performed using Gaussian 09.40
3. Results and discussion
3.1 Steady-state spectra and electronic excitation energy calculations
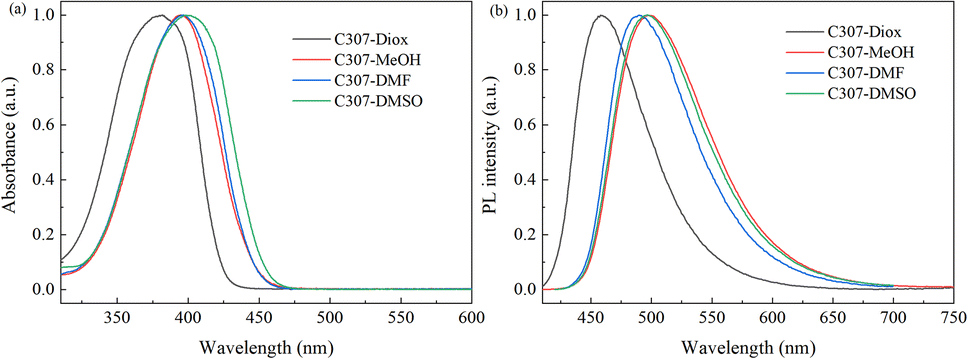
The normalized steady-state absorption and photoluminescence (PL) spectra of C307 in corresponding solvents are shown in Fig. 1. The absorption spectra reveal the broad bands centered at 381 nm, 394 nm, 395 nm, and 398 nm for C307 in Diox, MeOH, DMF, and DMSO, respectively. And the emission peaks of C307 in Diox, MeOH, DMF, and DMSO solvents locate at 458, 498, 490, and 496 nm, respectively. By contrast with each other, the Stokes shifts of C307 in MeOH, DMF, and DMSO are 104 nm, 95 nm, and 98 nm which are much larger than that of C307 in Diox (77 nm). The most plausible explanation for these large Stokes shifts is the redistribution of the probability density of the excited state wavefunction upon photoexcitation, which can be attributed to the presence of an electron donor (7-NHEt) and an electron acceptor (CF3) in C307 molecules. The molecular structure of C307 is depicted in Fig. 2.
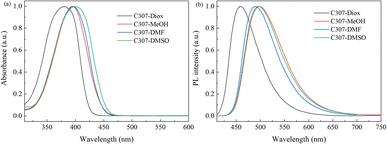 |
| | Fig. 1 Normalized steady-state absorption (a) and fluorescence (b) spectra of C307 in Diox, MeOH, DMF, and DMSO solvents. | |
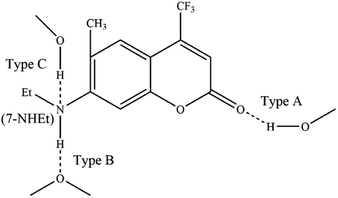 |
| | Fig. 2 Schematic representation of the C307 molecular structure and the hydrogen-bonded binding site. | |
Also, the electronic excitation energies and the corresponding oscillator strengths obtained from the TDDFT calculations on C307, C307–Diox, C307–MeOH, C307–DMF, and C307–DMSO in the low-lying electronic excited states are presented in Table 1. It can be seen that the S1 states of the hydrogen-bonded complexes correspond to the maximum oscillator strength, and that means the absorption maximums of the hydrogen-bonded complexes are all located in the S1 state. In addition, the fluorescence emission energies (nm) and corresponding oscillator strengths of the C307 and hydrogen-bonded complexes were also presented in Table 2. And the calculated results are commensurate with our steady-state absorption and PL spectra presented above.
Table 1 Calculated electronic excitation energies (nm) and corresponding oscillator strengths of the singlet excited states for the C307 monomer and hydrogen-bonded complexes with IEFPCM
| |
C307 |
C307–Diox |
C307–MeOH |
C307–DMF |
C307–DMSO |
| S1 |
349 (0.3486) |
382 (0.4556) |
395 (0.4456) |
399 (0.4642) |
402 (0.4632) |
| H → L 97.4% |
H → L 98.2% |
H → L 98.2% |
H → L 98.3% |
H → L 98.3% |
| S2 |
314 (0.0010) |
318 (0.0140) |
320 (0.0205) |
321 (0.0219) |
321 (0.0236) |
| S3 |
285 (0.0021) |
282 (0.0003) |
271 (0.0332) |
284 (0.0003) |
305 (0.0003) |
| S4 |
259 (0.0128) |
277 (0.0010) |
269 (0.0016) |
272 (0.0408) |
273 (0.0428) |
| S5 |
245 (0.0571) |
269 (0.0321) |
253 (0.1605) |
269 (0.0017) |
268 (0.0015) |
| S6 |
235 (0.0938) |
