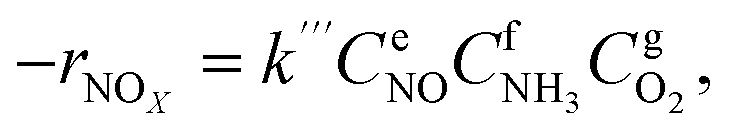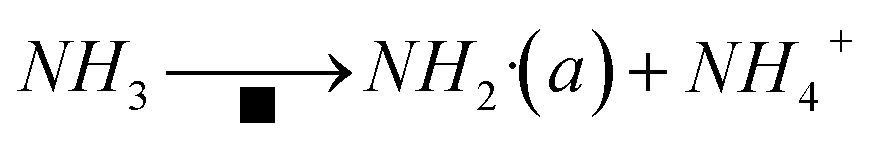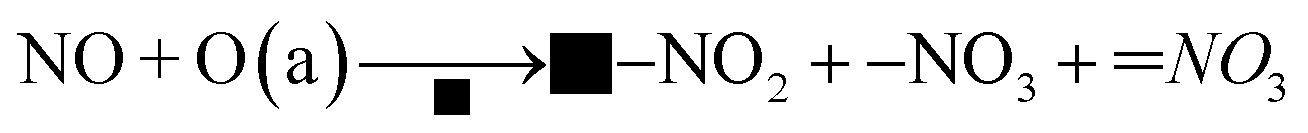DOI:
10.1039/D3RA03018K
(Paper)
RSC Adv., 2023,
13, 25989-26000
Different morphologies on Cu–Ce/TiO2 catalysts for the selective catalytic reduction of NOx with NH3 and DRIFTS study on sol–gel nanoparticles†
Received
6th May 2023
, Accepted 11th August 2023
First published on 1st September 2023
Abstract
The copper–cerium binary oxide catalysts supported by titanium dioxide with nanosphere core–shell structures, nanotube (TNT) core–shell structures, impregnation (imp) nanoparticles and sol–gel nanoparticles were prepared for NH3-SCR of NOx under medium-low temperature conditions. The effect of different morphologies on the Cu–Ce/TiO2 catalysts was comprehensively studied through physicochemical characterization. The results showed that the sol–gel nanoparticles exhibited 100% NOx reduction efficiency in the temperature range of 180–400 °C. Compared with the other catalysts, the sol–gel nanoparticle catalyst had the highest dispersion and lowest crystallinity, indicating that morphology played an important role in the NH3-SCR of the catalyst. The in situ DRIFTS study on the sol–gel nanoparticle catalyst shows that cerium could promote Cu2+ to produce abundant Lewis acid sites, which would significantly increase the adsorption reaction of ammonia on the catalyst surface, thereby promoting the occurrence of the Eley–Rideal (E–R) mechanism. With the Ce–Ti interaction on the atomic scale, the Ce–O–Ti structure enhanced the redox properties at a medium temperature. In addition, cerium oxide enhances the strong interaction between the catalyst matrix and CuO particles. Therefore, the reducibility of the CuO species was enhanced.
1. Introduction
With the in-depth study on DeNOx catalysts, many high-performance catalysts have been developed in recent years. Copper-cerium binary oxide catalysts supported by titanium dioxide (Cu–Ce/TiO2) have attracted extensive attention owing to their low-cost and high-efficiency.1–4 Solution impregnation and one-step hydrothermal methods were the common methods used to prepare medium-low temperature NH3-SCR catalysts.3,5–7
In the field of medium-low temperature NH3-SCR, catalysts prepared by sol–gel and two-step methods have rarely been reported. However, the two-step and sol–gel methods are already very mature in other fields.8–11 In our previous studies,11,12 various TiO2-based core–shell catalysts have been prepared using the two-step method, such as a novel porous CeO2@TiO2 core–shell catalyst and MnOx-CeO2@TiO2 core–shell nanorod catalyst. The TiO2 shell does not inhibit the catalytic activity of the catalyst and improves the physicochemical properties of the catalyst. The superior SO2 tolerance could be attributed to the unique core–shell structure that lowers the possibility of surface active sites being poisoned by SO2 and prevents the formation of ammonium sulfate species from covering the active site. In previous studies,2,13,14 the catalyst prepared by applying the sol–gel method had the characteristics of good particle dispersion, strong interaction among components, and low sample crystallinity. In addition, the defect sites, valence and acid–base properties of the catalyst elements can be adjusted through different morphologies.15 Therefore, the sol–gel method may be a good method for preparing an NH3-SCR catalyst at medium-low temperatures.
Herein, Cu–Ce/TiO2 catalysts with different morphologies were prepared by impregnation, sol–gel, two-step, and one-step hydrothermal methods. This study assessed the efficiency of catalysts on NOx removal in simulated flue gas. To investigate the physicochemical properties, the BET, TEM, XRD, Raman, NH3-TPD, HRTEM, XPS and H2-TPR techniques were used to characterize the catalysts. The in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) characterization method was used to study the Cu–Ce/TiO2 catalyst with a sol–gel nanoparticle morphology, and the specific reaction mechanism was analyzed. The intense interaction between Cu, Ce and Ti in the NH3-SCR reaction was also discussed.
2. Experimental section
2.1. Catalyst preparation
The chemicals used in this study were of analytical grade. The reagents used in this experiment were from Sinopharm Chemical Reagent Co., Ltd. The molar ratio of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ce:Ti in all catalysts was 1:2:38.
:Ce:Ti in all catalysts was 1:2:38.
2.1.1. Impregnation method. First, 120 ml deionized water, 1.7 g Ce(NO3)3·6H2O, 1.9 g Cu(NO3)2·3H2O and 6.0 g P25 were mixed under 180 rpm stirring at 25 °C for 6 h. Then, the resulting product was washed five times in deionized water and centrifuged. Finally, the obtained product was dried in an oven at 80 °C and calcined at 550 °C for 6 h. The catalyst was designated Cu–Ce/TiO2 (imp).
2.1.2. Sol–gel method. The catalyst was obtained using the sol–gel method. First, 34.0 g tetrabutyl titanate, 2.3 g Ce(NO3)3·6H2O, 36.9 g ethanol, 10.8 g deionized water, 18.0 g acetic acid and 0.6 g Cu(NO3)2·3H2O were mixed under 180 rpm stirring at ambient temperature. After stabilizing for four days at 25 °C, the sol turns into a gel. The gel was dried at 105 °C for 24 h to obtain a honeycomb solid. The obtained solid was pulverized and calcined in a muffle furnace at 550 °C for 5 h and was denoted as Cu–Ce/TiO2 (sol–gel).
2.1.3. Core-shell nanospheres. Two-step preparation of Cu–Ce@TiO2 core–shell nanostructured catalyst: The Cu–Ce (core) was prepared using a one-step hydrothermal method,16 and Cu–Ce@TiO2 was prepared using dynamic coating method.11The Cu–Ce (core) was synthesized using the following chemical materials: 50 ml deionized water, 8.1 g Cu(NO3)2·3H2O, 7.2 g Ce(NO3)3·6H2O and 300 ml glycol were mixed under 200 rpm stirring at ambient temperature for 6 h. The above solution was sealed in a Teflon-lined stainless-steel autoclave at 160 °C for 8 h. Finally, after centrifugation, the product was dried at 80 °C for 12 hours to form Cu–Ce (core).
In a typical synthesis, 2.6 g of Cu–Ce (core) was distributed uniformly in 100 ml of anhydrous ethanol. 0.3 g NH3 was slowly dropped into the above solution and then treated with ultrasonic for 30 min. Then, 133 ml tetrabutyl titanate was slowly added dropwise under continuous mechanical stirring at 45 °C for 24 h. The sediment was washed with deionized water and then dried at 105 °C for 18 h. Finally, the obtained solid product was calcined in a muffle furnace at 550 °C for 4 h. The catalyst was denoted as Cu–Ce@TiO2 (nanospheres).
2.1.4. Core–shell nanotubes. The Cu–Ce@TiO2(TNT) catalyst was prepared using the one-step hydrothermal method.5 First, 10.0 g P25, 2.6 g Ce(NO3)3·6H2O, 250 ml 10 M NaOH and 0.7 g Cu(NO3)2·3H2O were mixed under 280 rpm stirring at ambient temperature for 4 h. Then, the above mixture was sealed in a Teflon-lined stainless-steel autoclave at 130 °C for 12 h. The resulting product was treated with 1.5 L of 0.1 M HCl and then washed with deionized water until the pH was 7. Finally, the resulting powder was dried at 80 °C and calcined at 550 °C for 4 h. The catalyst was denoted as Cu–Ce@TiO2(TNTs).
2.2. Catalyst characterization
The surface area and pore size distribution of the catalysts were measured by Brunauer–Emmett–Teller (BET) (Micromeritics ASAP 2460, America) at −196 °C by N2 adsorption/desorption. The Raman spectra were recorded with a LabRam HR Evolution using a 633 nm laser Beam. The X-ray diffraction pattern (XRD) of the advanced ray diffractometer (Bruker D8, Germany) was used to study the crystal structure of the catalyst. A transmission electron microscope (TEM) and a high-resolution TEM (HR-TEM) instrument operated at 300 keV (JEM 2100 Plus, Japan) were used to evaluate the morphological characterization of the samples. The NH3 temperature-programmed desorption (NH3-TPD) and H2 temperature-programmed reduction (H2-TPR) were performed using a Tianjin XQ TP5080 autoadsorption apparatus equipped with a thermal conductivity detector (TCD). Before the experiment, each catalyst (150 mg) was placed in a nitrogen stream (35 ml min−1) at 550 °C for 1 h and then cooled to 25 °C in a nitrogen atmosphere. For the NH3-TPD experiment, the sample was pre-treated with NH3 atmosphere at 120 °C for 2 h and then purged by N2 for half an hour. Finally, the reactor temperature was increased to 850 °C (N2 atmosphere) at a rate of 10 °C min−1. For the H2-TPR experiment, the temperature was increased to 850 °C at a regular heating rate of 10 °C min−1 in a flow of N2 (35 ml min−1)/H2 (5 vol%). X-ray photoelectron spectroscopy (XPS) (Thermo Escalab 250Xi, America) was used to investigate the surface atomic states of the catalysts.
In situ diffuse reflection was used to study the reaction mechanism. Infrared Fourier transform spectroscopy (DRIFTS) spectra of adsorbed substances were investigated by applying an IR cell equipped with a KBr window (A BRUKER VERTEX 80v, Germany) and recorded in the range of 4000–800 cm−1. Before the adsorption, the wavenumber was set to 4 cm−1 and the number of scans was set to 64 times. In the IR cell, the sample was pre-treated with N2 at 600 °C for 3 h and then cooled to the temperature required for the experiment. After the temperature stabilizes, the infrared spectrum of the sample is scanned as the background value and subtracted from the background value in the subsequent response spectrum. The Kubelka–Munk function calculates the final spectrum signal. For NH3 adsorption, the sample was fixed in NH3 (800 ppm/60 ml min−1) for 2 h; then, the sample with N2 was purged for half an hour, and the DRIFT spectrum was collected simultaneously. The NO + O2 adsorption consists of two steps. First, the sample was exposed to 800 ppm NO (60 ml min−1) for 30 min and was then let in 7.5% O2 for 10 min. After purging the sample with N2 for 30 min, the DRIFT spectra were collected simultaneously.
For the NH3 + NO + O2 experiment, the sample was exposed to an NH3 atmosphere for half an hour and then purged the sample with N2 for half an hour at 200 °C. Afterward, NO + O2 was passed into the IR cell, and the changes in the spectrum were recorded over time. The pretreatment and background spectra were treated similarly. A similar method was used for the NO + O2 + NH3 experiment.
