
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization and properties of sulfate-modified silver carbonate with enhanced visible light photocatalytic performance†
Sara Ghaziab,
Benaissa Rhouta *a,
Claire Tenderob and
Francis Mauryb
*a,
Claire Tenderob and
Francis Mauryb
aIMED-Lab, Sciences and Technologies Faculty, Cadi Ayyad University, Avenue Abdelkrim Khattabi, Box 549, Marrakech, Morocco
bCIRIMAT, Université de Toulouse, CNRS-UPS-INP, ENSIACET, 4 allée Emile Monso, BP 44362, 31030 Toulouse, cedex 4, France. E-mail: b.rhouta@uca.ma
First published on 31st July 2023
Abstract
Sulfate-modified Ag2CO3 was successfully synthesized via a simple precipitation method. Its visible light photocatalytic performance against the removal of Orange G was found to be significantly enhanced in comparison with the one of pure Ag2CO3. While SO42−-Ag2CO3 ensured a removal efficiency of 100% of OG within 30 min, the unmodified Ag2CO3 exhibited a degradation threshold at hardly 60%. Likewise, the degradation rate constant in the presence of SO42−-Ag2CO3 photocatalyst was assessed to be twice that determined upon the involvement of pristine Ag2CO3. Furthermore, Total Organic Carbon (TOC) measurements evidenced the occurrence of a quasi-total mineralization of the dye pollutant upon the use of SO42−-Ag2CO3 photocatalyst. Scavenger experiments highlighted the dominant role of photo-induced h+ along with ˙O3− ozonide radicals in the OG photocatalytic oxidation mechanism. Reuse cycles revealed that the modification by SO42− is a promising route to improve the stability of silver carbonate against photocorrosion. All these improvements could be ascribed to electronic transfer from the upper SO42− HOMO to the lower Ag2CO3 conduction band.
1 Introduction
Great attention was recently paid to silver based photocatalysts such as Ag2O,1 AgX (X = Cl, I and Br),2 Ag3PO4 (ref. 3) and Ag2CO3.4 This is due to their high visible light absorption that yields an effective photocatalytic degradation of organic pollutants from aqueous media.5,6 Nevertheless, their photoactivity efficiency depends on the anion nature since for instance, Ag-oxosalt photocatalytic performances increased in the order Ag3AsO4 > Ag3PO4 > Ag2CO3 > Ag2SeO4 > Ag2SO4.7 However, this order seemed to be controversial in that a recent study reported that Ag2CO3 appeared more photoactive and photostable than Ag3PO4.8 Likewise, this order depended on the nature of the pollutant.8 Despite this, Ag3PO4, has been the most studied in photocatalysis with about 900 research papers dedicated to this compound compared to about 150 for Ag2CO3 in the same period.9 This highest interest for Ag3PO4 was likely due to its better thermal stability and higher photoactivity, which could be further enhanced by doping.In this context, several studies reported Ag3PO4 doping with Ni2+ (ref. 10) or Cu2+ (ref. 11) cations, and SO42− (ref. 12) or CO32− anions.13 Considering anion doping, it is difficult to claim the achievement of the doping of an Ag-oxosalt by a polyatomic anion since the authors do not clearly explain how 5 ions (S6+ and O2−) issued for instance from SO42− group were distributed in the crystalline matrix. A more generic term than doping could be activation or treatment of the photocatalyst. For example, a so-called doping by SO42− could be similar to a doping by sulfur. Anyway, in the particular case of Ag3PO4 doping by SO42−, it was suggested that an electronic transfer occurred between SO42− and PO43−, more precisely from atomic orbitals S 3s23p4 to P 3s23p3.12 The photocatalyst Ag2CO3 crystallizes in a monoclinic structure significantly different than the cubic structure of Ag3PO4. In addition, it presents an optical band gap (≈2.4 eV) in the same order of magnitude as the silver phosphate and thus is photoactive under visible light irradiation.4 Nonetheless, in comparison with Ag3PO4, it was less studied as photocatalyst, probably due to the fact that, as other silver oxosalts, it exhibited drawbacks related for instance to a lower transfer rate of charge carriers14 and a higher sensitivity to photocorrosion.15 By contrast with Ag3PO4, fewer strategies have been explored to improve the performance of Ag2CO3.
To overcome these issues, several routes have been proposed such as heterojunctions between Ag2CO3 and another semiconductor, which can promote high separation of the photo-generated electrons–holes pairs. This approach has been extended to nanocomposites with metal nanoparticles as Ag to produce Schottky junctions. For this purpose, beneficial heterojunctions were reported for TiO2/Ag2CO3,16 ZnO/Ag2CO3,17 Ag/Ag2CO3,18 Ag2O/Ag2CO3,19 Ag3PO4/Ag2CO3 (ref. 20) and AgX (X = Cl, I and Br)/Ag2CO3.21 Another way22 reported the effect of varying types and ratios of solvents upon the synthesis in tunning the size and morphology of Ag2CO3 and consequently improving its photocatalytic activity and photostability. In another route, it was reported that the immobilization of Ag2CO3 particles on supports with a large specific surface area such as graphene oxide,23 C3N4 (ref. 24–26) and some clay minerals like palygorskite27 was also a promising way. Surprisingly, in contrast to Ag3PO4, anionic modification of Ag2CO3 was not investigated. In particular, the route of modification by SO42− was not yet reported for improving photocatalytic properties of Ag2CO3.
Since both Ag-oxosalts differ in their crystallographic structure and anion nature (size, charge, composition), the response of these photocatalysts to a strategy will not necessarily be the same from one to another. The fact that SO42− presents an ionic radius of the same order of magnitude than CO3 (ref. 2–28) should facilitate bulk modification of Ag2CO3. However, the differences both in crystallographic structure between Ag2CO3 and Ag3PO4, and in charge transfer expected between the modifying anion (SO42−) and crystal lattice anion (CO32−), does not allow to think that the effects of the modification by SO42− should be similar for Ag2CO3 and Ag3PO4.12 These differences increase the interest of exploring Ag2CO3 modification with SO42− as a route to enhance photocatalytic properties and paving a new way to improve the stability against photocorrosion for practical applications.
In this paper, the photocatalytic performances were assessed (i) by measuring the degradation rate of a model pollutant under visible light irradiation (kinetic data), (ii) by analyzing the extent of mineralization (TOC analysis), (iii) by determining the nature of photogenerated oxidizing species responsible of the dye photocatalytic degradation through scavenger experiments (resulting in hypothesis on the reaction mechanism) and (iv) by studying the photo-stability in reuse experiments.
