
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrazinosulfonylation of aryl electrophiles: a straightforward approach for the synthesis of aryl N-aminosulfonamides
Chou-Yi Hsu a,
Ahmed Kareem Obaid Aldulaimi*b,
Mustafa humam samic,
Hala Bahird,
Ayat Hussein Adhabe and
Shelesh Krishna Saraswatf
a,
Ahmed Kareem Obaid Aldulaimi*b,
Mustafa humam samic,
Hala Bahird,
Ayat Hussein Adhabe and
Shelesh Krishna Saraswatf
aDepartment of Pharmacy, Chia Nan University of Pharmacy and Science, Tainan, Taiwan
bCollege of Food Sciences, Al-Qasim Green University, Babylon, Iraq. E-mail: ahmedaldulaimi1@gmail.com
cDepartment of Pharmacy, Al-Noor University College, Nineveh, Iraq
dMedical Technical College, Al-Farahidi University, Iraq
eDepartment of Pharmacy, Al-Zahrawi University College, Karbala, Iraq
fDepartment of ECE, GLA University, Mathura-281406, India
First published on 20th June 2023
Abstract
In recent years, the direct hydrazinosulfonylation of aryl electrophiles with SO2 and hydrazines has emerged as an efficient and versatile method for the synthesis of aryl N-aminosulfonamides. This method has the advantages of being operationally simple and requiring only readily available starting materials. This review article is an attempt to survey literature describing the preparation of aryl N-aminosulfonamides through the direct hydrazinosulfonylation of aryl electrophiles with SO2 and hydrazines, with special attention paid to the mechanistic features of the reactions. It can be used as a guide for chemists to apply the best hydrazinosulfonylation conditions in their work or serve as inspiration for future research related to the topic.
1. Introduction
Organosulfur compounds, which are organic molecules containing carbon–sulfur bonds, have played an important role in various fields, such as pharmaceuticals,1 agrochemicals,2 and materials science.3 Sulfonamides, the largest class of sulfur-containing motifs in pharmaceuticals, have up to 72 FDA-approved medicines.4 It is interesting to note that 25% of FDA-approved sulfur-containing drugs are derived from sulfonamide entities and are used to treat various types of diseases, such as HIV infection, cancer, glaucoma, edema, asthma, thrombosis, chronic prostatitis, seizures, and other conditions.4,5 The primary sulfonamides are the most important class of inhibitors acting on the metalloenzyme carbonic anhydrase (EC 4.2.1.1).6 Recently, the medicinal chemistry community has shown significant interest in N-amino-substituted sulfonamides, also known as N-aminosulfonamides or sulfonohydrazides, due to their diverse range of biological activities, including anticancer, antileishmanial, antifungal, and antibacterial activities.7 In addition to their biological importance, N-aminosulfonamides have been widely used as versatile building blocks in the assembly of various value-added chemicals (Fig. 1),8 such as sulfones, biaryls, 1,2-diphenylethenes, tetraaz-2-enes, benzo[b]thiophene 1,1-dioxides, and ((arylsulfonyl)methyl)diazene derivatives, because of their diverse reaction patterns.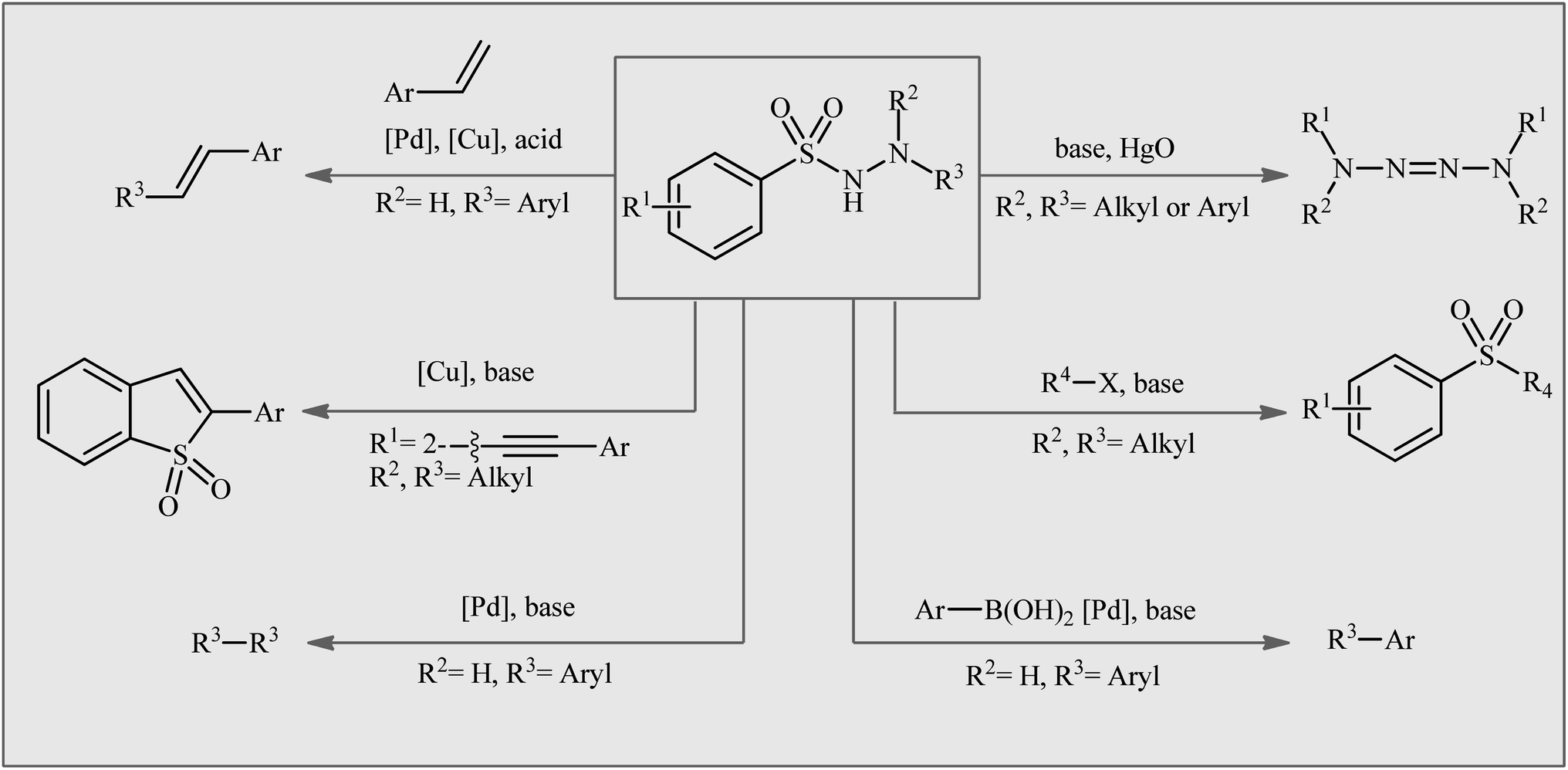 | ||
| Fig. 1 Selected examples of synthetic products from aryl N-aminosulfonamides. | ||
The traditional method for synthesizing N-aminosulfonamides is through the direct sulfonylation of hydrazines using sulfonyl chlorides.9 However, the irritating nature and moisture sensitivity of sulfonyl chlorides, as well as the production of a stoichiometric halogen waste stream, are considered serious drawbacks of this procedure. An alternative protocol involves the reaction of azodicarboxylate derivatives with various sulfonylating agents such as sulfonyl chlorides, sulfinates, sulfinic acid, and thiols.10 However, this method suffers from certain disadvantages, such as the shock sensitivity, thermal instability, and/or toxicity of azodicarboxylates,11 as well as inherent drawbacks of some sulfonylating agents, such as the poor storage stability and unpleasant odor of thiols. Therefore, the development of a convenient, efficient, and environmentally benign synthetic method for synthesizing these compounds from safe, simple, and easily available starting materials is still attractive and desirable.
To overcome the limitations mentioned above, a new method has recently emerged for the direct hydrazinosulfonylation of aryl electrophiles with sulfur dioxide (SO2) and hydrazines. This strategy, illustrated in Scheme 1, offers a versatile and powerful approach for forming C–SO2–N linkages. One of the key advantages of this method is that it eliminates the need for pre-prepared, unstable, irritating, and/or toxic sulfonylating reagents. This results in improved efficiency, atom and step economy, and minimized chemical waste. Although several reviews have covered different aspects of this topic,12 a comprehensive review of this exciting research area has not been published to date. This review aims to present the information in a more straightforward manner, making it more accessible to non-specialists.
 | ||
| Scheme 1 Direct hydrazinosulfonylation of aryl electrophiles. | ||
2. Hydrazinosulfonylation of aryl halides
The first report on the synthesis of aryl N-aminosulfonamides via the three-component coupling of aryl halides, hydrazines, and sulfur dioxide was published in a 2010 paper by Willis et al.13 They disclosed that using 10 mol% Pd(OAc)2 and 20 mol% PtBu3·HBF4, along with the base DABCO (1,4-diazabicyclo[2.2.2]octane), enabled the direct aminosulfonylation of (hetero)aryl iodides 1 with hydrazines 2 and DABCO·(SO2)2, abbreviated as DABSO, to yield (hetero)aryl N-aminosulfonamides 3 in modest to excellent yields (Scheme 2). The results showed that aryl iodides with electron-donating substituents (e.g., Me, OMe, OH) produced better yields than those with electron-withdrawing groups (e.g., CO2Me, CF3). For slow-reacting substrates (i.e., thienyl and electron-deficient aryl iodides), the use of 1.1 equivalents of DABSO without extra DABCO was more efficient. Using the same conditions as for electron-deficient aryl iodides, an (E)-configured alkenyl iodide was also employed, resulting in good isolated yield of the corresponding alkenyl aminosulfonamide. It is worth noting that this groundbreaking work not only demonstrated the first example of three-component coupling of aryl electrophiles, hydrazines, and SO2 but also introduced DABSO as a bench-stable and harmless source of gaseous SO2.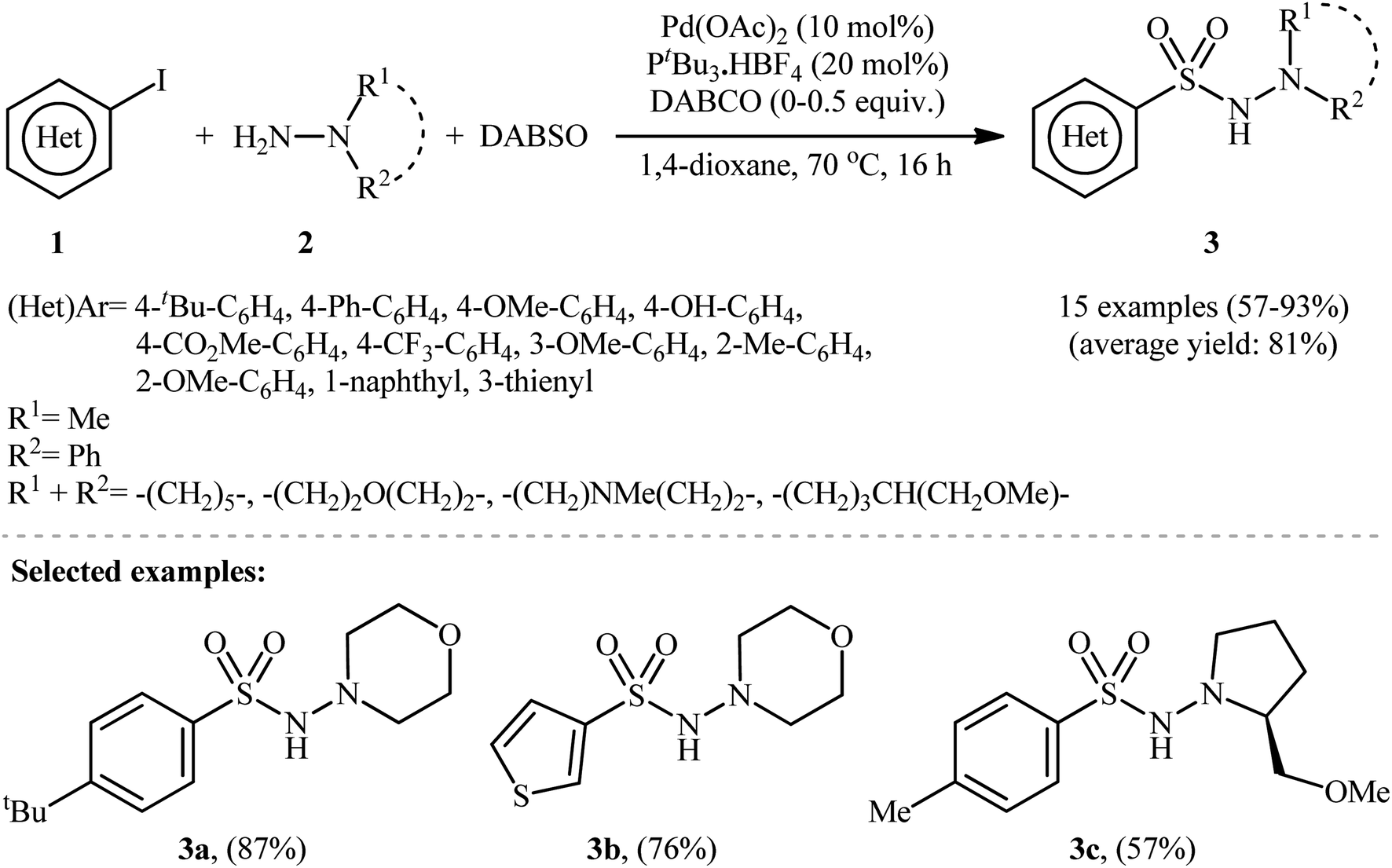 | ||
| Scheme 2 Pd-catalyzed hydrazinosulfonylation of (hetero)aryl iodides 1 with hydrazines 2 and SO2, developed by Willis. | ||
A possible catalytic cycle for this transformation was illustrated in Scheme 3.14 Initially, the oxidative addition of Pd(0) with aryl iodide 1 generates the Pd(II) species I. Then, the insertion of SO2 into the Pd–C bond gives the complex II. Subsequently, the nucleophilic attack of hydrazine 2 on this intermediate II produces complex III, which, after reductive elimination, yields the desired product 3 and regenerates Pd(0) species to enter the next catalytic cycle.