249 (0.1389) |
242 (0.0958) |
258 (0.0027) |
255 (0.1650) |
Table 2 Calculated fluorescence emission energies (nm) and corresponding oscillator strengths of the C307 monomer and hydrogen-bonded complexes with IEFPCM
| |
C307 |
C307–Diox |
C307–MeOH |
C307–DMF |
C307–DMSO |
| S1 |
424 (0.2534) |
459 (0.3465) |
487 (0.5337) |
492 (0.5325) |
495 (0.5334) |
| S2 |
363 (0.0299) |
363 (0.0510) |
359 (0.0514) |
360 (0.0533) |
361 (0.0565) |
| S3 |
302 (0.0034) |
302 (0.0002) |
286 (0.0440) |
302 (0.0006) |
324 (0.0000) |
3.2 Transient absorption spectra
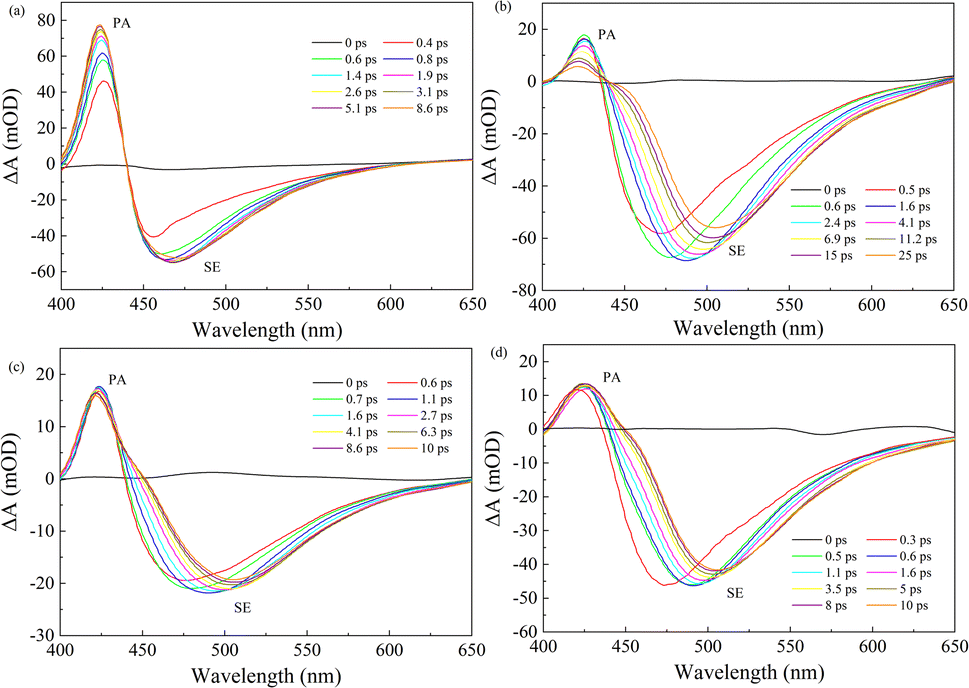
To further explore the effect of different solvents on the relaxation dynamics of the electronic excited state, the femtosecond transient absorption (fs-TA) measurements of C307 in Diox, MeOH, DMF, and DMSO solvents were carried out by photoexcitation at 400 nm. Fig. 3 displays the TA spectra of C307 in Diox, MeOH, DMF, and DMSO within 400–650 nm regions at the indicated time delays. The TA positive signals can be assigned to photo-induced absorption (PA), while the negative signals can be due to stimulated emission (SE). As compared to the C307–Diox complex, we observed a significant broadening of the negative signal band and a substantial red shift of the SE peaks in the C307–MeOH, C307–DMF, and C307–DMSO complexes. These findings suggest the presence of intermolecular reorganization or an intermediate state arising from the hydrogen-bonded interactions between C307 and the solvent molecules.
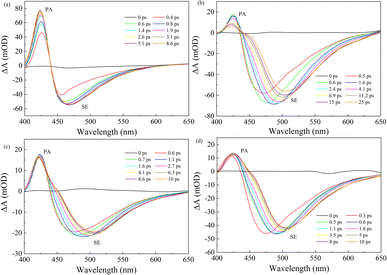 |
| | Fig. 3 Transient absorption spectra of C307 in Diox (a), MeOH (b), DMF (c), and DMSO (d) solvents at indicated delay times. | |
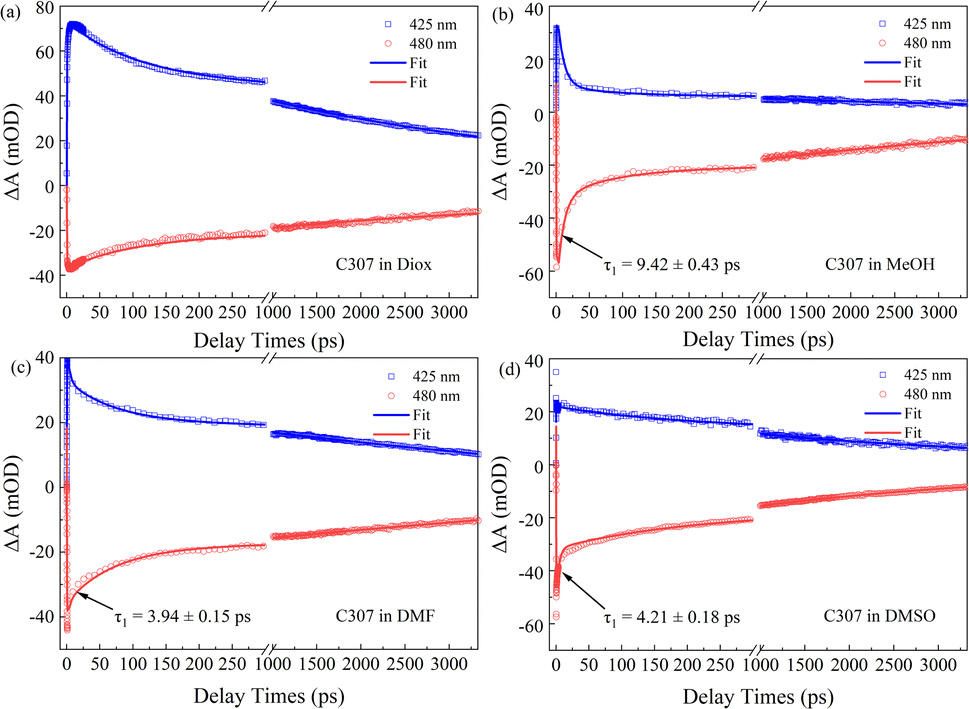
Fig. 4 presents the TA kinetic traces recorded at 425 nm and 480 nm for all the hydrogen-bonded complexes. Using a global analysis, the TA kinetics can be well fitted by a triexponential function for C307 in Diox: τrise ∼ 0.44 ps, τ2 ∼ 103.48 ps, and τ3 > 3.5 ns due to the limit of the delay line length. As is well known, τrise can be ascribed to a rapid formation of the ICT state, and the following slower time constants can be ascribed to the solvation process and fluorescence emission process, respectively.13 While the TA kinetics can be fitted by four-time constants for C307 in MeOH, DMF, and DMSO, as shown in Table 3, respectively. Obviously, an additional early time component emerges: τ1 ∼ 9.42 ps for the C307–MeOH complex; τ1 ∼ 3.94 ps for the C307–DMF complex, and τ1 ∼ 4.21 ps for the C307–DMSO complex. According to the results above, the additional time can be associated with the red-shift of PL peaks, which implied a transient ICT state converted into the twisted intramolecular charge transfer state of C307 in polar solvents. The results are consistent with the discussion below by scanning the first electronic excited state of the molecules.