2 Experimental
2.1 Synthesis of the photocatalysts
All reagents were of analytical grade and used without further purification. Pure Ag2CO3 was synthesized according to a method previously published.4,27 For Ag2CO3 treatment with SO42−, the method was adapted from that already reported for sulfate-doped Ag3PO4.12 In a typical procedure, 20 mL of a translucent mixture of AgNO3 (0.272 M) and Na2SO4 (0.0125 M) was prepared at room temperature in distilled water and stirred during 10 min for homogenization. Then 40 mL of Na2CO3 (0.068 M) was added dropwise under continuous stirring. The formed precipitate (i.e. SO42−-modified Ag2CO3) was collected by centrifugation, washed with distilled water and ethanol three times to remove reaction byproducts mainly NaNO3, Na2SO4 and traces of remaining reactants. The samples were dried at 60 °C overnight, then stored in the dark at room temperature before being used. With these synthesis conditions, the theoretical SO42− content of the photocatalyst cannot exceed 9 at% since this was the value of the SO42−/CO32− anion ratio used.2.2 Characterization techniques
The phase identification was carried out by X-ray diffraction (D8 Advance Diffractometer Bruker) with Cu Kα radiation source. The scanning was performed at room temperature in the 2θ range from 5° to 70° with a step size of 0.05° for 1 s. Infrared spectroscopy analysis was performed on a pellet of KBr–photocatalyst mixture (≈1 wt%) using a PerkinElmer FTIR in the range 400–4000 cm−1. The SEM microscopy observations were performed using a scanning electron microscope VEGATE-SCAN3 coupled with an EDAX analyzer for elemental analysis. UV-vis Diffuse Reflectance Spectra (DRS) were recorded with respect to PTFE as standard using a Cary 5000 UV-vis-NIR spectrophotometer for wavelengths ranging from 200 to 800 nm. The zeta potential of the synthesized Ag-oxosalt powders was measured at room temperature in an aqueous suspension using a Zetasizer apparatus from Nano ZS, Malvern instruments.2.3 Photocatalytic tests under visible light
The photocatalytic activity was evaluated by recording the temporal variation of Orange G (OG) concentration during its degradation under visible light irradiation in presence of the photocatalyst. OG is an anionic azo dye with the formula C16H10N2Na2O7S2. It was selected as model pollutant because it is frequently used as dye in textile industry, and it does not undergo photolysis under the conditions of this test. The visible light irradiation was provided by 4 light color/840 lamps (13 W each) coupled with a UV cut-off filter (λ < 400 nm). Practically, 25 mg of the photocatalyst was dispersed into a quartz photo-reactor containing 25 mL of OG (10−5 M) aqueous solution. It should be noted that such conditions were the same as the ones already established by Lakbita et al.27 to be optimal for the study of photocatalytic performances under visible light of Ag2CO3 supported palygorskite clay mineral in a similar slurry reactor. Afterwards, the suspension was stirred in the dark for 30 min to achieve adsorption–desorption equilibrium before to start the photocatalytic test by switching on the lamps. At regular time intervals, aliquots were picked up, centrifuged and OG concentration in supernatant was determined using UV-vis spectrophotometer from the intensity of the OG absorption band in the range 470–500 nm. It should be noted that a calibration curve in the Beer–Lambert linearity domain was checked beforehand in the low concentration range (10−7–10−4 M) that includes the test value (10−5 M). The experimental data reported here were the average of 3 photocatalytic tests and an error bar represented the dispersion of data.To check if the photocatalytic degradation of OG organic dye pollutant led to its partial or total mineralization, the Total Organic Carbon (TOC) corresponding to remaining organic dye plus possibly organic byproducts was determined after the test using a TOC analyzer (TOC-L CPH/CPN, Shimadzu). For this purpose, after the test, photocatalyst dispersion was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm during 5 min and the resulting supernatant was analyzed.
000 rpm during 5 min and the resulting supernatant was analyzed.
To further investigate the photocatalytic mechanism that would ensure the OG photocatalytic degradation, isopropanol (IPA) was utilized to quench hydroxyl radicals (˙OH),12 trichloromethane (TCM) served as scavenger of superoxide radical anions (˙O2−),29 indigo carmine (IC) used to trap the ozonide radicals (˙O3−)8 and ethylenediamine tetra-acetic acid disodium (EDTA) was used as a hole (h+) scavenger.30 Before adding Ag2CO3 or SO42−-Ag2CO3 powders and running photocatalysis experiment, 1 mM of each scavenger was added to the OG dye solution.31
3 Results and discussion
3.1 Bulk modification of Ag2CO3 by SO42−
As shown in Fig. 1, XRD patterns recorded on unmodified and SO42− modified Ag2CO3 appeared quite similar with several diffraction peaks amongst which the ones at 2θ around 18.32° (d100 = 4.85 Å), 18.54° (d020 = 4.78 Å), 20.5° (d110 = 4.32 Å), 32.74° (d![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 01 = 2.73 Å), 33.81° (d130 = 2.65 Å), 37.07° (d200 = 2.42 Å) and 39.60° (d031 = 2.27 Å). These peaks were indexed on the basis of the stable monoclinic Ag2CO3 phase (ICDD file no. 01-070-2184). This result confirmed that the Ag2CO3 treatment by SO42− had no effect on the crystallographic structure of the silver carbonate that remained monoclinic while several polytypes exist.27 Furthermore, the non-appearance of additional peaks in the SO42− modified Ag2CO3 pattern denoted the absence of any crystallized secondary phases as byproducts from the base reactions (e.g. Ag2SO4) or from side decomposition reactions (e.g. Ag2O, Ag). Regarding Ag2SO4, the corresponding main XRD peaks, expected at 31.13° (311) and 28.08° (040), were not present (Fig. 1). This is consistent with the fact that it is water-soluble and was therefore washed away from the final photocatalyst powder (see Section 2.1).
01 = 2.73 Å), 33.81° (d130 = 2.65 Å), 37.07° (d200 = 2.42 Å) and 39.60° (d031 = 2.27 Å). These peaks were indexed on the basis of the stable monoclinic Ag2CO3 phase (ICDD file no. 01-070-2184). This result confirmed that the Ag2CO3 treatment by SO42− had no effect on the crystallographic structure of the silver carbonate that remained monoclinic while several polytypes exist.27 Furthermore, the non-appearance of additional peaks in the SO42− modified Ag2CO3 pattern denoted the absence of any crystallized secondary phases as byproducts from the base reactions (e.g. Ag2SO4) or from side decomposition reactions (e.g. Ag2O, Ag). Regarding Ag2SO4, the corresponding main XRD peaks, expected at 31.13° (311) and 28.08° (040), were not present (Fig. 1). This is consistent with the fact that it is water-soluble and was therefore washed away from the final photocatalyst powder (see Section 2.1).
 | ||
| Fig. 1 X-ray diffraction patterns of pure Ag2CO3 and SO42− modified Ag2CO3 (left) and zoom in the 32.0°–34.5° angular region corresponding to ( 01) and (130) reflection planes of both Ag2CO3 and SO42−-Ag2CO3 (right). | ||
Further detailed analysis of the diffractograms interestingly revealed a slight shift of the peaks towards smaller 2θ angles. Indeed, in the 2θ region between 32.0° and 34.5°, ( 01) and (130) peaks were observed respectively at 32.74° and 33.81° for pure Ag2CO3 while they appeared at 32.64° and 33.69° for SO42− modified Ag2CO3 (Fig. 1b). The values of a, b, and c lattice parameters and β angle of unmodified- and SO42− modified Ag2CO3 samples, were calculated from all the XRD peaks, using the Unit Cell software.32 They were reported in Table S1 (ESI).† They revealed an increase of the lattice parameters for SO42−-modified Ag2CO3 with respect to unmodified Ag2CO3 corresponding to a volume expansion of the unit cell of ca. 1.0%, which was certainly due to the higher ionic radius of SO42− (0.218 nm) compared to the one of CO32− (0.189 nm).28 This finding ascertained that CO32− anions in the crystal lattice were substituted by modifying SO42− anions larger by 15% and hence confirmed the achievement of a bulk modification of Ag2CO3 by sulfate anions. This ruled out a simple surface functionalization by SO42− anions grafted on Ag2CO3 particles, which should not induce an expansion of the crystal lattice.