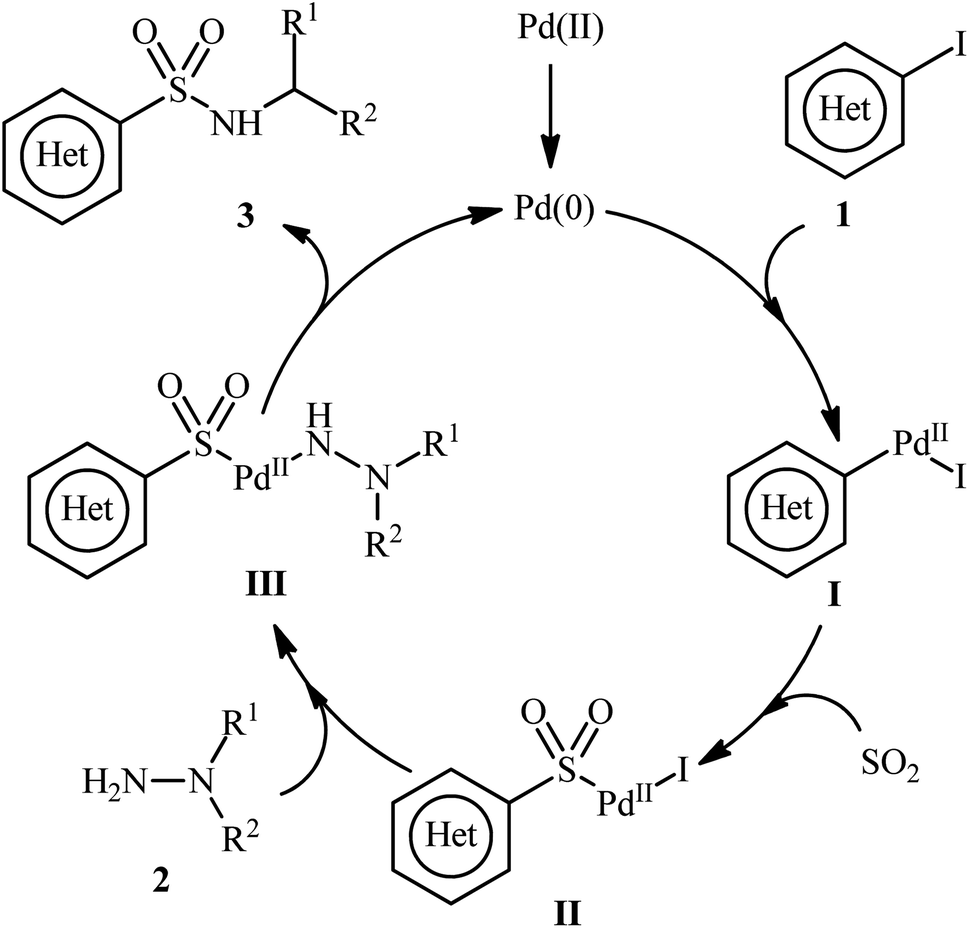 | ||
| Scheme 3 Proposed mechanism for the formation of (hetero)aryl N-aminosulfonamides 3. | ||
To demonstrate the robustness of their methodology further, the same research group successfully synthesized several N-, O-, S-heteroaromatic and olefinic N-aminosulfonamides from the corresponding heteroaryl and alkenyl iodides, respectively, using their standard conditions.15 They also revealed that the use of isolated DABSO is not mandatory for these transformations and that in situ generation of DABSO by bubbling SO2 gas into a DABCO solution is possible, as demonstrated in the synthesis of 4-methyl-N-morpholinobenzenesulfonamide (93%). Additionally, they demonstrated that hydrazine·SO2 complexes can act as both the N-nucleophile and SO2 source. Therefore, under standard conditions, the reaction of a series of (hetero)aryl iodides 4 with N-aminomorpholine·SO2 complex 5 produced the corresponding N-morpholino (hetero)arenesulfonamides 6 in good to excellent isolated yields ranging from 64% to 93% (as shown in Scheme 4).
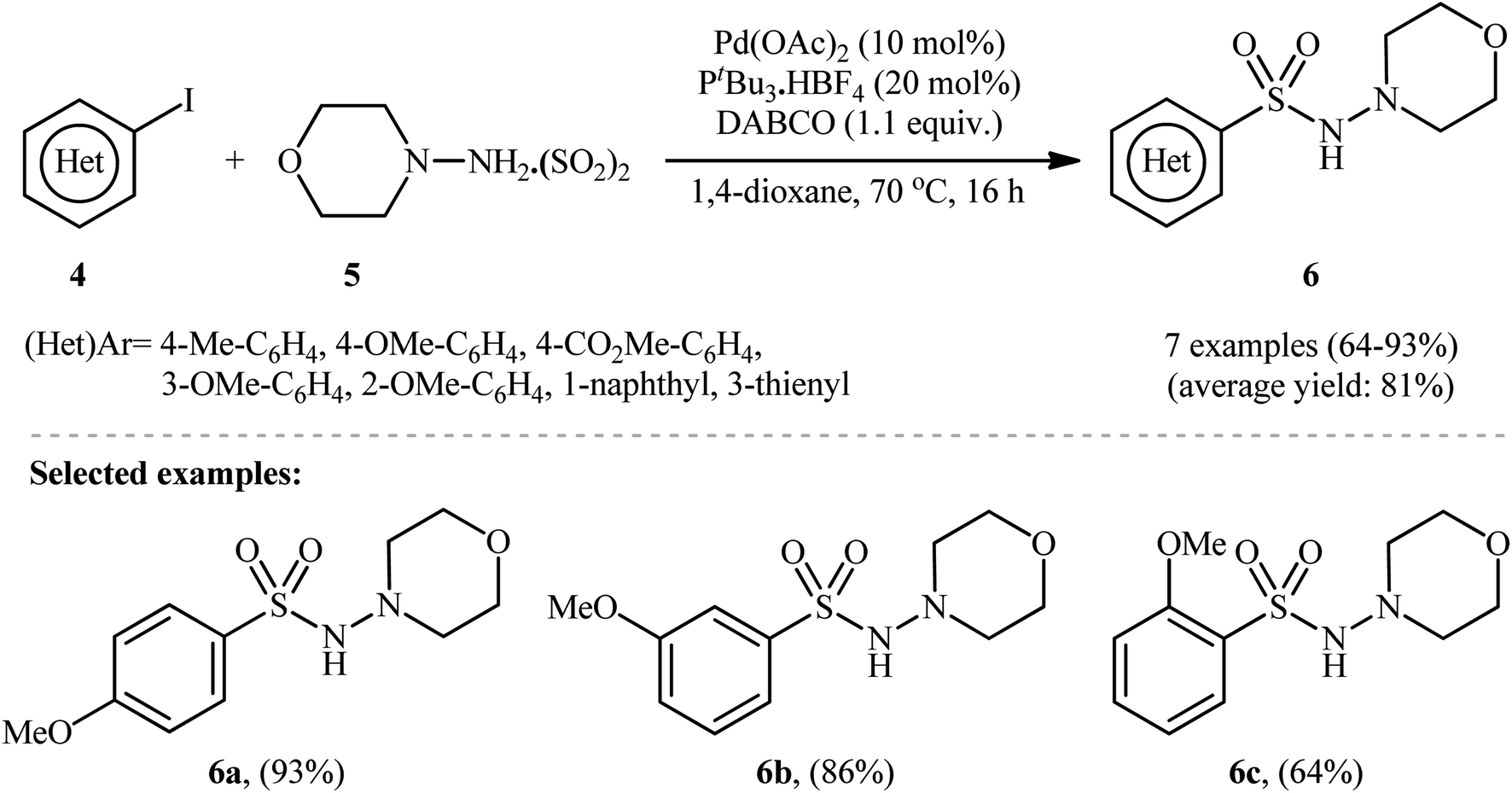 | ||
| Scheme 4 Pd-catalyzed hydrazinosulfonylation of (hetero)aryl iodides 4 with N-aminomorpholine·SO2 complex 5. | ||
In 2012, Ye and Wu developed an efficient and practical protocol for preparing aryl N-aminosulfonamides 9 by directly hydrazinosulfonylating various aryl iodides 7 with N,N-dialkyl/N-alkyl-N-aryl hydrazines 8, and potassium metabisulfite (K2S2O5) as a stable and inexpensive source of SO2 (Scheme 5).16 The reaction's best conversion efficiency was achieved with Pd(OAc)2 (5–10 mol%), PtBu3·HBF4 (20–30 mol%), and TBAB (1.5 equiv.) in 1,4-dioxane at 80 °C. The reaction tolerated a wide range of functional groups with different electronic characteristics, and under optimized conditions, it produced the desired aryl N-aminosulfonamide products in modest to high yields. Notably, aryl bromides 10 were also effective in this reaction, but required a higher reaction temperature (100 °C) to afford products 11 in satisfactory yields (Scheme 6). Overall, this study demonstrated significant scope of aryl halide reagents but a limited scope of the hydrazine substrate. It is important to mention that other inorganic sulfites, such as NaHSO3 and ZnSO3, were also effective sources of SO2 in this reaction but gave lower yields of the product. However, Na2SO5 proved to be completely ineffective. In a closely related investigation, the Wu laboratory disclosed a Pd-catalyzed hydrazinosulfonylation of aryl iodides with gaseous SO2, generated ex situ from Na2SO3, using similar reaction conditions (Pd(OAc)2, PtBu3·HBF4, Cs2CO3).17
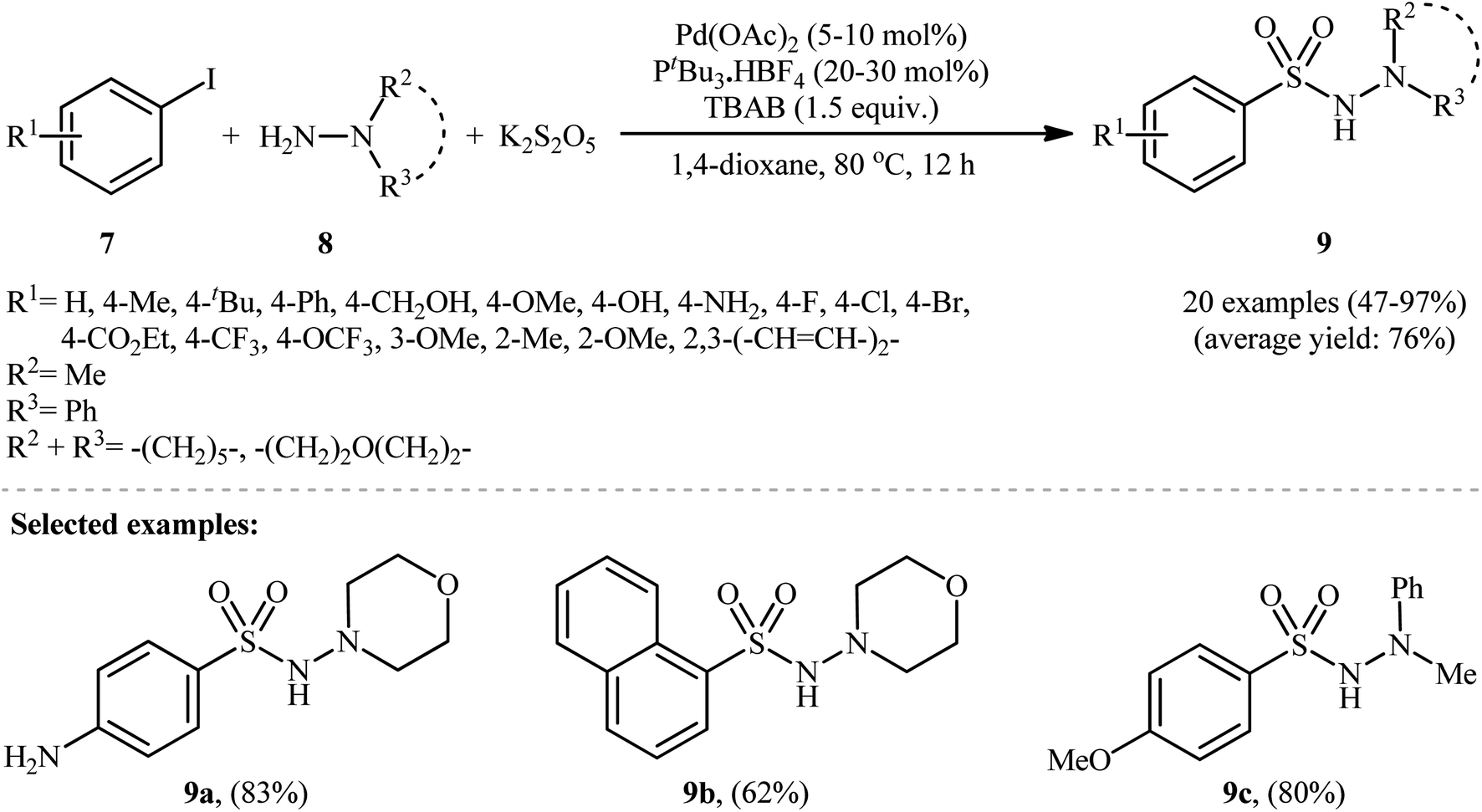 | ||
| Scheme 5 Pd-catalyzed aminosulfonylation of aryl iodides 7 with hydrazines 8 and K2S2O5. | ||
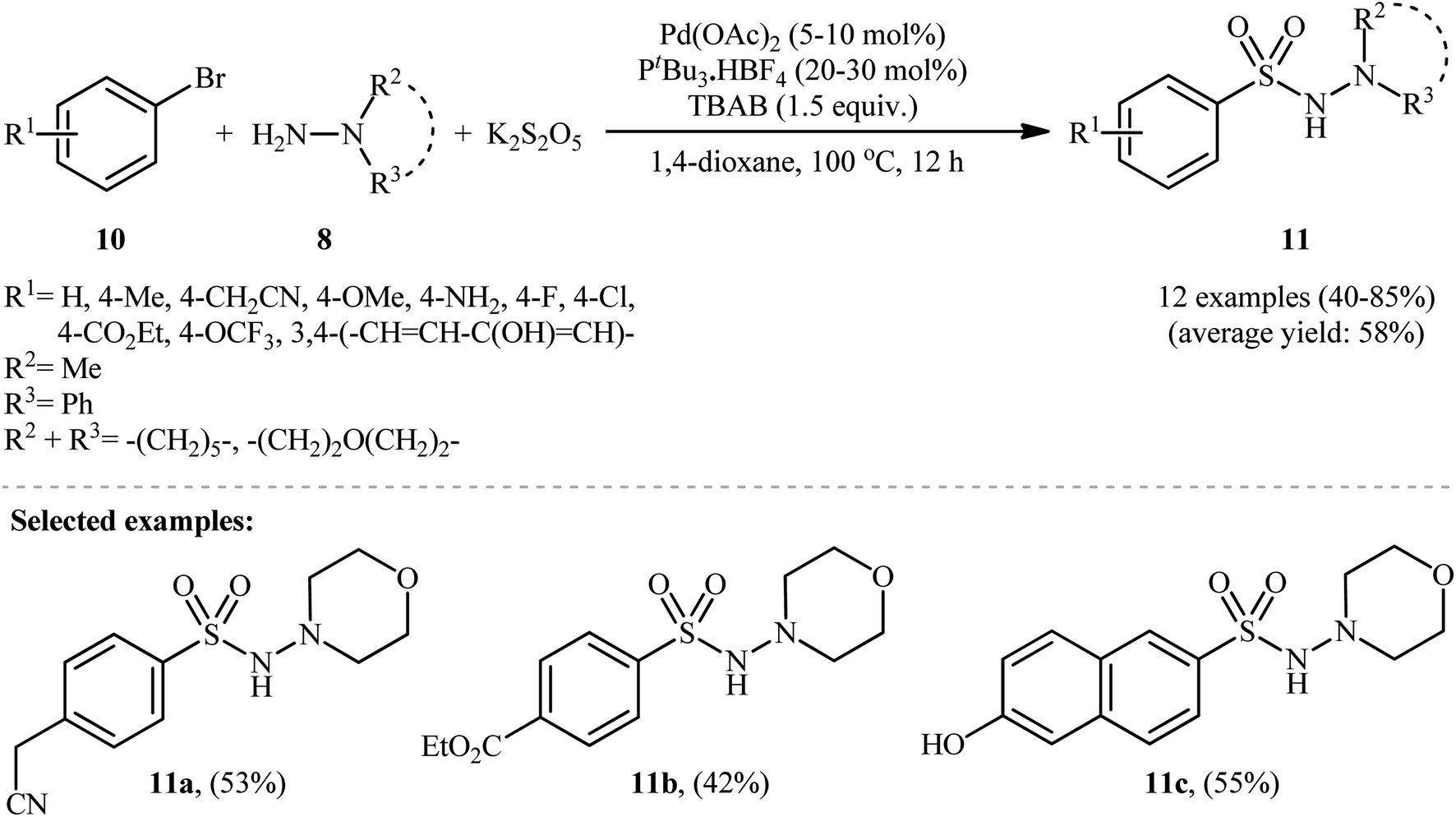 | ||
| Scheme 6 Pd-catalyzed aminosulfonylation of aryl bromides 10 with hydrazines 8 and K2S2O5. | ||
Li-Wu's research group aimed to design a milder and greener procedure for the synthesis of aryl N-aminosulfonamide derivatives using a transition-metal-free coupling reaction between aryl halides, hydrazines, and sulfur dioxide. They were successful in demonstrating that various functionalized aryl N-aminosulfonamides 14 could be obtained from the reaction of aryl bromides 12 with various N,N-dialkyl, N,N-diaryl, N-alkyl, N-aryl hydrazines 13 and DABSO under catalyst-free and ambient conditions.18 TBAI (tetrabutylammonium iodide) was used as an additive, and ultraviolet irradiation was applied to provide moderate to good yields of the target aryl N-aminosulfonamides (Scheme 7). The reaction system was also suitable for less reactive aryl chlorides, as exemplified by the formation of N-morpholinobenzenesulfonamide (51%) from chlorobenzene and morpholin-4-amine. The system was also capable of hydrazinosulfonylation of alkyl halides, resulting in moderate to high yields of alkyl N-aminosulfonamide products. Notably, N,N-diaryl hydrazines, which were unsuitable substrates under Willis' condition, were compatible with this photo-induced reaction.