2.3. Activity and kinetic measurements
The NOx conversion was measured in a fix-bed reactor (XQ WFS-3015, China) with a 4 ml sample of 40–60 mesh at 100 to 480 °C. The reaction gas comprised 500 ppm NO, 500 ppm NH3, 7.5% O2 and N2. Then, a flue gas analyzer (MRU Vario Plus, Germany) was used to detect the outlet gas. The gas hourly space velocity (GHSV) and total flow rate were 24000 h−1 and 1600 ml min−1, respectively.
The NOx conversion, N2 selectivity and NH3-SCR reaction rate were calculated using eqn (1)–(3), respectively:
| |
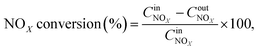 | (1) |
| |
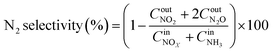 | (2) |
| |
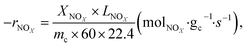 | (3) |
where −
rNOx is NO
x consumption rate.
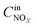
,
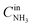
and
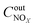
,
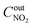
,
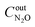
are concentrations of inlet gas (ppm) and outlet gas (ppm), respectively.
XNOx is the conversion of NO
x (%) (
eqn (1)),
LNOx is the flow rate of NO
x (ml min
−1), and
mc is the mass of the catalyst (g).
3. Results and discussion
3.1. Catalytic activities
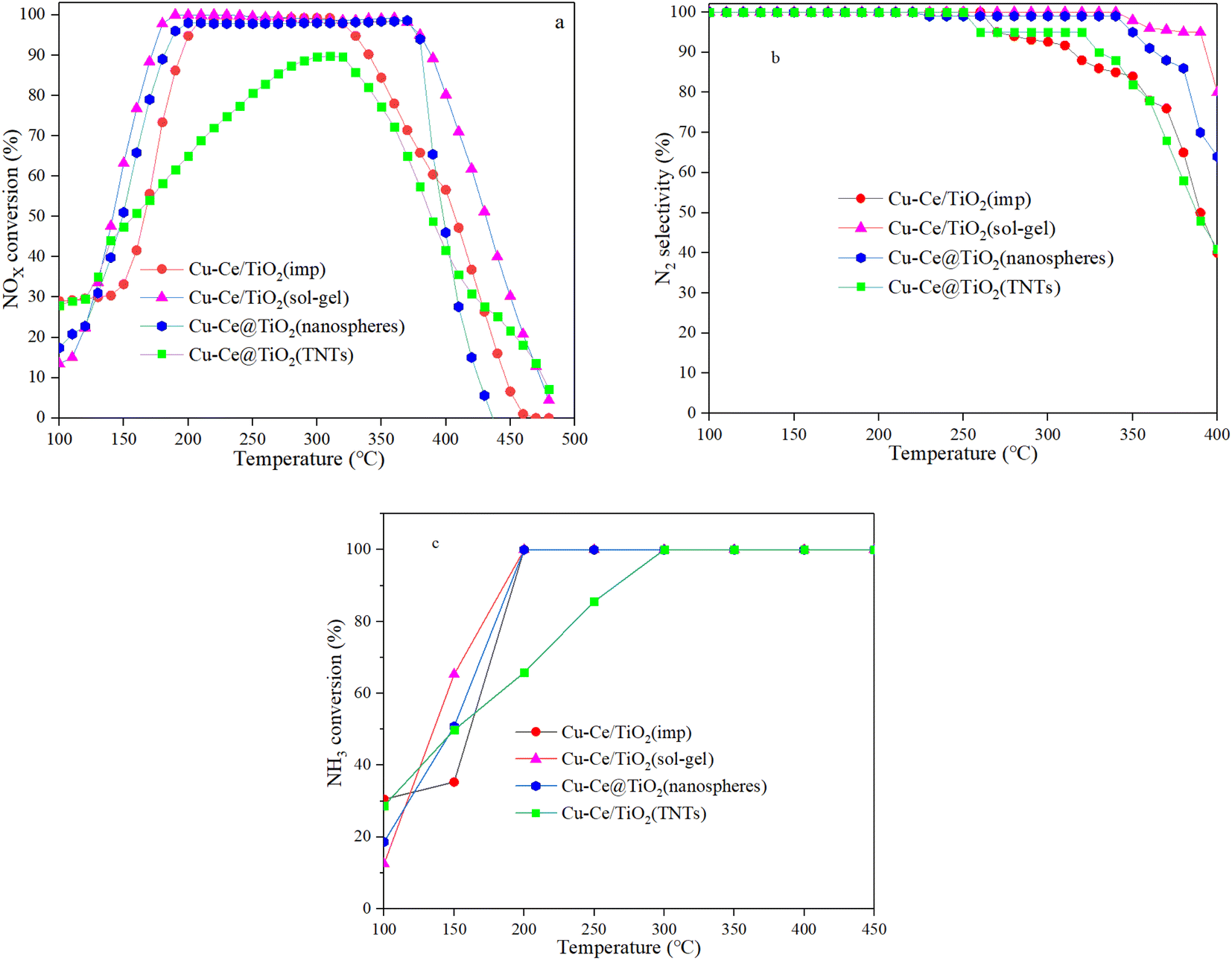
The SCR performance of all catalysts was investigated. From Fig. 1a, the temperature window of the catalysts with different morphologies increased in the order of Cu–Ce/TiO2(imp) < Cu–Ce@TiO2(nanospheres) < Cu–Ce@TiO2(TNTs) < Cu–Ce/TiO2(sol–gel). As the temperature increased, the NOx conversion decreased significantly, which might be due to the enhancement of the side reaction of NH3 oxidation.15 The N2 selectivity of all samples is shown in Fig. 1b. The Cu–Ce/TiO2 (sol–gel) catalyst exhibited a broader window of N2 selectivity. Furthermore, the NH3 conversion of all catalysts was investigated. As shown in Fig. 1c, the NH3 conversion of all catalysts increases with the temperature and reaches 100% at 300 °C. The rate of NH3 consumption also reflects the catalytic activity of the catalyst. These results indicate that different morphologies could effectively improve the NH3-SCR activity of Cu–Ce/TiO2 catalysts. In addition, the effects of H2O and SO2 on the NOx conversion over the catalysts at 310 °C are illustrated in Fig. S1.† The Cu–Ce/TiO2 (sol–gel) exhibited higher resistance to H2O and SO2, and 71% NOx conversion was preserved for about 8 h.
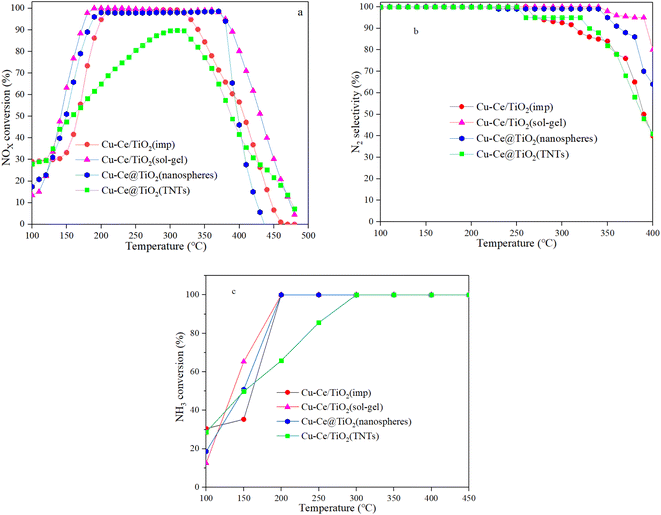 |
| | Fig. 1 a) NOx conversion, (b) N2 selectivity and (c) NH3 conversion of all catalysts. Reaction conditions: 500 ppm NO, NO/NH3: 1, 7.5% O2 and N2 in balance, and GHSV = 24000 h−1. | |
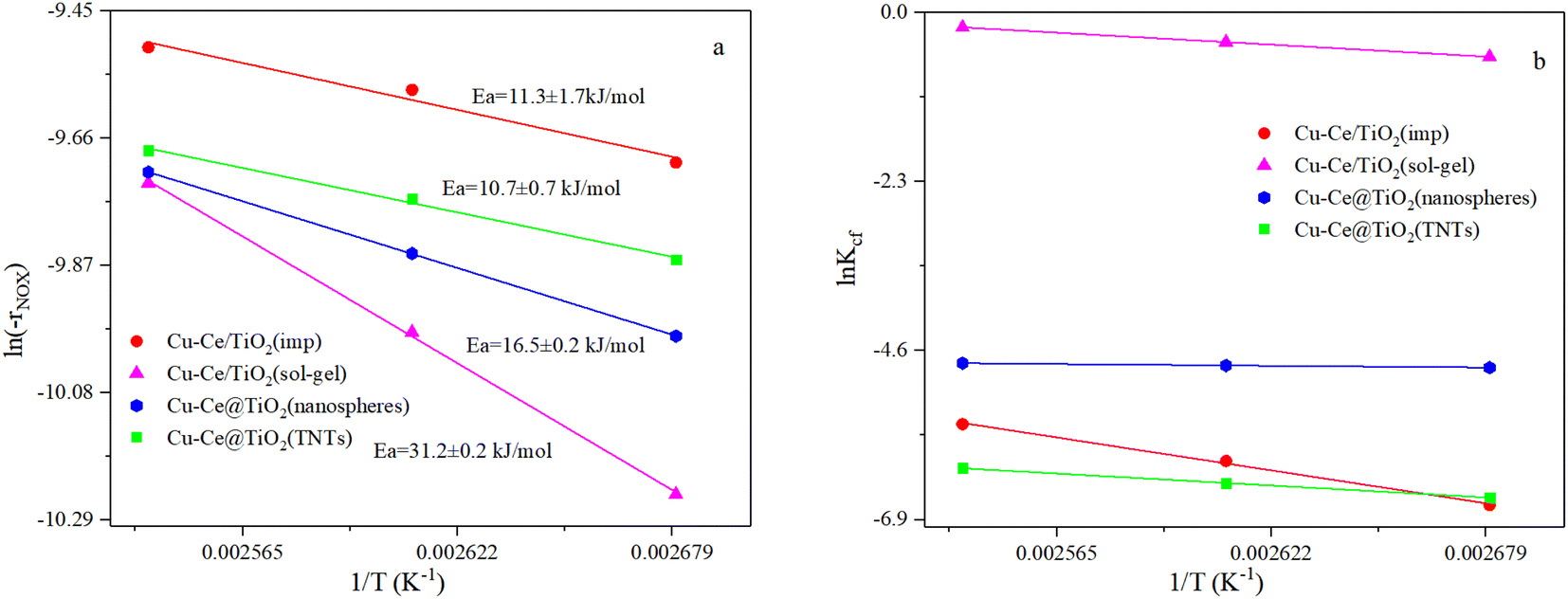
3.2. NH3-SCR kinetic tests
To precisely study the performance of the catalyst, a kinetic experiment was conducted.17 The potential impact of mass transfer limitations from film diffusion on the catalysts was first ruled out (Fig. S2a and b†). The Arrhenius plots are shown in Fig. 2a. In this graph, the slope was −Ea/R and the intercept was ln kcf, where Ea and kcf represent activation energy and collision frequency, respectively. In addition, the collision frequency diagram is shown in Fig. 2b. The NOx consumption equation for the NH3-SCR reaction is shown in eqn (4). The concentration, constant parameters and forward reaction rate were considered:18| |
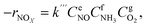 | (4) |
where constant k′′′ is an intrinsic reaction rate, rNOx is the observed NH3-SCR reaction rate (mol g−1 s−1), and CNO, CNH3 and CO2 are the concentrations of inlet gas (ppm). The exponents of e, f, and g were the reaction orders of NO, NH3, and O2, respectively. In this case, oxygen was in zero reaction order owing to the excessive concentration of oxygen.17 The Ea of the catalysts with different morphologies increased in the order of Cu–Ce@TiO2(TNTs) < Cu–Ce/TiO2(imp) < Cu–Ce@TiO2(nanospheres) < Cu–Ce/TiO2(sol–gel). Therefore, the morphology significantly affected the activation energy of the catalyst. Furthermore, the reaction rate increased with the enhancement of the activation energy at high temperature.19 Notably, the Cu–Ce/TiO2 (sol–gel) catalyst had the highest collision frequency, which could explain the largest collision probability of NH3 and NO on the catalyst surface (Fig. 2b).