To complete the crystallographic analysis, the two photocatalysts were characterized by FTIR spectroscopy. In addition to the presence of H2O molecules that was revealed by absorption bands around 3200 and 1650 cm−1, both FTIR spectra exhibited characteristic absorption bands of Ag2CO3 (Fig. 2). The two intense bands at 1442 and 1377 cm−1, as well as those at 879 and 697 cm−1 may be assigned to the distinctive vibrations of the planar CO32− groups, i.e. deformation and stretching, respectively.33 Additional bands were exclusively detected in SO42−-modified Ag2CO3 sample with bands at 1113, 1056, and 606 cm−1 attributed to the stretching mode of S–O and O–S–O bonds in SO42− tetrahedra, as reported for Ag2SO4 elsewhere.34 Except the bands of CO32− and SO42− anions (and H2O traces), no other absorption band was observed in the sulfate modified Ag2CO3 photocatalyst. This result further supported the beforehand mentioned statement issued from XRD analysis according to which on one hand a bulk modification of Ag2CO3 with SO42− anions was successfully achieved rather than a sulfate anion grafting on the surface of Ag2CO3 particles and on the other hand the absence of secondary phases formation.
 | ||
| Fig. 2 FTIR spectra of pure Ag2CO3 (bottom) and SO42− modified Ag2CO3 (top). The absorption bands were assigned according to literature data of carbonates and sulfates including Ag2CO333 and Ag2SO4.34 | ||
SEM analyses showed that pure Ag2CO3 and SO42−-Ag2CO3 samples exhibited the same microstructure with micron-size and monodisperse particles with pretty similar polyhedral shapes (Fig. 3). They appeared well crystallized, in the form of facetted rhombohedra with an average size of 0.3 μm according to the Dynamic Light Scattering (DLS) particle size distribution analysis (Fig. S1†). This microstructure was similar to that previously reported for silver carbonate synthesized by the same method.27 Therefore, SO42−-modification did not change the microstructure of Ag2CO3 particles (Fig. 2). This should be consistent with the incorporation into the Ag2CO3 crystal lattice of only a very small amount of SO42− anions. This assumption was further supported by EDS analysis showing the SO42−-Ag2CO3 sample was mainly composed of Ag, C, O. Very small peak, corresponding to sulfur, was detected in the energy range (Fig. 3d). Moreover, the SEM-EDS elemental mapping of the SO42−-Ag2CO3 powder spread on the sample holder did not reveal any evidence for S-rich regions, which additionally confirmed the absence of secondary phase such as Ag2SO4 (Fig. 3). Thus, the S amount in SO42−-Ag2CO3 sample was estimated to be around 1.8 atomic% fraction, which is way lower than the 9% allowed by the initial proportion of reagents and confirms the Ag2CO3 modification with SO42−.
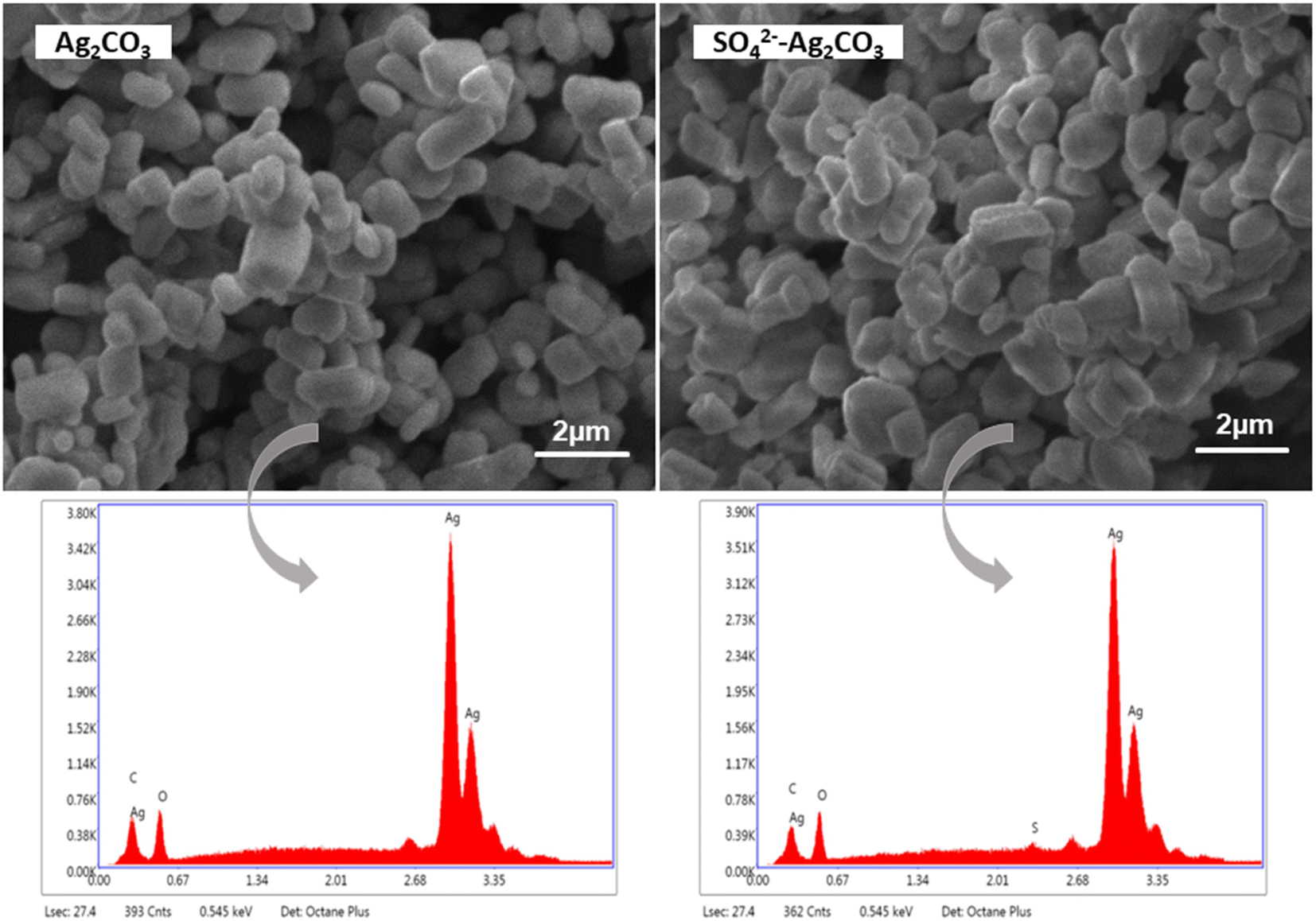 | ||
| Fig. 3 SEM micrographs of pure Ag2CO3 (left) and SO42− modified Ag2CO3 (right), and the EDS spectra corresponding to each image. | ||
UV-vis diffuse reflectance spectra recorded on pure Ag2CO3 and SO42− modified Ag2CO3 samples did not reveal significative differences. For both the samples, they displayed a high and sharp absorption below 500 nm, in the visible light range, denoting as expected their wide band gap semiconductor characters (Fig. 4a). In both the cases, there was no evidence of absorption band above 500 nm that could be due to plasmonic effect induced by Ag nanoparticles as reported elsewhere.35 The recorded UV-vis spectra herein were in good agreement with those previously reported for monoclinic Ag2CO3.4,8,15 The absorption edges, corresponding to the intersection of the extrapolation of the linear part of the absorption curve with the wavelength axis, were determined to be around 509 nm (2.44 eV) and 533 nm (2.33 eV) for pure Ag2CO3 and SO42−-modified Ag2CO3 samples, respectively (Fig. 4a).