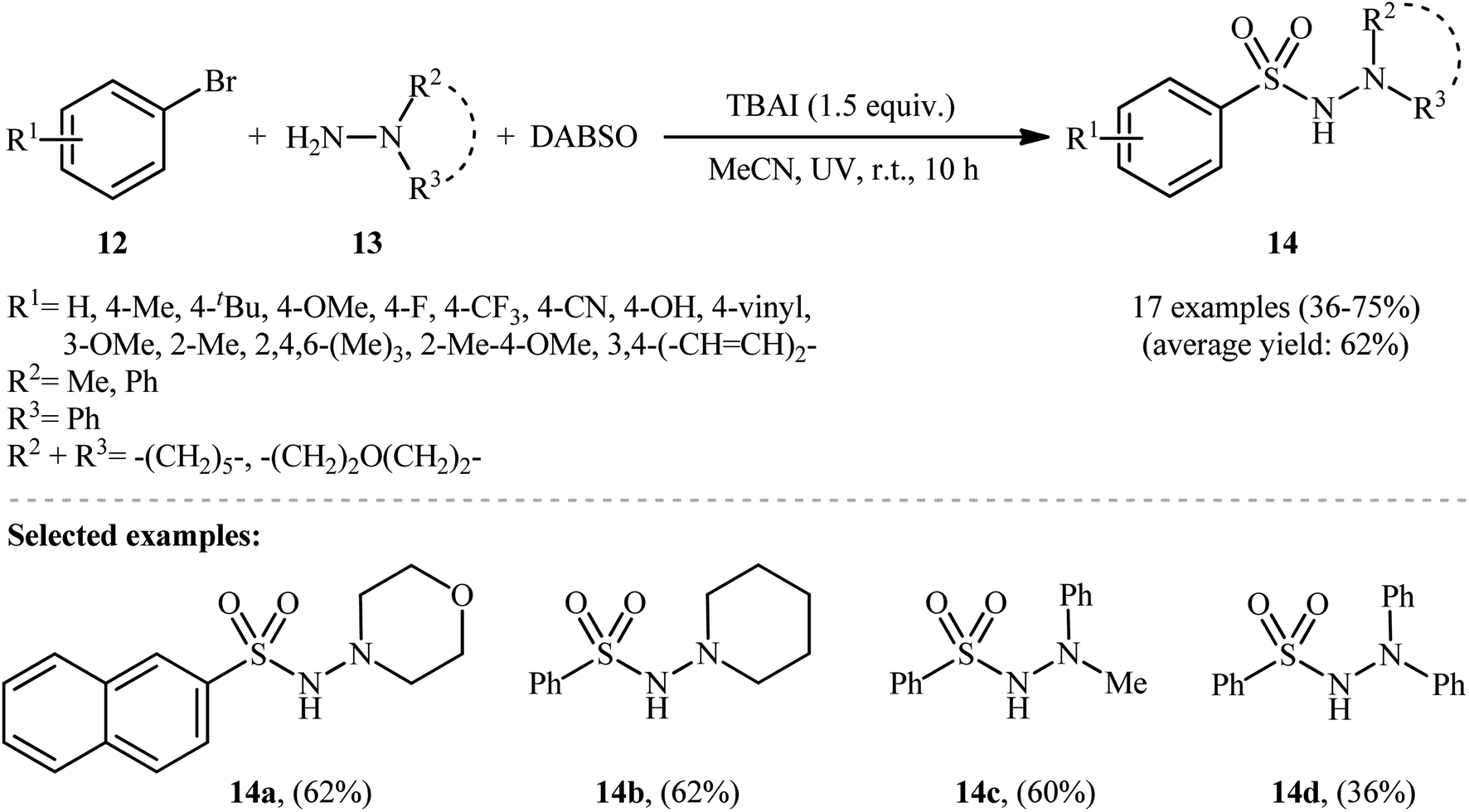 | ||
| Scheme 7 Photoinduced metal-free hydrazinosulfonylation of aryl bromides 12 with hydrazines 13 and DABSO. | ||
Based on a series of control experiments such as radical trapping, DFT calculation, and others, the authors suggested a mechanistic pathway for the formation of aryl N-aminosulfonamides 14, as shown in Scheme 8. Initially, a hydrazine·SO2 complex A was formed via the exchange of a sulfur dioxide molecule between DABSO and hydrazine 13. In parallel, the aryl radical and the bromine radical were generated via the homolytic cleavage of C–Br bond of aryl bromide 12 under ultraviolet irradiation. Complex IV reacted with the aryl radical to produce the radical species V, which then released hydrazine 13 to form the highly active intermediate VI. Subsequently, the integration of bromine radical with hydrazine 13 resulted in the formation of intermediate VII, which after deprotonation provided the radical VIII. Finally, radical VIII combined with intermediate VI to yield the desired product 14.
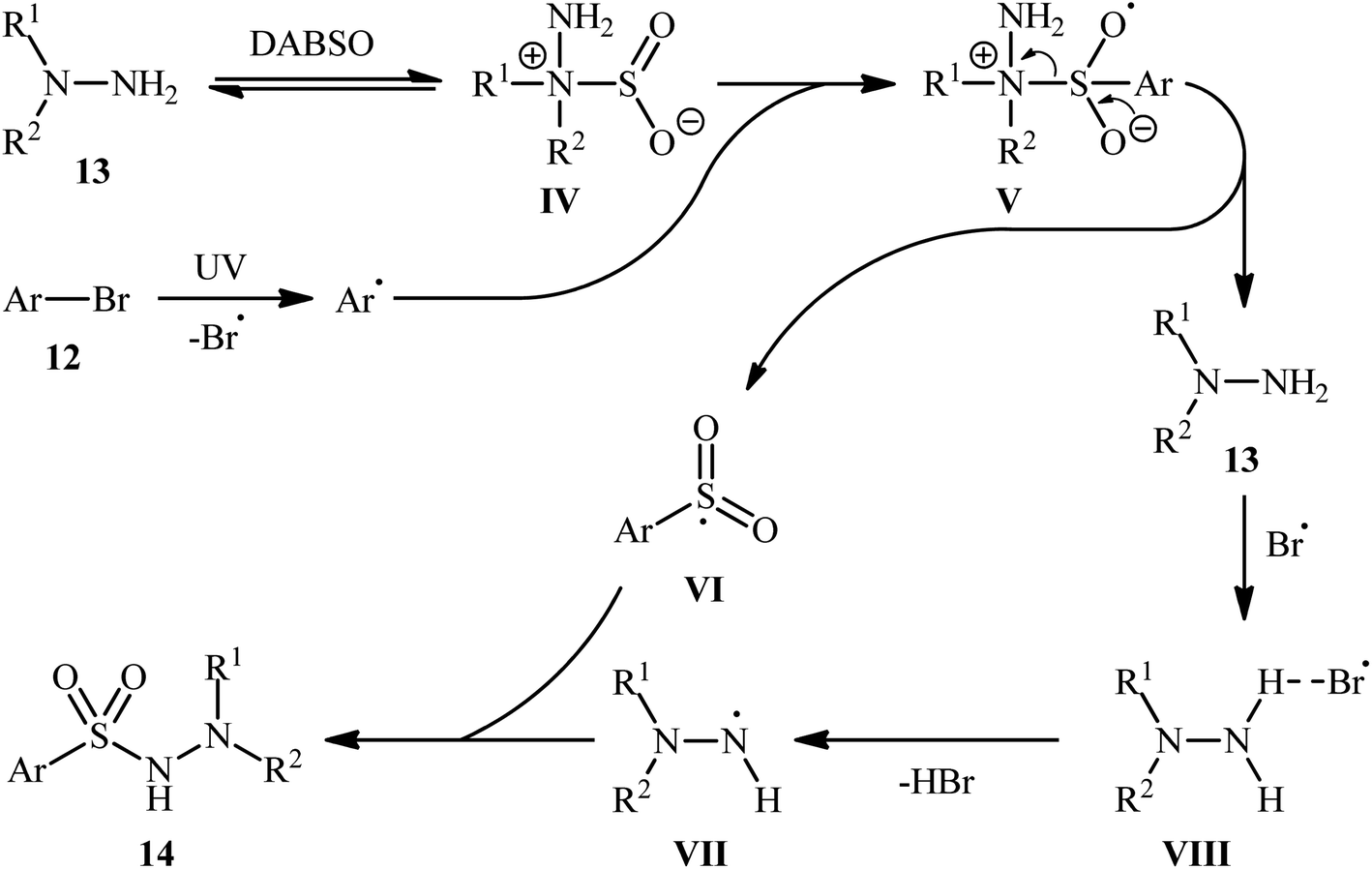 | ||
| Scheme 8 Proposed mechanism for the formation of aryl N-aminosulfonamides 14. | ||
Ding and colleagues recently made an interesting contribution to this field by reporting the direct synthesis of aryl N-aminosulfonamides 17 from simple arenes 15 through sequential C–H functionalization and aminosulfonylation steps (Scheme 9).19 First, aryl iodides A were formed by treating arenes 15 and N-iodosuccinimide (NIS) via Au-catalyzed C–H bond activation at room temperature. Next, hydrazines 16 and DABSO were added, and the mixture was heated with a palladium catalyst at 80 °C for 12 hours to complete the hydrazinosulfonylation reaction.
