
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(II) carboxylates and simple carboxamides: an inexpensive and modular catalyst system for the synthesis of PLLA and PLLA-PCL block copolymers†
Sarah Kirchhecker a,
Ngoc Nguyenb,
Stefan Reichertc,
Karola Lützowa,
Paul S. Eselem Bungua,
Axel Jacobi von Wangelinc,
Sebastian Sandlc and
Axel T. Neffe*ab
a,
Ngoc Nguyenb,
Stefan Reichertc,
Karola Lützowa,
Paul S. Eselem Bungua,
Axel Jacobi von Wangelinc,
Sebastian Sandlc and
Axel T. Neffe*ab
aInstitute of Active Polymers, Helmholtz-Zentrum Hereon, Kantstr. 55, 14513 Teltow, Germany. E-mail: axel.neffe@hereon.de
bInstitute for Technical and Macromolecular Chemistry, University of Hamburg, Bundesstrasse 55, 20146 Hamburg, Germany
cDepartment of Chemistry, University of Hamburg, Martin-Luther-King-Platz 6, 20146 Hamburg, Germany
First published on 7th June 2023
Abstract
The combination of inexpensive Fe(II) acetate with low molecular weight aliphatic carboxamides in situ generates an effective catalyst system for the ring opening polymerisation of lactones. PLLAs were produced in melt conditions with molar masses of up to 15 kg mol−1, narrow dispersity (Đ = 1.03), and without racemisation. The catalytic system was investigated in detail with regard to Fe(II) source, and steric and electronic effects of the amide's substituents. Furthermore, the synthesis of PLLA-PCL block copolymers of very low randomness was achieved. This commercially available, inexpensive, modular, and user-friendly catalyst mixture may be suitable for polymers with biomedical applications.
Introduction
Polymers which can degrade over short and medium time spans are important in many different areas of application. Regenerative medicine1,2 requires biocompatible materials which can be resorbed by the body after a defined amount of time. Inbuilt degradability can also improve the environmental impact of single-use packaging,3 when recycling is not possible or not practised.4,5 Aliphatic polyesters, which can degrade by hydrolysis, are an important class of degradable materials. The major synthetic routes are polycondensation of diols and dicarboxylic acids and ring-opening (co)polymerisation (ROP) of lactones such as lactide, glycolide, ε-caprolactone or para-dioxanone.6 The latter method generally achieves higher molar masses and narrower dispersities.Poly(L-lactic acid) (PLLA)7,8 and poly(ε-caprolactone) (PCL)9,10 are complimentary polymers with regard to their mechanical properties, drug permeabilities, and physiological degradation rates.11,12 Consequently, copolymers of the two monomers L-lactide and ε-caprolactone can provide materials with tailor-made properties for a wide range of applications. Precise control over the copolymerisation reaction enables the preparation of a range of different microstructures ranging from diblock to completely random distributions of the monomer units. Both starting materials and their copolymers are non-toxic and can be derived from biorenewable resources. Lactic acid (LA) for use in polymerisations can be produced by the fermentation of starch13,14 or non-edible lignocellulose15 and by chemical methods from cellulose16 or glycerol.17,18 Besides chemical manufacture, several syntheses of ε-caprolactone (CL) from biomass have been reported, such as via 5-hydroxymethylfurfural19 or enzymatic routes.20
ROP of lactones can be effected under various conditions with different catalysts, such as organocatalysts,21,22 enzymes,23 Lewis acids or bases,24 or metal–organic complexes.25 The latter class of catalysts, metal complexes, operate via a coordination-insertion mechanism and have been most widely employed.
Tin(II) octanoate (Sn(Oct)2) is the prototypical catalyst with a well-established efficacy in the ROP of many lactones.26,27 Despite numerous investigations into alternative metal catalysts that provide good control of sequence structures28,29 and relative stereochemistries,30 tin(II)octanoate remains the only commercially applied catalyst for the production of polylactide. Until very recently, it has not been surpassed under industrially relevant conditions.31,32 However, many tin compounds are toxic, severely limiting applications in biological settings such as the human body or ecosystems and requires careful removal of residuals.33–36 Tin(II)octanoate, in particular, is suspected of damaging fertility or the unborn child and is harmful to aquatic life with long lasting effects such that the general use raises questions. The problematic eco-profile of tin-containing chemicals and chemical reactions has prompted great efforts toward the design of alternative metal catalysts for polyester synthesis, such as zinc37–40 and iron.
Iron is one of the most abundant metals on earth. As an essential nutrient and component of many human proteins, such as enzymes, its availability in the body is regulated via several transport and storage mechanisms. The high natural abundance of iron results in much lower market prices than tin, and its production rates are high and stable and have a low environmental impact.41,42 Still, comparatively little research has been directed at using inexpensive iron catalysts for effective ROP reactions.43–51 Several ligands have been evaluated in coordination complexes with Fe(II) and Fe(III) ions. Only a few catalysts were demonstrated to give reasonably good activity and control over the molar mass of the obtained polymer.11,37,52 Many of these studies were performed in solution, sometimes using the toxic propylene oxide as solvent and precursor to the initiating species.53–57 Polymerisation in the melt is preferable with regard to processing and environmental impact. The use of simple iron(III) catalysts were reported for CL58 and LA,59,60 but in most cases, relatively high dispersities were obtained.61,62 Fe(II) acetate (Fe(OAc)2) and several other Fe(II) carboxylates were employed for the ROP of L-lactide ((L-LA)2) in the melt.63 They required high temperatures of 190–210 °C to proceed efficiently but could reach molar masses of up to 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 for Fe(OAc)2 and 150000 g mol−1 for Fe(II)isobutyrate. However, the dispersities were very broad, typically between 2.5 and 5. There was also evidence of racemisation at the required high reaction temperatures.
000 g mol−1 for Fe(OAc)2 and 150000 g mol−1 for Fe(II)isobutyrate. However, the dispersities were very broad, typically between 2.5 and 5. There was also evidence of racemisation at the required high reaction temperatures.