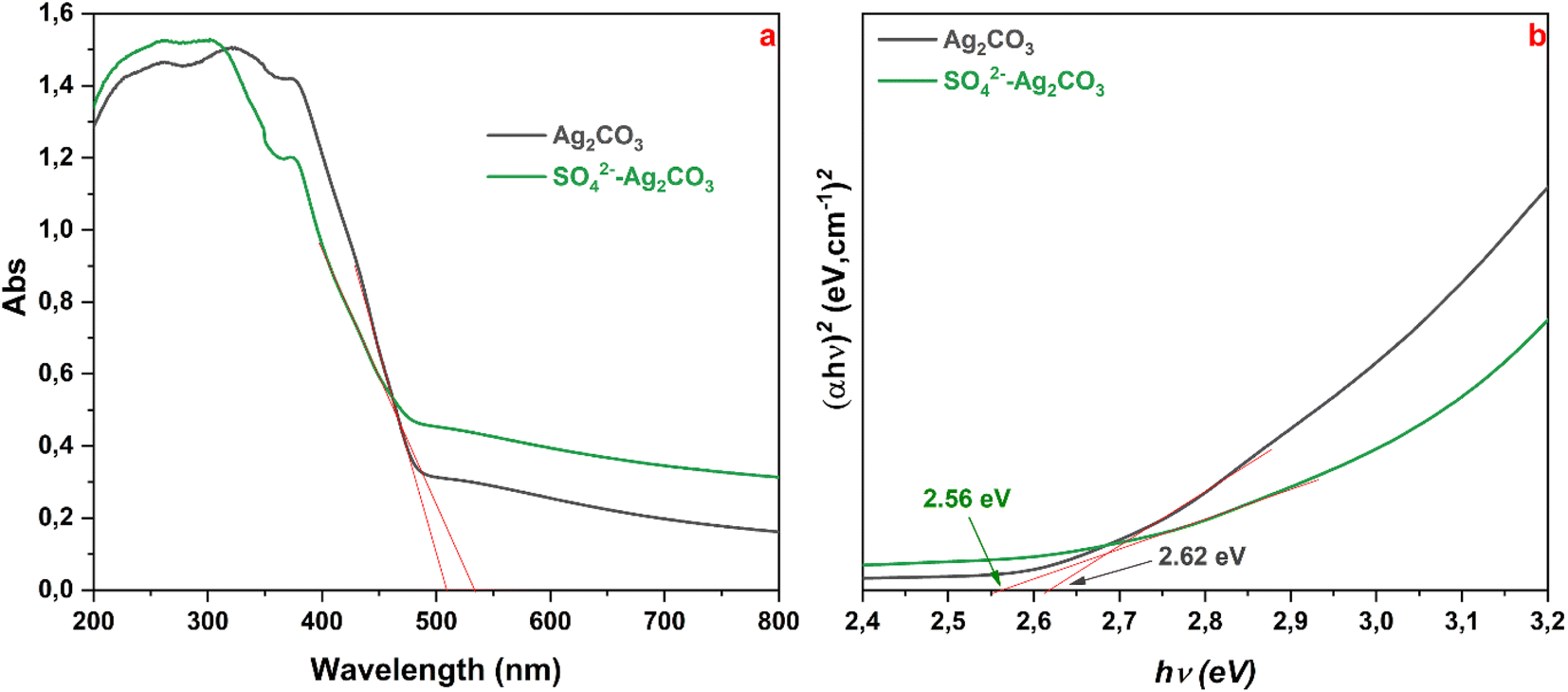 | ||
| Fig. 4 UV-vis DRS spectra of Ag2CO3 and SO42−-Ag2CO3 samples (a), and corresponding plot (αhν)2 vs. energy hν (b). Red straight lines are extrapolations to determine optical gap (absorption edge) from (a) and the Tauc band gap energy from (b). | ||
As both the Ag2CO3-based compounds are indirect semiconductors according to previously reported studies,4,7,8 the corresponding band gap energies (Eg) determined by the Tauc method4 were found to be about 2.62 and 2.56 eV, for unmodified and SO42− modified Ag2CO3 respectively (Fig. 4b). These Eg values were consistent with those of Ag2CO3 which were reported over a fairly wide range from 2.30 eV15 to 2.62 eV20 depending on the synthesis method, the crystallographic structure, the microstructure and purity (Table S2 in ESI†). In this respect, as beforehand evidenced that the treatment by SO42− did change neither the structure (Fig. 1) nor the microstructure of Ag2CO3, the slight decrease of Eg (≈2.3%) between both the band gap energies may result from the anionic modification effect. This SO42− effect contrasted with that exerted by Ag3PO4 doping by SO42− yielding rather to a slight increase of Eg as reported elsewhere.12 Therefore, SO42−-modified Ag2CO3 photocatalyst exhibited a slightly better absorption of the photons in the visible range. This means that, as more discussed below, more photo-generated electrons (e−) were transferred from the valence band (VB) to the conduction band (CB), leaving more holes (h+) in the VB that will be capable of oxidizing more organic species in primary reactions.
Furthermore, the valence band position (EVB) and the conduction band position (ECB) of SO42−-Ag2CO3 can be computed through the Mulliken electronegativity empirical equations:15
| EVB = χ − E0 + 0.5Eg | (1) |
| ECB = EVB − Eg | (2) |
3.2 Visible-light photocatalytic properties
The variation of OG dye concentration versus irradiation time under visible light in presence of pure Ag2CO3 and SO42−-Ag2CO3 photocatalysts was reported in Fig. 5. It is worth noting that keeping beforehand in the dark during 30 min the dispersion of the two photocatalysts in OG aqueous solution did not reveal OG adsorption onto the surface of the two photocatalysts due to electrostatic repulsions between anionic OG dye molecules and negatively charged surfaces of the photocatalysts. This was confirmed by the zeta potentials measured to be −23 mV for pure Ag2CO3 and −27 mV for SO42−-Ag2CO3, which was consistent with that reported elsewhere (−36 mV) for the pure silver carbonate.7 Furthermore, the concentration of OG solution free from photocatalyst appeared not decreasing during the whole duration of its exposure to visible light. This proved that OG dye did not undergo photolysis (Fig. 5a). | ||
| Fig. 5 Variation of the relative OG concentration vs. irradiation time over Ag2CO3 and SO42−-Ag2CO3 (a). Plots of −ln(Ct/C0) vs. irradiation time for the two photocatalysts (b). Proportion of Total Organic Carbon (TOC) removal analyzed upon OG photocatalytic degradation under visible light irradiation during 30 min (c). The errors bars in (a) and (c) correspond to the data dispersion from 2 experiments. | ||
In the presence of pure Ag2CO3, OG concentration considerably decreased during the first 20 minutes of irradiation to reach an OG degradation plateau after a removal of ca. 60% (Fig. 5a) beyond which no more dye photocatalytic degradation occurred. This striking behavior could be ascribed either to (i) surface Ag2CO3 poisoning by byproducts issued from photocatalytic reactions, or (ii) to photocorrosion of the photocatalyst induced by premature Ag2CO3 decomposition under light irradiation. The second assumption seemed to be of minor impact since the catalyst has been found still active in reuse experiments as further discussed in the Section 3.3. The observation of photocatalytic degradation threshold has already been reported upon assays involving pure Ag2CO3 against Methyl Orange (MO) anionic dye,7,8 and Rhodamine B (RhB)7,8,18 and Methylene Blue (MB)7 cationic dyes despite their electrostatic attraction by negatively Ag2CO3 surfaces. In contrast, the most salient result from the use of SO42−-Ag2CO3 photocatalyst was that the relative OG concentration continuously decreased during the photocatalytic test to reach remarkably a total removal of the dye pollutant within 30 min (Fig. 5a). This denoted its relative better photostability, i.e. a higher resistance to both surface poisoning and photo-corrosion issues as below evidenced in Section 3.3.