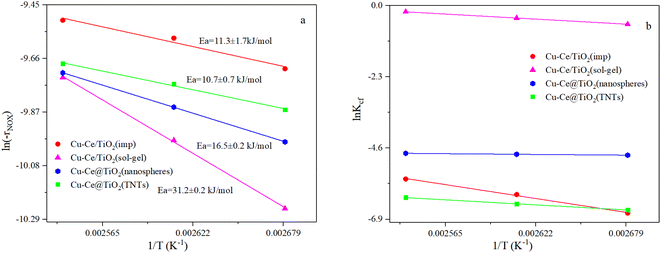 |
| | Fig. 2 (a and b) Arrhenius and collision frequency plots of all catalysts. Ea and Kcf denote energy activation and collision frequency, respectively. Reaction conditions: 500 ppm NO; NH3/NO: 1; 7.5 vol% O2; balanced by N2, GHSV: 360000 h−1. | |
3.3. BET results
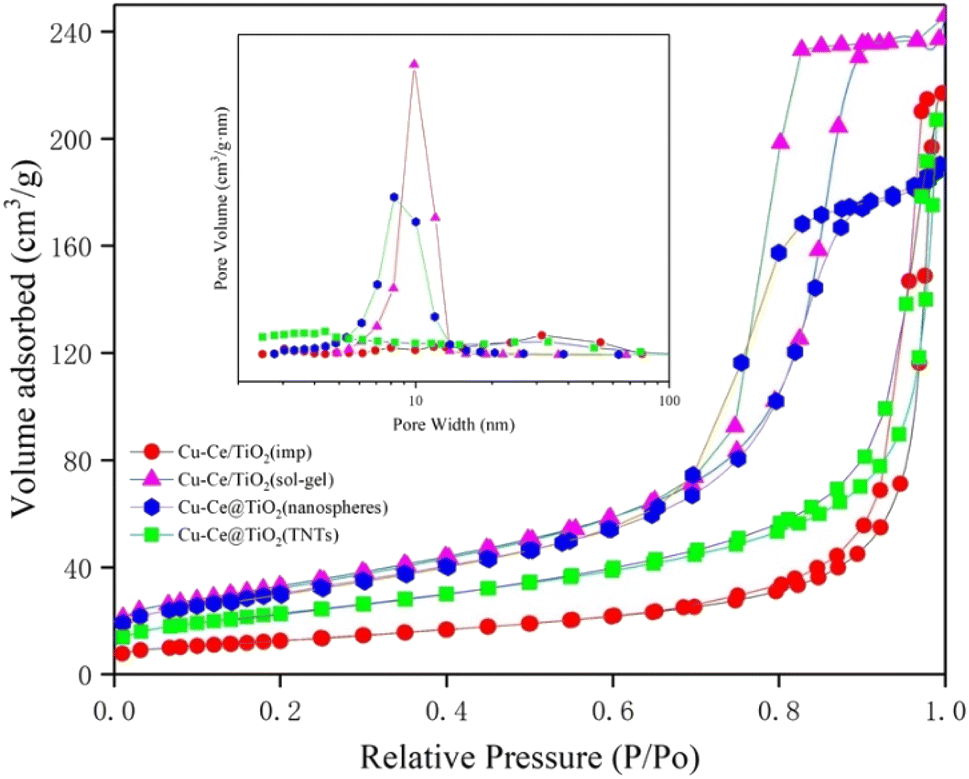
The specific surface area, pore volume, and pore size of the samples are summarized in Table S1.† The results indicated that the BET surface area and pore volume of the Ce/TiO2(sol–gel) were the highest of all catalysts. Fig. 3 shows the pore size distribution curves and N2 adsorption–desorption isotherms of the catalysts. According to the definition of the International Union of Pure and Applied Chemistry (IUPAC) classification, the curves with significant hysteresis loops in the P/P0 range of 0.7 to 1 in Fig. 3 can be classified as type IV, indicating that the sample has a large number of mesoporous properties of particle accumulation.11,20 The Cu–Ce/TiO2 (sol–gel) catalyst with a larger surface area was beneficial in improving the adsorption and activation of the reactants.21 In addition, Fig. 3 shows that Cu–Ce/TiO2 (sol–gel) and Cu–Ce@TiO2 (nanospheres) have H2 type hysteresis loops, while Cu–Ce/TiO2 (imp) and Cu–Ce@ TiO2 (TNTs) exhibit an H3 type hysteresis loop.22 From the inset in Fig. 3, the pore size distribution of Cu–Ce/TiO2 (sol–gel), Cu–Ce@TiO2 (nanospheres) is concentrated at 10 nm, which confirms that these two catalysts have typical mesoporous structures. The abundant mesoporous structure could provide channels for reaction gas to improve the catalytic performance.23 The Cu–Ce/TiO2 (sol–gel) exhibited the highest absorption capacity under relatively high pressure, which might be attributed to the largest average pore volume.24,25 As shown in Table S1,† the average pore diameters of Cu–Ce/TiO2 (imp), Cu–Ce/TiO2 (sol–gel), Cu–Ce@TiO2 (nanospheres) and Cu–Ce@TiO2 (TNTs) are 28.59 nm, 12.32 nm, 10.69 nm and 15.54 nm, respectively. In addition, Table S1† shows that Cu–Ce/TiO2 (sol–gel) has the largest specific surface area among all the catalysts, which is beneficial for improving catalytic performance. Generally, the porous structure and larger specific surface area could improve the catalytic performance by increasing the adsorption of reactants and providing more active sites.15 The results are consistent with the catalyst activity, as depicted in Fig. 1c.
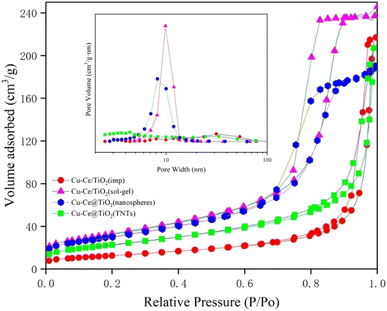 |
| | Fig. 3 Nitrogen adsorption–desorption isotherms and the corresponding size distribution curves (inset) of all catalysts. | |
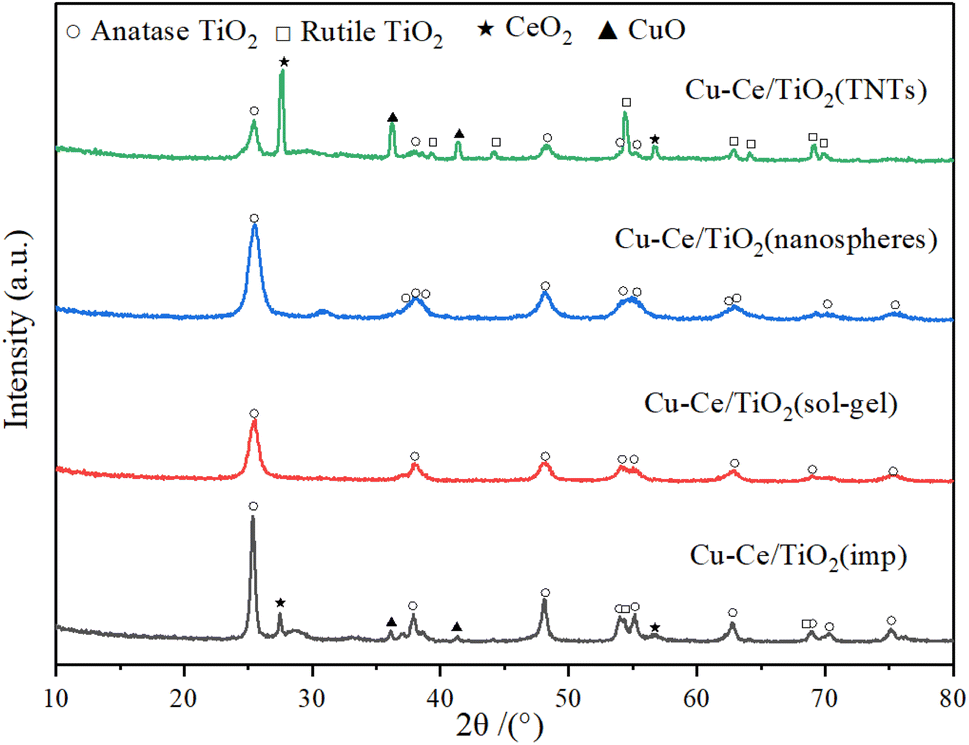
3.4. XRD analysis
The X-ray diffraction patterns of the catalysts are illustrated in Fig. 4. The characteristic peaks of ordered CuO (JCPDS 78-0428), CeO2 (JCPDS 81-0792), rutile (JCPDS 87-0920) and anatase (JCPDS 83-2243) could be formed in Cu–Ce/TiO2 (imp) and Cu–Ce@TiO2 (TNTs) catalysts. The crystalline sizes of TiO2, CuO and CeO2 in the catalysts were calculated using the Scherrer equation (Table S2†). In addition, the sharpness and relative intensity of the anatase and rutile peaks for Cu–Ce/TiO2 (imp) and Cu–Ce@TiO2 (TNTs) catalysts are more significant than those of the two other catalysts (Fig. 4). The low activity of Cu–Ce/TiO2 (imp) and Cu–Ce@TiO2 (TNTs) catalysts might be due to the existence of the rutile phase.26 Compared with Cu–Ce@TiO2 (TNTs), CeO2 and CuO peaks were unobserved on Cu–Ce@TiO2 (nanospheres) (Fig. S3†). This is mainly because CeO2 and CuO combined well and were wrapped by TiO2, which could reduce the intensity of the diffraction peaks of CeO2 and CuO.2,27 Furthermore, the Cu–Ce/TiO2 (sol–gel) catalyst demonstrated only pure anatase peaks, which was mainly attributed to the high dispersion and amorphous phase of copper and cerium.