Recently, iron complexes with N–O donor ligands consisting of guanidines fused with benzoate esters were demonstrated to give faster rates than their zinc analogues and to catalyse lactide polymerisation at similar or even higher rates than Sn(Oct)2 under industrial conditions.32,37 The N–O ligand chelates the Fe via the N of guanidine and carbonyl O of the benzoate ester. Very recently, the same catalyst was successfully applied to the copolymerisation of LA and CL.11
It is important to note that in copolymerisation, the reaction rates of each monomer differ with different catalysts, and the rates directly impact the copolymer sequence structure. With large reactivity differences between the different monomers, fully random copolymers cannot be produced; rather, gradient structures are obtained. Sn(Oct)2 and other catalysts that allow rapid transesterifications between polyester chains can provide more randomized structures at long reaction times, but mostly with higher dispersities.64 For the synthesis of block copolymers, high transesterification rates are undesirable. Fully random copolymers of (L-LA)2 and CL are very difficult to prepare due to the significantly different reactivities with most catalysts. However, careful stereoelectronic ligand design can reduce LA polymerisation rates and produce nearly fully random copolymers.65–67 Catalysts of easily available, inexpensive, non-toxic metals that effectively catalyse the ROP of lactones under mild conditions and provide good control over molar mass, dispersity, and randomness are still in great demand. In the extension of the rather few literature reports on iron-catalysed ROP, our group has reported the successful polymerisation of morpholine-2,5-dione with Fe(OAc)2,68 and the beneficial effect of carboxamides as non-polymerisable catalytic adjuncts (NPCA) in the polymerisation of L-lactide by Fe(OAc)2.69 The addition of 10 wt% of the NPCA carboxamide (vs. the carbonyl units of the monomer) allowed for effective polymerisation at lower temperatures and with narrower dispersities than with Fe(OAc)2 alone. Our previous studies suggest that the carboxamide coordinates to the Fe centre via the carbonyl oxygen as a transient ligand (Scheme 1). In fact, the addition of N-methyl pyrrolidone (NMP), a cyclic tertiary amide, has been shown to enhance the catalytic activity of iron in cross-coupling reactions which enables the formation of catalytically active monomeric ferrates by coordination.70–73 The crucial role of the carboxamide was further investigated in this study by systematic variation of the amide substituents to assess the contribution of electronic and steric factors. In comparison, the effect of a defined Fe(II) carboxylate consisting of a sterically demanding pivalate ligand, was investigated.74
 | ||
| Scheme 1 Illustration of a hypothetical iron(II) complex bearing L-lactide, carboxamide, and alkoxide ligands. | ||
The resulting polyesters were analysed by NMR, MALDI-ToF-MS, GPC, and polarimetry. The optimised catalytic system was then applied to the synthesis of PLLAs of different defined molar masses and the copolymerisation of (L-LA)2 and CL.
Experimental
Materials
L-Lactide and 4-N,N-dimethylaminoacetophenon (TCI, Eschborn, Germany), 1,8-octanediol (Sigma-Aldrich, Munich, Germany), N-tert-butylacetamide (Fluorochem, Hadfield, UK), and acetanilide (Merck, Darmstadt, Germany) were purified by recrystallisation (twice) from THF. 2-Chloro-N,N-diethylacetamide (Sigma-Aldrich, Munich, Germany), and 1,3-dimethyl-2-imidazolidone (ABCR, Karlsruhe, Germany) were dried over CaH2 at room temperature under stirring for 24 h, and after separation from the hydride degassed with nitrogen. N-Ethylacetamide, N,N-diethylacetamide, N,N-dimethylacetamide and tetramethylurea (Merck, Darmstadt, Germany) were dried over CaH2 at room temperature for 16 h, followed by distillation and storage under N2. Urea (Merck, Darmstadt, Germany or BASF, Ludwigshafen, Germany), was used as received or recrystallised from dry ethanol and stored under N2 until use. ε-Caprolactone (99%, Sigma-Aldrich, Munich, Germany) was distilled under vacuum and stored under N2 over molecular sieves. 1-Hexanol (Acros Organics, Nidderau, Germany; 99%, extra dry) was used as received. Iron(II) acetate (99.995%, Sigma-Aldrich, Munich, Germany) was stored under N2 atmosphere.Methods
1H- and 13C-NMR spectra were recorded at 298 K on an Avance Neo 700 MHz spectrometer equipped with a triple resonance inverse Prodigy cryoprobe (Bruker, Rheinstetten, Germany; software Topspin version 4) in CDCl3. The determination of the number average molar mass Mn from the 1H spectra was performed by comparing the integrals of the CH2–O group of the initiator (4.11–3.96 ppm) with the CH protons of the lactic acid unit (5.33–4.86 and 4.37–4.22 ppm).GPC measurements were carried out using chloroform as an eluent at 35 °C with a flow rate of 1 mL min−1 in the presence of 0.2 wt% toluene as internal standard. The 1260 Infinity II PSS Security GPC system (Polymer Standards Service, Mainz, Germany) was equipped with an isocratic pump, a degasser, an autosampler, a column-heating compartment, a UV and a RI detector. Two further detectors were used: a BI-MwA light scattering (Brookhaven Instruments, Holtsville, New York) and Security DVD 1260 viscometer (Polymer Standards Service GmbH, Mainz, Germany, PSS), which were used for universal calibration to determine absolute polymer molar mass. Size separation was achieved using a VS Lux, 10 μm, 50 mm × 8 mm ID pre-column and two SDV 10 μm, 300 mm × 8.0 mm ID analytical, linear XL columns (Polymer Standards Service GmbH, Mainz, Germany, PSS). Polymer molar masses were evaluated with the help of WINGPC UniChrom software Build 9050 (Polymer Standards Service GmbH, Mainz, Germany, PSS) using a universal calibration, which was obtained by applying polystyrene standards with Mn between 580 g mol−1 and 975000 g mol−1 (Polymer Standards Service GmbH, Mainz, Germany).
To narrow down the molar mass heterogeneity of the PLLA-urea sample, preparative fractions were collected from the GPC eluent using the 1260 Infinity II fraction collector (Polymer Standards Service, Mainz, Germany). For the preparative fractionation experiment, a solution (8 wt%) was made by dissolving 160 mg in 20 mL chloroform. 100 μL of this solution was injected into the GPC column, and five fractions of 2 mL each were collected between 16.5 and 26.5 min elution times. The solvent was removed by evaporation, and the obtained fractions were dried overnight under a vacuum at room temperature. Sample recovery of approximately 90 wt% was achieved.
Differential scanning calorimetry (DSC) measurements were performed on a Netzsch DSC 204, Selb, Germany. The experiments were carried out under continuous nitrogen flow by heating a sample from room temperature up to 200 °C. This was followed by two cycles of cooling to −70 °C and heating to 200 °C at 10 K min−1.
The specific rotation [α]D of polymer solutions in chloroform was measured at a concentration of 5 mg mL−1 in chloroform at 22 °C, using a P-2000 polarimeter (Jasco, Groß Umstadt, Germany).
Mass spectra were recorded on an UltrafleXtreme MALDI-ToF-MS spectrometer (Bruker, Bremen, Germany) equipped with a Smartbeam-II laser. trans-2-[3-(4-tert-Butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) was used as matrix and NaI or NaTFA to ionisation by Na+ addition. Polymer (5 mg mL−1 in THF or CHCl3), matrix (10 mg mL−1 in THF) and sodium ion source (10 mg mL−1 in THF) were mixed in a 1:10:1 or 5:10:1 volume ratio and then 1 μL pipetted on the matrix target for analysis. Some samples were also measured on a Bruker UltrafleXtreme but using DHB as matrix without an additional sodium source.
Synthesis
Elementary analysis: found: C: 46.76%, H: 7.08%; calc. for C10H18FeO4: C: 46.54%, H: 7.03%.
1H-NMR (700 MHz, CDCl3) δ = 5.17–5.14 (q, J = 7.1 Hz, 200H, CHα), 4.37–4.33 (q, J = 6.7 Hz, 2H, CHα next to initiator), 4.11 (tt, J = 10.8, 4.3 Hz, 4H, CH2O initiator), 1.86–1.47 (m, 647H, CH3 plus CH2 and CH3 of initiator) ppm;
13C-NMR (176 MHz, CDCl3) δ = 169.74, (CHCOO), 69.14, (OCHCO), 65.70 (OCH2CH2), 29.15, (OCH2CH2), 28.55, (2 CH2CH2CH2CH2O), 25.7,77 (2 CH2CH2CH2CH2O), 16.78 (CHCH3) ppm.