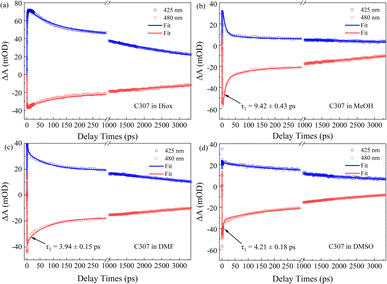 |
| | Fig. 4 The TA kinetic traces and fitting curves of C307 in Diox (a), MeOH (b), DMF (c), and DMSO (d) at wavelength 425 nm and 480 nm. | |
Table 3 Lifetimes of C307 in Diox, MeOH, DMF, and DMSO obtained by global fitting
| Solute |
Solvent |
τrise (ps) |
τ1 (ps) |
τ2 (ps) |
τ3 (ns) |
| C307 |
Diox |
0.44 ± 0.01 |
— |
103.48 ± 2.75 |
>3.5 |
| MeOH |
0.37 ± 0.01 |
9.42 ± 0.43 |
74.06 ± 11.95 |
>3.5 |
| DMF |
0.31 ± 0.01 |
3.94 ± 0.15 |
72.60 ± 9.15 |
>3.5 |
| DMSO |
0.23 ± 0.01 |
4.21 ± 0.18 |
178.88 ± 24.23 |
>3.5 |
Moreover, an obvious red shift accounting for the existence of TICT states within early 20 ps time windows can be shown for C307 in the polar solvents including MeOH, DMF, and DMSO, in contrast to C307 in nonpolar solvent such as Diox. Essentially, these significant spectral differences can be associated with the hydrogen bonding interactions between C307 and solvents. Thus, the following quantum chemical calculations will reveal the underlying factors responsible for the aforementioned phenomenon.
3.3 Quantum chemical calculation
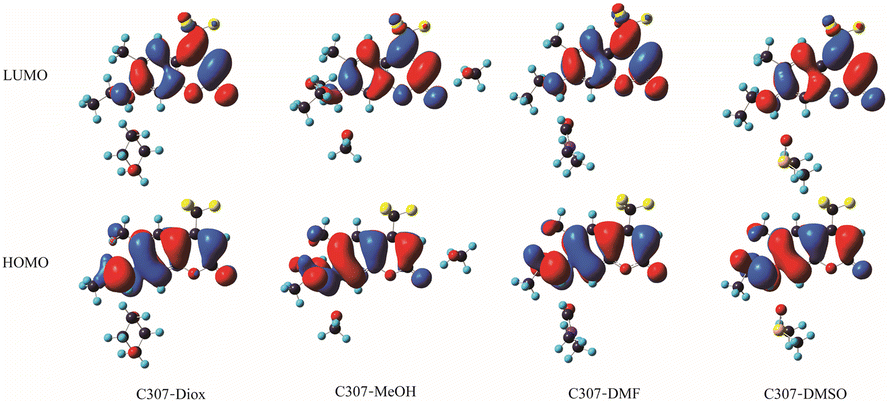
Based on Table 1 presented above, one can see that the S1 state which exhibits the largest oscillator strength corresponds to the orbital transition from the highest occupied orbital (HOMO) to the lowest unoccupied orbital (LUMO). To gain further insight into the ICT process, the frontier molecular orbitals (FMOs) of all the hydrogen-bonded complexes are exhibited in Fig. 5. The electrons are mainly distributed on the left 7-NHEt group in the HOMO and redistributed on the CF3 group in the LUMO, which indicates that ICT occurs from the 7-NHEt group to the CF3 group in C307 complexes once the electrons populated from HOMO to LUMO under photoexcitation. These findings are in excellent agreement with the results obtained from our analyses by the steady-state/transient spectroscopy.