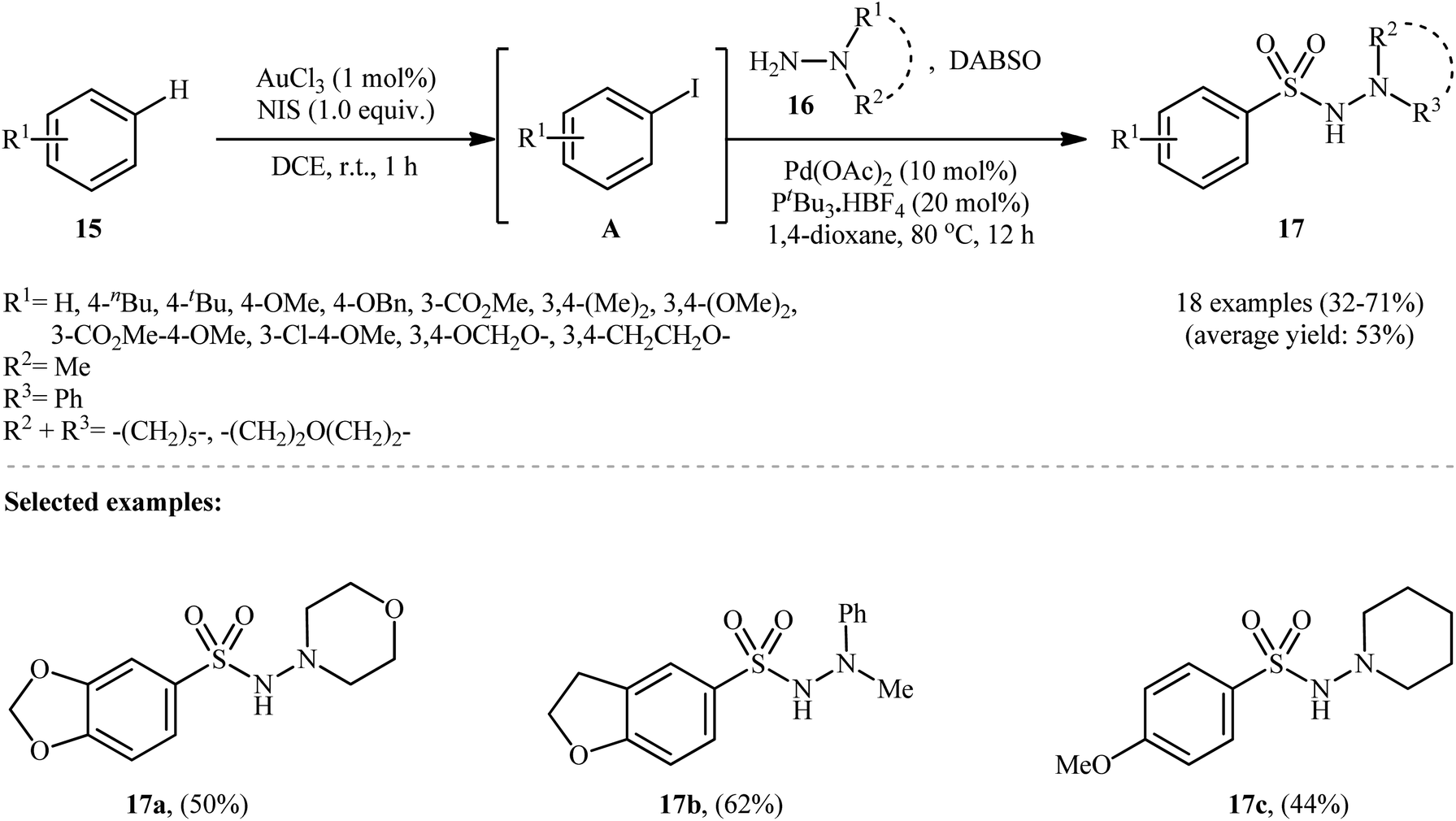 | ||
| Scheme 9 Synthesis of aryl N-aminosulfonamides 17 via an Au(III) and Pd(II) cocatalyzed reaction of arenes 15, sulfur dioxide, and hydrazines 16. | ||
3. Hydrazinosulfonylation of arylboronic acids
In 2012, Ye and Wu conducted a study on the synthesis of aryl N-aminosulfonamides via the three-component coupling of arylboronic acids, sulfur dioxide, and hydrazines.20 To optimize the reaction conditions, they chose 4-methylphenylboronic acid and morpholin-4-amine as the model reactants and screened various parameters, such as catalyst, ligand, additive, and solvent. The results showed that the combination of 5 mol% of Pd(OAc)2 and 1.5 equiv. of TBAB was the most suitable catalytic system for this transformation, and 1,4-dioxane was found to be the most appropriate solvent among various organic solvents (such as DMF, MeCN, DCE, tBuOH, 1,4-dioxane, toluene). Under the optimized conditions, a range of aryl boronic acids 18 coupled with a series of hydrazines 19 and SO2 (DABSO was used as a sulfur dioxide surrogate) to produce the corresponding aryl N-aminosulfonamides 20 in moderate to quantitative yields within 12 h (Scheme 10). The catalytic system was also compatible with alkenyl boronic acids as substrates. Interestingly, the substituents of chloro and bromo on the aromatic ring were retained under the standard conditions, providing potential opportunities for further product manipulation. However, this catalytic system did not work with heteroaryl boronic acids, and no reaction occurred when hydrazines were replaced with amines (such as piperidine and p-toluidine), which was consistent with the results reported by Willis. According to the authors, this may be due to the difference in nucleophilicity and basicity between amines and hydrazines. The proposed mechanism for this transformation is similar to the one depicted for hydrazinosulfonylation of aryl halides in Scheme 3. To the best of our knowledge, this is the first and only reported example of hydrazinosulfonylation of arylboronic acids thus far, and further studies are needed to explore the scope and limitations of this reaction.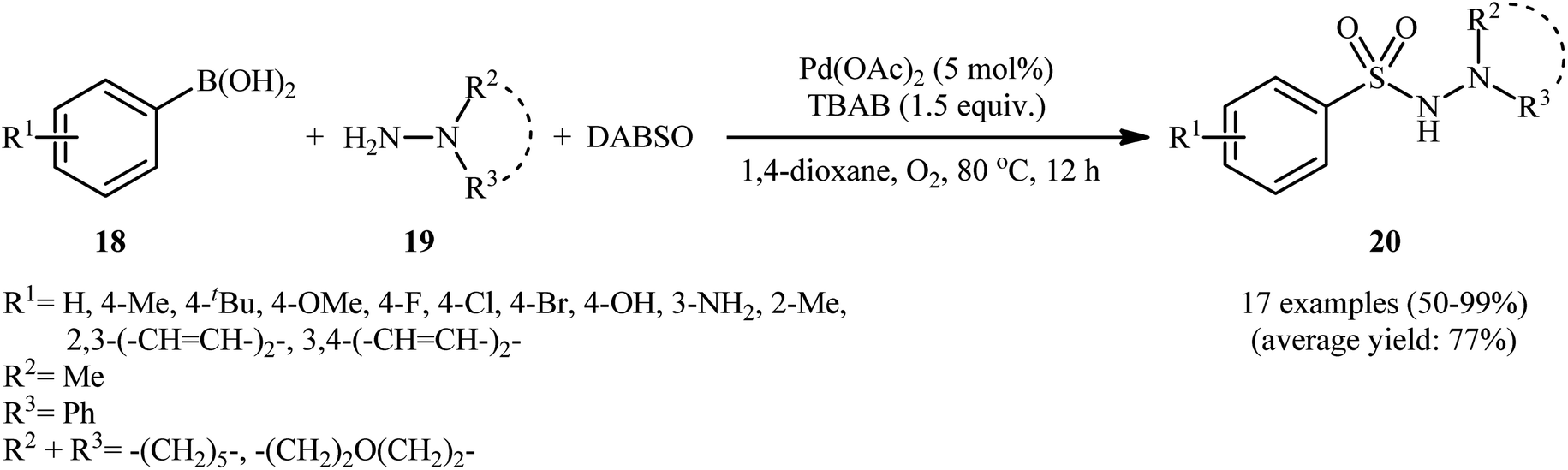 | ||
| Scheme 10 Pd-catalyzed hydrazinosulfonylation of arylboronic acids 18 with hydrazines 19 and SO2. | ||
4. Hydrazinosulfonylation of aryldiazonium salts
Aryldiazonium salts are efficient, readily available, and inexpensive substitutes for conventional aryl halides, and are frequently used as coupling partners in many cross-coupling reactions.21In 2014, Li, Wu, and colleagues demonstrated the usefulness of aryldiazonium tetrafluoroborates as aryl coupling partners in hydrazinosulfonylation reactions.22 The reaction proceeds without the need for any catalyst or additive, and involves the treatment of aryldiazonium tetrafluoroborates 21 with hydrazines 22 and DABSO at room temperature, yielding the corresponding aryl N-aminosulfonamides 23 in moderate to excellent yields within minutes (Scheme 11). The system can accommodate the presence of various important functional groups, including fluoro, chloro, bromo, nitro, ester, and ether functionalities, which may provide potential opportunities for further product derivation. However, the methodology was found to be ineffective for anilines and aliphatic amines, which is similar to previous reports.
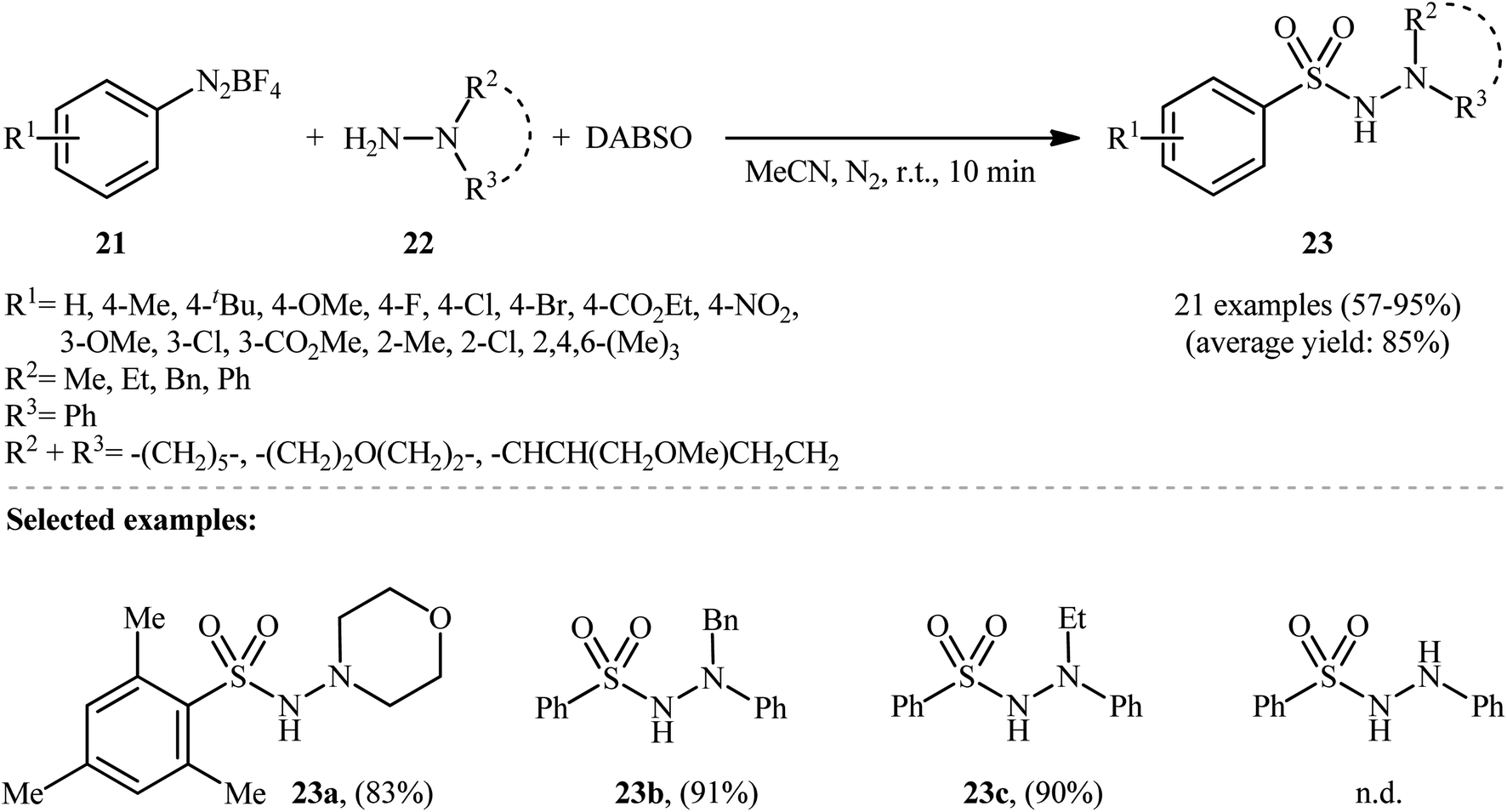 | ||
| Scheme 11 Catalyst-free hydrazinosulfonylation of aryldiazonium tetrafluoroborates 21 with hydrazines 22 and DABSO. | ||
Based on several control experiments and theoretical calculations, the authors proposed that this hydrazinosulfonylation reaction proceeds through the mechanistic pathway shown in Scheme 12. The reaction starts with the generation of the hydrazine–SO2 complex IX, which is formed through the barrierless exchange of sulfur dioxide between DABSO and hydrazine 22. Subsequently, the electrostatic interaction between this complex A and the arydiazonium cation 21 forms complex X, which after homolytic cleavage of the N–S bond and a single-electron transfer, gives rise to the arydiazonium radical XI, SO2, and radical cation intermediate XII. Next, the release of a molecule of nitrogen from the arydiazonium radical XI yields the aryl radical XIII, which after trapping of SO2, provides the radical intermediate XIV. Finally, the reaction of this radical XIV with the hydrazinium radical XV (generated from deprotonation of the radical cation intermediate XII) leads to the observed product 23.
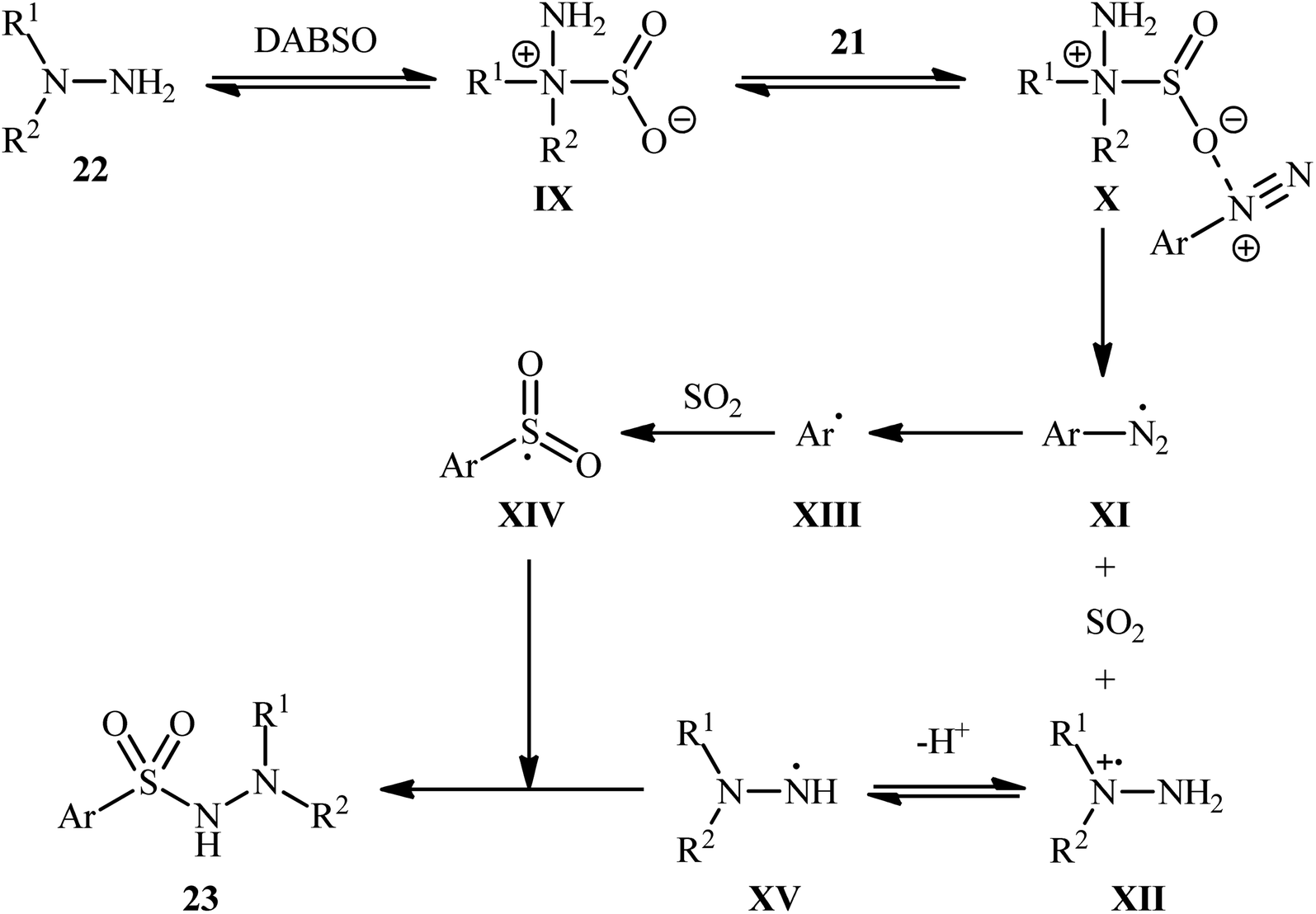 | ||
| Scheme 12 Proposed reaction mechanism for the formation of aryl N-aminosulfonamides 23. | ||
Subsequently, an innovative research group discovered that aromatic amines (24) can be easily converted into their corresponding aryl N-aminosulfonamides (26) by treatment with hydrazines (25) and DABSO in the presence of 1.2 equivalents of tBuONO and 1.2 equivalents of BF3·OEt2 in MeCN at 30 °C (Scheme 13).23 To establish the general applicability of this metal-free hydrazinosulfonylation reaction, various anilines with either electron-donating or electron-withdrawing substituents in the p-, m-, or o-positions were used. Interestingly, the outcome of the reaction was almost independent of the steric and electronic factors of the substituents. Although a series of heteroaromatic amines were also subjected to the reaction, they were mainly sluggish to participate in this protocol.