For both the pristine photocatalyst and SO42−-modified photocatalysts, OG dye photocatalytic degradation obeyed a pseudo first-order kinetics during approximately the first 15 min of visible light illumination. The rate constant assessed in this time range was about 5.1 × 10−2 min−1 for pure Ag2CO3 and 7.8 × 10−2 min−1 for SO42−-Ag2CO3 (Fig. 5b). This result denoted that the photocatalytic activity of the SO42−-modified sample was ca. 1.5 times faster than that of pure Ag2CO3.
The mineralization of OG, i.e. its ability to be converted into H2O and CO2 upon photocatalytic degradation, was also investigated. Fig. 5c depicted the total organic carbon (TOC) removal analyzed in the supernatant aqueous solution after a 30 min photocatalytic test using each photocatalyst. In presence of Ag2CO3, only around 52% of the TOC was removed, otherwise the proportion of OG and eventually its organic byproducts originated from photocatalytic reactions which was able to be mineralized. It should be noted that this OG amount was of the same magnitude order than that (≈60%) previously assessed by in situ UV-VIS spectrophotometry during the photocatalyst test. Nevertheless, it is worth noting that upon the use of SO42−-Ag2CO3, almost 94% of the TOC was removed, indicating a relatively huge mineralization of OG dye. Interestingly, it should be noted that this amount was fairly close to the 100% of OG photo-catalytically degraded assessed above by UV-VIS spectrophotometry.
3.3 Effect of SO42− modification on Ag2CO3 stability
The above presented results highlighted that modification of Ag2CO3 by SO42− could be an efficient route to enhance the photocatalytic activity under visible light with (i) a higher degradation rate, (ii) an almost complete decomposition of the organic pollutant and (iii) a high efficiency of mineralization of the OG dye. However, the effect of SO42− anions on both the stability of the photocatalyst and the photocatalytic oxidation mechanism needed to be investigated for further improvement and practical applications.To investigate the behavior of pure and sulfate-modified Ag2CO3 photocatalysts towards photo-corrosion issue, cycling experiments were performed. Fig. 6a reported the relative variation of OG concentration versus irradiation time under visible light recorded in presence of both the photocatalysts during three successive runs of 30 min each one. As previously mentioned, when using pure Ag2CO3, the dye photocatalytic degradation curve reached a plateau after 20 min of irradiation in the 1st cycle and the same behavior tended to be exhibited during the 2nd and 3rd cycles. It should be noted that as the cycles number increased, the maximal amount of OG removed decreased from approximately 60% at the end of the 1st cycle, to 45 and 35% after the 2nd and 3rd ones respectively. Accordingly, the corresponding rate constants deduced over the first 15 minutes, labelled R15, also rapidly decreased from 5.1 × 10−2 min−1 for the 1st cycle to 2.6 × 10−2 and 1.7 × 10−2 min−1 for the 2nd and 3rd ones, respectively (Fig. 6b). This steady decline of rate constants corresponded to an efficiency loss with respect to the 1st cycle of 50 and 65% for the 2nd and 3rd ones respectively. These results ascertained the poor stability of pure Ag2CO3 in agreement with elsewhere reported studies.8,15 The variation of the OG half-degradation time (t1/2) versus cycling recorded upon the use of Ag2CO3 (Fig. 6) showed that t1/2 of about 14 min for the 1st cycle strongly increased to 35 min for the 2nd one while it cannot be determined after 3 cycles since the total amount of OG removed did not exceed 35% (Fig. 6c). These results further supported previous deductions (Section 3.2) regarding the low stability and/or deactivation, by being sensitive to photo-corrosion and/or poisoning issues respectively, of the unmodified Ag2CO3.
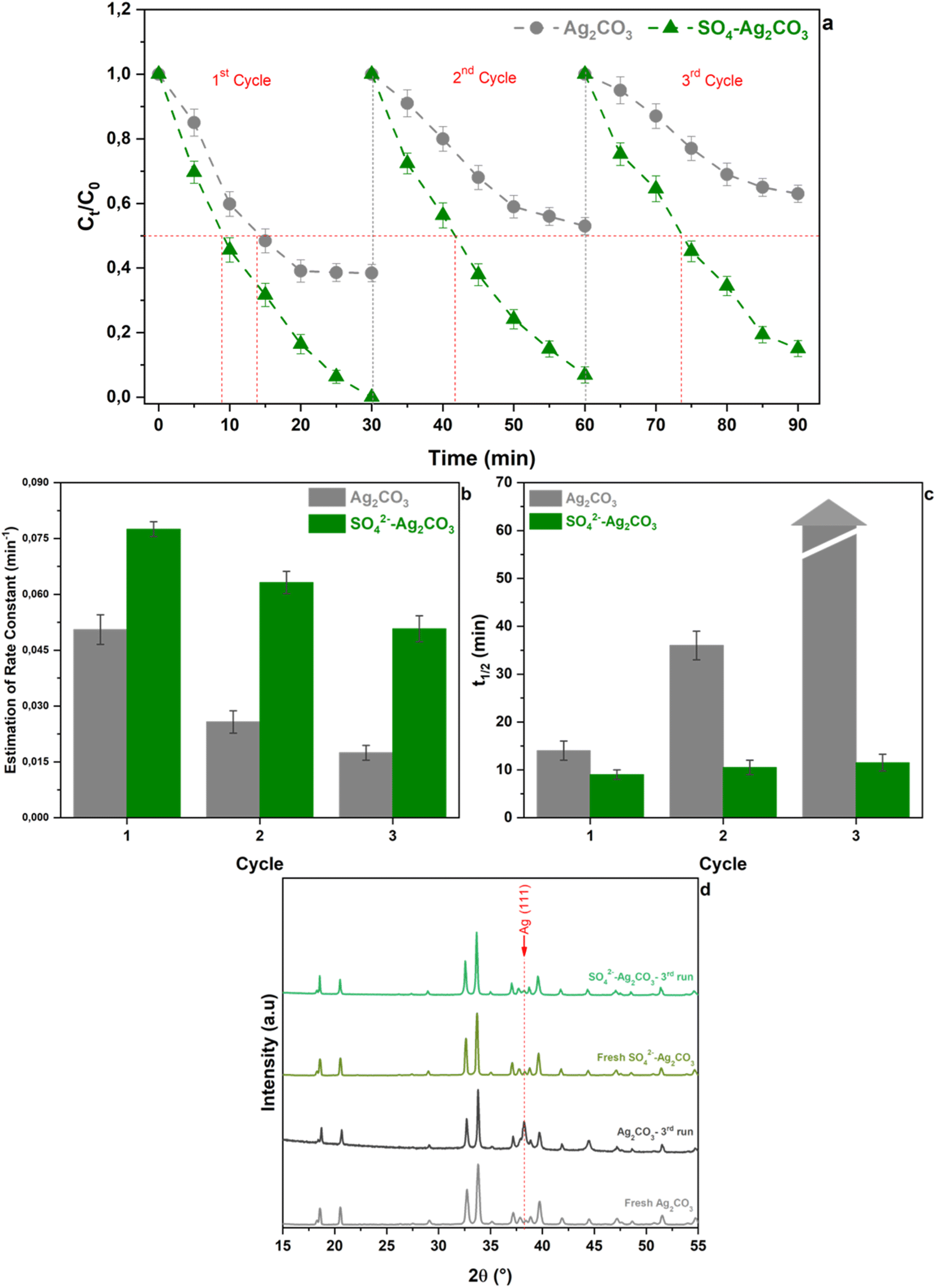 | ||
| Fig. 6 Cycling runs for OG photodegradation under visible light irradiation over pure Ag2CO3 and SO42−-Ag2CO3 (a). Rate constant (b) and half-photodegradation time of OG (t1/2) versus cycling of pure Ag2CO3 and SO42−-Ag2CO3 photocatalysts (c). X-ray diffraction patterns of unmodified Ag2CO3 and SO42− modified Ag2CO3 after the 3rd cycle (d). The errors bars in (a)–(c) correspond to the data dispersion from 2 experiments. | ||
By contrast, the OG concentration variation curves versus cycling, recorded in presence of SO42−-Ag2CO3 photocatalyst, showed a continuous and very noticeable OG photocatalytic degradation versus irradiation time under visible light regardless the cycling run number (Fig. 6a). In fact, while OG dye was completely removed after 30 min during the 1st cycle, denoting 100% of removal efficiency, the efficiency remained greater than 90% and 80% over the same period upon the 2nd and the 3rd cycles, respectively. Likewise, the corresponding R15 rate constants slightly decreased from 7.8 × 10−2 min−1 after the 1st cycle to 6.3 × 10−2 min−1 after the 2nd and to 5.1 × 10−2 min−1 after the 3rd cycle, which corresponded to a thorough efficiency loss of about 35% being slighter than that (65%) previously computed for pure Ag2CO3 (Fig. 6b). The better stability of SO42−-Ag2CO3 photocatalyst was also illustrated throughout the assessment of t1/2 values versus cycling. Fig. 6c revealed that t1/2 remained quite of the same magnitude order around 10 min regardless the cycle run number. Furthermore, powder XRD carried out on unmodified Ag2CO3 sample, recovered after the 3rd cycle, revealed the appearance of a very intense peak ascribed to (111) metallic silver (Ag) (Fig. 6d). This observation denoted that bare Ag2CO3 underwent photo-corrosion issue. Nevertheless, it is worth noting that such Ag peak was very hardly observed in SO42−-Ag2CO3 after the same run of photocatalysis test. The whole of these results concorded for proving that Ag2CO3 modification by SO42− likely appeared to be an effective and efficient way to improve the resistance of Ag2CO3 against photo-corrosion as well as poisoning issues, and thus to enhance photocatalytic activity.
3.4 Photocatalytic mechanism
In order to explore the photocatalytic mechanism of both Ag2CO3 and SO42−-Ag2CO3 photocatalysts, reactive species trapping experiments were performed to elucidate the main oxidants responsible of OG degradation under visible light irradiation, including hydroxyl radical (˙OH), superoxide radicals (˙O2−), ozonide radicals (˙O3−) and photoinduced holes (h+). The obtained results, gathered in Fig. 7(a) and (b), did not reveal significant differences in the behavior of both the investigated Ag2CO3 based photocatalysts towards the radicals trapping agents above-mentioned. Indeed, the addition of TCM, as ˙O2− scavenger, had no noticeable effect on OG photocatalytic degradation since the efficiency almost remained constant in both the cases like before using the trapper. This likely stood for the noninvolvement of ˙O2− in OG degradation. The use of IPA, as hydroxyl radical trapper, yielded a small decrease in the photocatalytic degradation efficiency of OG within 30 min from initially almost 62% and 100% to around 58% and 82% over unmodified and modified Ag2CO3 respectively. Hence, ˙OH radicals seemed to play a minor role in OG photocatalytic degradation. Nevertheless, it is noteworthy that using IC quencher of ˙O3− provoked a relatively huge decrease of the OG degradation efficiency estimated at almost 40% in both the cases. This denoted that the contribution of ˙O3− radicals in OG photocatalytic degradation may be substantial. The most salient effect was that of the addition of EDTA as h+ trapper which thoroughly made both the Ag2CO3 based photocatalysts unactive towards the OG removal. This likely ascertained that photoinduced holes (h+) was the major reactive species responsible of OG photocatalytic oxidation over both the investigated photocatalysts.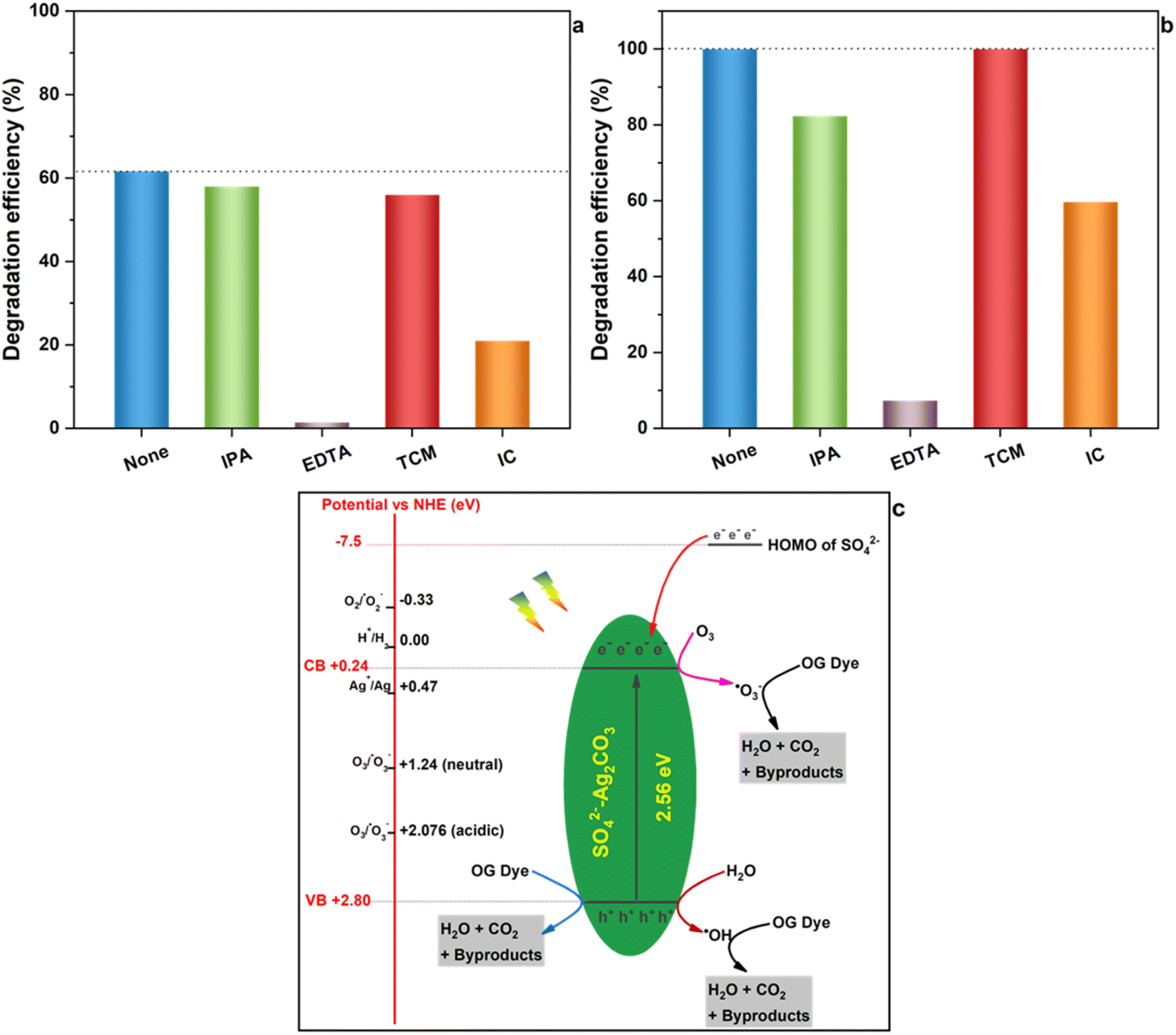 | ||
| Fig. 7 Photocatalytic degradation of OG by Ag2CO3 (a) and SO42−-Ag2CO3 (b) under visible light irradiation in the presence of scavengers: IPA, EDTA, TCM and IC. Schematic illustration of the plausible mechanism of OG degradation in presence of SO42−-Ag2CO3 photocatalyst (c). | ||
These results were further supported by the values of redox potentials of the top of valence band (VB) and the bottom of conduction band (CB) beforehand computed for both the Ag2CO3 based photocatalysts studied herein (Section 3.1) that were reported in Fig. 7c on which were also placed potentials of different reactive species in photocatalysis. In fact, the potential of photoelectrons on CB is too positive (0.24 eV) to be able to permit the reduction of dissolved O2 in aqueous medium into ˙O2− species whose potential is −0.33 eV. Therefore, ˙O2− entities cannot come into play in the OG photocatalytic degradation over Ag2CO3 based photocatalysts, and must be excluded as also emphasized by W. Jiang et al.8 This deduction was in contrast to results reported by several authors4,34 proposing the involvement of ˙O2− in the photodegradation of rhodamine B and/or Methylene Orange and/or Methylene Blue over silver carbonate although the potential of photoelectrons they estimated was 0.29 eV and 0.37 eV respectively, i.e. too positive with respect to the negative one characterizing O2/˙O2− couple. Nevertheless, the ozone, able to be provided from dissolved oxygen upon solution illumination,8 present an electrode potential in neutral and acidic medium of 1.24 eV and 2.076 eV respectively so that it can be reduced by photoelectrons generated herein (0.24 eV) to yield the formation of oxidative ˙O3− radicals which assured OG photocatalytic degradation onto both the investigated photocatalysts. Furthermore, taking into account the strongly oxidative electrode potential of photo-holes (h+) herein estimated of about 2.80 eV, these photoinduced entities assured direct OG degradation. Likewise, h+ specie could also hardly oxidize H2O to provide ˙OH radical whose the potential is close to its own (2.8 eV). This denoted that a part of h+ may indirectly participate through a minor amount of ˙OH radical in the OG photodegradation onto both the studied photocatalysts. In light of all these results, the OG photocatalytic degradation under visible light seemed to be achieved according to on the whole a similar mechanism illustrated in Fig. 7c onto unmodified and sulfate modified Ag2CO3. The plausible mechanism likely mainly implied photo-holes acting as latent oxidative entities along with to a relatively lesser extent ˙O3− reactive radicals and finally a minor participation of ˙OH hydroxyl radicals. Nonetheless, in view of the so proposed mechanism, one can ask how Ag2CO3 modification by SO42− improved photostability as well as photocatalytic efficiency beforehand evidenced (Section 3.2 and 3.3)?