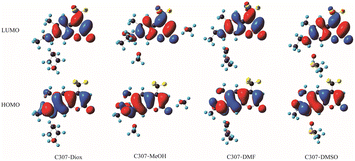 |
| | Fig. 5 Frontier molecular orbitals in the hydrogen-bonded complexes C307–Diox, C307–MeOH, C307–DMF, and C307–DMSO. | |
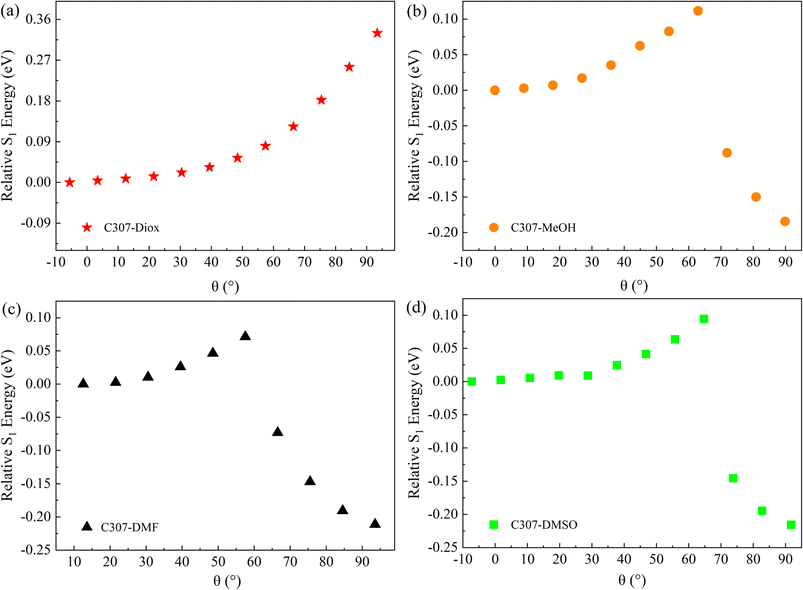
Besides, to explore the formation mechanism of TICT states, the CAM-B3LYP functional and TZVP basis sets combining with the SMD solvent model are utilized to calculate the potential energy surfaces of C307 in different solvents. The figures of relative energy of the S1 state for the hydrogen-bonded complexes versus the dihedral angle (θ) of the 7-NHEt group relative to the benzene ring plane are shown in Fig. 6. The relative S1 energy reaches its maximum at a certain angle and reaches its minimum as the dihedral angle approaches 90° for C307 in MeOH, DMF, and DMSO. However, the relative energy of the S1 state of C307 in Diox increases monotonously with the increment of the dihedral angle. The results imply that the formation of TICT states for C307 is favored in weak polar solvents (i.e. MeOH) and polar solvents (i.e. DMF, and DMSO), which is consistent with our experimental results.
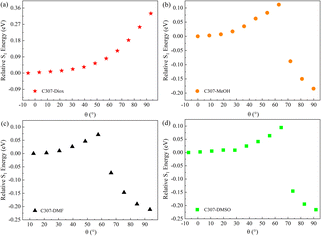 |
| | Fig. 6 The potential energy surfaces of C307 in Diox (a), MeOH (b), DMF (c), and DMSO (d) solvents at the S1 state. | |
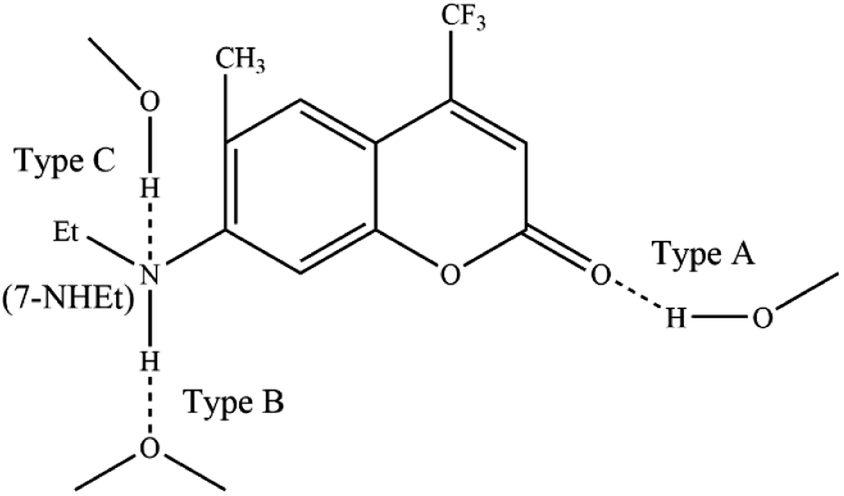
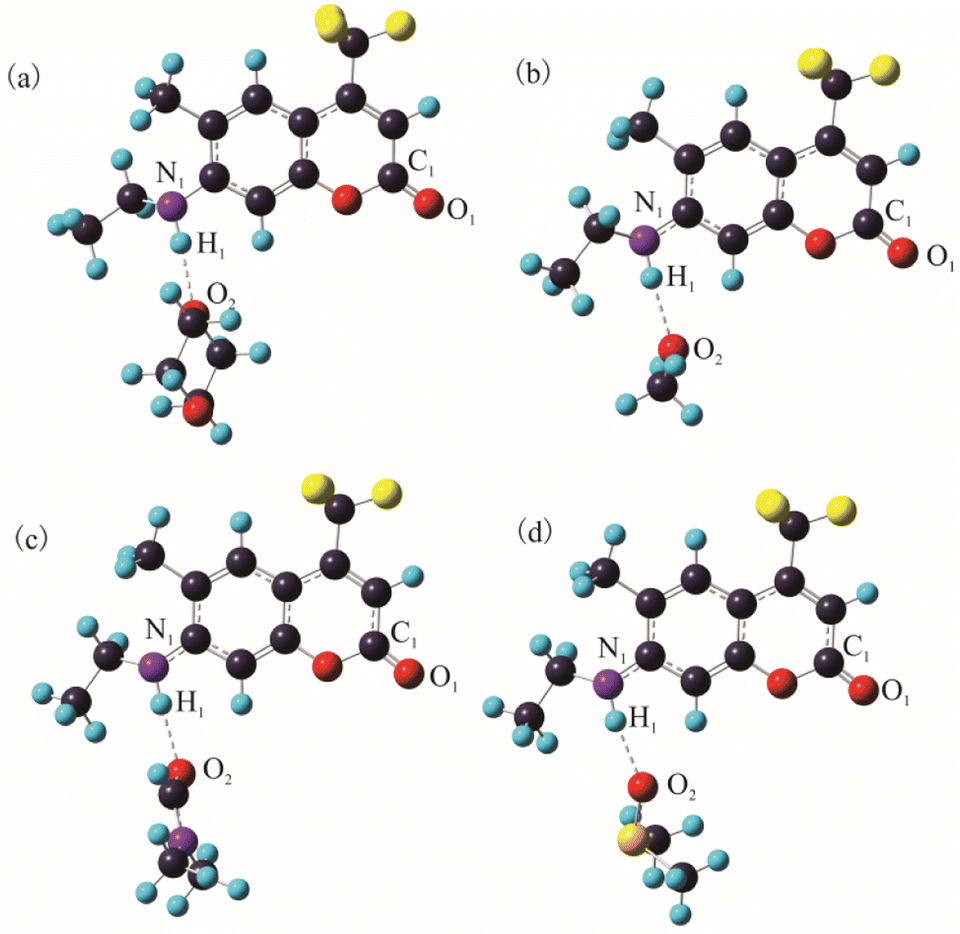
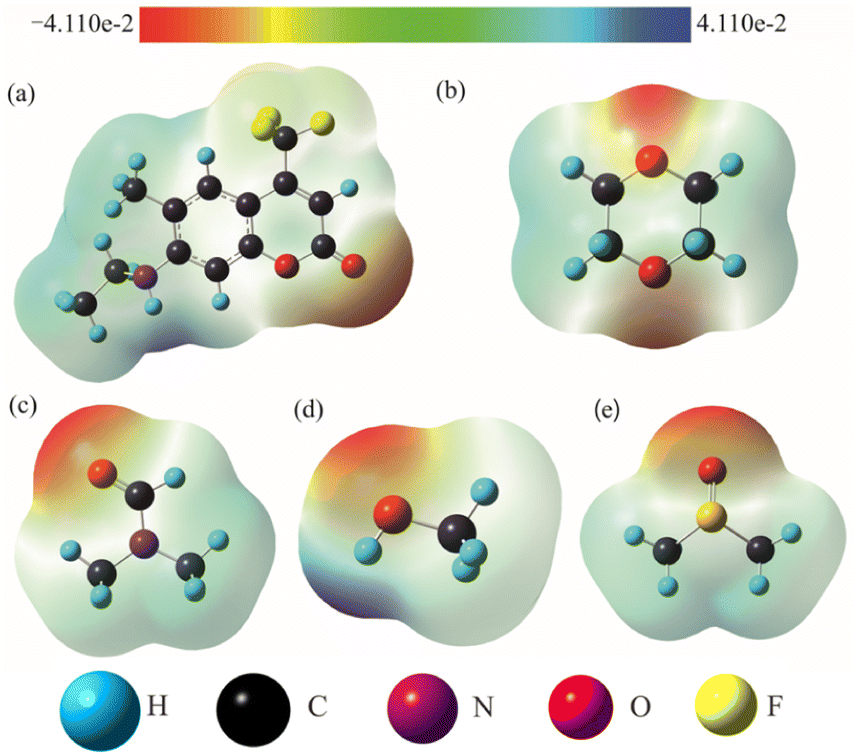
As is well known, hydrogen bond as a key factor can significantly affect the formation of the TICT state.26 In Fig. 2, the C307 molecule exhibits three potential positions at which can form hydrogen bonds as follows: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–O through carbonyl oxygen (type A); N–H⋯O through amino hydrogen (type B); and N⋯H–O through amino nitrogen (type C). Consequently, hydrogen bonds can be formed at position B between C307 and solvents including Diox, DMF, and DMSO, while three kinds of hydrogen bonds can be formed at all positions for C307 in MeOH. The optimized geometric configurations in the ground state of all the hydrogen-bonded complexes are shown in Fig. 7. The electrostatic potential (ESP) analysis provides an initial assessment of the reactive sites within a molecule, including the specific regions where intermolecular hydrogen bonds can form and their corresponding interactions. Fig. 8 presents the electrostatic potential diagram, which serves to further validate the previously discussed findings. And the lengths of the intermolecular bonds in the ground state and excited state for all the complexes are exhibited in Table 4. In the ground state, the lengths of hydrogen bonds at position B are as follows: 2.045 Å, 1.998 Å, 1.973 Å, and 2.045 Å, while those in the ICT state are 1.790 Å, 1.860 Å, 1.818 Å, and 1.873 Å for C307 in Diox, MeOH, DMF, and DMSO, respectively. It indicates that the hydrogen bonds N–H⋯O are strengthened in the ICT state. The excited state hydrogen bond dynamics can also be observed from the infrared spectrum of C307 monomer and complexes, as shown in Table 5. It is worth noting that three kinds of hydrogen bonds can be formed for C307 in MeOH at all positions, where the length of the hydrogen bond CO⋯H–O at position A was 1.892 Å in the ground state and 1.810 Å in the ICT state, while the length of hydrogen bond N⋯H–O at position C was calculated to be 1.965 Å in the ground state and 3.560 Å in the ICT state. Therefore, a possible explanation for the formation of the TICT state in MeOH is that not only strengthened hydrogen bonds at position B but also the hydrogen bonds at position A and position C play a significant role. And the detailed reasons accounting for TICT in MeOH are as follows: the electron density on the 7-NHEt group decreases under the photoexcitation, and