1H-NMR (700 MHz, CDCl3) δ = 5.16 (q, J = 7.1 Hz, CH, LA–LA dyad, 40H), 5.11 (q, J = 7.2 Hz, LA-CL dyad; partially overlapping with LA–LA dyad, ∼2H), 4.35 (q, Ha next to initiator, 1H), 4.21–4.11 (m, O–CH2, CL–LA dyad, 4H, overlapping with CH2O of hexanol next to a LA (if existing)), 4.05 (t, J = 6.7 Hz, O–CH2, CL–CL dyad, 88H), 2.39 (t, J = 7.5 Hz, CO–CH2, LA–CL dyad, 2H), 2.30 (t, J = 7.5 Hz, CL–CL dyad, 88H), 1.66–1.61 (m, 182H), 1.59–1.56 (m, 118H), 1.50–1.46 (m, CH2 hexanol, 8H), 1.41–1.35 (m, 88H, CH3), 1.31–1.26 (CH2 hexanol, 8H), 0.88 (t, J = 6.8 Hz, 3H, CH3 of hexanol) ppm.
13C-NMR (176 MHz, CDCl3) δ = 173.67, 169.67, 69.13, 64.64, 64.27, 34.24, 31.54, 28.48, 25.65, 24.70, 22.65, 20.66, 16.77, 14.12 ppm.
1H-NMR (700 MHz, CDCl3) δ = 5.39–5.07 (m, 92H), 4.98–4.55 (m, 135H), 4.39–4.22 (m, 2H), 4.20–4.07 (m, 2H), 3.49 (bs, 2H), 3.28 (m, 1H), 2.67 (bs, 1H), 1.77–1.38 (m, 304H), 1.39–1.24 (m, 8H), 1.14 (t, J = 7.3 Hz, 2H), 0.89 (t, J = 7.0 Hz, 3H) ppm; 13C-NMR (176 MHz, CDCl3) δ = 169.61, 169.54, 169.47, 169.33, 166.52, 166.44, 166.41, 166.36, 77.21, 77.03, 76.84, 72.47, 69.33, 69.22, 69.12, 69.02, 68.96, 60.95, 60.88, 60.82, 60.76, 60.67, 16.75, 16.70, 16.65, 16.61, 15.83, 13.97 ppm.
Results & discussion
Optimisation of catalyst system
|
|||||||
|---|---|---|---|---|---|---|---|
| Polymer | Conv. [%] | Ini % (NMR) | Mn NMR [g mol−1] | Mn MALDI [g mol−1] | Mw MALDI [g mol−1] | Đ MALDI | 2n/(2n + 1) [%] |
| a Reaction conditions: 105 °C, 3 h, 10 mol% carboxamide w.r.t. LA, initiator: 1/104 equiv. 1,8-octanediol (target molar mass: 15 kg mol−1).b MALDI-ToF-MS values were determined separately for the octanediol- and water-initiated species using the fractionated samples.c Some (L-LA)2 was lost from the reaction by sublimation to the neck of the flask. Conv.: conversion, ini %: percent of product molecules in which the initiator was incorporated, Mn: number-average molar mass, Mw: weight-average molar mass, Đ: dispersity, 2n/(2n + 1): relative content of polymers having 2n LA units, calculated from MALDI-ToF-MS spectra; polymers having 2n + 1 LA units could only be produced by transesterification. | |||||||
| PLLA69 | 0 | — | — | — | — | — | — |
| PLLA-EAA | 77 | 74 | 8790 | 8655 | 9000 | 1.04 | 66 |
| PLLA-DEAA | 93 | 94 | 11280 |
9780 | 10120 |
1.03 | 79 |
| PLLA-DMAA | 93 | 99 | 9424 | 7745 | 7989 | 1.03 | 68 |
| PLLA-tBuAA | 44 | 75 | 4040 | 2395 | 3440 | 1.44 | 58 |
| PLLA-AcAn | 29 | 83 | 4160 | 2600 | 2990 | 1.15 | 67 |
| PLLA-DMAAP | 15 | 79 | 2440 | 1665 | 1825 | 1.10 | 69 |
| PLLA-ClDEAA | 39 | 89 | 4980 | 2670 | 3350 | 1.26 | 62 |
| 4045 | — | — | — | — | |||
| PLLA-ureab | 88c | 45 | 5490 | 6120 | 6255 | 1.02 | 78 |
| 1572 | 2503 | 2652 | 1.06 | — | |||
| PLLA-TMU | 87 | 91 | 9885 | 9470 | 9820 | 1.04 | 68 |

MALDI-ToF-MS analyses (Fig. 1 and ESI†) displayed two series, 2n and 2n + 1. The major polymer is the octanediol-initiated polymer (+Na+). The series of lower intensity has a mass difference of 72 g mol−1, which corresponds to one LA unit. The ROP of the lactide dimer leads to a polymer of even numbers of LA units (2n). Transesterification, however, can happen at any LA unit in the polymer chain, leading to the formation of PLLAs with uneven numbers of LA repeating units (2n + 1). The ratio of the intensities between these two series (2n/(2n + 1)) can therefore be used as a measure for the transesterification activity of the catalyst and the reaction conditions. The values are listed in Table 1, along with the number and weight average molar masses and dispersity (Đ) determined from the MALDI-ToF-MS spectra. In the lower molar mass range, some cyclics and methanol-terminated linear oligomers were detected, as well as H2O-initiated species, if these were long enough.
 | ||
| Fig. 1 Representative MALDI-ToF-MS spectrum. (A) Full spectrum of PLLA-DEAA. (B) excerpt showing the two series (2n and (2n + 1)). | ||
N-Ethyl acetamide (EAA, 1, entry 1)69 gave moderately high conversion (77%), initiator incorporation (74%), a low dispersity (1.04), and a molar polymer mass of ∼9000 g mol−1. Highest conversions (>90%), highest molar masses (>9000), lowest dispersities (1.03), and highest degree of initiator incorporations (>94%) were observed with the tertiary carboxamides DEAA (2), DMAA (3), and TMU (9), respectively (entries 2, 3, and 9). Employment of the bulky tert-butyl derivative (4), the anilide (5), the acetophenone derivative (6), or the chloro acetamide (7) afforded low conversions (15–44%), lower molar masses (<5000 g mol−1), and higher dispersities (>1.1–1.44). The unique stereoelectronic properties of urea (8) – as a primary amide derivative with a more basic oxygen atom donor – showed high monomer conversion. However, minor amounts of the (L-LA)2 monomer sublimed to the neck of the reaction flask, not participating in the further reaction. In the other polymerisations, this also occurred, but the refluxing of the other, in contrast to urea liquid NPCAs, washed back any sublimated (L-LA)2. Initiator incorporation was the lowest, with urea as NPCA (45%). The highest 2n/(2n + 1) ratio, and therefore the lowest transesterification activity was achieved with DEAA and urea (78–79%), while all other amides gave lower values (58–69%).