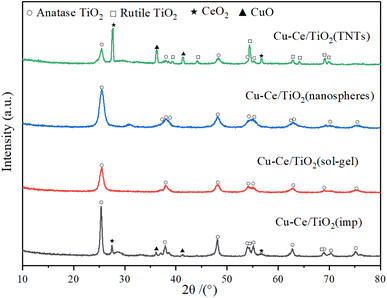 |
| | Fig. 4 XRD patterns of all catalysts. | |
3.5. TEM images
The morphologies and structures of Cu–Ce/TiO2 (imp), Cu–Ce/TiO2 (sol–gel), Cu–Ce@TiO2 (nanospheres) and Cu–Ce@TiO2 (TNTs) were studied using TEM. Fig. 5a and b shows that catalysts have well-dispersed nanoparticle morphologies. As shown in Fig. 5c and d, the catalysts have spherical and nanotube structures, respectively. As shown in Fig. 5e, there are four kinds of lattice fringes. The measured lattice spacings of 0.35 nm, 0.32 nm, 0.18 nm and 0.26 nm were consistent with the (101), (110), (220) and (110) planes of anatase, rutile, CeO2 and CuO, respectively.28–30 These results indicated that a large amount of TiO2, CuO and CeO2 crystal phases were formed in the Cu–Ce/TiO2 (imp) catalyst, which was consistent with the XRD results. As shown in Fig. 5f, no planes corresponding to CuO and CeO2 are observed in Cu–Ce/TiO2 (sol–gel), only the anatase phase (101) plane is found to be consistent with the lattice fringes identified as 0.35 nm.30 The HRTEM image of the Cu–Ce@TiO2 (nanospheres) catalyst is shown in Fig. 5g. The clear structure of the nanosphere was covered by the core–shell structure of the TiO2 layer.31,32 As shown in Fig. 5e, CuO and CeO2 particles are mainly entrapped into the inner cavity of the TiO2 (TNTs). The measured lattice spacings were consistent with the (110) and (111) planes of CuO and CeO2, which are 0.27 nm and 0.30 nm, respectively.2 The results agreed well with the XRD results.
 |
| | Fig. 5 TEM and HRTEM images of four catalysts: (a and e) Cu–Ce/TiO2 (imp), (b and f) Cu–Ce/TiO2 (sol–gel), (c and g) Cu–Ce@TiO2 (nanospheres) and (d and h) Cu–Ce@TiO2 (TNTs). | |
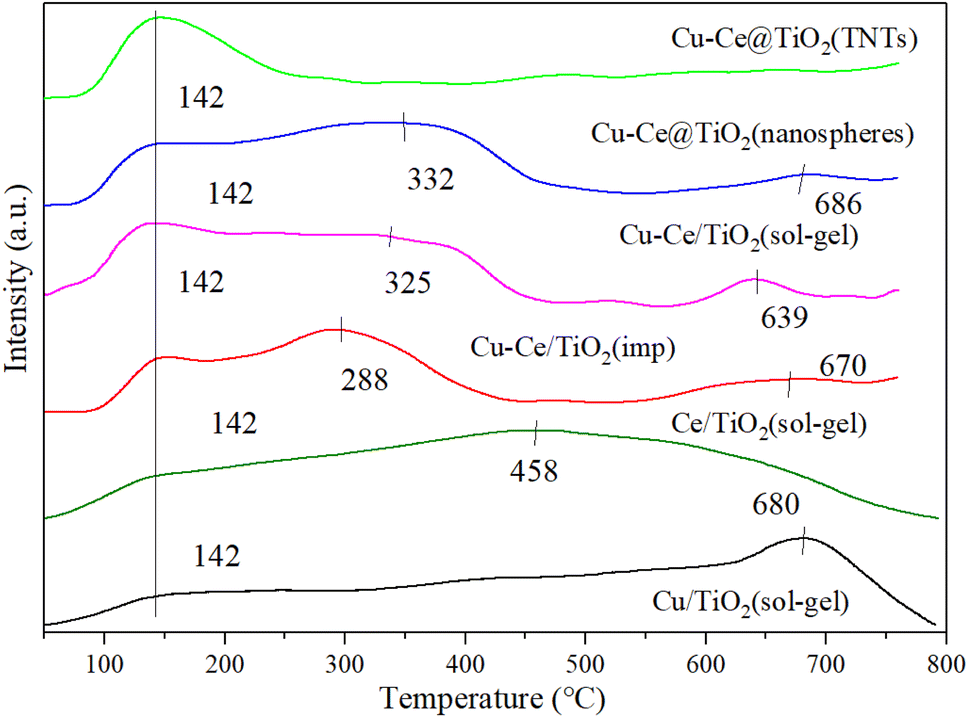
3.6. NH3-TPD
Generally, the NH3-SCR reaction was affected by the adsorption and activation of NH3.33 As shown in Fig. 6, the surface acidity of the catalyst is explored by the NH3-TPD characterization. A broad peak around 142 °C could be observed for all catalysts, which was attributed to the desorption of NH3 from weakly acidic sites.12 For all catalysts, the desorption peaks at 288 °C, 325 °C and 332 °C corresponded to the NH4+ released from the acid site.1,27,34 Moreover, the desorption peaks at 458 °C, 639 °C, 670 °C, 680 °C and 686 °C were assigned to the NH3 desorbed from the acid sites.34–36 Compared to the Cu/TiO2 (sol–gel) or Ce/TiO2 (sol–gel) catalyst, the desorption peaks of the bimetallic catalyst changed, indicating that a reaction occurred between Cu and Ce. As depicted in Table S3,† Cu–Ce/TiO2 (sol–gel) catalyst showed the highest amount of acid sites. In addition, the acidity of the Cu–Ce@TiO2 (TNT) catalyst is lower than that of Cu/TiO2 (sol–gel) or Ce/TiO2 (sol–gel), indicating that the structure of the catalyst was changed. In contrast, the acidity of catalyst Cu–Ce/TiO2 (sol–gel) is approximately equal to the sum of Cu/TiO2 (sol–gel) and Ce/TiO2 (sol–gel), indicating that the catalyst structure is well preserved. From Table S5,† the quantity of acid sites in catalyst Cu–Ce/TiO2 (sol–gel) is significantly higher than that in catalysts Cu/TiO2 (sol–gel) and Ce/TiO2 (sol–gel). In addition, the proportion of Brønsted acid is also significantly better than the other two catalysts. This enables the catalyst to have better activity at high temperatures.
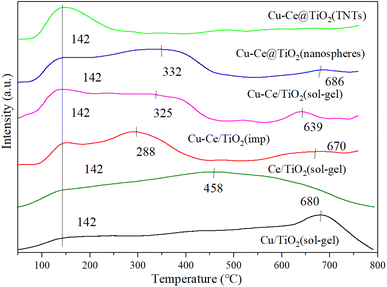 |
| | Fig. 6 NH3-TPD results for all catalysts. | |
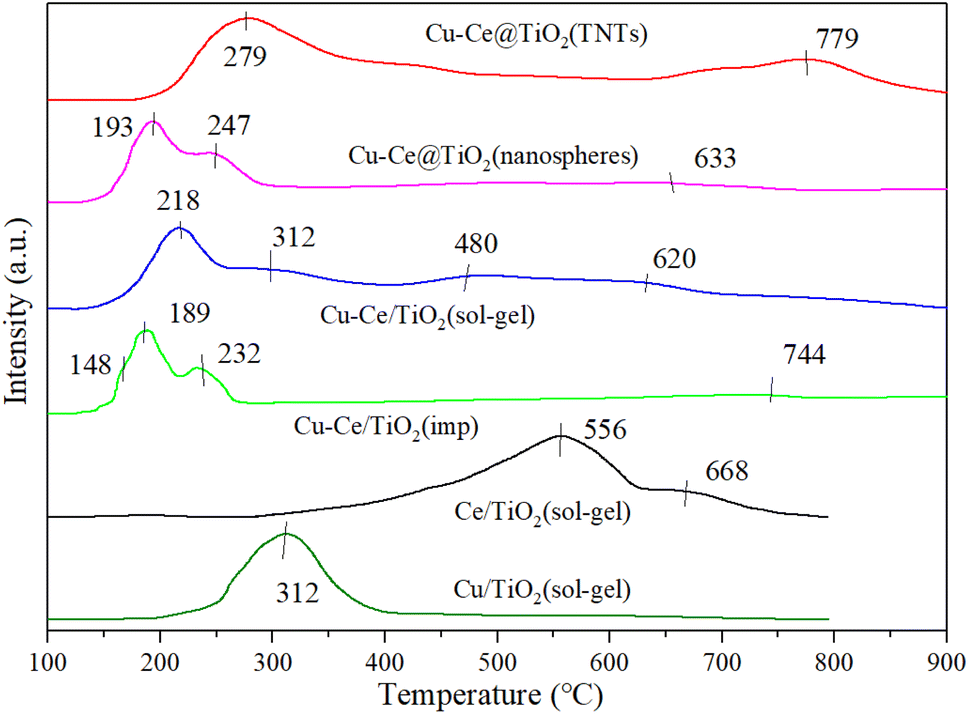
3.7. H2-TPR
Fig. 7 shows the H2-TPR characterization results of all samples. For Cu/TiO2 (sol–gel) and Ce/TiO2 (sol–gel) catalysts, strong reduction peaks appeared at 312 °C and 568 °C, respectively. The peaks at 568 °C and 668 °C were attributed to the stepwise reduction of Ce4+ to Ce3+ alongside strong interactions with TiO2.21,37 From previous reports,34,37,38 less overall interaction existed in the reduction of weakly magnetic Cu2+ and catalyst substrate, resulting in the peak at 148 °C. Moreover, the isolated Cu2+ ions could generate a reduction of peak (148 °C). It was also concluded that the peaks at 189 °C, 193 °C, 218 °C, 232 °C, 247 °C, 279 °C and 312 °C were attributed to the reduction of small-sized CuO particles (Cu2+ → Cu+ → Cu0). The H2-TPR results showed that bulk and dimensional (two- or three-) clusters of CuO were the main components of Cu–Ce/TiO2 (imp) and Cu–Ce@TiO2 (nanosphere) catalysts.39 Fig. 7 shows that the sharpest peaks (except Cu–Ce@TiO2 (TNTs)) moved to lower temperatures (232 °C to 189 °C, 312 °C to 218 °C and 247 °C to 193 °C), which is mainly attributed to the interaction between CuO and CeO2–TiO2 substrates. Most importantly, the dispersion of CuO was enhanced under the reaction CuO and CeO2–TiO2 substrates, thereby improving the medium-low temperature reduction performance of CuO.36
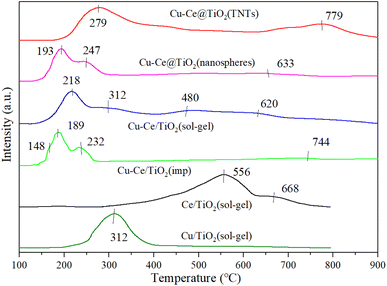 |