the hydrogen bond at position A/B strengthens accompanied by the weakened hydrogen bond at position C, which induce the twisting of 7-NHEt group leading to the formation of TICT state. The findings for C307 in MeOH are consistent with the reports for C500 in MeOH by Cong et al.26 However, they suggested that the change of hydrogen bond plays a vital role in the formation of the TICT state due to the similar polarity of MeOH in contrast to Diox. Here, the existence of the TICT state for C307 in DMF and DMSO allows us to confidently deduce that the polarity of solvents has a pronounced influence on the formation of the TICT state in addition to the strengthened hydrogen bond, because the TICT feature cannot occur for C307 in nonpolar Diox. The polarity of the solvent not only plays a significant role in stabilizing the charge-separated TICT state but also influences the ICT to TICT mechanism by modifying the barrier height. In the case of C307, it needs to overcome barrier heights of 0.11, 0.07, and 0.09 eV to access the TICT state in MeOH, DMF, and DMSO solvents, respectively. However, the barrier height for C307 in Diox continues to increase, making it impossible to surmount the energy barrier and form the TICT state. These observations align with the results obtained from transient absorption spectra, providing further evidence for the influence of solvent polarity on the excited-state dynamics of C307.41,42
O⋯H–O through carbonyl oxygen (type A); N–H⋯O through amino hydrogen (type B); and N⋯H–O through amino nitrogen (type C). Consequently, hydrogen bonds can be formed at position B between C307 and solvents including Diox, DMF, and DMSO, while three kinds of hydrogen bonds can be formed at all positions for C307 in MeOH. The optimized geometric configurations in the ground state of all the hydrogen-bonded complexes are shown in Fig. 7. The electrostatic potential (ESP) analysis provides an initial assessment of the reactive sites within a molecule, including the specific regions where intermolecular hydrogen bonds can form and their corresponding interactions. Fig. 8 presents the electrostatic potential diagram, which serves to further validate the previously discussed findings. And the lengths of the intermolecular bonds in the ground state and excited state for all the complexes are exhibited in Table 4. In the ground state, the lengths of hydrogen bonds at position B are as follows: 2.045 Å, 1.998 Å, 1.973 Å, and 2.045 Å, while those in the ICT state are 1.790 Å, 1.860 Å, 1.818 Å, and 1.873 Å for C307 in Diox, MeOH, DMF, and DMSO, respectively. It indicates that the hydrogen bonds N–H⋯O are strengthened in the ICT state. The excited state hydrogen bond dynamics can also be observed from the infrared spectrum of C307 monomer and complexes, as shown in Table 5. It is worth noting that three kinds of hydrogen bonds can be formed for C307 in MeOH at all positions, where the length of the hydrogen bond CO⋯H–O at position A was 1.892 Å in the ground state and 1.810 Å in the ICT state, while the length of hydrogen bond N⋯H–O at position C was calculated to be 1.965 Å in the ground state and 3.560 Å in the ICT state. Therefore, a possible explanation for the formation of the TICT state in MeOH is that not only strengthened hydrogen bonds at position B but also the hydrogen bonds at position A and position C play a significant role. And the detailed reasons accounting for TICT in MeOH are as follows: the electron density on the 7-NHEt group decreases under the photoexcitation, and the hydrogen bond at position A/B strengthens accompanied by the weakened hydrogen bond at position C, which induce the twisting of 7-NHEt group leading to the formation of TICT state. The findings for C307 in MeOH are consistent with the reports for C500 in MeOH by Cong et al.26 However, they suggested that the change of hydrogen bond plays a vital role in the formation of the TICT state due to the similar polarity of MeOH in contrast to Diox. Here, the existence of the TICT state for C307 in DMF and DMSO allows us to confidently deduce that the polarity of solvents has a pronounced influence on the formation of the TICT state in addition to the strengthened hydrogen bond, because the TICT feature cannot occur for C307 in nonpolar Diox. The polarity of the solvent not only plays a significant role in stabilizing the charge-separated TICT state but also influences the ICT to TICT mechanism by modifying the barrier height. In the case of C307, it needs to overcome barrier heights of 0.11, 0.07, and 0.09 eV to access the TICT state in MeOH, DMF, and DMSO solvents, respectively. However, the barrier height for C307 in Diox continues to increase, making it impossible to surmount the energy barrier and form the TICT state. These observations align with the results obtained from transient absorption spectra, providing further evidence for the influence of solvent polarity on the excited-state dynamics of C307.41,42
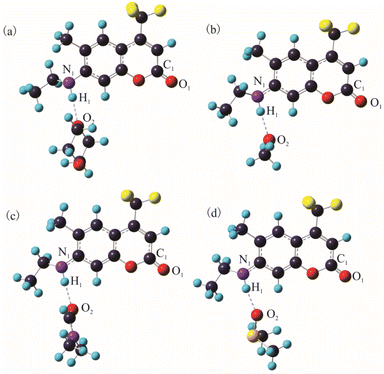 |
| | Fig. 7 The optimized hydrogen-bond complex C307–Diox (a), C307–MeOH (b), C307–DMF (c), and C307–DMSO (d) in the S0 state at the B3LYP/TZVP/IEF-PCM level. | |
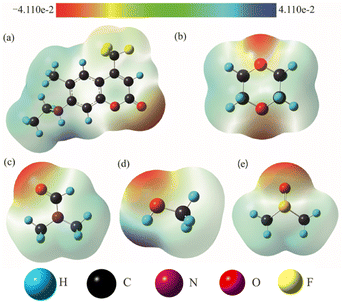 |
| | Fig. 8 The ESP maps of C307 (a), Diox (b), DMF (c), MeOH (d), and DMSO (e) molecules. | |
Table 4 Bond lengths (Å) of the ground and excited states of C307 monomer and hydrogen-bonded complexes with four solvents
| Molecule |
C307 |
C307–Diox |
C307–MeOH |
C307–DMF |
C307–DMSO |
| S0 |
S1 |
S0 |
S1 |
S0 |
S1 |
S0 |
S1 |
S0 |
S1 |
| C1–O1 |
1.202 |
1.212 |
1.208 |
1.221 |
1.214 |
1.230 |
1.214 |
1.230 |
1.214 |
1.231 |
| N1–H1 |
1.012 |
1.011 |
1.016 |
1.023 |
1.017 |
1.026 |
1.019 |
1.029 |
1.021 |
1.032 |
| Type A |
— |
— |
— |
— |
1.892 |
1.810 |
— |
— |
— |
— |
| Type B |
— |
— |
2.089 |
1.915 |
2.035 |
1.891 |
2.003 |
1.844 |
1.955 |
1.802 |
| Type C |
— |
— |
— |
— |
1.965 |
3.560 |
— |
— |
— |
— |
Table 5 Infrared spectra of hydrogen bond formation sites (cm−1) of the C307 monomer and hydrogen-bonded complexes in four solvents in S0 state and S1 state
| |
Parameters |
C307 |
C307–Diox |
C307–MeOH |
C307–DMF |
C307–DMSO |
| S0 |
C1–O1 |
1805 |
1772 |
1737 |
1736 |
1734 |
| N1–H1 |
3543 |
3484 |
3450 |
3418 |
3385 |
| S1 |
C1–O1 |
1781 |
1753 |
1716 |
1716 |
1715 |
| N1–H1 |
3580 |
3364 |
3318 |
3245 |
3187 |
3.4 Different excited state deactivation mechanisms of C307 hydrogen-bonded complexes
The deactivation mechanisms of the excited state for C307 in both nonpolar and polar solvents have been proposed, and schematic diagrams illustrating these mechanisms are exhibited in Fig. 9. Through analysis of the steady-state/transient absorption spectra combining with PL spectra, significant hydrogen bonding effects are observed in different solvents. For C307 in Diox, the maximum PL peak centered at 458 nm is attributed to the formation of an ICT state within a lifetime of approximately 0.44 ps, as illustrated in the left panel of Fig. 9. However, the faster formation lifetimes of ICT states are observed: τrise ∼ 0.37 ps for C307 in MeOH; τrise ∼ 0.31 ps for C307 in DMF; and τrise ∼ 0.23 ps for C307 in DMSO. Once in the ICT state, the electron density decreases on the 7-NHEt group with the increase in the strength of hydrogen bond N–H⋯O, leading to the twisting of the 7-NHEt group and the formation of TICT state in weak polar and polar solvents. The formation lifetimes of TICT states are ∼9.42 ps, ∼3.94 ps, and 4.21 ps in C307–MeOH, C307–DMF, and C307–DMSO complexes, respectively. Fluorescence is emitted immediately after the TICT state, and the duration of this process is typically in the nanosecond range. The specific mechanism is depicted in the right panel of Fig. 9.
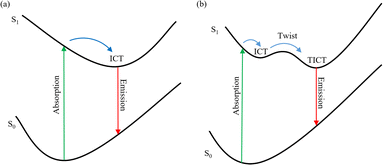 |
| | Fig. 9 Schematic diagrams illustrating the excited-state deactivation mechanisms for C307 in nonpolar (a), and polar (b) solvents. | |
4. Conclusions
In conclusion, we have interrogated the effect of hydrogen bond and the polarity of solvent on the behavior of electronic excited state in C307 systems which are dissolved in the distinct solvents, employing steady-state photoluminescence spectroscopy and steady-state/ultrafast transient absorption spectroscopy combining with quantum chemical calculations. The results obtained in this study clearly demonstrate the crucial role of solvent polarity and intermolecular hydrogen bond strength in determining the dynamics of the electronic excited state. Specifically, the formation of the TICT state was observed in solvents such as MeOH, DMF, and DMSO, where the strengthened hydrogen bonding interactions and inherent solvent polarity promote this phenomenon. In contrast, in the nonpolar solvent Diox, the strengthened hydrogen bonding occurs exclusively at position B, resulting in a stable ICT state with a lifetime exceeding 3.5 ns. Furthermore, global fitting analysis of the TA experiments revealed additional time constants in the picosecond timescales, indicating the conversion from an intermediate ICT state to a stable TICT state in polar solvents. These experimental findings are in line with the results obtained from TDDFT calculations, providing further support to the proposed deactivation mechanisms of the excited state. The comprehensive understanding gained from the combination of experimental and theoretical results strongly validates the proposed deactivation mechanisms of the excited state. This work significantly contributes to the field by providing insightful knowledge into the photochemistry and photophysics of the C307 system in solvents with varying polarity and hydrogen bonding characteristics, with a particular emphasis on the dynamics of the excited state. Overall, this research deepens our understanding of the excited-state dynamics of C307 and offers valuable insights into the photochemical and photophysical properties of this system in solvents exhibiting distinct polarity and hydrogen bonding behavior.
Author contributions
Jing Ge: conceptualization, formal analysis, supervision, writing – original draft, review & editing. Xue-Dong Zhang: methodology, data curation, software, visualization, writing – original draft. Zhi-Biao Li: methodology, investigation. Bing-Qian Xue: investigation, visualization. Xi-Lin Bai: methodology, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Project supported by the National Natural Science Foundation of China (NSFC) (No. 21903050), Shanxi Scholarship Council of China Scientific (No. 2021-092), and Fund Program for the Scientific Activities of Selected Returned Overseas Professionals in Shanxi Province (20220024).
Notes and references
- Y. Rout, C. Montanari, E. Pasciucco, R. Misra and B. Carlotti, J. Am. Chem. Soc., 2021, 143, 9933–9943 CrossRef CAS PubMed.
- A. M. El-Zohry, E. A. Orabi, M. Karlsson and B. Zietz, J. Phys. Chem. A, 2021, 125, 2885–2894 CrossRef CAS PubMed.
- P. Xing, Y. Niu, R. Mu, Z. Wang, D. Xie, H. Li, L. Dong and C. Wang, Nat. Commun., 2020, 11, 1573 CrossRef CAS.
- P. Wiggins, J. A. G. Williams and D. J. Tozer, J. Chem. Phys., 2009, 131, 091101 CrossRef.
- B. Abulimiti, R. S. Zhu, J. Y. Long, Y. Q. Xu, Y. Z. Liu, A. Y. Ghazal, M. H. Yang and B. Zhang, J. Chem. Phys., 2011, 134, 13274–13279 CrossRef.
- P. Zhou, P. Song, J. Liu, K. L. Han and G. He, Phys. Chem. Chem. Phys., 2009, 11, 9440–9449 RSC.
- C. Wang, Q. Qiao, W. Chi, J. Chen, W. Liu, D. Tan, S. McKechnie, D. Lyu, X. F. Jiang and W. Zhou, et al., Angew. Chem., Int. Ed., 2020, 59, 10160–10172 CrossRef CAS PubMed.
- X. Liu, Q. Qiao, W. Tian, W. Liu, J. Chen, M. J. Lang and Z. Xu, J. Am. Chem. Soc., 2016, 138, 6960–6963 CrossRef CAS.
- P. Verma and H. Pal, J. Phys. Chem. A, 2012, 116, 4473–4484 CrossRef CAS PubMed.
- J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC.
- F. Han, W. Liu, L. Zhu, Y. Wang and C. Fang, J. Mater. Chem. C, 2016, 4, 2954–2963 RSC.
- J. Ge, X. D. Zhang, Y. Peng and X. L. Bai, RSC Adv., 2023, 13, 4924–4931 RSC.
- D. De, M. Sajjan, J. Narayanan, J. R. Bellare and A. Datta, J. Phys. Chem. B, 2013, 117, 2106–2112 CrossRef CAS PubMed.
- K. Dobek, J. Karolczak and J. Kubicki, Dyes Pigm., 2014, 100, 222–231 CrossRef CAS.
- R. K. Venkatraman and A. J. Orr-Ewing, J. Am. Chem. Soc., 2019, 141, 15222–15229 CrossRef CAS PubMed.
- Y. Z. Liu, B. F. Tang, H. Shen, S. Zhang and B. Zhang, Opt. Express, 2010, 18, 5791 CrossRef CAS PubMed.
- S. Halder, R. Aggrawal, V. K. Aswal, D. Ray and S. K. Saha, J. Mol. Liq., 2021, 332, 114532 CrossRef.
- Y. Q. Sun, M. Chen, J. Liu, X. Lv, J. F. Li and W. Guo, Chem. Commun., 2011, 47, 11029–11031 RSC.
- K. Das, B. Jain and H. S. Patel, J. Phys. Chem. A, 2006, 110, 1698–1704 CrossRef CAS PubMed.
- S. Nad and H. Pal, J. Phys. Chem. A, 2003, 107, 501–507 CrossRef CAS.
- Y. Li, C. Sun, J. Han, Q. Zhou, B. Cao, H. Yin and Y. Shi, J. Lumin., 2020, 221, 670–675 Search PubMed.
- R. K. Venkatraman, S. Kayal, A. Barak, A. J. Orr-Ewing and S. Umapathy, J. Phys. Chem. Lett., 2018, 9, 1642–1648 CrossRef CAS PubMed.
- Y. Li, B. Xu, P. Song, F. Ma and M. Sun, J. Phys. Chem. C, 2017, 121, 12546–12561 CrossRef CAS.
- N. N. Ayarea, S. Sharmaa, K. K. Sonigara, J. Prasad, S. S. Soni and N. Sekar, J. Photochem. Photobiol., A, 2020, 394, 112466 CrossRef.
- M. M. Jadhav, J. V. Vaghasiya, D. S. Patil, S. S. Soni and N. Sekar, New J. Chem., 2018, 42, 5267–5275 RSC.
- L. Cong, H. Yin, Y. Shi, M. Jin and D. J. Ding, RSC Adv., 2015, 5, 1205–1212 RSC.
- A. K. Singh, G. Ramakrishna, H. N. Ghosh and D. K. Palit, J. Phys. Chem. A, 2004, 108, 2583–2597 CrossRef CAS.
- N. Barman and K. Sahu, RSC Adv., 2014, 4, 58299–58306 RSC.
- M. A. Ramegowda, Spectrochim. Acta, Part A, 2015, 137, 99 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Q. Zhang, H. Zheng, Z. Geng, S. Jiang, J. Ge, K. Fan, S. Duan, Y. Chen, X. Wang and Y. Luo, J. Am. Chem. Soc., 2013, 135, 12468–12474 CrossRef CAS PubMed.
- D. Hait and M. Head-Gordon, J. Phys. Chem. Lett., 2021, 12, 4517–4529 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Comput. Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- X. Zhao, Y. Liu, L. Zhou, Y. Li and M. Chen, J. Lumin., 2010, 130, 1431–1436 CrossRef CAS.
- R. D. Senanayake and D. B. Lingerfelt, J. Phys. Chem. C, 2019, 123, 14734–14745 CrossRef CAS.
- D. Yang, Y. Yang and Y. Liu, Comput. Theor. Chem., 2012, 997, 42–48 CrossRef CAS.
- R. Büchner, V. Da-Cruz, M. Fondell, E. J. Mascarenhas, A. Pietzsch and A. Föhlisch, J. Phys. Chem. B, 2021, 125, 2372–2379 CrossRef PubMed.
- Y. F. Liu, D. P. Yang, D. H. Shi and J. F. Sun, J. Comput. Chem., 2011, 32, 3475–3484 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- J. S. Yang, C. K. Lin, A. M. Lahoti, C. K. Tseng, Y. H. Liu, G. H. Lee and S. M. Peng, J. Phys. Chem. A, 2009, 143, 4868–4877 CrossRef PubMed.
- F. J. Liu, S. Q. Ma, Z. Y. Lu, A. Nangia, M. Duan, Y. M. Yu, G. C. Xu, Y. Mei, M. Bietti and K. N. Houk, J. Am. Chem. Soc., 2022, 144, 6802–6812 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Zhi-Biao Li,
Bing-Qian Xue and
Xi-Lin Bai*
,
Zhi-Biao Li,
Bing-Qian Xue and
Xi-Lin Bai*