For the carboxamides 1–3 and 8-9, which showed promising results in the Fe-catalysed ROP, additional GPC and optical rotation measurements were performed (Table 2). Molar masses data determined by GPC are generally higher than those determined by MALDI-ToF-MS. Likely, the actual molar mass lies in between. However, the dispersity values from both measurements are in agreement. Carboxamides 1–3 and 9 with high initiator incorporation produced PLLAs with very low dispersities of 1.03–1.04. Additives 4–7 led to slightly higher dispersity values. The molar mass distribution for PLLA synthesised with urea 8 as NPCA was bimodal, still, when each peak was analysed separately by MALDI-ToF MS after fractionation by preparative GPC, the dispersities were very narrow (Table 1). In terms of molar masses, DEAA effected the highest polymer molar masses, followed by TMU, EAA and DMAA. The molar masses achieved with urea as NPCA were lower due to the bimodality and the sublimation of monomer during the reaction.
| Polymer | Mn (GPC) [g mol−1] | Mw (GPC) [g mol−1] | Đ (GPC) | [α]D22[°] |
|---|---|---|---|---|
| a Some (L-LA)2 monomer was lost from the melt reaction by sublimation to the neck of the flask. Mn: number-average molar mass, Mw: weight-average molar mass, Đ: dispersity, [α]D22: specific optical rotation at 589 nm and 22 °C. | ||||
| PLLA-EAA | 14100 |
14700 |
1.04 | −153 |
| PLLA-DEAA | 14500 |
15000 |
1.04 | −154 |
| PLLA-DMAA | 12200 |
13000 |
1.06 | −150 |
| PLLA-ureaa | 3240 (bimodal) | 5600 | 1.72 | −147 |
| PLLA-TMU | 12500 |
14400 |
1.16 | −151 |
The specific optical rotation [α]D22 was measured to determine the degree of racemisation during the reaction. The specific optical rotation of commercial PLLA is −155 to −153° at 22 °C.76,77 Our previous report showed that by running the polymerisation at 105 °C, racemisation of (L-LA)2 could be prevented, but some racemisation was observed at 140 °C.69 This was confirmed by our measurements for amides 1–3 and 9. However, PLLA-urea showed a slightly lower value of −147°, which could indicate that some racemisation occurred, possibly due to urea's basic character.
Overall, the tertiary carboxamide DEAA was determined to be the most efficient NPCA of the tested series giving high molar masses, low dispersity, slightly lower initiator incorporation than the related DMAA, and significantly lower transesterification. The results with DMAA, a tertiary amide with shorter alkyl substituents, mainly differed in the amount of transesterification. These results indicate that the catalytic activity of iron is enhanced by the coordination of small, electron-donating carboxamides. Noteworthy, both DEAA and DMAA are used as solvents and are, therefore, inexpensive and widely available.
Further, the optimum amount of DEAA for the polymerisations was investigated. Reactions with 5 mol% of carboxamide (with regards to each LA carbonyl unit) only reached 50% conversion after 3 h. 10 and 20 mol% of DEAA gave very similar results in conversion and initiator incorporation. GPC analysis showed a slightly higher dispersity of 1.13 at increased NPCA loading. Increasing the amount of NPCA has no beneficial effect on the outcome of the reaction.
The addition of urea (8) to the Fe-catalysed ROP of LA produced polymers with a bimodal distribution (by GPC and MALDI-ToF-MS (Fig. S3.8A and B†)). NMR analysis indicated the formation of at least two polymer species (low initiator incorporation, 45%). The polymer sample was therefore fractionated by GPC, and the individual fractions were analysed by MALDI-ToF-MS (Fig. S3.8C in the ESI†). After fractionation, an end group analysis confirmed that the main polymer fraction is the octanediol-initiated PLLA with a molar mass Mn of 6120 g mol−1 and a dispersity Đ of 1.02. The second and smaller fraction was H2O-initiated PLLA. MALDI-ToF-MS analysis indicated a third minor polymer series (GPC showed a tiny third peak), which was not assigned. Purification of the urea by repeated recrystallisation from dry ethanol and drying under a vacuum did not improve the result. Further, sublimation of the LA monomer was observed.
For comparison, Fe(II) pivalate (Fe(OPiv)2), with a sterically demanding carboxylate and a defined polymeric structure, was freshly prepared and tested for (L-LA)2 polymerisation with the different carboxamides under the same conditions as above (Table S4.1†).74 The reaction rates with Fe(II) pivalate were much faster, so the polymerisations were stopped after 1 h in order to compare the effects of the different NPCAs. Within the series of carboxamides, the trends for monomer conversion, initiator incorporation, and calculated molar masses of the polymers were very similar for Fe(OAc)2 and Fe(OPiv)2. Again DEAA gave the best results in terms of conversion and initiator incorporation. Interestingly, dimethylurea, which was used here instead of tetramethylurea, gave an even lower initiator incorporation than urea. Overall, the enhanced reaction rates observed with Fe(OPiv)2 support the hypothesised role of the amide to form monomeric iron complexes by coordination.
Freshly prepared Fe(OPiv)2 is sensitive to air and moisture and requires handling under inert conditions. Similar treatment should be followed when using freshly prepared Fe(OAc)2, while commercial samples of Fe(OAc)2 are more stable due to contamination with undefined amounts of hydrates, such as Fe(OAc)2·4H2O, hydroxides like Fe(OH)(OAc)2, Fe(OAc)3 and other impurities.79,80 It is therefore expected that an enhanced activity would be obtained with freshly prepared Fe(OAc)2 and dry reagents under rigorous exclusion of air and moisture.63
000 g mol−1 under these reaction conditions. The specific optical rotation showed slightly lower [α]D22 values for all polymerisations performed at longer reaction times, independent of the temperature. The largest value (−150°) indicates no or minor racemisation and was measured for PLLA-DEAA-15k, which was only reacted for 8 h.
| Polymer | Target mass | Initiator [%] | Reaction time [h] | Temp [°C] | Mn (NMR) [g mol−1] | Mn (GPC) [g mol−1] | Mw (GPC) [g mol−1] | Đ (GPC) | Mn (MALDI) [g mol−1] | Mw (MALDI) [g mol−1] | Đ (MALDI) | 2n: (2n + 1) [%] | [α]D22 [°] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a All polymers were synthesised using 1/160 equiv. Fe(OAc)2, 10 mol% DEAA as the amide adjunct and an appropriate amount of 1,8-octanediol (ranging from 1/34 eq. to 1/694 equiv.) as initiator to reach the targeted mass. Temp: reaction temperature, Mn number-average molar mass, Mw: weight-average molar mass, Đ: dispersity, 2n/(2n + 1): relative content of polymers having 2n LA units, calculated from MALDI-ToF-MS spectra, [α]D22: specific optical rotation at 589 nm and 22 °C. | |||||||||||||
| PLLA-DEAA-5k | 5k | 99 | 22 | 105 | 3700 | 4700 | 4900 | 1.06 | 3800 | 3950 | 1.04 | 59 | −136 |
| PLLA-DEAA-15k | 15k | 90 | 8 | 105 | 12400 |
15000 |
18500 |
1.81 | 9170 | 9650 | 1.05 | 60 | −150 |
| PLLA-DEAA-25k | 25k | 73 | 22 | 105 | 22500 |
16800 |
24700 |
1.47 | 10730 |
11750 |
1.10 | 53 | −148 |
| PLLA-DEAA-50k | 50k | 45 | 22 | 140 | 24400 |
12700 |
27700 |
2.18 | — | — | — | — | −147 |
| PLLA-DEAA-100k | 100k | 22 | 22 | 140 | 29800 |
9200 | 27900 |
3.03 | — | — | — | — | −148 |
Copolymers
000 g mol−1 (see S6 in the ESI†).