| | Fig. 7 H2-TPR results of all catalysts. | |
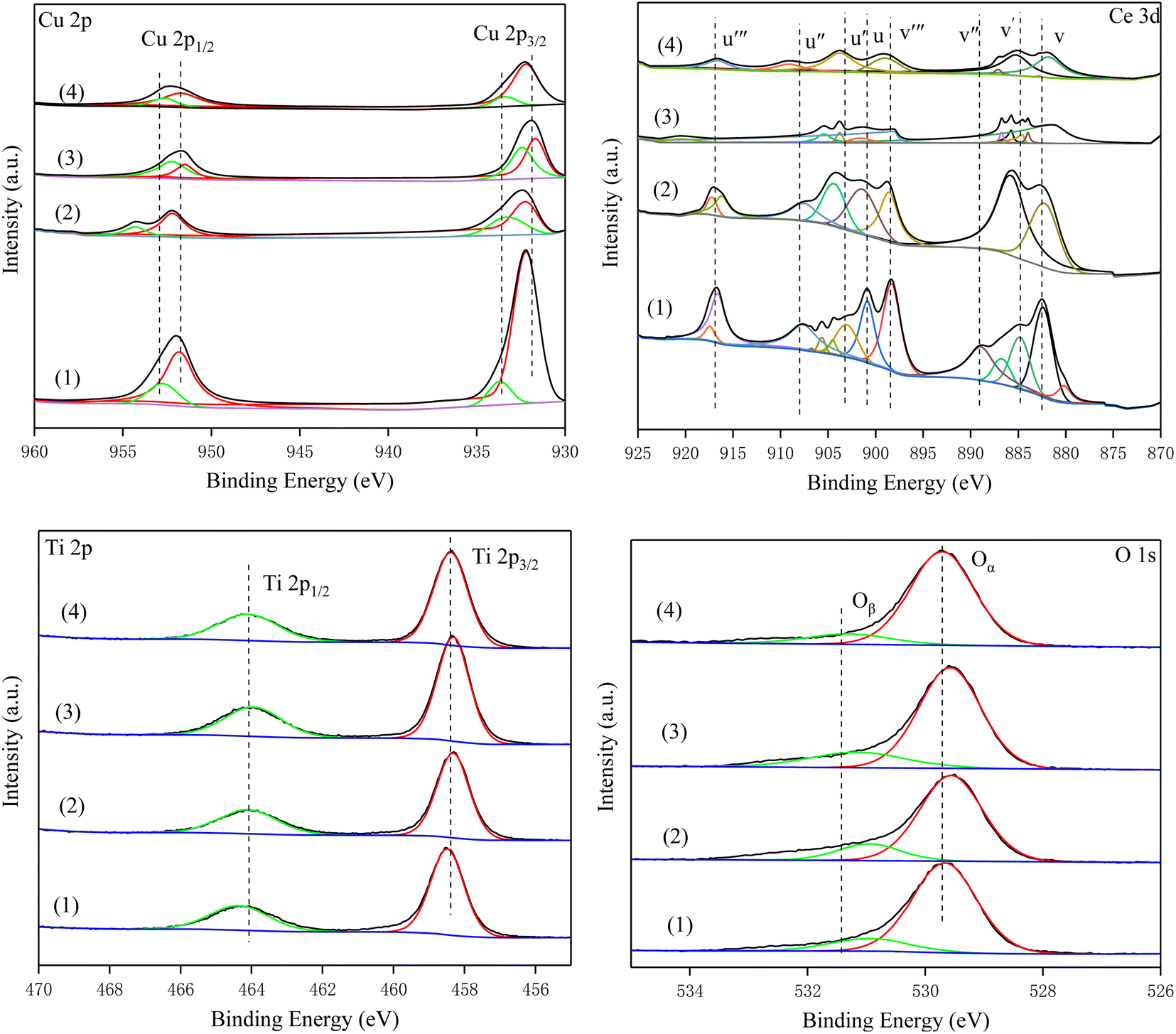
3.8. XPS results
To investigate the surface composition and oxidation state of the elements of the catalyst, all samples were characterized by XPS. Fig. 8 shows the Cu 2p spectra of all catalysts. The peaks at 932 eV (Cu2+) and 952.4 eV (Cu+) were Cu 2p3/2 and Cu 2P1/2, respectively.24,40,41 The peaks at 934.2 eV and the corresponding satellite peaks were assigned to Cu2+, and the peaks located at 932.5 eV were attributed to Cu+. Compared with Cu–Ce/TiO2 (imp) and Cu–Ce/TiO2 (TNTs) catalysts, the surface relative atomic number of Cu2+ in Cu–Ce/TiO2 (sol–gel) and Cu–Ce@TiO2 (nanospheres) are much higher than that of Cu+ (Table S4†). It is worth mentioning that the increase in Cu2+ significantly promoted the SCR activity of the catalyst, which was in accordance with previous studies.36
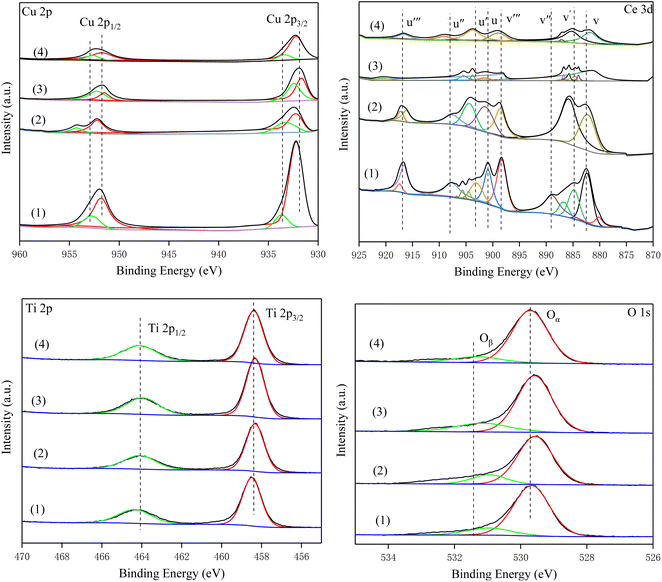 |
| | Fig. 8 XPS high-resolution scans of the Cu 2p, Ce 3d, Ti 2p and O 1s peaks for the catalysts. (1) Cu–Ce/TiO2 (imp), (2) Cu–Ce/TiO2 (sol–gel), (3) Cu–Ce@TiO2 (nanospheres) and (4) Cu–Ce@TiO2 (TNTs). | |
The Ce 3d orbits of all catalysts are composed of two multiple peaks (v and u) and can be completely divided into 8 peaks (Fig. 8). The peaks marked v′ and u′ components were attributed to 3d3/2 and 3d5/2 features of Ce3+ species, while v/v′′/v′′′ and u/u′′/u′′′ were assigned to the Ce4+ 3d3/2 and Ce4+ 3d5/2 species.42,43 Furthermore, compared with other catalysts, Cu–Ce/TiO2 (sol–gel) exhibits a higher relative atomic fraction of Ce4+ (Table S4†).
In the Ti 2p spectra, the peaks of the four catalysts were consistent. The peak near 458.8 eV was TiO2, and Ti4+ was mainly present on the surface of the catalysts. As shown in Table S4,† the Ti content in Cu–Ce/TiO2 (imp) and Cu–Ce/TiO2 (sol–gel) are lower than that in Cu–Ce@TiO2 (nanospheres) and Cu–Ce@TiO2 (TNTs). The results demonstrated that TiO2 encapsulated Cu–Ce in Cu–Ce@TiO2 (nanospheres) and Cu–Ce@TiO2 (TNTs) catalysts, which was similar to the core–shell morphology results of HRTEM.
From the XPS spectra, the O 1s spectra of all catalysts can be separated into two peaks. The peaks at 529.8 eV and 531.7 eV were defined as the lattice oxygen (Oα) peak and the weakly adsorbed oxygen (Oβ) peak, respectively.44,45 According to previous studies,46,47 Oα had weakly active chemical properties because of lower mobility than Oβ. From Table S4,† the relative atomic fraction of Oβ (36.44%, Cu–Ce/TiO2 (sol–gel)) is much greater than that of others. The presence of abundant Oβ could affect the promotion of the conversion of NO to NO2, which in turn promoted the “fast SCR” process.48
| | |
2NH3 + NO + NO2 = 3H2O + 2N2
| (5) |
3.9. In situ-DRIFTs studies
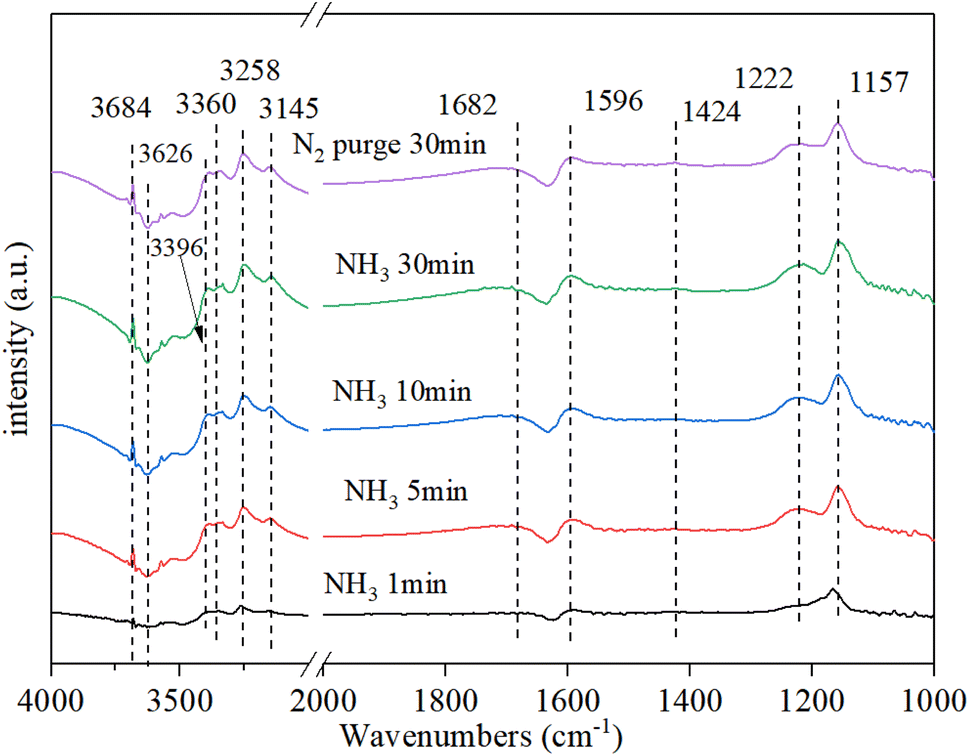
3.9.1. NH3 adsorption. Fig. 9 shows that the NH3 adsorption on Cu–Ce/TiO2 (sol–gel) spectra was obtained by DRIFT. Strong bands were observed at 1157 cm−1, 1222 cm−1, and 1596 cm−1 owing to coordinated asymmetric and symmetric deformations of NH3 at Lewis acid sites on the surface.49 The band at 1423 cm−1 was attributed to the asymmetric deformation of NH4+ at the Brønsted acid site.50 There was a clear negative peak around 1630 cm−1, which was likely to be water desorbed by ammonia adsorption. Moreover, a clear negative peak was the NH3 adsorbed on the Lewis site. In the N–H region, some coordinated NH3 bands and one NH2 group band, which corresponded to Lewis acid sites, were observed at 3360 cm−1, 3258 cm−1, 3145 cm−1 and 3396 cm−1, respectively.17,51 In addition, the strong and negative bands of the O–H groups occupied by NH3 adsorption were detected at 3684 cm−1 and 3626 cm−1, respectively.52,53 After the N2 purge, the intensity of each peak did not change significantly, indicating that the adsorption of NH3 was stable.
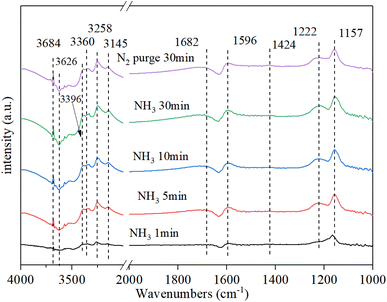 |
| | Fig. 9 In situ DRIFT spectra of Cu–Ce/TiO2 (sol–gel). Reaction conditions: 800 ppm NH3 and 200 °C. | |
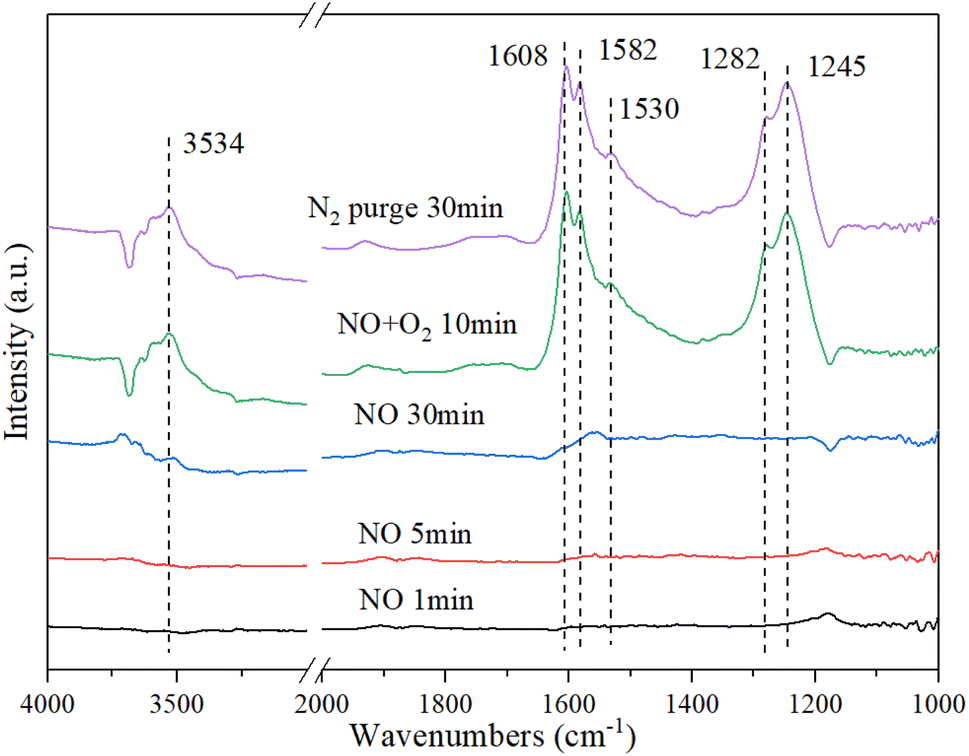
3.9.2. NO + O2 Co-adsorption. Fig. 10 shows the DRIFT spectrum of NO + O2 co-adsorption. As shown in Fig. 10, the bands at 1245 cm−1, 1282 cm−1, 1530 cm−1, 1582 cm−1, 1608 cm−1, and 3534 cm−1 are detected for Cu–Ce/TiO2 (sol–gel) catalyst. Compared with the single adsorption of NO, the addition of O2 affected the adsorption of NO on the surface of the catalyst. When O2 was introduced, four new additional bands of 1608 cm−1, 1582 cm−1, 1282 cm−1 and 1245 cm−1 appeared. The surface nitro substance or nitrate on the active site of the transition metal of the catalyst could produce bands in the range of 1650 cm−1–1500 cm−1.54,55 The bands at 1582 cm−1, 1530 cm−1, and 1245 cm−1 were classified as bidentate or bridged nitrate.4,56,57 In addition, bands of monodentate nitrate (1282 cm−1 and 1608 cm−1) were detected.