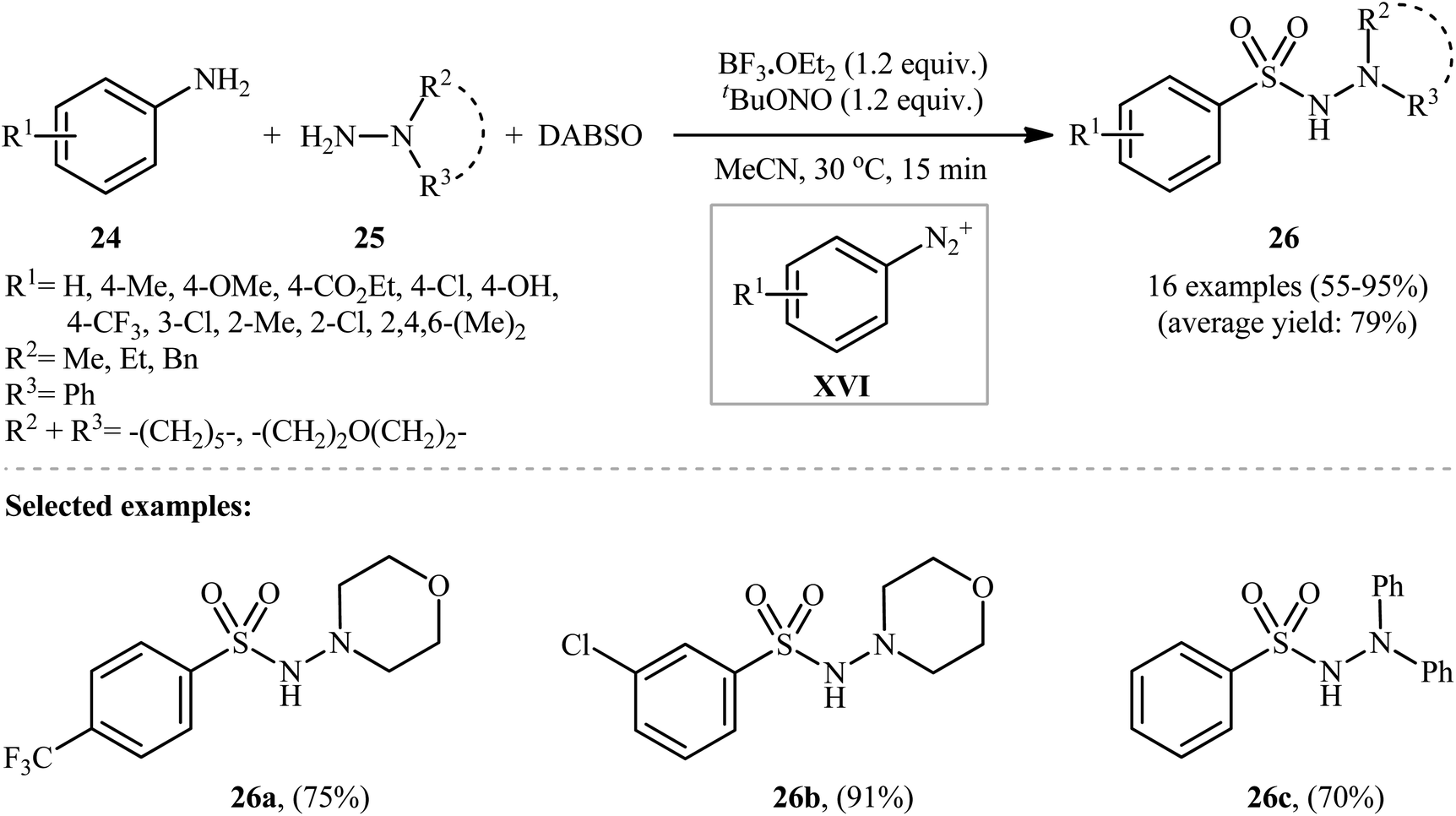 | ||
| Scheme 13 Aminosulfonylation of anilines 24 with hydrazines 25 and SO2. | ||
Mechanistically, after the formation of the aryldiazonium ion XVI, the reaction proceeds along a similar pathway as described in Scheme 11. Shortly afterwards, the authors elegantly applied their methodology to synthesize 1-(2,3-dihydrobenzofuran-3-yl)-methanesulfonohydrazides (29) in high yield through a three-component reaction of 2-(allyloxy)anilines (27), morpholin-4-amine (28), and SO2 (Scheme 14).24 According to the authors, this radical process includes the in situ formation of an aryl radical, followed by intramolecular 5-exo-cyclization and insertion of sulfur dioxide.
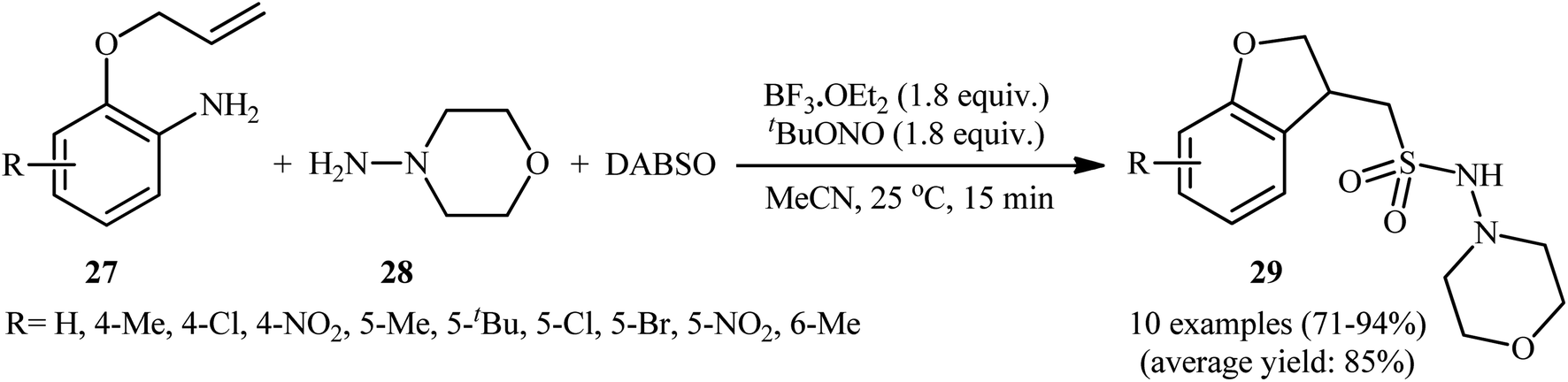 | ||
| Scheme 14 Wu's synthesis of 1-(2,3-dihydrobenzofuran-3-yl)-methanesulfonohydrazides 29. | ||
In a related study, Wu and colleagues demonstrated that 1-aryl-triazene derivatives can serve as the aryl source for the synthesis of N-aminosulfonamides.25 They found that treating a diverse set of 1-(hetero)aryl-triazenes 30 with morpholin-4-amine 28 and SO2 in the presence of 1.5 equiv. BF3·OEt2 in MeCN provided the corresponding N-morpholino (hetero)arenesulfonamides 31 in satisfactory yields (Scheme 15). The results proved that the electronic nature of the substituents on the phenyl rings had strong effects on the efficiency of the reaction. Generally, the aryl-triazenes bearing electron-donating groups afforded better yields compared to electron-withdrawing ones. Notably, 1,1-dimethylhydrazine and phenylhydrazine did not take part in the reaction and therefore no other types of substituted hydrazines were examined in the protocol. Therefore, the reaction, appears to be limited to only morpholin-4-amine as hydrazine substrate. It should be mentioned that when the reaction was carried out in the absence of a hydrazine counterpart and BF3·OEt2 was replaced with CuCl2, the corresponding sulfonamides were obtained as unexpected products.
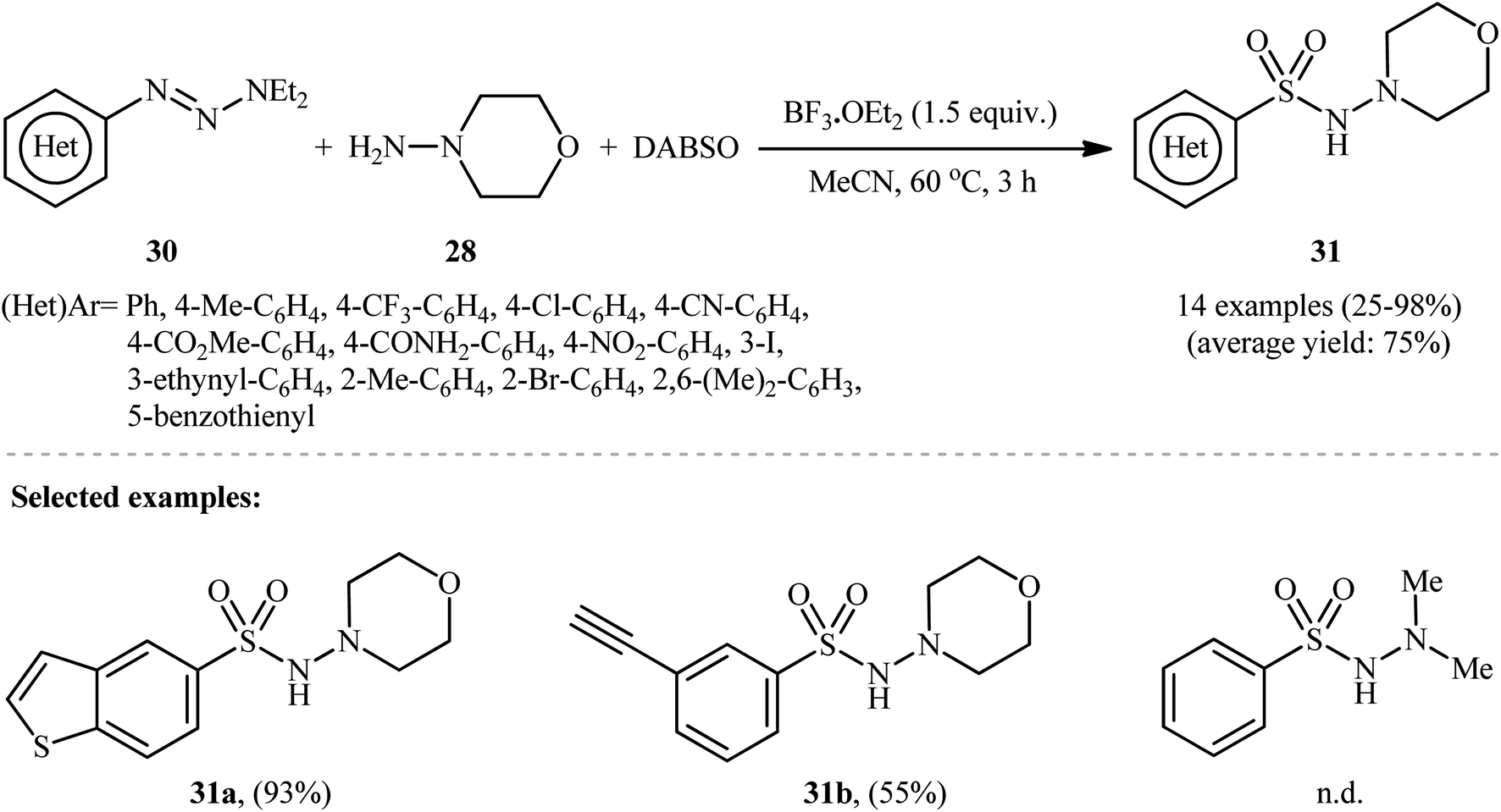 | ||
| Scheme 15 BF3·OEt2-mediated hydrazinosulfonylation of 1-(hetero)aryl-triazenes 30 with morpholin-4-amine 28, and DABSO. | ||
The plausible mechanism for the formation of N-morpholino (hetero)arenesulfonamides 31 involves the generation of aryl diazonium fluoride XVII through the activation of 1-aryltriazene molecule 30 by boron trifluoride. This reactive species then converts into a sulfonyl radical in the presence of SO2, followed by free-radical addition with in situ generated hydrazino radical XVIII to deliver the desired product 31 (Scheme 16).
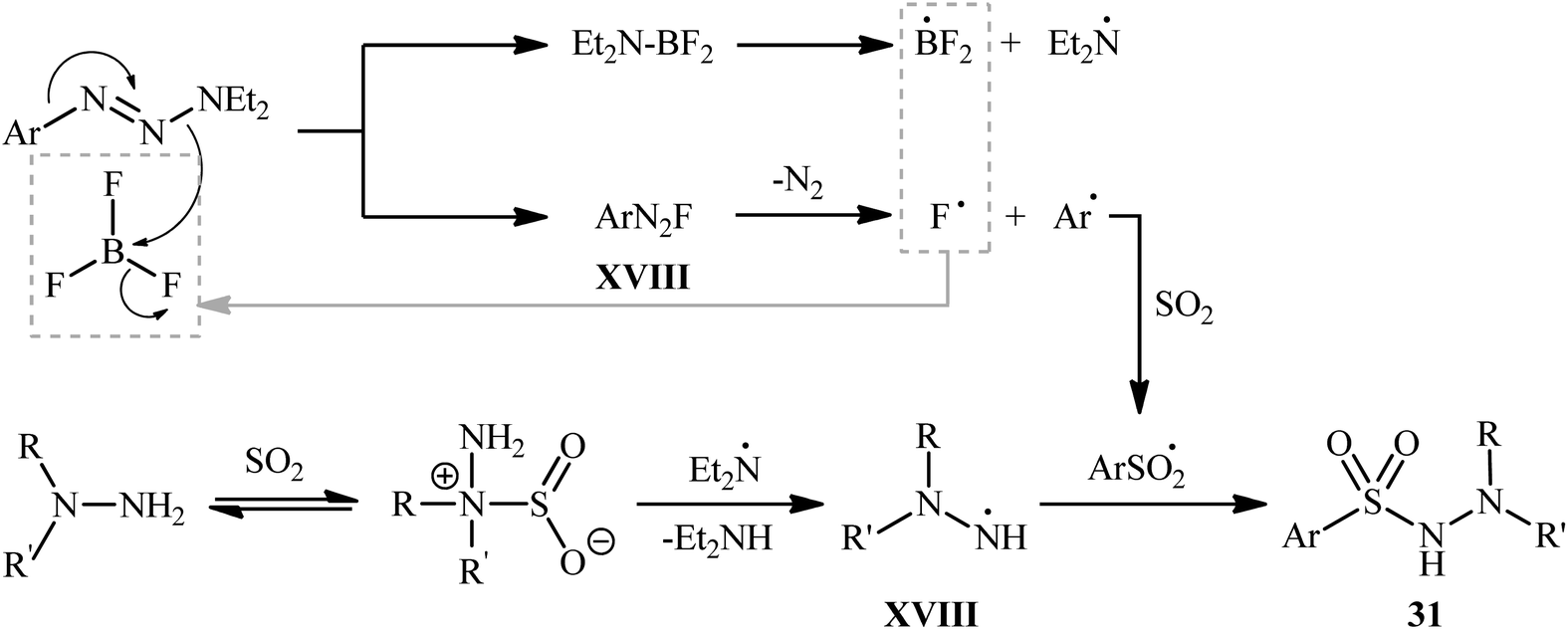 | ||
| Scheme 16 Proposed reaction pathway for the formation of N-morpholino (hetero)arenesulfonamides 31. | ||
Taking inspiration from these works, Han and colleagues have developed an efficient protocol for synthesizing aryl N-aminosulfonamides 34 through denitrogenative aminosulfonylation of aryl hydrazines 32 with N-alkyl hydrazines 33 and K2S2O5 under an air atmosphere and catalyst-free conditions (Scheme 17).26 Although various aryl hydrazines bearing both electron-withdrawing and electron-donating groups were well-tolerated under the reaction conditions, the scope of hydrazine substrates is mainly limited to N,N-dialkyl and N-alkyl-N-aryl hydrazines. The reaction completely failed in the case of unsubstituted hydrazine, 2-aminoisoindoline-1,3-dione, and 1H-benzo[d]1–3 triazol-1-amine, and even trace amounts of the corresponding products were not detected in the reaction mixture.