 | ||
| Scheme 2 Copolymerisation of LA and CL using the Fe(OAc)2-amide catalyst system. | ||
Lactide conversion was much slower with 1-hexanol than with octanediol as the initiator (Table 4). The target molar mass of 10k was achieved within 24 h and without racemisation of the lactide units. The dispersity was slightly higher than with octanediol (1.13 vs. 1.04). PCL-DEAA-hex had a much higher dispersity than PLLA-DEAA-hex (1.85, by GPC) and a lower molar mass (around half that of the target molar mass).
| Polymer | Target mass | Initiator [%] | Reaction time [h] | Temp [°C] | Mn (NMR) | Mn (GPC) | Mw (GPC) | Đ (GPC) | Mn (MALDI) | Mw (MALDI) | Đ (MALDI) | 2n/(2n + 1) | [α]D22 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Initiator incorporation could not be calculated; Mn of hexanol-initiated PCL. Temp: reaction temperature, Mn: number-average molar mass, Mw: weight-average molar mass, Đ: dispersity, 2n/(2n + 1): relative content of polymers having 2n LA units, calculated from MALDI-ToF-MS spectra, [α]D22: specific optical rotation at 589 nm and 22 °C. | |||||||||||||
| PLLA-DEAA-hex | 10k | 75 | 24 | 105 | 9866 | 9500 | 11900 |
1.13 | 5400 | 5600 | 1.02 | 71% | −151 |
| PCL-DEAA-hex | 10k | n.d.a | 19 | 140 | 3600a | 4000 | 7400 | 1.85 | 2800 | 3200 | 1.13 | — | — |
The catalyst activity was determined by the turnover number TON after 1 h reaction time: for (L-LA)2:90 (105 °C); for CL: 6 (105 °C), 61 (140 °C). It has been reported that the reaction rates of monomers can differ in the presence of other comonomers.12,13,81 Especially with catalysts based on Al, Mg and Zn, the rate of homopolymerisation of CL is often much faster than that of (L-LA)2.82 Here, this was not the case. The reaction rate of (L-LA)2 was much faster than that of CL in the homopolymerisations. The 1:1 mixture of both monomers showed the same conversion rate for L-(LA)2, but a lower rate for CL (87% after 24 h at 105 °C; nearly complete after 3 h at 140 °C). Copolymerisation at 140 °C is very likely to involve racemisation of lactic acid units. GPC, MALDI-ToF-MS and NMR analyses of the two homo-polymers are given in Table 4. The lactide homo-polymerisation took significantly longer with 1-hexanol than with octanediol, leading to lower incorporation of the initiator (75% vs. 93%). While the bifunctionality of the latter may play a role, the used 1-hexanediol may also have contained some water, which would simultaneously reduce the initiator incorporation and thus, the rate of reaction. The dispersity Đ of the hexanol-initiated polymer was higher than that derived from octanediol (1.13 vs. 1.04, by GPC). Optical rotations showed no significant racemisation in both cases. The caprolactone polymer PCL-DEAA-hex displayed much higher dispersity than the lactide polymer PLLA-DEAA-hex (1.85 vs. 1.13, by GPC) and a lower molar mass. It has been reported that enhanced transesterification takes place with less reactive monomers, as this reaction is energetically favoured (also see below).11 This may be the reason for the lower mass and higher dispersity found.
Copolymerisation reactions were performed with equimolar amounts of the two monomers under different reaction conditions and addition sequences to investigate the resulting copolymer architectures (Table 5). The target molar mass was kept at 10000 g mol−1, and the reactions were allowed to proceed to completion. Where possible, the reaction temperature was kept at 105 °C to prevent racemisation of (L-LA)2, which led to very long reaction times for CL. Therefore, some experiments were also conducted at 140 °C. The copolymers were analysed by NMR and GPC (Table 5). The copolymer microstructure was analysed by 1H-NMR (see Fig. S.7†). The molar ratios of LA and CL incorporation were calculated from the integrals of the lactate methine resonance and the ε-CH2 resonance of CL in the polymers. The ratios of LA–CL hetero-dyads to homo-dyads (CL–CL, LA–LA) can be best calculated from the CL–CL and CL–LA dyads, as these are better separated.
| Polymer | Reaction time | Temp [°C] | Conv LA [%] | Conv CL [%] | Molar ratio LA:CL |
(LA–CL) rel. molar fraction | lLA | lCL | R | Mn (GPC) | Mw (GPC) | Đ (GPC) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The target molar mass was 10000 g mol−1. The monomer feed was calculated to be equimolar in lactic acid and CL. The initiator was 1-hexanol (1/412 w.r.t. to LA + CL, Fe(OAc)2 was 1/160 equiv. of the cyclic monomers), amide: DEAA, 0.15 mol% (w.r.t. carbonyl units of the monomers). The reactions were allowed to continue until all (nearly all) monomer was used up, as assessed by 1H-NMR.b CL was added after 4 h, when (L-LA)2 conv. was 99%.c (L-LA)2 was added after 24 h. Some (L-LA)2 sublimed to the neck of flask and was lost from the reaction.d (L-LA)2 was added after 4 h and temperature decreased to 105 °C. Temp: reaction temperature, Conv.: conversion of monomer, calculated from NMR, l: average block length of respective monomer; R: randomness; Mn: number-average molar mass, Mw: weight-average molar mass, Đ: dispersity, 2n/(2n + 1): relative content of polymers having 2n LA units, calculated from MALDI-ToF-MS spectra, [α]D22: specific optical rotation at 589 nm and 22 °C. |
||||||||||||
| PLLA-grad-PCL-105 | 7 d | 105 | >99 | 97 | 45:55 |
0.100 | 9.0 | 11 | 0.20 | 3300 | 6400 | 1.94 |
| PLLA-grad-PCL-140 | 48 h | 140 | 92 | 91 | 42:58 |
0.168 | 5.0 | 6.9 | 0.30 | 4000 | 12900 |
3.22 |
| PLLA-b-PCL-105 | 7 d | 105 | >99 | 91b | 50:50 |
0.097 | 10.3 | 10.3 | 0.19 | 2500 | 7300 | 2.92 |
| PCL-b-PLLA-140 | 48 h | 140 | 86c | >99 | 27:73 |
0.028 | 19.3 | 52.1 | 0.07 | 5000 | 9100 | 1.82 |
| PCL-b-PLLA-140-105 | 30 h | 140/105 | 80d | 97 | 47:53 |
0.021 | 44.3 | 50.2 | 0.04 | 7100 | 10400 |
1.47 |
From these values the number average block lengths of each monomer can be calculated:83,84
 | (1) |
 | (2) |
The randomness R of a polymer can be calculated from:
 | (3) |
For a completely random polymer, R approaches 1, with values above 0.8 indicating significant randomness. Pure block copolymers have R = 0.
Random copolymerisations were attempted at 105 and 140 °C (entries 1 and 2, Table 5). A reaction time of 7 days was required for full conversion at 105 °C. The incorporation of both monomers was very similar and slightly lower for the lactide. The randomness was found to be low, which may indicate gradient structures in both cases due to the much higher reactivity of the lactide. The R-value of PLLA-ran-PCL-140 and the dispersity were higher than those of PLLA-ran-PCL-105, which supports the notion of active transesterification at higher temperatures. Alternative catalysts, such as Sn(Oct)2, produce much higher randomized structures than the Fe(OAc)2/DEAA catalyst via transesterification.64
Catalysts, which can only start block copolymerisations with one of the monomers, have been reported.85,86 We, therefore, performed reactions starting from (L-LA)2 (entry 3) and CL (entries 4 and 5). Incorporation of both monomers into the copolymer could be achieved via sequential addition starting with either monomer. However, due to the slow reaction rate of CL at 105 °C, synthesis of PLLA-b-PCL-105 took seven days. The randomness of PLLA-b-PCL-105 was similar to that of PLLA-grad-PCL-105 due to enhanced transesterification. At extended reaction times, transesterification becomes competitive with the slow CL incorporation, as also observed in a recent study with iron-guanidine catalysts.11 PCL-b-PLLA block co-polymers with very low R values are given in entries 4 and 5. For PCL-b-PLLA-140-105, the temperature was lowered to 105 °C when the lactide was added after 4 h. Incorporation of lactide into PCL-b-PLLA-140 was lower as some of the (L-LA)2 sublimed from the reaction mixture. In all cases, the R values of PCL-b-PLLA are much lower than those of PLLA-b-PCL, possibly due to an initial faster incorporation of CL and lower transesterification.
The target molar mass for all reactions in Table 5 was 10000 g mol−1, however, this mass was mostly not reached. The lowest mass was obtained from PLLA-grad-PCL-105, and the highest polymer masses from PCL-b-PLLA-140-105 (Mn = 7100 g mol−1, Mw = 10400 g mol−1, by GPC). The latter stepwise procedure also afforded the far lowest dispersity of the co-polymers (Đ = 1.47).
DSC analysis was performed on some selected examples of copolymers. The results are summarised in Table S11.01.† Both the PLLA (163–168 °C) and PCL (33–51 °C) related melting transitions are relatively low, which indicates a small crystallite size, while the melting enthalpy indicated a low degree of crystallinity. In fact, in PLLA-b-PCL, no crystallisation of the PLLA was observed.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O resonance between 170.8 ppm (ref. 11) and 171.1 ppm.67 We observed the carbonyl signal of CL–LA–CL at 171.0 ppm in the spectra of the statistical copolymer and PLLA-b-PCL which confirmed that transesterification took place (Fig. 2). The signal is not present in the co-polymer that was initiated with the CL monomer.
O resonance between 170.8 ppm (ref. 11) and 171.1 ppm.67 We observed the carbonyl signal of CL–LA–CL at 171.0 ppm in the spectra of the statistical copolymer and PLLA-b-PCL which confirmed that transesterification took place (Fig. 2). The signal is not present in the co-polymer that was initiated with the CL monomer.
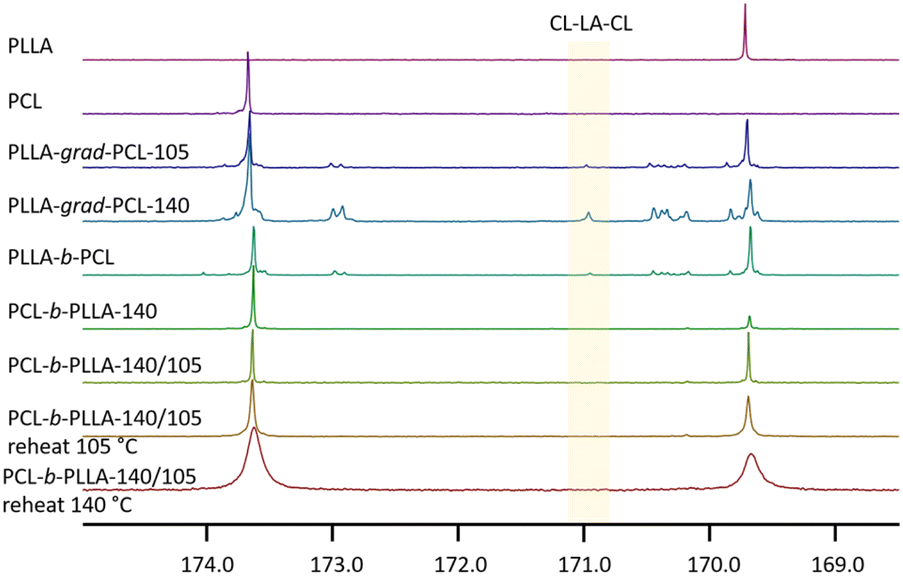 | ||
| Fig. 2 13C-NMR carbonyl regions of the L-lactide and CL homopolymers, copolymers, and block copolymers (in ppm). | ||
Further, we studied potential transesterification reactions on the pre-formed copolymers under standard conditions. A mixture of PCL-b-PLLA-140-105, the copolymer displaying the lowest randomness value R, the catalyst Fe(OAc)2 and the carboxamide additive DEAA were heated for more than 2 days at 105 °C and 140 °C, respectively. No differences were detected in the 13C-NMR spectra before and after reaction. Our observations that transesterification was only observed in the block copolymers starting with lactide incorporation but was absent in the CL-initiated copolymers and when treating the final polymers under standard reaction conditions may be indicative of an active transesterification pathway during the slow CL insertion steps. The absence of transesterification in the addition sequence CL–(L-LA)2 makes the Fe(OAc)2/DEAA catalyst system ideally suited for the synthesis of PCL-PLLA block copolymers.
The synthesis of highly blocky copolymers in the melt is not an easy task, Sn(Oct)2 under similar conditions produces copolymers of much higher randomness.88 In our case, the sequential addition of monomer can produce real block copolymers. Where necessary, the polymerisation sequence can also be started with (L-LA)2, followed by CL at 140 °C, which should shorten the reaction times.
Conclusions
In conclusion, a simple and inexpensive catalyst system was generated in situ from Fe(II) acetate and 10 mol% of small aliphatic tertiary carboxamides. The catalyst is effective for the ROP of L-lactide, achieving PLLAs with very narrow dispersities (Đ = 1.03) up to a molar mass of 15000 g mol−1 under melt conditions. When the polymerisation is performed at 105 °C and the reaction time is kept short enough, no racemisation of L-lactide occurs. Carboxamides with two small aliphatic substituents having a +I-effect seem to be most active in supporting the ROP by Fe(II) carboxylates.
Furthermore, the optimised catalyst system was applied to the synthesis of PLLA-PCL copolymers, where it was found to be very useful for the synthesis of block copolymers of very low randomness due to its low transesterification activity, and the difference in reactivities of the two monomers. Starting from PCL, the catalyst can produce highly blocky copolymers with moderate dispersity at a target molar mass of 10 kg mol−1. The block-copolymerisations could be performed in the melt and without intermediate purification steps. This represents an inexpensive and simple way of synthesising PLLAs and block copolymers for use in applications such as the biomedical field and may have a lower environmental impact than the use of tin catalysts.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Regine Apostel and Susanne Schwanz for technical assistance. This work was financially supported by the Helmholtz Association of German Research Centers through program-oriented funding.References
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- V. Izraylit, P. J. Hommes-Schattmann, A. T. Neffe, O. E. Gould and A. Lendlein, Eur. Polym. J., 2020, 136, 109908 CrossRef CAS.
- V. Martino, R. Ruseckaite and A. Jiménez, J. Therm. Anal. Calorim., 2006, 86, 707–712 CrossRef CAS.
- C. Alberti, N. Damps, R. R. Meißner and S. Enthaler, ChemistrySelect, 2019, 4, 6845–6848 CrossRef CAS.
- C. Alberti, N. Damps, R. R. Meißner, M. Hofmann, D. Rijono and S. Enthaler, Adv. Sustainable Syst., 2020, 4, 1900081 CrossRef CAS.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef PubMed.
- L. Ranakoti, B. Gangil, S. K. Mishra, T. Singh, S. Sharma, R. A. Ilyas and S. El-Khatib, Materials, 2022, 15, 4312 CrossRef CAS PubMed.
- N.-A. A. B. Taib, M. R. Rahman, D. Huda, K. K. Kuok, S. Hamdan, M. K. B. Bakri, M. R. M. B. Julaihi and A. Khan, Polym. Bull., 2022, 80, 1179–1213 CrossRef.
- R. Dwivedi, S. Kumar, R. Pandey, A. Mahajan, D. Nandana, D. S. Katti and D. Mehrotra, J. Oral Biol. Craniofac. Res., 2020, 10, 381–388 CrossRef PubMed.
- N. Raina, R. Pahwa, J. K. Khosla, P. N. Gupta and M. Gupta, Polym. Bull., 2021, 79, 7041–7063 CrossRef.
- R. D. Rittinghaus, J. Zenner, A. Pich, M. Kol and S. Herres-Pawlis, Angew Chem. Int. Ed. Engl., 2022, 61, e202112853 CrossRef CAS PubMed.
- I. D'Auria, M. Lamberti, R. Rescigno, V. Venditto and M. Mazzeo, Heliyon, 2021, 7, e07630 CrossRef PubMed.
- R. A. Auras, L.-T. Lim, S. E. Selke and H. Tsuji, Poly (lactic acid): synthesis, structures, properties, processing, and applications, Wiley, 2011 Search PubMed.
- E. T. Vink, K. R. Rabago, D. A. Glassner, B. Springs, R. P. O'Connor, J. Kolstad and P. R. Gruber, Macromol. Biosci., 2004, 4, 551–564 CrossRef CAS PubMed.
- E. Cubas-Cano, C. González-Fernández, M. Ballesteros and E. Tomás-Pejó, Biofuels, Bioprod. Biorefin., 2018, 12, 290–303 CrossRef CAS.
- M. Dusselier and B. F. Sels, in Selective Catalysis for Renewable Feedstocks and Chemicals, ed. K. M. Nicholas, Springer International Publishing, Cham, 2014, pp. 85–125, DOI:10.1007/128_2014_540.
- N. Razali and A. Z. Abdullah, Appl. Catal., A, 2017, 543, 234–246 CrossRef CAS.
- S. Li, W. Deng, Y. Li, Q. Zhang and Y. Wang, J. Energy Chem., 2019, 32, 138–151 CrossRef.
- T. Buntara, S. Noel, P. H. Phua, I. Melian-Cabrera, J. G. de Vries and H. J. Heeres, Angew Chem. Int. Ed. Engl., 2011, 50, 7083–7087 CrossRef CAS PubMed.
- S. Schmidt, C. Scherkus, J. Muschiol, U. Menyes, T. Winkler, W. Hummel, H. Groger, A. Liese, H. G. Herz and U. T. Bornscheuer, Angew Chem. Int. Ed. Engl., 2015, 54, 2784–2787 CrossRef CAS PubMed.
- J. U. Pothupitiya, N. U. Dharmaratne, T. M. M. Jouaneh, K. V. Fastnacht, D. N. Coderre and M. K. Kiesewetter, Macromolecules, 2017, 50, 8948–8954 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS PubMed.
- J. Engel, A. Cordellier, L. Huang and S. Kara, ChemCatChem, 2019, 11, 4983–4997 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
- J. W. Leenslag and A. J. Pennings, Makromol. Chem., 1987, 188, 1809–1814 CrossRef CAS.
- S. Kaihara, S. Matsumura, A. G. Mikos and J. P. Fisher, Nat. Protoc., 2007, 2, 2767–2771 CrossRef CAS PubMed.
- H. Li, R. M. Shakaroun, S. M. Guillaume and J. F. Carpentier, Chem. –Eur. J., 2020, 26, 128–138 CrossRef CAS PubMed.
- Y. Wang, Z. Jia, J. Jiang, X. Mao, X. Pan and J. Wu, Macromolecules, 2019, 52, 7564–7571 CrossRef CAS.
- J. Hu, C. Kan, H. Wang and H. Ma, Macromolecules, 2018, 51, 5304–5312 CrossRef CAS.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew Chem. Int. Ed. Engl., 2020, 59, 21778–21784 CrossRef CAS PubMed.
- R. D. Rittinghaus, P. M. Schäfer, P. Albrecht, C. Conrads, A. Hoffmann, A. N. Ksiazkiewicz, O. Bienemann, A. Pich and S. Herres-Pawlis, ChemSusChem, 2019, 12, 2161–2165 CrossRef CAS PubMed.
- A. Stjerndahl, A. Finne-Wistrand, A. C. Albertsson, C. M. Backesjo and U. Lindgren, J. Biomed. Mater. Res., 2008, 87, 1086–1091 CrossRef CAS PubMed.
- H. R. Kricheldorf, Chemosphere, 2001, 43, 49–54 CrossRef CAS PubMed.
- H. Rüdel, Ecotoxicol. Environ. Saf., 2003, 56, 180–189 CrossRef PubMed.
- K. Fent, Crit. Rev. Toxicol., 2008, 26, 3–117 CrossRef.
- P. M. Schäfer and S. Herres-Pawlis, ChemPlusChem, 2020, 85, 1044–1052 CrossRef PubMed.
- D. J. Darensbourg and O. Karroonnirun, Macromolecules, 2010, 43, 8880–8886 CrossRef CAS.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- R. Platel, L. Hodgson and C. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef PubMed.
- L. Rüttinger, P. van Ackern, T. Lepold, R. Vogt and A. Auberger, Impacts of climate change on mining, related environmental risks and raw material supply, Final Report, 2020, http://www.umweltbundesamt.de/publikationen Search PubMed.
- Y. Champouret, O. H. Hashmi and M. Visseaux, Coord. Chem. Rev., 2019, 390, 127–170 CrossRef CAS.
- J. A. Stewart, P. McKeown, O. J. Driscoll, M. F. Mahon, B. D. Ward and M. D. Jones, Macromolecules, 2019, 52, 5977–5984 CrossRef CAS.
- U. Herber, K. Hegner, D. Wolters, R. Siris, K. Wrobel, A. Hoffmann, C. Lochenie, B. Weber, D. Kuckling and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2017, 2017, 1341–1354 CrossRef CAS.
- Z. Yaghobinia, Z. Ahmadi and M. Abdouss, J. Vinyl Addit. Technol., 2019, 25, 215–224 CrossRef CAS.
- J. Dou, D. Zhu, W. Zhang, R. Wang, S. Wang, Q. Zhang, X. Zhang and W.-H. Sun, Inorg. Chim. Acta, 2019, 488, 299–303 CrossRef CAS.
- K. Delle Chiaie, A. Biernesser, M. Ortuño, B. Dereli, D. Iovan, M. Wilding, B. Li, C. Cramer and J. Byers, Dalton Trans., 2017, 46, 12971–12980 RSC.
- O. J. Driscoll, C. H. Hafford-Tear, P. McKeown, J. A. Stewart, G. Kociok-Kohn, M. F. Mahon and M. D. Jones, Dalton Trans., 2019, 48, 15049–15058 RSC.
- X. Xu, H. Lu, G. Luo, X. Kang and Y. Luo, Inorg. Chem. Front., 2021, 8, 1005–1014 RSC.
- D. S. McGuinness, E. L. Marshall, V. C. Gibson and J. W. Steed, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3798–3803 CrossRef CAS.
- P. Marin, M. J. L. Tschan, F. Isnard, C. Robert, P. Haquette, X. Trivelli, L. M. Chamoreau, V. Guérineau, I. Del Rosal and L. Maron, Angew. Chem., Int. Ed., 2019, 58, 12585–12589 CrossRef CAS PubMed.
- R. Duan, C. Hu, X. Li, X. Pang, Z. Sun, X. Chen and X. Wang, Macromolecules, 2017, 50, 9188–9195 CrossRef CAS.
- Y. Liang, R.-L. Duan, C.-Y. Hu, L.-L. Li, X. Pang, W.-X. Zhang and X.-S. Chen, Chin. J. Polym. Sci., 2018, 36, 185–189 CrossRef CAS.
- M. Cozzolino, V. Leo, C. Tedesco, M. Mazzeo and M. Lamberti, Dalton Trans., 2018, 47, 13229–13238 RSC.
- S. Impemba, F. Della Monica, A. Grassi, C. Capacchione and S. Milione, ChemSusChem, 2020, 13, 141–145 CrossRef CAS PubMed.
- O. J. Driscoll, C. K. C. Leung, M. F. Mahon, P. McKeown and M. D. Jones, Eur. J. Inorg. Chem., 2018, 2018, 5129–5135 CrossRef CAS.
- R. R. Gowda and D. Chakraborty, J. Mol. Catal. A: Chem., 2009, 301, 84–92 CrossRef CAS.
- B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 4384–4393 CrossRef PubMed.
- J. L. Gorczynski, J. Chen and C. L. Fraser, J. Am. Chem. Soc., 2005, 127, 14956–14957 CrossRef CAS PubMed.
- C. S. Hege and S. M. Schiller, Green Chem., 2014, 16, 1410–1416 RSC.
- Y. N. Lei, Y. B. Zhu, C. F. Gong, J. J. Lv, C. Kang and L. X. Hou, J. Mater. Sci. Mater. Med., 2014, 25, 273–282 CrossRef PubMed.
- M. Stolt and A. Södergård, Macromolecules, 1999, 32, 6412–6417 CrossRef CAS.
- A. T. Neffe, V. Izraylit, P. J. Hommes-Schattmann and A. Lendlein, Nanomaterials, 2021, 11, 1472 CrossRef CAS PubMed.
- T. Shi, W. Luo, S. Liu and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 611–617 CrossRef CAS.
- M. Florczak and A. Duda, Angew Chem. Int. Ed. Engl., 2008, 47, 9088–9091 CrossRef CAS PubMed.
- N. Nomura, A. Akita, R. Ishii and M. Mizuno, J. Am. Chem. Soc., 2010, 132, 1750–1751 CrossRef CAS PubMed.
- T. Naolou, A. Lendlein and A. T. Neffe, Eur. Polym. J., 2016, 85, 139–149 CrossRef CAS.
- T. Naolou, A. Lendlein and A. T. Neffe, Front. Chem., 2019, 7, 346 CrossRef CAS PubMed.
- K. Ding, F. Zannat, J. C. Morris, W. W. Brennessel and P. L. Holland, J. Organomet. Chem., 2009, 694, 4204–4208 CrossRef CAS.
- S. Sandl and A. Jacobi von Wangelin, Angew Chem. Int. Ed. Engl., 2020, 59, 5434–5437 CrossRef CAS PubMed.
- J. D. Sears, S. B. Munoz 3rd, S. L. Daifuku, A. A. Shaps, S. H. Carpenter, W. W. Brennessel and M. L. Neidig, Angew Chem. Int. Ed. Engl., 2019, 58, 2769–2773 CrossRef CAS PubMed.
- S. B. Munoz 3rd, S. L. Daifuku, J. D. Sears, T. M. Baker, S. H. Carpenter, W. W. Brennessel and M. L. Neidig, Angew Chem. Int. Ed. Engl., 2018, 57, 6496–6500 CrossRef PubMed.
- I. L. Eremenko, M. A. Kiskin, I. G. Fomina, A. A. Sidorov, G. G. Aleksandrov, V. N. Ikorskii, Y. G. Shvedenkov, Y. V. Rakitin and V. M. Novotortsev, J. Cluster Sci., 2005, 16, 331–351 CrossRef CAS.
- D. Noda, A. Tahara, Y. Sunada and H. Nagashima, J. Am. Chem. Soc., 2016, 138, 2480–2483 CrossRef CAS PubMed.
- PLLA, Sigma Aldrich catalogue, https://www.sigmaaldrich.com/DE/de/product/aldrich/764698) Search PubMed.
- N. Yui, P. J. Dijkstra and J. Feijen, Makromol. Chem., 1990, 191, 481–488 CrossRef CAS.
- B. Weber, R. Betz, W. Bauer and S. Schlamp, Z. Anorg. Allg. Chem., 2011, 637, 102–107 CrossRef CAS.
- E. Wildermuth, H. Stark, G. Friedrich, F. L. Ebenhöch, B. Kühborth, J. Silver and R. Rituper, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a14_591.
- D. L. Perry, Handbook of Inorganic Compounds, Taylor & Francis, US, 2nd edn, 2011 Search PubMed.
- K. Phomphrai, P. Chumsaeng, P. Sangtrirutnugul, P. Kongsaeree and M. Pohmakotr, Dalton Trans., 2010, 39, 1865–1871 RSC.
- H.-Y. Chen, B.-H. Huang and C.-C. Lin, Macromolecules, 2005, 38, 5400–5405 CrossRef CAS.
- J. Fernandez, A. Etxeberria and J. R. Sarasua, J. Mech. Behav. Biomed. Mater., 2012, 9, 100–112 CrossRef CAS PubMed.
- I. R. Herbert, in NMR Spectroscopy of Polymers, ed. R. N. Ibbett, Springer Netherlands, Dordrecht, 1993, pp. 50–79, DOI:10.1007/978-94-011-2150-7_2.
- V. Simic, S. Pensec and N. Spassky, Macromol. Symp., 2000, 153, 109–121 CrossRef CAS.
- R. I. Kim, G. Lee, J.-H. Lee, J. J. Park, A. S. Lee and S. S. Hwang, ACS Appl. Polym. Mater., 2021, 3, 4708–4716 CrossRef CAS.
- J. Kasperczyk and M. Bero, Makromol. Chem., 1991, 192, 1777–1787 CrossRef CAS.
- K. Nalampang, R. Molloy and W. Punyodom, Polym. Adv. Technol., 2007, 18, 240–248 CrossRef CAS.
- M. Mir, N. Ahmed and A. U. Rehman, Colloids Surf., B, 2017, 159, 217–231 CrossRef CAS PubMed.
- P. Gentile, V. Chiono, I. Carmagnola and P. V. Hatton, Int. J. Mol. Sci., 2014, 15, 3640–3659 CrossRef CAS PubMed.
- J. Sun, J. Walker, M. Beck-Broichsitter and S. P. Schwendeman, Drug Deliv. Transl. Res., 2022, 12, 720–729 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra03112h |
| This journal is © The Royal Society of Chemistry 2023 |