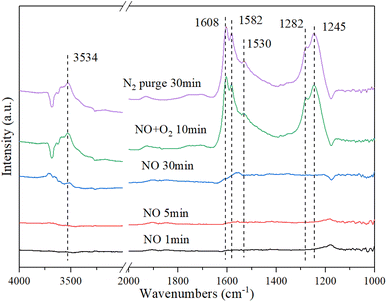 |
| | Fig. 10 In situ DRIFT spectra of Cu–Ce/TiO2 (sol–gel). Reaction condition: 800 ppm NO (30 min), followed by the introduction of O2 at 200 °C. | |
When NO was introduced for 30 min, a band of nitrosyl (1530 cm−1) appeared. Specifically, the formation of this band (3534 cm−1) represented the formation of a large number of –OH groups on the catalyst surface, and these –OH groups came from H2O molecules in the adsorption-reaction process.58 When NO was adsorbed on the catalyst, it recombines to produce NO2 using eqn (6):
NO and NO2 could react with acid sites on the catalyst surface to form H2O using eqn (7):
| | |
NO + NO2 + 2H+ → 2NO+ + H2O
| (7) |
In summary, there were numerous acid sites on the catalyst surface, which promoted the reaction in eqn (7). In the presence of O2, the new bands were generated at 1608 cm−1 and 1245 cm−1, which were formed by the formation of bridge nitrates on the catalyst. The strength of the groups on the surface of the catalyst increased significantly with the introduction of O2. The amounts of nitrate and nitrosyl formed on the CeO2 were improved considerably. In addition, the oxidation performance of the catalyst surface increased with the addition of O2, which could be seen from the stronger –OH peak at 3534 cm−1. Therefore, more NO could be bonded to the catalyst surface and react in the adsorption process.
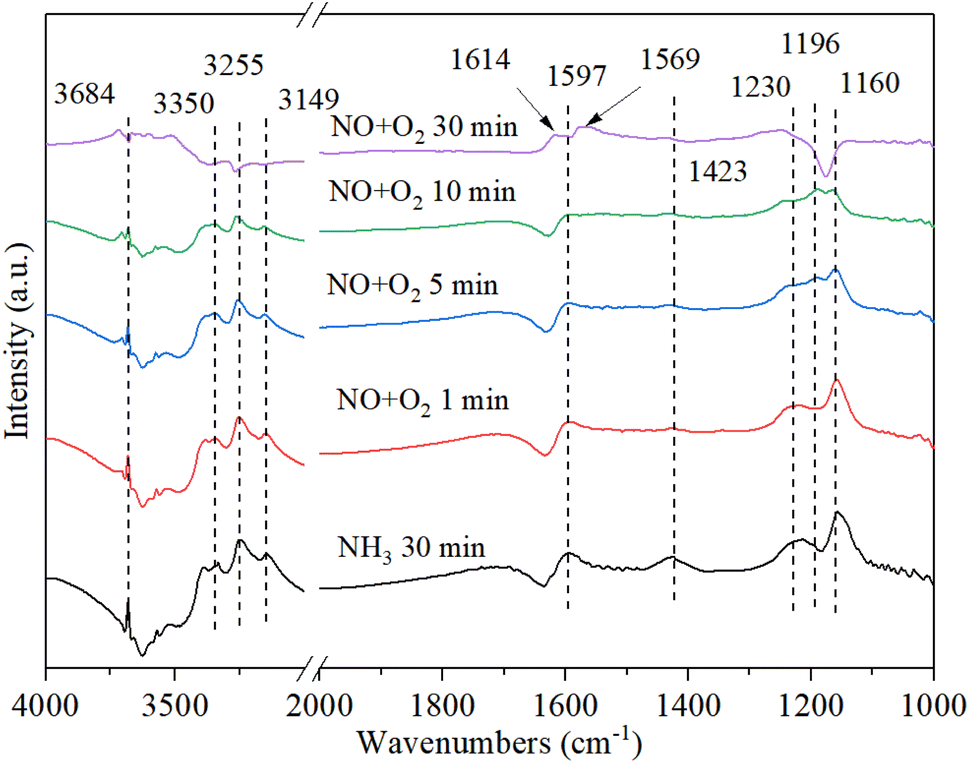
3.9.3. The reaction between nitrogen oxides and pre-adsorbed ammonia. Fig. 11 shows the DRIFT spectrum of Cu–Ce/TiO2 (sol–gel) catalyst and pre-adsorbed NH3 when exposed to NO + O2 at 200 °C. The adsorbed NH3 decreased after the introduction of NOx owing to the reaction between NOx and adsorbed NH3. With the introduction of NO + O2, the new bands (1614 cm−1, 1569 cm−1 and 1196 cm−1) corresponding to NOx species appeared, and the species of NH3 (1160 cm−1) and NH4+ (1597 cm−1 and 1423 cm−1) were gradually disappeared.50 The negative peak at 1614 cm−1 changed to a positive peak, indicating that the ammonia species adsorbed by the Lewis acid site gradually reacted with NOx to form NH4NOx. To investigate the type of mechanism, NO, and O2 were introduced again after blocking ammonia. However, the band of 1196 cm−1 gradually disappeared, indicating that the band corresponding to the NOx species was unstable. However, the nitrite/nitrate band on the catalyst became dominant, and the NH3-related bands of 3684 cm−1, 3350 cm−1, 3255 cm−1 and 3149 cm−1 (N–H region) disappeared, which confirmed that the reaction of ad-NOx and ad-NH3 was accelerated by cerium oxide.47,59 It was suggested that NH4NOx species were formed. Because the NH4NOx species decomposed into N2 and H2O, the N2 selectivity was enhanced.
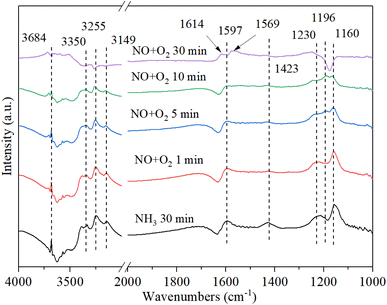 |
| | Fig. 11 In situ DRIFT spectra of Cu–Ce/TiO2 (sol–gel) were pretreated with 800 ppm NH3 (30 min) at 200 °C, and then 800 ppm NO and 7.5% O2 were introduced. | |
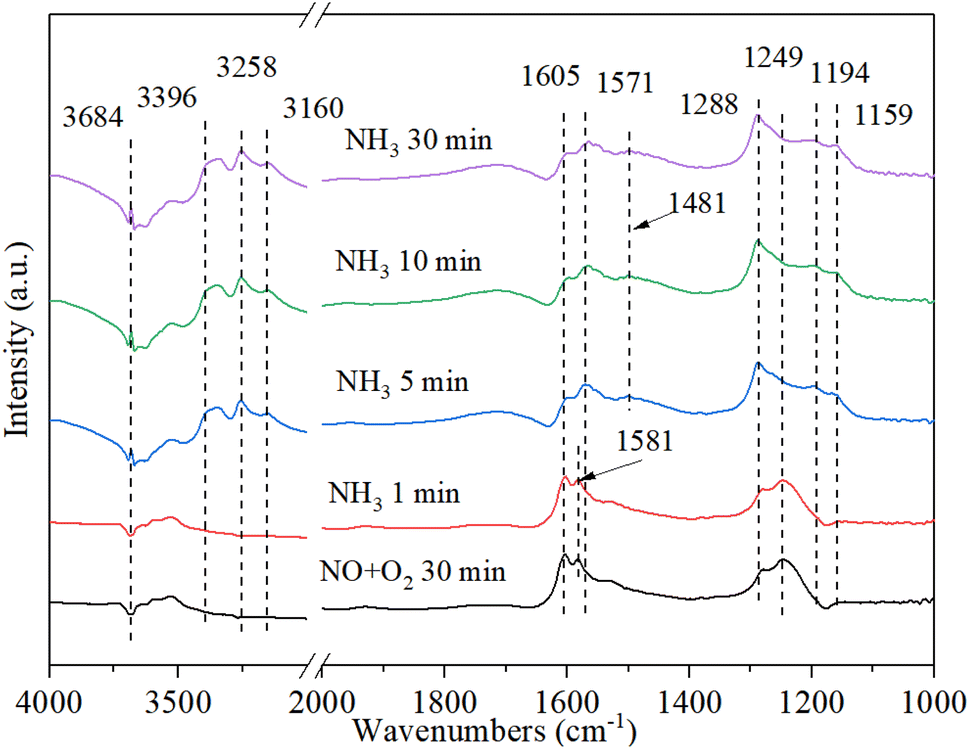
3.9.4. The reaction between ammonia and pre-adsorbed nitrogen oxides. Fig. 12 shows the DRIFT spectrum of Cu–Ce/TiO2 (sol–gel) catalyst and pre-adsorbed NO + O2 when exposed to NH3 at 200 °C. The bands at 1581 cm−1 and 1249 cm−1 rapidly decreased, indicating that part of the nitrate reacted with ammonia to form NH4NO2/NH4NO3. Therefore, the inhibition of active sites by nitrate on the catalyst might inhibit the activity of NH3-SCR. In addition, after purging with NH3 for 5 minutes, the Lewis acid bonded NH3 and Brønsted acid bonded NH4+ appeared on the catalyst at 3396 cm−1, 3258 cm−1, 3160 cm−1, 1571 cm−1, 1194 cm−1, 1159 cm−1 and 1481 cm−1 59. The new band appeared within 5 minutes after the introduction of NH3. However, the NOx species band did not disappear, indicating that NOx and NH3 could coordinate to different acid sites.56 The above phenomenon indicated that the NOx species adsorbed on Cu–Ce/TiO2 (sol–gel) was consumed by reacting with gaseous NH3, which was in accordance with the Eley–Rideal (E–R) mechanism.15
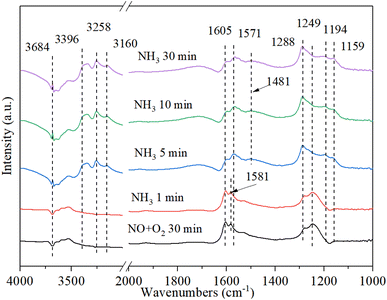 |
| | Fig. 12 In situ DRIFT spectra of Cu–Ce/TiO2 (sol–gel) were pretreated with 800 ppm NO and O2 (30 min) at 200 °C, and then 800 ppm NH3 and 7.5% O2 were introduced. | |
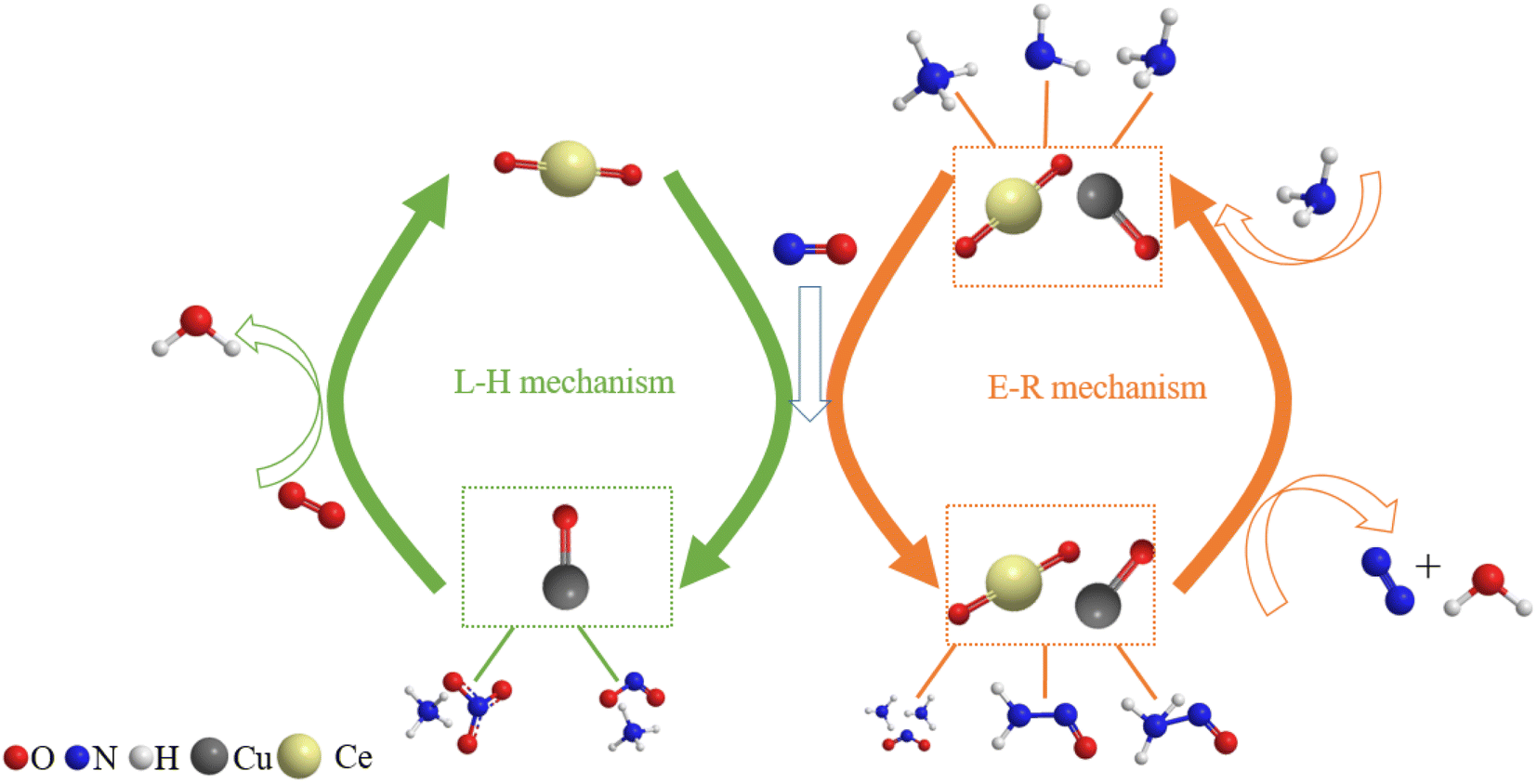
3.10. Discussion
The amide NH2 was an intermediate in the oxidation of NH3 to N2, which was easily formed by hydrogen abstraction conversion from coordinating ammonia on Lewis acid sites.49,53 From the results of DRIFTS, the Brønsted acid sites on the Cu–Ce/TiO2 catalyst were used as NH3 reservoirs. Specifically, the NH3 could migrate to the Lewis acid sites to provide additional NH3 for the SCR reaction, which could form NH4NO3. Therefore, the Lewis acid sites mainly contributed to NH3 activation (<400 °C). The Brønsted acid site was essential for the binding and dispersion of metal ions.60 From the peak intensity of nitrite shown in Fig. 10 and 12, the adsorption of nitrite on Cu–Ce/TiO2 (sol–gel) was weak, resulting in a small number of nitrite species that could participate in the reaction. Consequently, this phenomenon could weaken the L–H mechanism. In contrast, the E–R mechanism could be enhanced, indicating that cerium oxide could promote the oxidation of NH3. Therefore, the formation of NH2, which could react with gaseous NO, was promoted.15,61 During the SCR reaction of reducing NOx with NH3, the L–H and E–R mechanisms occurred simultaneously under medium-low temperatures, but the E–R mechanism might be dominant. Based on analysis of the in situ DRIFT results, this research proposed a reaction for the Cu–Ce/TiO2 (sol–gel) catalyst as follows:| |
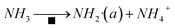 | (8) |
Moreover, there was another reaction pathway on the Cu–Ce/TiO2 (sol–gel) catalyst:
| |
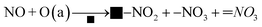 | (11) |
| | |
■–NO2 + NH+4 → NH4NO2
| (12) |
| |
■–NO3 + ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NO3 + NH+4 → NH4NO3 NO3 + NH+4 → NH4NO3
| (13) |
| | |
NH4NO2 → NH2NO → N2 + H2O
| (14) |
| | |
NH4NO3 → NH2NO → N2 + H2O
| (15) |
Furthermore, various NO2 and NO are adsorbed on the surface of the Cu–Ce/TiO2 (sol–gel) (Fig. 10). The substance reacts as follows:62
| | |
■–NO2 + 2NH3 → ■−NO2[NH3]2
| (17) |
| | |
■–NO2[NH3]2 + NO → ■ → 2N2 + 3H2O
| (18) |
where ■ denotes the catalyst surface.
Finally, the reaction mechanism diagram was drawn, showing the changes in active sites and immediate products in the reaction process of NH3-SCR (Fig. 13). These findings could provide new perspectives for medium-low NH3-SCR mechanisms and DeNOx catalysts design.
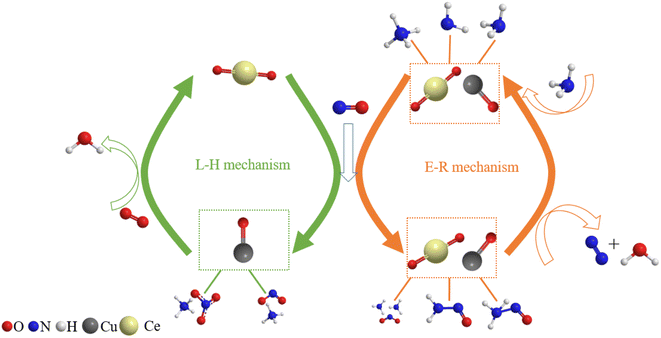 |
| | Fig. 13 Proposed reaction scheme for NH3-SCR over Cu–Ce/TiO2 (sol–gel) catalyst. | |
4. Conclusions
Catalysts with different morphologies of Cu–Ce/TiO2 were explored in this study. The sol–gel nanoparticle catalyst exhibited excellent catalytic activity at 200–400 °C. Moreover, different morphologies could significantly affect the catalyst activation energy and collision frequency. From the characterization test study, the catalysts with different morphologies had different phases of CeO2, CuO and TiO2. The results of XPS and XRD demonstrated that the solid structural interaction of oxides (Ce, Cu and Ti included) generates metal oxides with high dispersibility and adsorbed oxygen with high mobility. The Cu–Ce/TiO2 (sol–gel) was confirmed to have the highest activation energy and collision frequency. This phenomenon suggested that Cu–Ce/TiO2 (sol–gel) had the highest redox cyclability and the largest amounts of Cu2+ and Ce4+. It is worth mentioning that NH3 was preferentially adsorbed on Ti sites, and abundant Lewis acid sites formed by cerium oxide could facilitate the production of highly dispersed Cu2+ ions. Therefore, the oxidation and activation of NOx and NH3 were enhanced, resulting in the promotion of the acid cycle of NH3-SCR. Specifically, it was proposed that the reaction between Cu2+-NOx, nitrate-Ce3+ sites and NH3–Ti3+ was the main reaction route of the catalyst for NH3-SCR. Moreover, Ce promoted the production of isolated Cu2+, which suppressed the formation of the CuO species. The active oxygen generated by the strong interaction of metal oxides could promote the formation of nitrate oxidized by Cu2+-NOx. Furthermore, Ce enhanced the redox cycle by promoting the electron transfer effect of the Cu sites.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The funding of this research was supported by the Social Development of Jiangsu Province-General Project (Grant No. BE2020754), National Key Research and Development Plan (Grant No. 2022YFC3701600) and the National Nature Science Foundation of China (Grant No. 41877469 and 41771498).
References
- X. Wang, Y. Liu, Q. Ying, W. Yao and Z. Wu, Appl. Catal., A, 2018, 562, 19–27 CrossRef CAS.
- G. Yao, L. Wu, T. Lv, J. Li, Y. Huang, K. Dong and X. Li, Open Chem., 2018, 16, 1–8 CAS.
- W. Zhang, Y. Tang, C. Lu, J. Zou, M. Ruan, Y. Yin, M. Qing and Q. Song, Ultrason. Sonochem., 2021, 72, 105466 CrossRef CAS PubMed.
- Z. M. Liu, Y. Yi, J. H. Li, S. I. Woo, B. Y. Wang, X. Z. Cao and Z. X. Li, Chem. Commun., 2013, 49, 7726–7728 RSC.
- X. Zhou, Z. Chen, Z. Guo, H. Yang, J. Shao, X. Zhang and S. Zhang, J. Hazard. Mater., 2021, 405, 124177 CrossRef CAS PubMed.
- X. Chen, H. Wang, S. Gao and Z. Wu, J. Colloid Interface Sci., 2012, 377, 131–136 CrossRef CAS PubMed.
- X. Liu, P. Jiang, Y. Chen, Y. Wang, Q. Ding, Z. Sui, H. Chen, Z. Shen and X. Wu, Chem. Eng. J., 2021, 421 CAS.
- M. Maulidiyah, P. E. Susilowati, N. K. d. Mudhafar, L. O. A. Salim, D. Wibowo, M. Z. Muzakkar, I. Irwan, Z. Arham and M. Nurdin, Biointerface Res. Appl. Chem., 2022, 12, 1628–1637 CAS.
- N. Mozaffari, V. Vambol, Y. Hamzah, A. E. D. Mahmoud, N. Mozaffari, N. A. Khan, S. Vambol, N. Khan and A. Vinod, Biointerface Res. Appl. Chem., 2022, 12, 718–731 CAS.
- D. Wibowo, M. Z. Muzakkar, M. Maulidiyah, M. Nurdin, S. K. M. Saad and A. A. Umar, Biointerface Res. Appl. Chem., 2022, 12, 1421–1427 CAS.
- D. Ma, L. Yang, B. Huang, L. Wang, X. Wang, Z. Sheng and F. Dong, New J. Chem., 2019, 43, 15161–15168 RSC.
- B. Huang, D. Yu, Z. Sheng and L. Yang, J. Environ. Sci., 2017, 55, 129–136 CrossRef CAS.
- A. Gurbani, J. L. Ayastuy, M. P. Gonzalez-Marcos and M. A. Gutierrez-Ortiz, Int. J. Hydrogen Energy, 2010, 35, 11582–11590 CrossRef CAS.
- A. Gurbani, J. L. Ayastuy, M. P. Gonzalez-Marcos, J. E. Herrero, J. M. Guil and M. A. Gutierrez-Ortiz, Int. J. Hydrogen Energy, 2009, 34, 547–553 CrossRef CAS.
- L. Han, S. Cai, M. Gao, J. Y. Hasegawa, P. Wang, J. Zhang, L. Shi and D. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS PubMed.
- Q. Fang and X. Liang, RSC Adv., 2012, 2, 5370–5375 RSC.
- L. Wang, W. Li, S. J. Schmieg and D. Weng, J. Catal., 2015, 324, 98–106 CrossRef CAS.
- X. Hu, M. Yang, D. Fan, G. Qi, J. Wang, J. Wang, T. Yu, W. Li and M. Shen, J. Catal., 2016, 341, 55–61 CrossRef CAS.
- P. Atkins, J. de Paula and J. DePaula, Physical Chemistry, W. H. Freeman, 2002 Search PubMed.
- M. Kong, Q. Liu, L. Jiang, W. Tong, J. Yang, S. Ren, J. Li and Y. Tian, Chem. Eng. J., 2019, 370, 518–526 CrossRef CAS.
- J. Liu, X. Li, Q. Zhao, J. Ke, H. Xiao, X. Lv, S. Liu, M. Tadé and S. Wang, Appl. Catal., B, 2017, 200, 297–308 CrossRef CAS.
- K. S. W. Sing and R. T. Williams, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef CAS.
- A. Xie, Y. Tang, X. Huang, X. Jin, P. Gu, S. Luo, C. Yao and X. Li, Chem. Eng. J., 2019, 370, 897–905 CrossRef CAS.
- Q. Yan, S. Chen, C. Zhang, Q. Wang and B. Louis, Appl. Catal., B, 2018, 238, 236–247 CrossRef CAS.
- M. Luo, J. Ma, J. Lu, Y. Song and Y. Wang, J. Catal., 2007, 247, 127 CrossRef CAS.
- P. Forzatti, Catal. Today, 2000, 62, 51–65 CrossRef CAS.
- Z. Liu, Q. Wang, J. Wu, H. Zhang, Y. Liu, T. Zhang, H. Tian and S. Zeng, ACS Appl. Mater. Interfaces, 2021, 13, 35804–35817 CrossRef CAS PubMed.
- W. Xiang, J. Liu, M. Chang and C. Zheng, Chem. Eng. J., 2012, 200, 91–96 CrossRef.
- Z. Wang and R. Yu, Adv. Mater., 2019, 31 Search PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53–229 CrossRef CAS.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- K. B. Zhou, X. Wang, X. M. Sun, Q. Peng and Y. D. Li, J. Catal., 2005, 229, 206–212 CrossRef CAS.
- N. Y. Topsoe, Science, 1994, 265, 1217–1219 CrossRef CAS.
- Y. Zeng, S. Zhang, Y. Wang and Q. Zhong, J. Colloid Interface Sci., 2017, 496, 487–495 CrossRef CAS PubMed.
- D. Wang, Y. Jangjou, Y. Liu, M. K. Sharma, J. Luo, J. Li, K. Kamasamudram and W. S. Epling, Appl. Catal., B, 2015, 165, 438–445 CrossRef CAS.
- M. Luo, Y. Song, J. Lu, X. Wang and Z. Pu, J. Phys. Chem. C, 2007, 111(34), 12686–12692 CrossRef CAS.
- Z. Liu, R. Zhou and X. Zheng, Catal. Commun., 2008, 9, 2183–2186 CrossRef CAS.
- Z. Liu, R. Zhou and X. Zheng, J. Mol. Catal. A: Chem., 2007, 267, 137–142 CrossRef CAS.
- J. Liu, X. Li, Q. Zhao, D. Zhang and P. Ndokoye, J. Mol. Catal. A: Chem., 2013, 378, 115–123 CrossRef CAS.
- T. Ghodselahi, M. A. Vesaghi, A. Shafiekhani, A. Baghizadeh and M. Lameii, Appl. Surf. Sci., 2008, 255, 2730–2734 CrossRef CAS.
- X. Q. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martinez-Arias and M. Fernandez-Garcia, J. Phys. Chem. B, 2005, 109, 19595–19603 CrossRef CAS.
- S. Mahammadunnisa, P. M. K. Reddy, N. Lingaiah and C. Subrahmanyam, Catal. Sci. Technol., 2013, 3, 730–736 RSC.
- G. Praline, B. E. Koel, R. L. Hance, H. I. Lee and J. M. White, J. Electron Spectrosc. Relat. Phenom., 1980, 21, 17–30 CrossRef CAS.
- M. J. Guittet, J. P. Crocombette and M. Gautier-Soyer, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63(12), 125117 CrossRef.
- W. Tian, H. Yang, X. Fan and X. Zhang, J. Hazard. Mater., 2011, 188, 105–109 CrossRef CAS PubMed.
- D. Dogu, K. E. Meyer, A. Fuller, S. Gunduz, D. J. Deka, N. Kramer, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2018, 227, 90–101 CrossRef CAS.
- M. Y. Mihaylov, V. R. Zdravkova, E. Z. Ivanova, H. A. Aleksandrov, P. St Petkov, G. N. Vayssilov and K. I. Hadjiivanov, J. Catal., 2021, 394, 245–258 CrossRef CAS.
- Y. Geng, X. Chen, S. Yang, F. Liu and W. Shan, ACS Appl. Mater. Interfaces, 2017, 9, 16952–16959 Search PubMed.
- G. Ramis, L. Yi, G. Busca, M. Turco, E. Kotur and R. J. Willey, J. Catal., 1995, 157, 523–535 CrossRef CAS.
- J. Zawadzki and M. Wiśniewski, Carbon, 2003, 41, 2257–2267 CrossRef CAS.
- L. A. Chen, J. H. Li and M. F. Ge, Environ. Sci. Technol., 2010, 44, 9590–9596 CrossRef CAS PubMed.
- D. Sun, Q. Liu, Z. Liu, G. Gui and Z. Huang, Appl. Catal., B, 2009, 92, 462–467 CrossRef CAS.
- G. Ramis, L. Yi and G. Busca, Catal. Today, 1996, 28, 373–380 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev.: Sci. Eng., 2000, 42, 71–144 CrossRef CAS.
- H. Sjovall, E. Fridell, R. J. Blint and L. Olsson, Top. Catal., 2007, 42–43, 113–117 CrossRef.
- L. Chen, J. Li and M. Ge, J. Phys. Chem. C, 2009, 113, 21177–21184 CrossRef CAS.
- D. Wang, L. Zhang, K. Kamasamudram and W. S. Epling, ACS Catal., 2013, 3, 871–881 CrossRef CAS.
- H. Y. Huang and R. T. Yang, Langmuir, 2001, 17, 4997–5003 CrossRef CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- C. Paolucci, I. Khurana, A. A. Parekh, S. C. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Science, 2017, 357, 898–903 CrossRef CAS.
- D. Damma, P. Ettireddy, B. Reddy and P. Smirniotis, Catalysts, 2019, 9(4), 349 CrossRef CAS.
- R. Jin, Y. Liu, Z. Wu, H. Wang and T. Gu, Chemosphere, 2010, 78, 1160–1166 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc,
Xinyue Chub,
Zhipeng Zhuang*de and
Xiongbo Chene
*bc,
Xinyue Chub,
Zhipeng Zhuang*de and
Xiongbo Chene
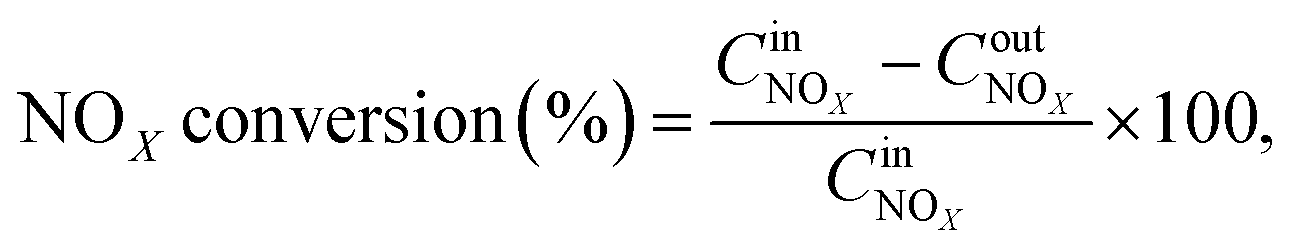
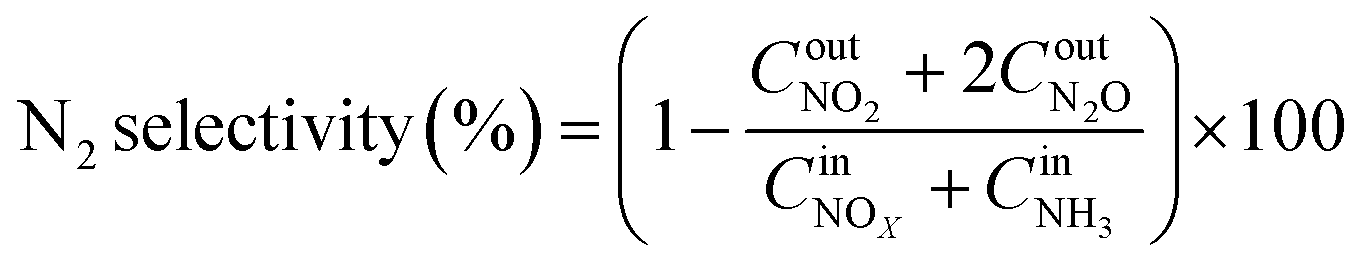
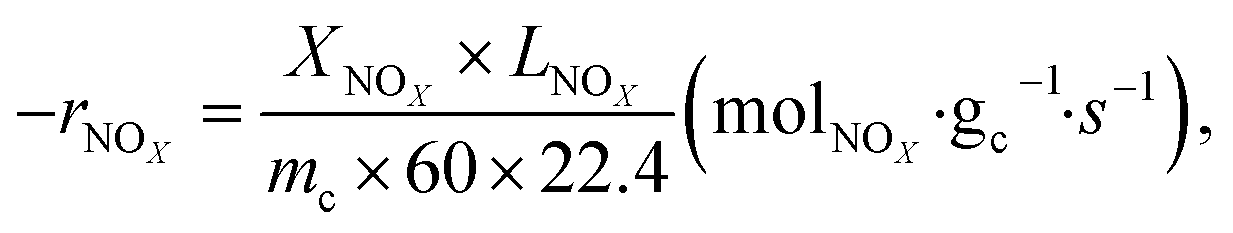
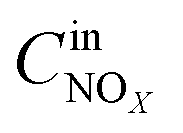 ,
, 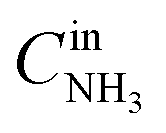 and
and 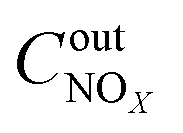 ,
, 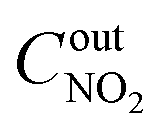 ,
, 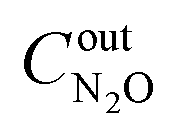 are concentrations of inlet gas (ppm) and outlet gas (ppm), respectively. XNOx is the conversion of NOx (%) (eqn (1)), LNOx is the flow rate of NOx (ml min−1), and mc is the mass of the catalyst (g).
are concentrations of inlet gas (ppm) and outlet gas (ppm), respectively. XNOx is the conversion of NOx (%) (eqn (1)), LNOx is the flow rate of NOx (ml min−1), and mc is the mass of the catalyst (g).