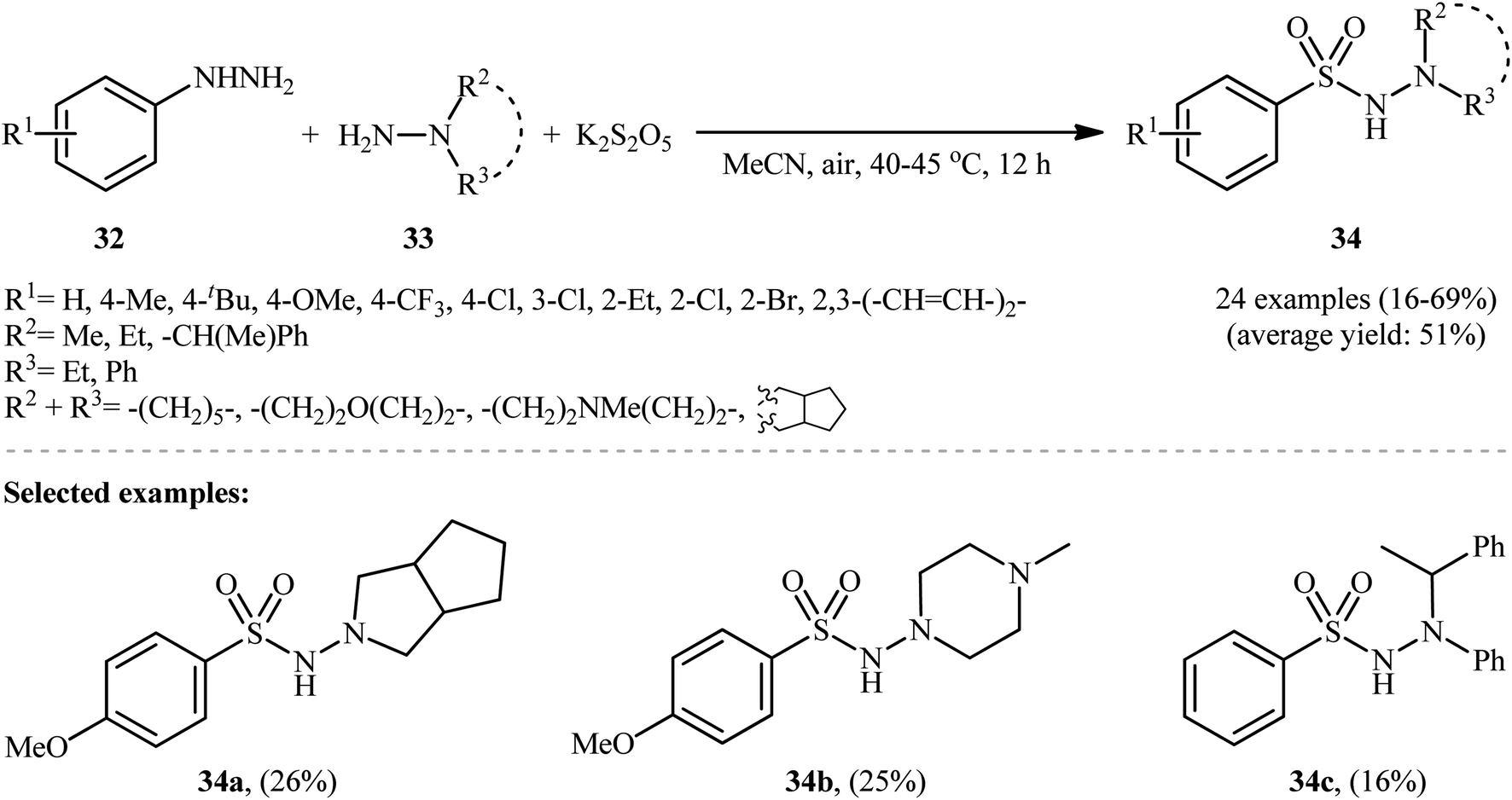 | ||
| Scheme 17 Han's synthesis of aryl N-aminosulfonamides 34. | ||
The mechanistic course of this transition-metal-free reaction sequence is shown in Scheme 18, and involves the initial formation of aryl radical C from arylhydrazine 32 via two-step deprotonation through intermediates XIX and XX with the release of N2. Concurrently, the reaction between the other two substrates, N-alkyl hydrazine 33 and K2S5O5, gives hydrazine–SO2 complex XXII, which reacts with aryl radical XXI to produce radical cation XXIII and anion intermediate XXIV. Subsequently, intermediate XXIII undergoes a deprotonation to afford intermediate XXV, while intermediate XXIV experiences an oxidation to generate intermediate XXVI. Finally, cross-coupling reaction happens between XXVI and XXV, to provide the expected product 34.
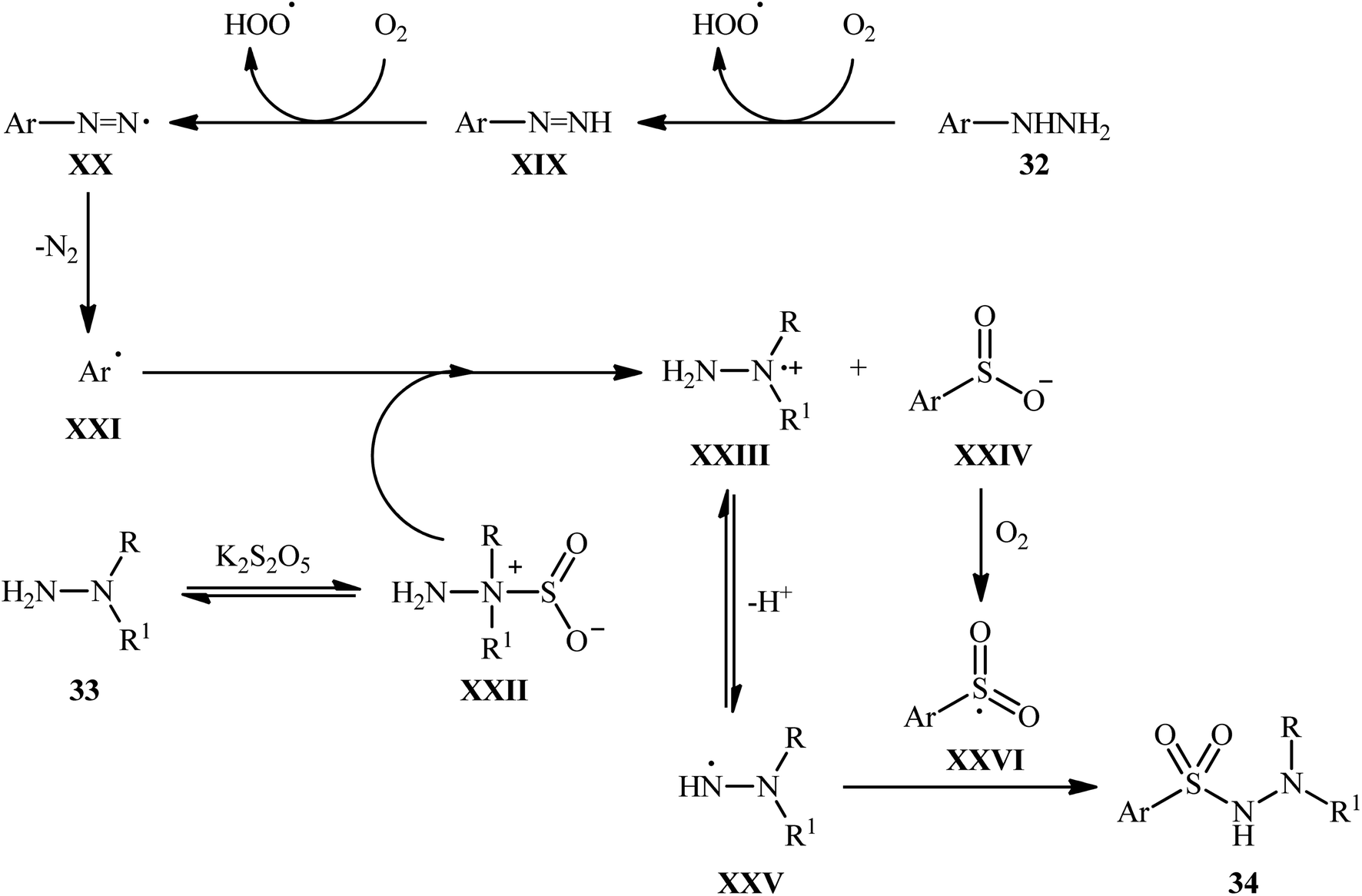 | ||
| Scheme 18 Mechanism that accounts for the formation of aryl N-aminosulfonamides 34. | ||
In an innovative study, Wang et al. described the synthesis of aryl N-aminosulfonamides 37 under catalyst- and additive-free conditions through coupling of aryldiazonium tetrafluoroborates 35 and rongalite 36 at room temperature (Scheme 19).27 In this reaction aryldiazonium salts served as the precursor of both the aryl and hydrazine units and rongalite played a dual role; the SO2 source and the reducing reagent.28 DMSO was found to be the best solvent for the reaction and, among several solvents tested, 1,4-dioxane, toluene, and DCE were found to be less effective. Apparently, the outcome of this coupling was also dependent on the ratios of the two reactants, with the optimum ratio of 36/35 found to be 2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0. According to the authors, this reaction proceeds through a radical pathway as shown in Scheme 20.
:1.0. According to the authors, this reaction proceeds through a radical pathway as shown in Scheme 20.
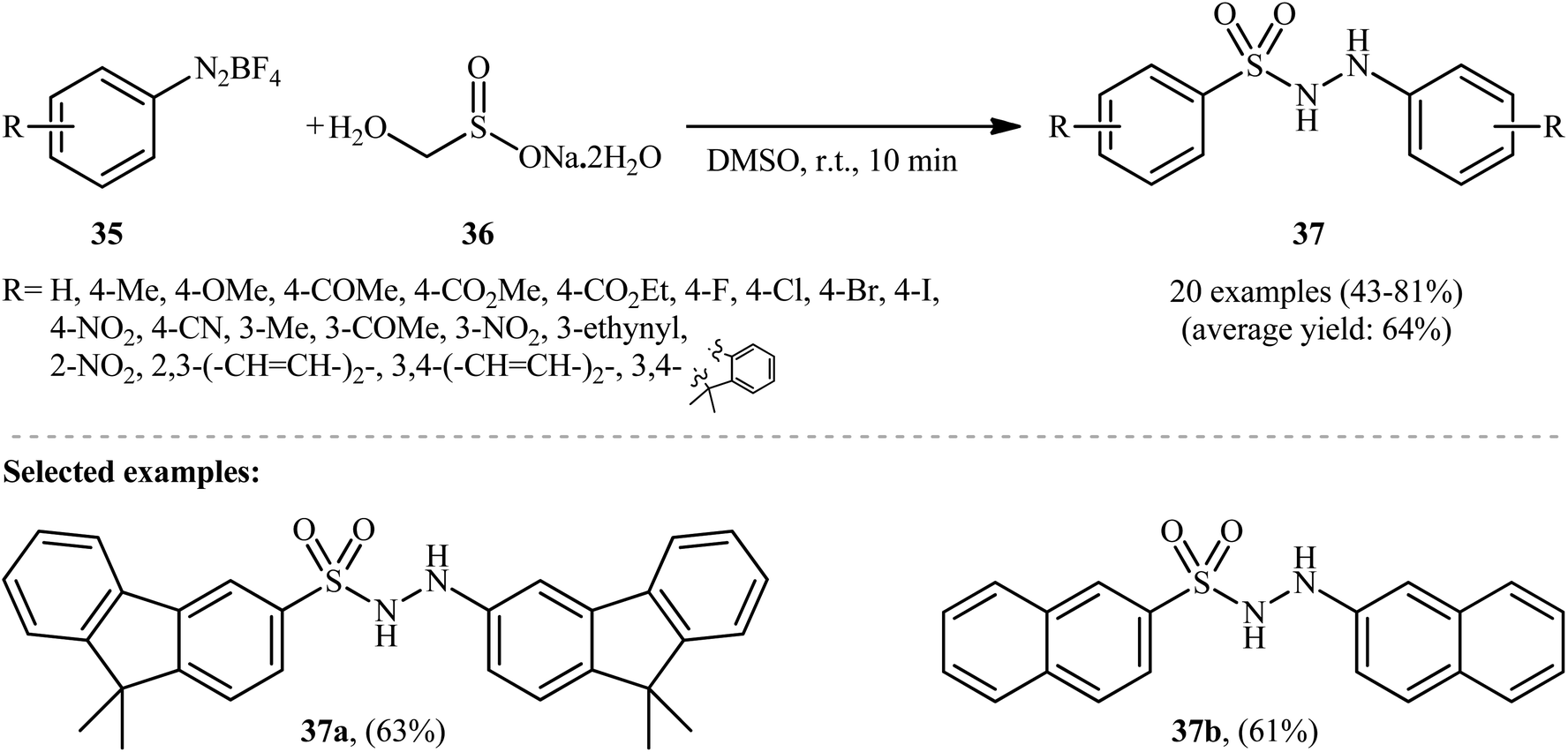 | ||
| Scheme 19 Catalyst-free coupling of aryldiazonium tetrafluoroborates 35 and rongalite 36. | ||
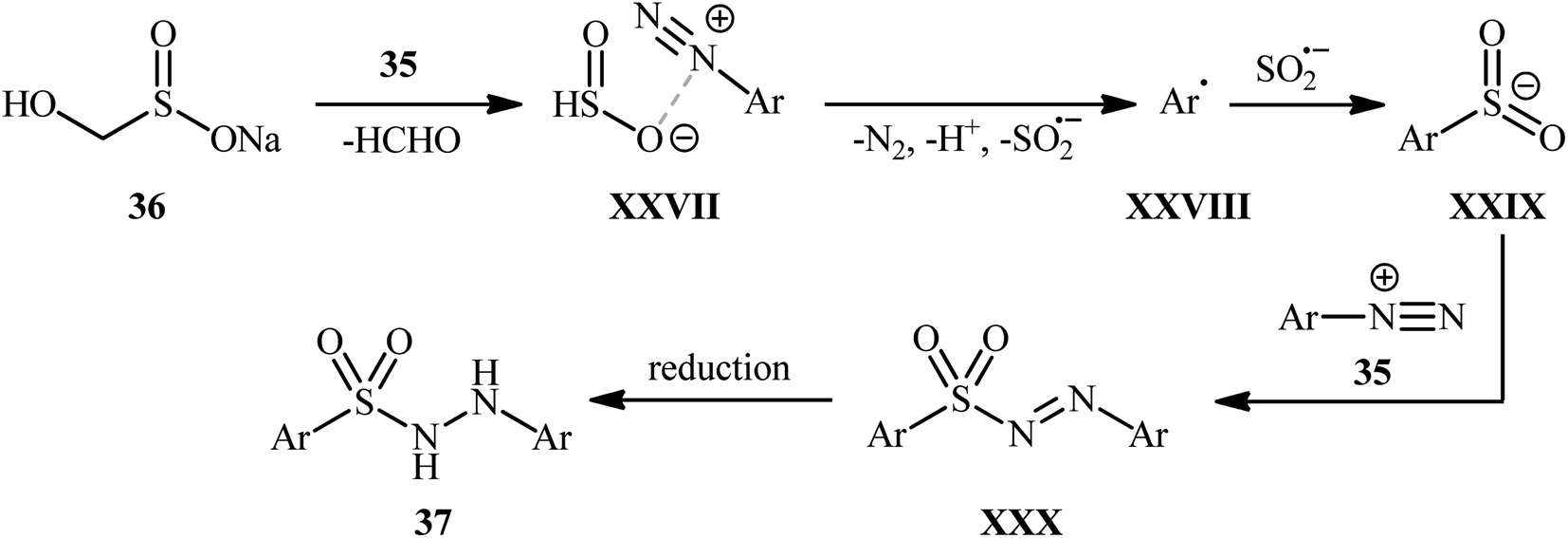 | ||
| Scheme 20 Possible mechanism for the formation of aryl N-aminosulfonamides 37. | ||
5. Hydrazinosulfonylation of aryl nonaflates
The application of aryl nonaflates, more stable and reactive alternative of triflates, as aryl sources in hydrazinosulfonylation reactions has been barely investigated. In fact, to the best of our awareness, only one example on direct hydrazinosulfonylation of aryl nonaflates was reported in literature till date. In this study, An, Xia, and Wu revealed that the treatment of various aryl nonaflates 38 bearing o/m/p substituents with hydrazines 39 and DABSO in the presence of the Pd(OAc)2/Xantphos/TBAB system in dioxane at 80 °C slowly afforded the corresponding aryl N-aminosulfonamides 40 in modest to good yields (Scheme 21).29 Other palladium catalysts such as PdCl2, Pd(PPh3)4 and Pd(CO2CF3)2 were also effective in this reaction but gave lower yield of product. Notably, the reaction temperature was highly important for the efficiency of this transformation and better results were achieved by performing the reaction at 80 °C. Yield outcomes significantly decreased with either increasing or decreasing the temperature. The results indicated that this coupling reaction works better with electron rich than with electron poor substituents in the phenyl ring periphery of aryl nonaflates (67% yield for 4-tBu-substituted substrate compared to 36% for the 4-F derivative). Mechanistically, it was suggested that this reaction proceeds through the similar pathway depicted in Scheme 3.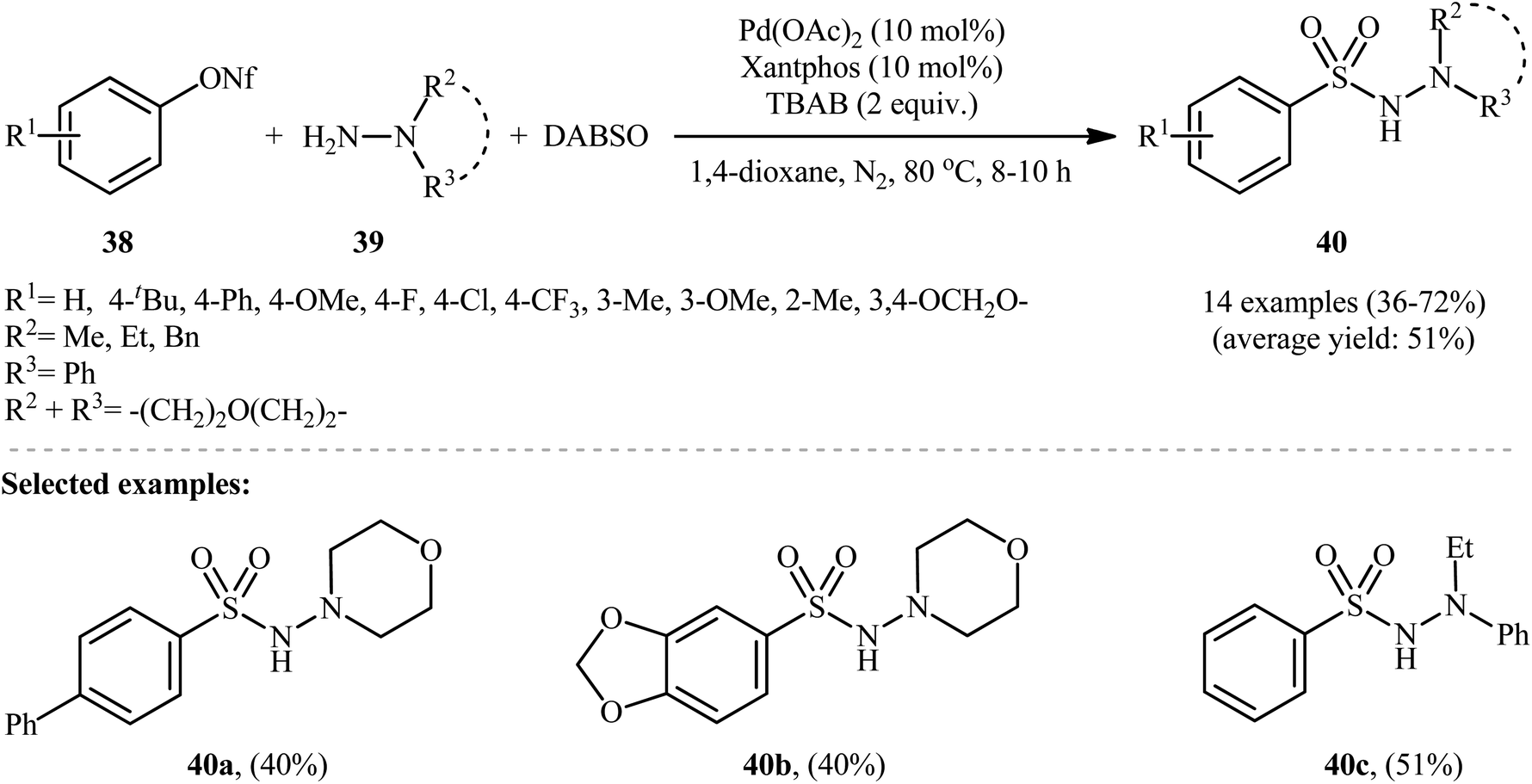 | ||