One beneficial key role of SO42− anions would be their ability to contribute with 32 valence electrons to the lattice of the sulfate-modified Ag2CO3 photocatalyst, instead of 24 electrons for CO32− anions, providing therefore a surplus of 8 valence electrons. As reported by Bishop et al.,36 the Highest Occupied Molecular Orbital (HOMO) of SO42− is located at around −7.5 eV, so that sulfate electrons can be transferred to lower AgCO3 conduction band. Thus, these excess electrons provided by SO42− together with those photogenerated in AgCO3 upon illumination can reduce more ozone to produce more ˙O3− reactive entities which can act as additional reactive species as evidenced above by IC trapping experiments. Consequently, the OG photocatalytic degradation could speed up as proved by corresponding higher rate constant with respect to that of the pristine Ag2CO3 above-computed in Section 3.2.
Another possible key effect of SO42− on the improvement of Ag2CO3 photostability, beforehand evidenced upon photocatalytic tests (Section 3.2) and cycling experiments (Section 3.3), may be its capacity of electrons transfer to Ag+ higher than that permitted by CO32−. In fact, as widely reported, primary mechanism of photocorrosion of pure Ag2CO3 could be attributed to the reduction of Ag+ cations, issued from Ag2CO3 solubilizing, by photoinduced e−, which produced metallic Ag and, over time, a progressive Ag2CO3 depletion into Ag cations resulting at the end in the photocatalyst degradation.2,4,15 Considering an isolated Ag+–anion interaction, the fractional number of electrons transferred from the anion to Ag+ cation can be estimated, according to the Pearson model37 (details reported in the ESI†), to be about 0.29 and 0.21 for SO42− and CO32− respectively. This indicated that an interaction of SO42− with Ag+ cation was 38% greater than that of CO32−. Such an effect can contribute to improve the stability of the photocatalyst because Ag+ would be less reactive towards photoelectrons to form metal Ag, hence yielding photocorrosion issue to get mitigated.
4 Conclusion
Ag2CO3 modification with sulfate was achieved according to a simple precipitation method. This was confirmed by XRD that revealed a volume expansion of the monoclinic Ag2CO3 unit cell due to the substitution of CO32− groups by the larger SO42− anions. This was hence consistent with a bulk modification rather than a simple surface grafting of SO42− ions on Ag2CO3 particles. However, Ag2CO3 modification with SO42− did not significantly affect the Ag2CO3 band gap. The evaluation of the photocatalytic degradation of OG dye under visible light revealed that the sulfate modified Ag2CO3 was more active than bare Ag2CO3 leading to the achievement of a quasi-total mineralization within 30 min of irradiation. In addition, evidence for poisoning and/or photocorrosion issues observed for pure Ag2CO3 did not occur for the modified photocatalyst. The active species trapping experiments revealed that the OG photocatalytic degradation seemed to be assured over both the photocatalysts by photoinduced holes (h+) along with ˙O3− radicals, while ˙OH radicals would play a minor role. Improvements allowed by Ag2CO3 modification by SO42−, in terms of increase of photocatalytic rate and enhancement of Ag2CO3 stability, could be due to valence electrons added to photoinduced electrons, as a result of their transfer from SO42− HOMO to Ag2CO3 conduction band, which may together assure the formation of much active ˙O3− radicals entities. This proposed mechanism ought to be further validated by using more sophisticated characterization technique such as ESR and PL analyses and DFT calculations. On the whole, in view of the simple synthesis route of SO42− modified Ag2CO3 photocatalyst and its promising performances, in term of enhancement of photoactivity and photostability under visible light at laboratory scale, this study should be carried on by evaluating its photocatalytic properties in an outdoor solar pilot. This will increase its prospect of application in the field of tertiary treatment of wastewater.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The support from the “Programme de soutien aux Centres d’Etudes Doctorales, Franco-Moroccan cooperation” is gratefully acknowledged. The authors further thank the Center of Analysis and Characterization of Cadi Ayyad University, Marrakesh.References
- Y. Wang, N. Bi, H. Zhang, W. Tian, T. Zhang, P. Wu and W. Jiang, Colloids Surf., A, 2020, 585, 124105 CrossRef CAS.
- Y. Wang, H. Liu, B. Wu, T. Zhou, J. Wang, J. Zhou, S. Li, F. Cao and G. Qin, J. Alloys Compd., 2019, 776, 948–953 CrossRef CAS.
- U. Sulaeman, F. Febiyanto, S. Yin and T. Sato, Catal. Commun., 2016, 85, 22–25 CrossRef CAS.
- H. Dong, G. Chen, J. Sun, C. Li, Y. Yu and D. Chen, Appl. Catal., B, 2013, 134–135, 46–54 CrossRef CAS.
- G. Li, Y. Wang and L. Mao, RSC Adv., 2014, 4, 53649–53661 RSC.
- J. Li, W. Fang, C. Yu, W. Zhou, L. zhu and Y. Xie, Appl. Surf. Sci., 2015, 358, 46–56 CrossRef CAS.
- X. Jin, I. Y. Kim, Y. K. Jo, J. L. Bettis Jr, H.-J. Koo, M.-H. Whangbo and S.-J. Hwang, J. Phys. Chem. C, 2013, 117, 26509–26516 CrossRef CAS.
- W. Jiang, Y. Zeng, X. Wang, X. Yue, S. Yuan, H. Lu and B. Liang, Photochem. Photobiol., 2015, 91, 1315–1323 CrossRef CAS PubMed.
- Document search – Web of Science Core Collection, https://www.webofscience.com/wos/woscc/basic-search, (accessed November 9, 2022) Search PubMed.
- L. Song, Z. Chen, T. Li and S. Zhang, Mater. Chem. Phys., 2017, 186, 271–279 CrossRef CAS.
- H. El Masaoudi, I. Benabdallah, B. Jaber and M. Benaissa, Chem. Phys., 2021, 545, 111133 CrossRef CAS.
- W. Cao, Z. Gui, L. Chen, X. Zhu and Z. Qi, Appl. Catal., B, 2017, 200, 681–689 CrossRef CAS.
- J. Luo, Y. Luo, Q. Li, J. Yao, G. Duan and X. Liu, Colloids Surf., A, 2017, 535, 89–95 CrossRef CAS.
- H. Wang, J. Li, P. Huo, Y. Yan and Q. Guan, Appl. Surf. Sci., 2016, 366, 1–8 CrossRef CAS.
- G. Dai, J. Yu and G. Liu, J. Phys. Chem. C, 2012, 116, 15519–15524 CrossRef CAS.
- Y. Wang, P. Ren, C. Feng, X. Zheng, Z. Wang and D. Li, Mater. Lett., 2014, 115, 85–88 CrossRef CAS.
- Z. Xiang, J. Zhong, S. Huang, J. Li, J. Chen, T. Wang, M. Li and P. Wang, Mater. Sci. Semicond. Process., 2016, 52, 62–67 CrossRef CAS.
- N. Yu, R. Dong, J. Liu, K. Huang and B. Geng, RSC Adv., 2016, 6, 103938–103943 RSC.
- W.-K. Jo, S. Kumar, P. Yadav and S. Tonda, Appl. Surf. Sci., 2018, 445, 555–562 CrossRef CAS.
- W. Fa, P. Wang, B. Yue, F. Yang, D. Li and Z. Zheng, Chin. J. Catal., 2015, 36, 2186–2193 CrossRef CAS.
- J. Dostanić, D. Lončarević, V. Đorđević, S. P. Ahrenkiel and J. M. Nedeljković, J. Photochem. Photobiol., A, 2017, 336, 1–7 CrossRef.
- X. Yang, R. Li, Y. Wang, K. Wu, S. Chang and H. Tang, Ceram. Int., 2016, 42, 13411–13420 CrossRef CAS.
- C. Dong, K.-L. Wu, X.-W. Wei, X.-Z. Li, L. Liu, T.-H. Ding, J. Wang and Y. Ye, CrystEngComm, 2013, 16, 730–736 RSC.
- N. Tian, H. Huang, Y. He, Y. Guo and Y. Zhang, Colloids Surf., A, 2015, 467, 188–194 CrossRef CAS.
- C. Feng, L. Tang, Y. Deng, J. Wang, J. Luo, Y. Liu, X. Ouyang, H. Yang, J. Yu and J. Wang, Adv. Funct. Mater., 2020, 30, 2001922 CrossRef CAS.
- C. Feng, Z.-P. Wu, K.-W. Huang, J. Ye and H. Zhang, Adv. Mater., 2022, 34, 2200180 CrossRef CAS.
- O. Lakbita, B. Rhouta, F. Maury, F. Senocq, M. Amjoud and L. Daoudi, Appl. Surf. Sci., 2019, 464, 205–211 CrossRef CAS.
- M. C. Simoes, K. J. Hughes, D. B. Ingham, L. Ma and M. Pourkashanian, Inorg. Chem., 2017, 56, 7566–7573 CrossRef CAS PubMed.
- A. Bouziani, M. Yahya, Y. Naciri, A. Hsini, M. A. Khan, M. Sillanpää and G. Celik, Surf. Interfaces, 2022, 34, 102328 CrossRef CAS.
- K. Perumal, S. Shanavas, T. Ahamad, A. Karthigeyan and P. Murugakoothan, J. Environ. Sci., 2023, 125, 47–60 CrossRef.
- X. Bu, C. Chen, X. Zhao, Q. Huang, X. Liao, H. Fan, P. Wang, H. Hu, Y. Zhang and Z. Huang, Appl. Surf. Sci., 2022, 588, 152887 CrossRef CAS.
- T. J. B. Holland and S. A. T. Redfern, Mineral. Mag., 1997, 61, 65–77 CrossRef CAS.
- H. Zeng, Z. Yu, L. Shao, X. Li, M. Zhu, Y. Liu, X. Feng and X. Zhu, Desalination, 2020, 491, 114558 CrossRef CAS.
- E. Tomaszewicz, M. Kurzawa and L. Wachowski, J. Mater. Sci. Lett., 2002, 21, 547–549 CrossRef CAS.
- M. Rehan, A. Barhoum, T. A. Khattab, L. Gätjen and R. Wilken, Cellulose, 2019, 26, 5437–5453 CrossRef CAS.
- D. M. Bishop, M. Randič and J. R. Morton, J. Chem. Phys., 2004, 45, 1880–1885 CrossRef.
- R. G. Pearson, Inorg. Chem., 1988, 27, 734–740 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03120a |
| This journal is © The Royal Society of Chemistry 2023 |