| Scheme 21 Pd-catalyzed coupling reaction of aryl nonaflates 38, hydrazines 39, and SO2. | ||
6. Hydrazinosulfonylation of arylsilanes
The first and only example of the three-component coupling reactions between aryl silanes, hydrazines, and sulfur dioxide was reported in 2014 by Wang and colleagues,30 who showed that the treatment of aryltriethoxysilanes 41 with hydrazines 42 and DABSO in the presence of a combination of Cu(OAc)2, X-phos and CsF under the O2 atmosphere afforded the aryl N-aminosulfonamide derivatives 43 in moderate to good yields, ranging from 52% to 78% (Scheme 22). Besides aryltriethoxysilanes, alkyl and alkenyl triethoxysilanes were also successfully served as coupling partners in this reaction; however, no reaction occurred when amines or anilines were utilized as the replacement for hydrazines. Subsequently, the authors considerably improved the efficiency of their methodology by simple replacing aryltriethoxysilanes with diethoxydiarylsilanes without any modification in conditions. Other common copper catalysts such as CuF, CuI, CuSCN, CuBr2, CuCl2, Cu(OTf)2, and Cu2O were also found to promote these coupling reactions, albeit at lower efficiencies. It is worth noting that the presence of CsF as a fluoride source is crucial for the success of this transformation. No product was observed in the absence of CsF. Replacing CsF with some other fluorides (e.g., TBAF, NaF, KF, LiF) led to much lower yields or even no desired product at all. Unfortunately, no comment was made by the authors regarding the possible mechanistic cycle of this conversion. However, it seems the reaction proceeds through a sulfonyl fluoride intermediate.31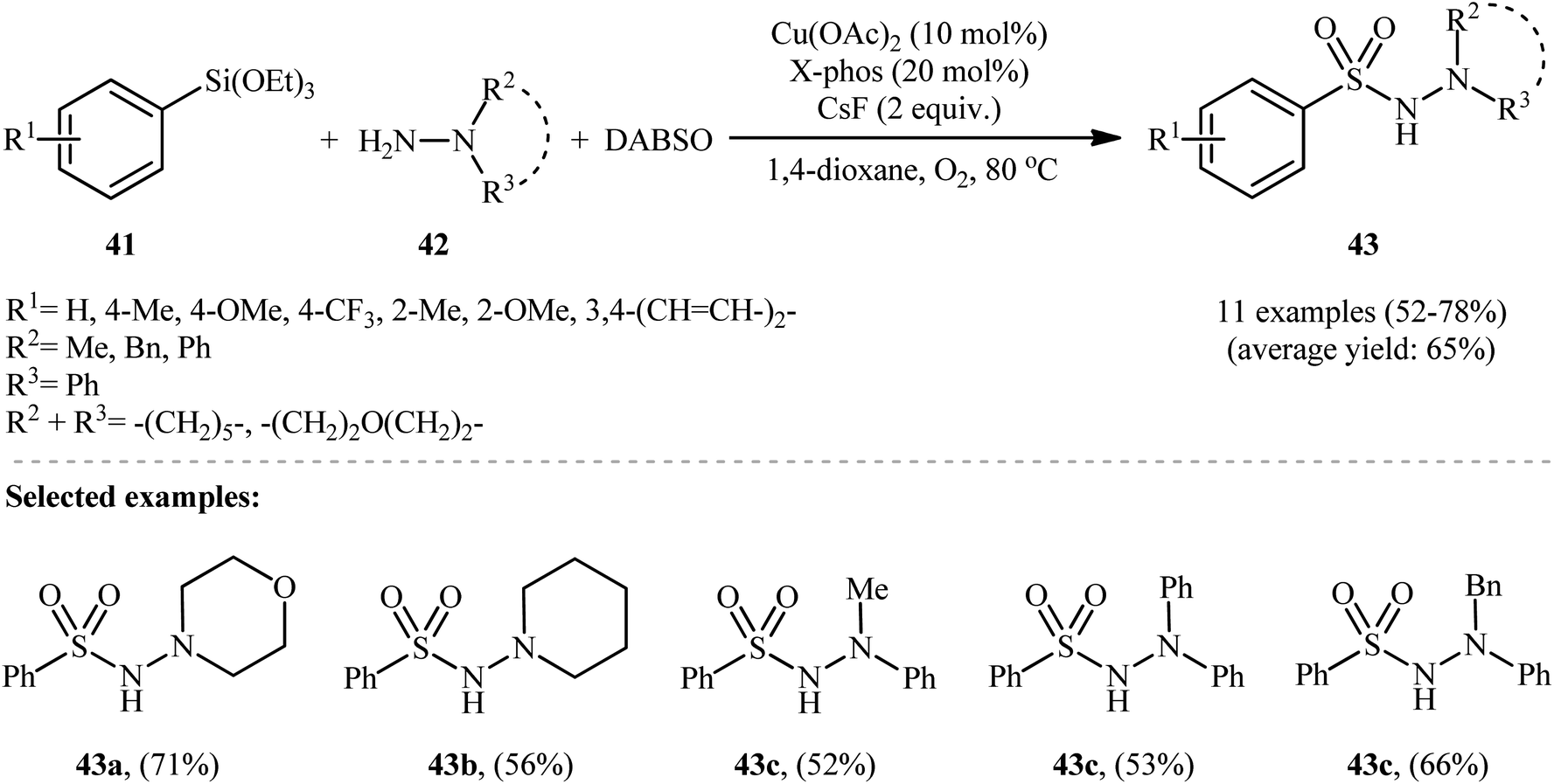 | ||
| Scheme 22 Cu-catalyzed three-component reaction of aryltriethoxysilanes 41, hydrazines 42 and SO2. | ||
7. Hydrazinosulfonylation of diaryliodonium salts
The idea to use diaryliodonium salts as aryl components for three-component coupling of aryl electrophiles, hydrazines, and SO2 was exploited in 2017 by Manolikakes and co-workers.32 By choosing diphenyliodonium triflate and 4-aminomorpholine as model reactants and DABSO as sulfur dioxide source upon irradiation with visible light, quite some photocatalysts were screened, including [Ru(bpy)3]Cl2·6H2O, [Ir(ppy)3], [Ir(ppy)2(dtbbpy)]PF6, eosin Y, and two perylene dyes, choosing eventually the perylenediimide (PDI1) in only 2 mol%. The optimization of the reaction conditions also indicated that a 1:1 mixture of DMSO:MeCN was more effective reaction medium than either DMSO alone or MeCN alone. Interestingly, replacing DABSO with the K2S2O5/trifluoroacetic acid (TFA) system led to the similar results. With these optimized reaction conditions, a library of aryl N-aminosulfonamides 46 were obtained in relatively poor to good yields from the corresponding diaryliodonium salts 44 and hydrazines 45 (Scheme 23).
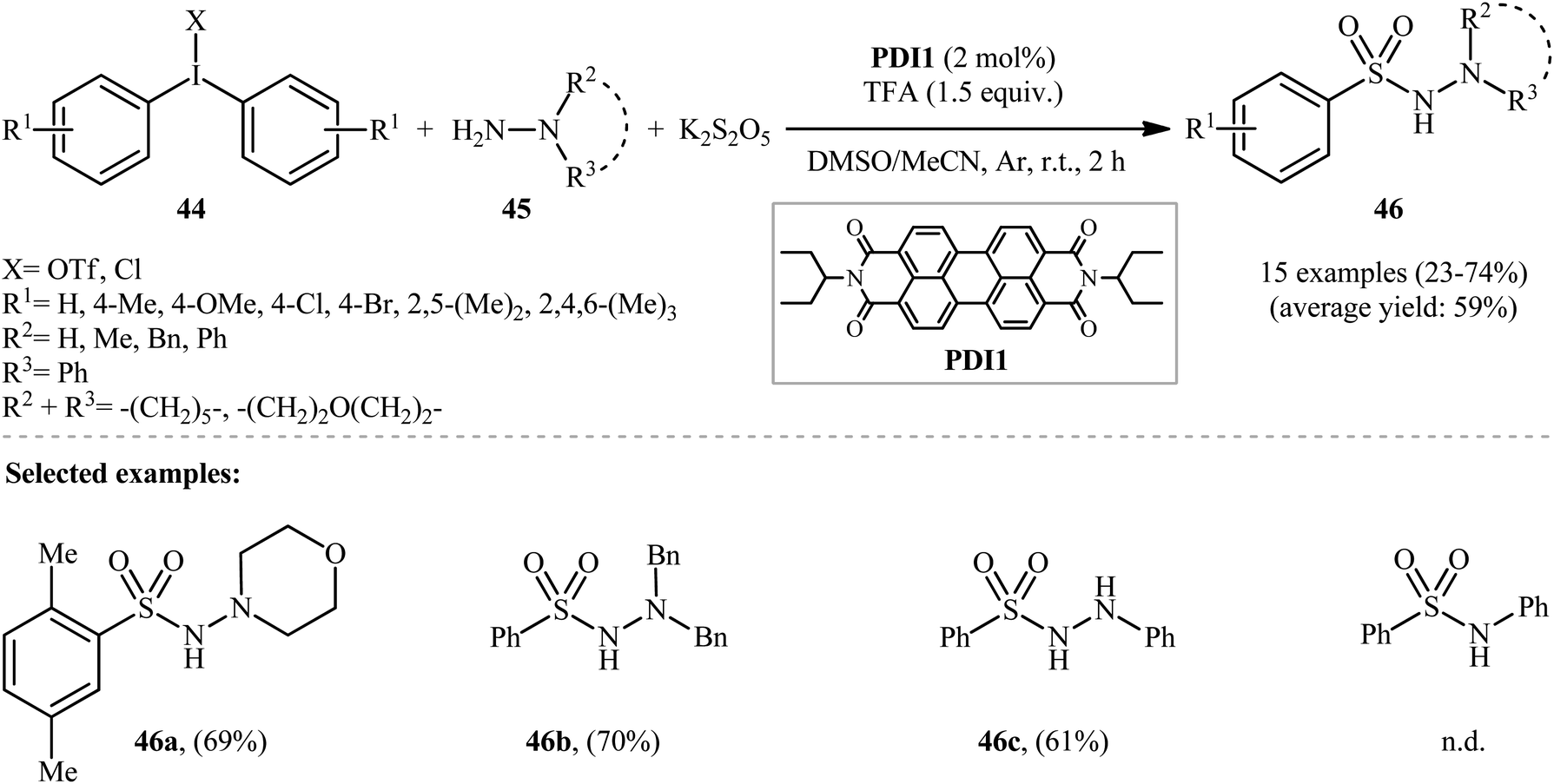 | ||
| Scheme 23 Visible-light photoredox-catalyzed hydrazinosulfonylation of diaryliodonium salts 44 with hydrazines 45 and SO2. | ||
Mechanistically, the authors suggested that the reaction proceeds through the following key steps (Scheme 24): (i) photoexcitation of the ground state photocatalyst (PDI) by visible light to form the excited state photocatalyst (PDI*); (ii) reductive quenching of PDI* with the hydrazine–SO2 complex XXXI (generated from hydrazine 45 and SO2) to produce the radical cation XXXII and the reduced catalyst PDI˙−; (iii) deprotonation of intermediate XXXII to afford radical adduct XXXIII; (iv) electron-transfer from PDI˙− onto diaryliodonium salt 44 to yield the reduced species XXXIV and the regenerated catalyst PDI in its ground state; (v) the fragmentation of intermediate XXXIV to generate aryl radical XXXV; and (vi) free-radical addition of XXXV with the hydrazine–SO2 adduct XXXIII to give the desired product 46.
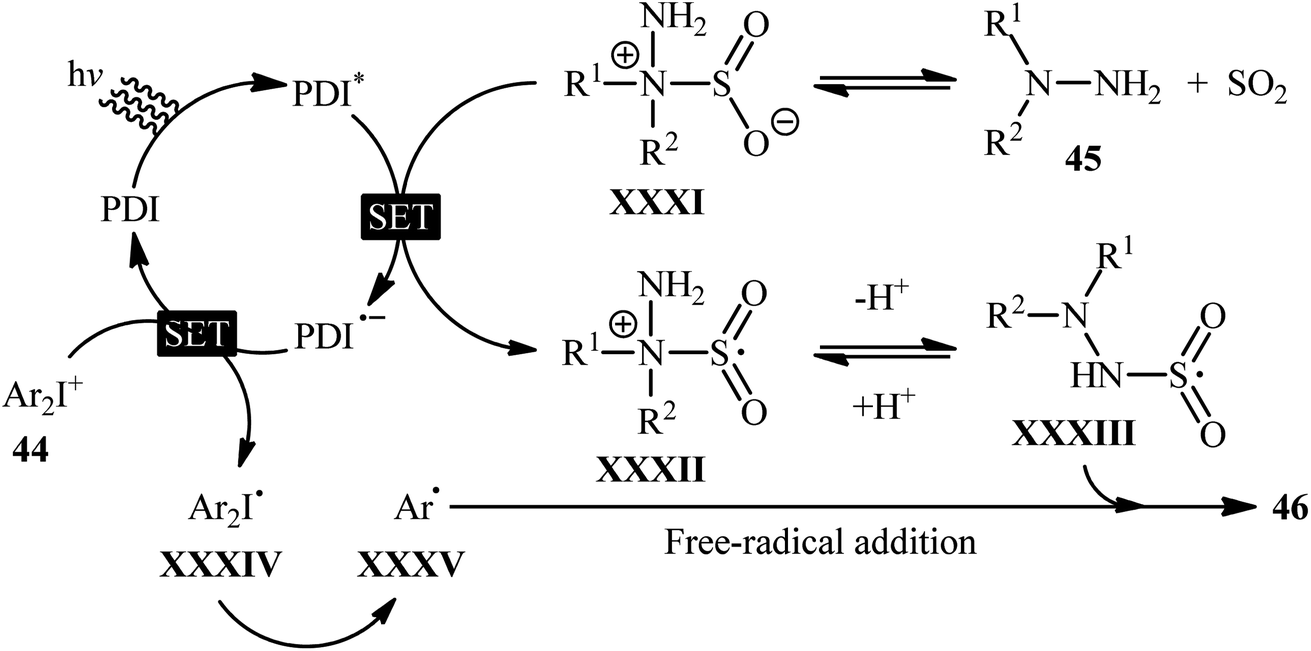 | ||
| Scheme 24 Proposed mechanism for the reaction in Scheme 23. | ||
8. Conclusion
In conclusion, the direct hydrazinosulfonylation of aryl electrophiles with SO2 and hydrazines has emerged as a promising strategy for the synthesis of aryl N-aminosulfonamides, offering advantages over conventional sulforylating agents that are often unstable, malodorous, or toxic. Despite the significant advances made in this field, several challenges remain to be addressed. For instance:(a) The majority of metal-catalyzed reactions covered in this review were conducted in the presence of palladium-based catalysts. Given cost and safety concerns, the development of related processes employing inexpensive and less toxic catalysts such as copper and nickel would be highly desirable.
(b) The existed methodologies largely limited to the use of 1,1-di-alkyl substituted hydrazines (mainly, morpholine). Therefore, of course, further research should be focused on development of hydrazinosulfonylation reactions with much broader substrate scope.
(c) Monosubstituted hydrazines were unsuitable substrates in the majority of reaction conditions described above. Thus, development of catalytic systems which allow the use of these substrates in hydrazinosulfonylation reactions will be desirable.
(d) The number of reported examples on the applicability of common aryl electrophiles (e.g., arylboronic acids and arylsilanes) in this page of aryl N-aminosulfonamides synthesis are narrow and there is future need to study the scope and limitations of these electrophiles.
(e) The applicability of other aryl electrophiles, such as non-toxic aryl fluorosulfates should be explored.
We hope that this review will be beneficial to stimulate scientists to further research and thinking on exciting field, and may ultimately lead to the discovery of new and useful compounds for various applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledged from Prof. Esmail Vessally for his useful comments and help.References
- M. Feng, B. Tang, H. S. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed.
- (a) P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed; (b) J. Yu and X. Jiang, Adv. Agron., 2023, 2, 3–14 CrossRef.
- (a) X. Zhang, K. Chen, Z. Sun, G. Hu, R. Xiao, H. M. Cheng and F. Li, Energy Environ. Sci., 2020, 13, 1076–1095 RSC; (b) Z. Shadike, S. Tan, Q. C. Wang, R. Lin, E. Hu, D. Qu and X. Q. Yang, Mater. Horiz., 2021, 8, 471–500 RSC.
- K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- C. Zhao, K. P. Rakesh, L. Ravidar, W. Y. Fang and H. L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- (a) F. Carta, C. T. Supuran and A. Scozzafava, Future Med. Chem., 2014, 6, 1149–1165 CrossRef CAS PubMed; (b) M. Abdoli, S. Giovannuzzi, C. T. Supuran and R. Žalubovskis, J. Enzyme Inhib. Med. Chem., 2022, 37, 1568–1576 CrossRef CAS; (c) M. Abdoli, V. De Luca, C. Capasso, C. T. Supuran and R. Žalubovskis, J. Enzyme Inhib. Med. Chem., 2023, 38, 2163243 CrossRef PubMed.
- (a) C. Kannigadu, J. Aucamp and D. D. N'Da, Chem. Biol. Drug Des., 2022, 100, 267–279 CrossRef CAS PubMed; (b) A. Khushal, A. Mumtaz, W. A. Shadoul, S. H. M. Zaidi, H. Rafique, A. Munir, A. Maalik, S. J. A. Shah, A. Baig, W. Khawaja and M. Al-Rashida, BioMed Res. Int., 2022, 5293349 CAS; (c) J. Liu, F. Li, Y. Wang, H. Zhang, Y. Li and Z. Li, Med. Chem. Res., 2020, 29, 495–503 CrossRef CAS; (d) P. Li, J. Zhou, Y. Liu and X. Wang, Phosphorus, Sulfur Silicon Relat. Elem., 2020, 195, 976–980 CrossRef CAS; (e) M. M. Shaaban, H. M. Ragab, K. Akaji, R. P. McGeary, A. E. A. Bekhit, W. M. Hussein, J. L. Kurz, B. H. Elwakil, S. A. Bekhit, T. M. Ibrahim and M. A. Mahran, Bioorg. Chem., 2020, 105, 104386 CrossRef CAS PubMed; (f) J. Liu, F. Li, Y. Wang, H. Zhang, Y. Li and Z. Li, Med. Chem. Res., 2020, 29, 495–503 CrossRef CAS.
- (a) D. M. Lemal, F. Menger and E. Coats, J. Am. Chem. Soc., 1964, 86, 2395–2401 CrossRef CAS; (b) D. Zheng, Y. Kuang and J. Wu, Org. Biomol. Chem., 2015, 13, 10370–10375 RSC; (c) J. B. Liu, H. Yan, H. X. Chen, Y. Luo, J. Weng and G. Lu, Chem. Commun., 2013, 49, 5268–5270 RSC; (d) J. B. Liu, L. Nie, H. Yan, L. H. Jiang, J. Weng and G. Lu, Org. Biomol. Chem., 2013, 11, 8014–8017 RSC; (e) C. S. Richards-Taylor, D. C. Blakemore and M. C. Willis, Chem. Sci., 2014, 5, 222–228 RSC; (f) Y. Luo, X. Pan, C. Chen, L. Yao and J. Wu, Chem. Commun., 2015, 51, 180–182 RSC.
- (a) C. S. Rooney, E. J. Cragoe, C. C. Porter and J. M. Sprague, J. Med. Pharm. Chem., 1962, 5, 155–167 CrossRef CAS PubMed; (b) J. Chen, Y. Zhang, W. Hao, R. Zhang and F. Yi, Tetrahedron, 2013, 69, 613–617 CrossRef CAS; (c) N. Zhao, Y. Li, Y. Wang and J. Wang, J. Sulfur Chem., 2006, 27, 427–432 CrossRef CAS; (d) S. Noda and S. Tanimori, Tetrahedron Green Chem, 2023, 1, 100001 CrossRef.
- (a) J. Wen, W. Wei, D. Yang, Y. Fan, L. Fu and H. Wang, Synth. Commun., 2015, 45, 1574–1584 CrossRef CAS; (b) W. Y. Chan and C. Berthelette, Tetrahedron Lett., 2002, 43, 4537–4540 CrossRef CAS; (c) B. Zhou and J. Xu, Org. Biomol. Chem., 2016, 14, 4918–4926 RSC; (d) J. Xu, B. Zhou and X. Yang, Synthesis, 2016, 49, 1632–1640 CrossRef.
- A. Berger and K. D. Wehrstedt, J. Loss Prev. Process Ind., 2010, 23, 734–739 CrossRef CAS.
- (a) K. Hofman, N. W. Liu and G. Manolikakes, Eur. J. Chem., 2018, 24, 11852–11863 CrossRef CAS PubMed; (b) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691–705 RSC; (c) D. Zeng, M. Wang, W. P. Deng and X. Jiang, Org. Chem. Front., 2020, 7, 3956–3966 RSC.
- B. Nguyen, E. J. Emmett and M. C. Willis, J. Am. Chem. Soc., 2010, 132, 16372–16373 CrossRef CAS PubMed.
- M. Wang and X. Jiang, Chem. Rec., 2021, 21, 3338–3355 CrossRef CAS PubMed.
- E. J. Emmett, C. S. Richards-Taylor, B. Nguyen, A. Garcia-Rubia, B. R. Hayter and M. C. Willis, Org. Biomol. Chem., 2012, 10, 4007–4014 RSC.
- S. Ye and J. Wu, Chem. Commun., 2012, 48, 10037–10039 RSC.
- W. Li, H. Li, P. Langer, M. Beller and X. F. Wu, Eur. J. Org. Chem., 2014, 3101–3103 CrossRef.
- Y. Li, D. Zheng, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 574–578 RSC.
- S. Ye, H. Wang, Q. Xiao, Q. Ding and J. Wu, Adv. Synth. Catal., 2014, 356, 3225–3230 CrossRef CAS.
- S. Ye and J. Wu, Chem. Commun., 2012, 48, 7753–7755 RSC.
- A. Roglans, A. Pla-Quintana and M. Moreno-Manas, Chem. Rev., 2006, 106, 4622–4643 CrossRef CAS PubMed.
- D. Zheng, Y. An, Z. Li and J. Wu, Angew. Chem., Int. Ed. Engl., 2014, 53, 2451–2454 CrossRef CAS PubMed.
- D. Zheng, Y. Li, Y. An and J. Wu, Chem. Commun., 2014, 50, 8886–8888 RSC.
- Y. An, D. Zheng and J. Wu, Chem. Commun., 2014, 50, 11746–11748 RSC.
- W. Li, M. Beller and X. F. Wu, Chem. Commun., 2014, 50, 9513–9516 RSC.
- Y. Wang, B. Du, W. Sha, H. Mei, J. Han and Y. Pan, Org. Chem. Front., 2017, 4, 1313–1317 RSC.
- M. Wang, B. C. Tang, J. G. Wang, J. C. Xiang, A. Y. Guan, P. P. Huang, W. Y. Guo, Y. D. Wu and A. X. Wu, Chem. Commun., 2018, 54, 7641–7644 RSC.
- M. Wang, B. C. Tang, J. C. Xiang, X. L. Chen, J. T. Ma, Y. D. Wu and A. X. Wu, Org. Lett., 2019, 21, 8934–8937 CrossRef CAS PubMed.
- Y. An, H. Xia and J. Wu, Org. Biomol. Chem., 2016, 14, 1665–1669 RSC.
- X. Wang, L. Xue and Z. Wang, Org. Lett., 2014, 16, 4056–4058 CrossRef CAS.
- A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, Chem. Sci., 2017, 8, 1233–1237 RSC.
- N. W. Liu, S. Liang and G. Manolikakes, Adv. Synth. Catal., 2017, 359, 1308–1319 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |