DOI:
10.1039/D3RA02965D
(Paper)
RSC Adv., 2023,
13, 19757-19769
Exploitation of expired cellulose biopolymers as hydrochars for capturing emerging contaminants from water†
Received
4th May 2023
, Accepted 24th June 2023
First published on 3rd July 2023
Abstract
Expired chemicals pose a potential environmental threat to humans and living organisms. Herein, we proposed a green approach whereby expired cellulose biopolymers were converted to hydrochar adsorbents and tested for removing the emerging pharmaceutical contaminants of fluoxetine hydrochloride and methylene blue from water. A thermally stable hydrochar was produced with an average particle size of 8.1 ± 1.94 nm and a mesoporous structure that exhibited a larger surface area than the expired cellulose by 6.1 times. The hydrochar was efficient in removing the two contaminants with efficiencies that reached above 90% under almost neutral pH conditions. Adsorption exhibited fast kinetics and regeneration of the adsorbent was successful. The adsorption mechanism was hypothesized in view of the Fourier Transform Infra-Red (FTIR) spectroscopy and pH effect measurements to be mainly electrostatic. A hydrochar/magnetite nanocomposite was also synthesized, and its adsorption behavior for both contaminants was tested and it revealed an enhanced percent removal relative to the bare hydrochar by 27.2% and 13.1% for FLX and MB, respectively. This work supports the strategies for zero waste management and the circular economy.
1. Introduction
Biopolymers are gaining interest worldwide for their environmentally friendly properties owing to their well-known biodegradability, non-toxicity and biocompatibility.1 However, the issue of biodegradability has been debated in recent years because it was found that even the most known commercial biopolymers such as polylactic acid (PLA) degrade slowly over time under ordinary conditions such that 80% of PLA was degraded in compost over a period of 80 days,2 while the disposed biopolymers in landfills may create methane emissions which are more hazardous than CO2. Also, the supply of biopolymers far exceeds the demand for composting.3 Thus, our approach to manipulate biopolymer waste is to convert it into the more porous and economic hydrochar using hydrothermal treatment (HTT) processes to utilize it in wastewater treatment.4 HTT proved to be more efficient than conventional pyrolysis in increasing the oxygenated group content in the created activated carbon. In addition, the procedure for HTT does not require chemical treatment since it is water-based which makes this approach greener than the conventional one.5 The biopolymer deployed in this study is cellulose, which is the most abundant biopolymer on earth and is a main constituent in plants and tree barks. It is formed of a linear polysaccharide chain of cellobiose or disaccharide D glucose having β-1,4 linkages with strong inter- and intramolecular hydrogen bonds.6 The life-cycle assessment of composting the cellulose acetate present in organic waste streams showed that the formed compost was of low quality due to improper biodegradation. Besides, the presence of cellulose acetate in organic waste streams had a negative effect on the environment by contributing to global warming and stratospheric ozone depletion through its degradation which creates methane emissions.7 On the other hand, the eco-costs of woven cotton textile at 70 dtex also showed the highest values compared to nylon, acryl, PET, and ESTANE®.8 This demonstrates that the production of biopolymers and their waste disposal may possibly contribute to environmental ecotoxicity which should encourage scientists to work on mitigating such harmful effects arising from either the production or disposal phases or both. In this work, we applied HTT on expired cellulose to produce hydrochar (Cel-HTT), then utilized Cel-HTT in removing the pharmaceutical Fluoxetine HCl (FLX) and the organic dye methylene blue (MB) from water for the sake of controlling the harmful effects of biopolymers disposal and contaminants release into the environment. These two contaminants were chosen as examples of emerging contaminants that are produced in wastewater effluents of the pharmaceutical industries and known for their persistence and resistance to degradation.
FLX is a selective serotonin re-uptake inhibitor for treating depression. It was detected in US surface waters at concentrations of 0.012 μg L−1, and in surface, drinking and wastewater of China at concentrations of 0.4–91 ng L−1.9 It was also found in the sewage treatment plant effluents of Canada at 0.038–0.099 μg L−1 (ref. 10) and in the UK wastewater in the range of 4.9–175.9 ng L−1.11 Chronic exposure to this pharmaceutical leads to bioaccumulation in the muscles and liver of aquatic organisms.12 Similarly, organic dyes are non-biodegradable and are stable in the presence of heat, light and chemicals besides being carcinogenic and mutagenic.13 MB, in particular, causes gastrointestinal pain, nausea, diarrhea, fever, and elevation of blood pressure.14 The concentration of MB in textile wastewater from Saudi Arabia was estimated to be 1.08 mg L−1.15
Cellulose-derived activated carbon has been previously applied for the removal of dyes from aqueous solutions. For example, industrial cellulose waste was chemically and thermally treated at 500–800 °C to obtain a nitrogen-doped carbon. The cellulose thermally treated at 800 °C removed 337.8 mg g−1 of methyl orange at pH 7.1 and 20 °C.16 This high adsorption capacity could be due to the high temperature applied in the preparation of the activated carbon from cellulose. Also, microwave carbonized cellulose was successful in the removal of diclofenac from aqueous solution where adsorption followed Langmuir isotherm with a maximum adsorption capacity of 27.3 mg g−1 at pH 5.85 and 25 °C.17 Cellulose powder hydrochar was rarely applied in literature for water treatment and usually biomass wastes containing cellulose were mainly applied for heavy metal removal.18 However, cellulose was hydrothermally treated in the presence of acid activated montmorillonite to form an adsorbent efficient for removing MB from aqueous solutions. Adsorption followed Redlich-Peterson isotherm while kinetics followed the pseudo-second-order model. The maximum adsorption capacity for this nanocomposite was about 138 mg g−1 which was owed to the contribution of the montmorillonite in the adsorption process.19 In another study, a carboxylate-functionalized bamboo hydrochar gave a maximum adsorption capacity of 1155.57 mg g−1 at 30 °C for MB according to Langmuir model,20 while sugarcane bagasse hydrochar activated by phosphoric acid and sodium hydroxide gave a Langmuir maximum adsorption capacity of 357.14 mg g−1 at 30 °C for MB.21
To the best of our knowledge, this is the first report to tackle the issue of expired chemicals and their conversion to a hydrochar. FLX and MB are the first contaminants to be studied for their removal onto such expired chemicals. The expired biopolymers and produced hydrochar are thoroughly characterized using various techniques and compared to the fresh biopolymers. Various adsorption parameters are investigated while adsorption kinetics and regeneration are studied. In view of these studies, the mechanism of adsorption is discussed. For facile collection of the hydrochar, magnetic hydrochar is synthesized by incorporating magnetite nanoparticles into the hydrochar for the purpose of enhancing the surface area of the hydrochar, and the bionanocomposite is then tested for its adsorption performance.
2. Materials and methods
2.1. Materials
Expired cellulose powder (chemically prepared ashless powder) originally obtained from W&R Balston, Ltd Genuine Whatman, England was supplied by the chemical store at the department of Chemistry in the American University in Cairo, Egypt. For comparison, fresh cellulose powder for column chromatography was purchased from Loba Chemie, pvt Ltd. Fluoxetine HCl (purity 100.3%) was cordially provided by Amoun Pharmaceuticals, Egypt. Methylene blue (NLT 85%) was purchased from Advent CHEMBIO Ltd™, India. Sodium hydroxide pellets (purity >97%) were obtained from Fisher, Germany, HCl (37% by vol.) of analytical grade was obtained from Acros, Germany, ethanol absolute (99.9%) was obtained from Fisher, Germany, and orthophosphoric acid (85%) and acetone (extra pure) were brought from Piochem, Egypt. The magnetic precursors anhydrous ferric chloride (97% purity) and ferrous sulfate heptahydrate (98% purity) were purchased from Fisher, Germany. NaCl (purity ≥99.5%) was obtained from Fisher Scientific, Loughborough, UK.
2.2. Physicochemical properties of expired cellulose
Bulk density was determined by weighing 0.5 g of the expired cellulose powder and placing it in a 10 mL measuring cylinder to first obtain the volume (V) and substitute it in (eqn S1 in ESI†) to calculate the density. For molecular weight, it was determined using Ubblohde U-shaped viscometer where cellulose concentrations (C) of 0.25, 0.5, 0.8, and 1 g/100 mL were prepared in orthophosphoric acid and allowed to flow between the two marks of the viscometer, then the flow times were measured using a mobile stopwatch. Specific viscosity (ηsp) was then calculated based on eqn S2 in ESI† and intrinsic viscosity was determined from the intercept of the plot between ηsp/C and C (Fig. S1 of ESI†). A two-tailed t-test of two samples with unequal variance was used to assess the significance of the measured physicochemical properties.
2.3. Preparation of Cel-HTT
HTT was performed by dispersing 10 g of the expired cellulose powder in 100 mL of distilled water then placing it in an autoclave at 230 °C while stirring for 4 h. The instrument was left to cool, and the solid product was collected and washed with consecutive cycles of distilled water and acetone. Finally, the washed powder was dried in air.
2.4. Preparation of Cel-HTT/M nanocomposite
A weight of 0.5 g of the prepared Cel-HTT was dispersed in 30 mL of distilled water and stirred for 15 min. Masses of 1.5 g ferrous sulfate heptahydrate and 1.75 g anhydrous ferric chloride were added and stirred for 20 min. To form the magnetite nanoparticles, a volume of 6 mL of 30% NaOH was added dropwise. The formed nanocomposite was washed three times with distilled water and left to dry under air. Also, bare magnetite was prepared using the same above method without the hydrochar.
2.5. Characterization
To determine the functional groups of the prepared Cel-HTT, FTIR measurements (Thermo Scientific, Nicolet 380, Germany) were performed by KBr disc method in the wavelength range of 4000–650 cm−1 and the spectrum was compared to those of the expired and fresh cellulose. Raman spectroscopy was also performed on the samples in the solid state using ProRaman-L High Performance Raman Spectrometer, Enwave optronics, Inc., Irvine, CA, USA, in the range of 100–3000 cm−1. The thermal stabilities of Cel-HTT, expired and fresh cellulose were tested by Thermogravimetric analysis (TGA Q50, Germany) in the range of 25–750 °C with a ramp of 10 °C min−1. To investigate the crystalline structure of Cel-HTT, expired and fresh cellulose, X-ray Diffraction (XRD) measurements were performed (Bruker, D8 Discover, USA) at 2θ range of 10–60°. Transmission Electron Microscope (TEM) (JEOL, JEM-2100, Japan) images were also taken for both the expired cellulose and Cel-HTT after sonicating the powder for 5 min and placing it on a carbon-coated copper grid. To determine the carbon content in Cel-HTT and the expired cellulose, Energy Dispersive X-ray (EDX) (Neoscope JCM-6000 Plus, JEOL Benchtop SEM, Japan) measurement was performed after gold sputtering (Hummer 8.0, USA) at 15 mA for 3 min. Porosity was also estimated from Brunauer–Emmett–Teller (BET) measurements (ASAP 2020-Micromeritics equipment) after nitrogen degassing at 40 °C for 8 h. Pore sizes, BET surface areas and BJH pore volumes were determined for fresh cellulose, expired cellulose, and Cel-HTT. Magnetization of the Cel-HTT/M was measured using Lakeshore 7410 vibrating sample magnetometer (VSM), USA and compared to a bare magnetite prepared in a similar way.
2.6. Removal studies for FLX and MB onto Cel-HTT
Adsorption experiments were performed in a batch mode that was carried out in a rotary shaker at a speed of 60 rpm using 15 mL falcon tubes. They were conducted by weighing a mass of 20 mg of the prepared Cel-HTT powder and placing it in 15 mL of a 50 ppm solution of FLX or MB at room temperature (25 ± 2 °C). To determine the optimum pH for adsorption, 50 ppm FLX and MB solutions were prepared at different pHs (3.7–9 in case of FLX and 3.7–10 for MB) adjusted by either 0.5 M HCl or 1%w/v NaOH, and adsorption was tested using a dose of 1.33 g L−1 for a contact time of 4 h. After adsorption, the final pH for FLX and MB solutions was measured to determine the point-of-zero charge (PZC) of Cel-HTT. The optimum pH was then applied in the subsequent adsorption experiments. The effect of contact time was determined at 25, 50, and 75 ppm of FLX or MB using the same dose of 1.33 g L−1. That dose was also applied in the determination of the effect of initial concentration in the range from 10 to 100 ppm. Finally, the effect of adsorbent dose was investigated by applying doses from 0.33 to 2.67 g L−1 on 50 ppm FLX or MB. After performing the adsorption experiment, Cel-HTT powder was separated from the FLX or MB solutions using two cycles of ultracentrifugation, each at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min. The supernatant was then collected and measured on a UV/VIS spectrophotometer (Pg instruments, T80+ UV/VIS spectrometer, UK) at 228 nm for FLX and 665 nm for MB. The obtained absorbances were then substituted in a pre-prepared calibration curve to determine the equilibrium adsorption concentration Ce. That latter value was subsequently substituted in eqn S3 and S4 in the ESI† to determine the percent removal and equilibrium adsorption capacity qe, respectively.22,23
000 rpm for 5 min. The supernatant was then collected and measured on a UV/VIS spectrophotometer (Pg instruments, T80+ UV/VIS spectrometer, UK) at 228 nm for FLX and 665 nm for MB. The obtained absorbances were then substituted in a pre-prepared calibration curve to determine the equilibrium adsorption concentration Ce. That latter value was subsequently substituted in eqn S3 and S4 in the ESI† to determine the percent removal and equilibrium adsorption capacity qe, respectively.22,23
Kinetic modeling was also investigated using pseudo-first-order (eqn S5 in ESI†),24 pseudo-second-order (eqn S6 in ESI†)25 and intra-particle diffusion models (eqn S7 in ESI†).26 To predict equilibrium parameters, Freundlich (eqn S8 in ESI†) and Langmuir (eqn S9 in ESI†) isotherms were plotted.27,28
In addition, regeneration of Cel-HTT was performed after FLX and MB adsorption using ethanol absolute. Adsorption was conducted at pH 7.5 on 25 ppm FLX or MB using a dose of 1.33 g L−1 till reaching equilibrium, then the particles were washed with 50 mL ethanol absolute for 30 min, dried and weighed again. Regeneration took place for four consecutive cycles.
To understand the adsorption mechanism, FTIR measurements were conducted on the adsorbent after adsorption using 1.33 g L−1 Cel-HTT on 25 ppm FLX or MB for 1 h at pH 7.5. To investigate the adsorption capacity of Cel-HTT/M, percent removal of FLX and MB was assessed at an initial concentration of 50 ppm, adsorbent dose of 1.33 g L−1, pH 7.5, and room temperature (25 ± 2 °C). Also, the effect of the addition of NaCl salt at different concentrations (0%, 0.75%, 1.5%, and 3%) was studied for the adsorption of FLX and MB on magnetic hydrochar at an initial concentration of 50 ppm, adsorbent dose of 1.33 g L−1 and pH 7.5 at 25 ± 2 °C.
3. Results and discussion
3.1. Physicochemical properties
As shown in Table 1, the bulk density for expired cellulose is significantly less than that of fresh cellulose (p < 0.05) by about 14%. However, the specific and intrinsic viscosities for the expired cellulose are not significantly different from those of the fresh cellulose (p > 0.05). The reason behind the change in bulk density could be attributed to the breaking up of the polymeric structure of fresh cellulose as it expired, thus leading to a reduction in particle size and consequent decrease in bulk density.
Table 1 Bulk density and intrinsic viscosity of expired cellulose in comparison to fresh cellulose
| Type of material |
Bulk density (g cm−3) |
Specific viscosity at 1 g% |
Intrinsic viscosity (L g−1) |
| Expired cellulose |
0.192 ± 0.015 |
0.25 |
0.3651 |
| Fresh cellulose |
0.224 ± 0.012 |
0.22 |
0.3205 |
3.2. Characterization
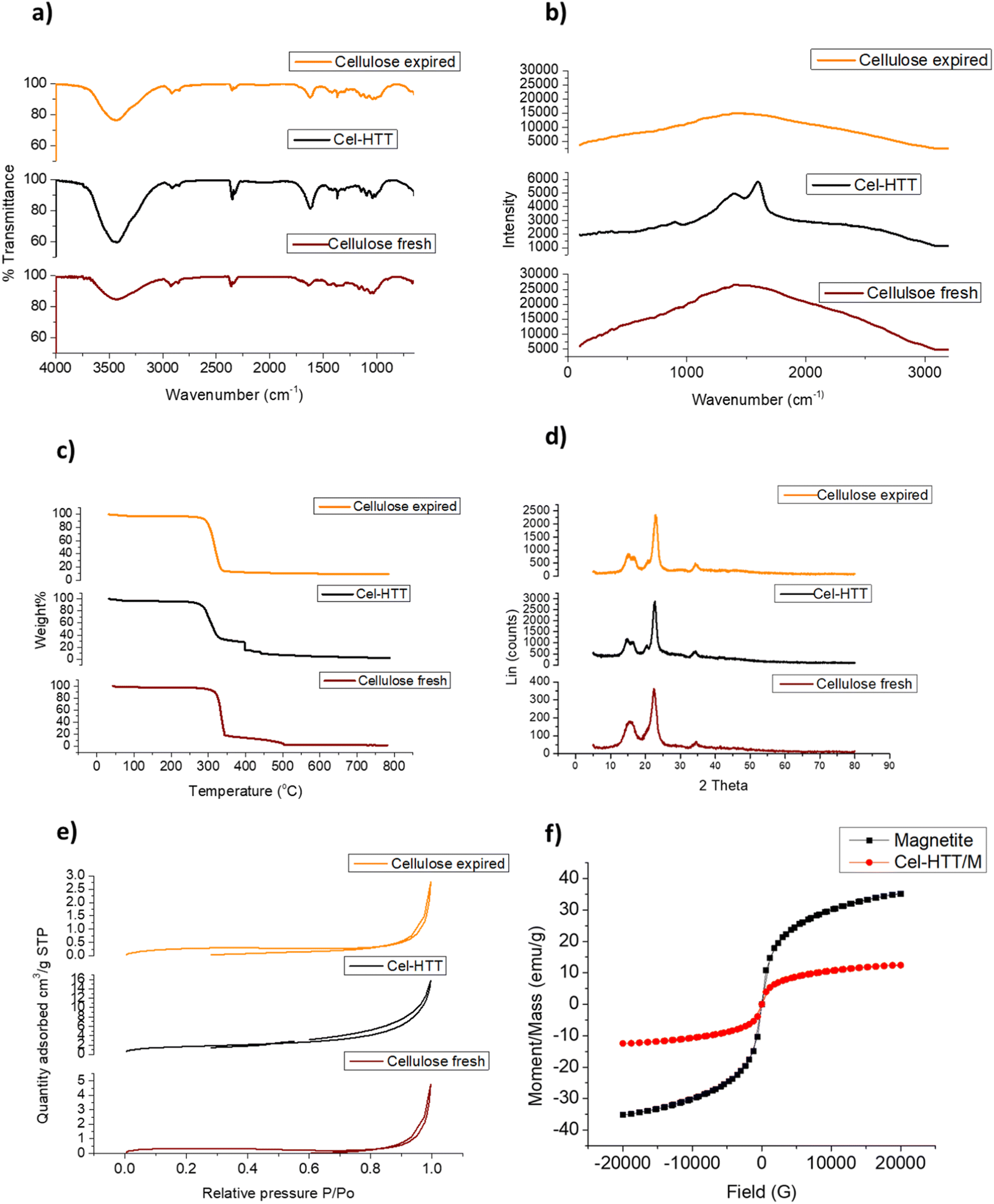
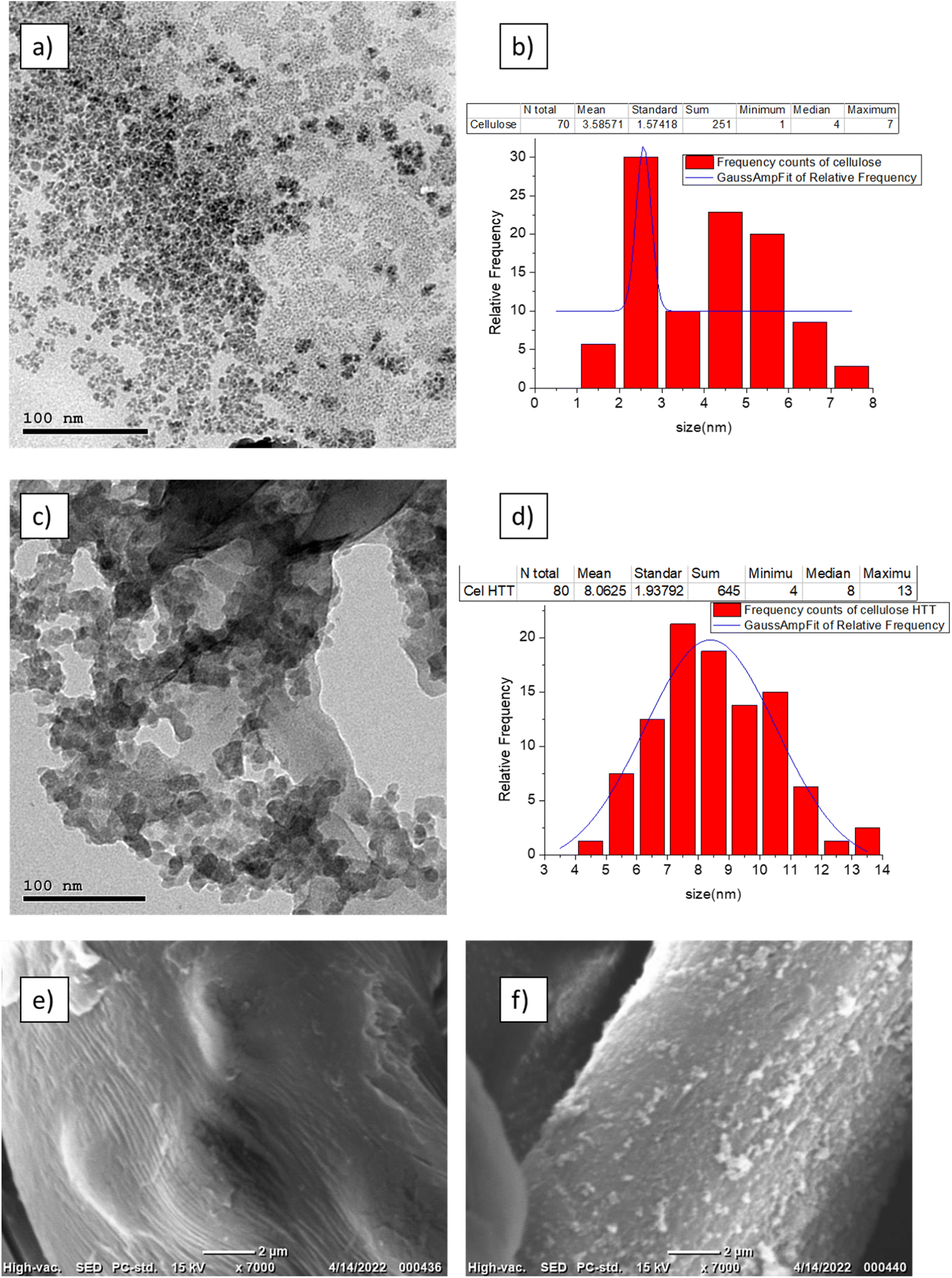
FTIR peaks for the fresh cellulose, expired cellulose, and Cel-HTT are presented in Fig. 1a. The expired cellulose exhibited peaks at 3454, 2920, 1635, and 1456–983 cm−1 which can be assigned to OH stretching, C–H stretching, OH bending from absorbed water molecules, and stretching and bending vibrations of CH2, –CH, –OH and C–O bonds of cellulose, respectively. The spectrum of expired cellulose is similar to that of fresh cellulose which was reported to show the same characteristic peaks.29–31 Again, similar spectrum is shown for Cel-HTT which demonstrates the main polysaccharide functional groups. However, there is a clear increase in the intensity of the peaks in the order of fresh cellulose < expired cellulose < Cel-HTT which could be a result of the degradation of cellulose and its carbonization as it got expired or as it was converted to hydrochar,32 except for the peak at 1635 cm−1 in Cel-HTT which is assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak and has an increased intensity than the expired cellulose indicating a carbonyl or carboxyl formation during HTT which were possibly formed by dehydration involving the hydroxyl groups.32–34 The Raman spectrum (Fig. 1b) confirms a hydrochar structure as confirmed from the formation of D and G bands where G/D ratio was about 1.17 close to what was reported in literature indicating the formation of many defects or the formation of several functional groups on the structure of the hydrochar.35 In addition, the intensity of the Raman peak of expired cellulose is lower than that of fresh cellulose, possibly due to the degradation of cellulosic fibres.36 The TGA profiles (Fig. 1c) show that both Cel-HTT and the expired cellulose maintain high thermal stabilities up till 300 °C as they lose only about 10% of their weight, while Cel-HTT exhibits a slightly enhanced thermal stability over expired cellulose and fresh cellulose at the temperature range of 300–450 °C. This behavior is similar to that previously reported for raw cellulose and hydrothermally treated cellulose where the former underwent a major mass loss at 320 °C and 450 °C, which was encountered at higher temperatures in case of the hydrothermally treated cellulose.37 XRD patterns of expired cellulose and Cel-HTT (Fig. 1d) show the same characteristic peaks of fresh cellulose at about 14.5°, 16.5°, 22.5°, and 35.19° where the peak at about 22.5° indicates the presence of crystalline cellulose.38,39 The peak intensities for Cel-HTT are higher than their counterparts for the fresh and expired cellulose. This indicates an increase in the number of atoms/molecules due to phase change which could be the reason behind the higher thermal stability exhibited by Cel-HTT relative to the expired and fresh cellulose.40 The carbonization of fresh cellulose into expired cellulose is confirmed by the carbon content of expired cellulose which is comparable to the hydrochar Cel-HTT, as estimated by EDX analysis (Table 2). The porosity test for expired cellulose and Cel-HTT (Table 2) shows an increase in the BET surface area and pore volume of Cel-HTT over those of expired cellulose by 6.1 and 5.9 times, respectively, as a result of the HTT process which resulted in a mesoporous structure for Cel-HTT with pore sizes ranging between 2-50 nm as confirmed by the BET isotherm plots (Fig. 1e) which display a type V isotherm with H3 hysteresis type indicating slit like pores.41 The pore volume distribution curves (Fig. S2†) indicate that the majority of pores are in the mesoporous range. The increase in specific surface area of Cel-HTT is further confirmed by SEM analysis (Fig. 2e and f) which illustrates the rougher surface of Cel-HTT relative to expired cellulose. Regarding the fresh cellulose, it shows a comparable BET surface area to the expired one (Table 2), however double its BJH pore volume which implies a deformed porous structure for the expired cellulose relative to the fresh cellulose. Magnetization of Cel-HTT to form Cel-HTT/M enhanced the specific surface area by about 25 times and the pore volume by about 5 times, while the pore size decreased by about 4 times owing possibly to deformation in the pore shape and structure induced by the presence of magnetite. Similar findings were reported in literature where raw cellulose showed a surface with no cracks or trenches unlike that of the cellulose hydrochar,42 while the TEM histograms of expired cellulose and Cel-HTT (Fig. 2b and d, respectively) show that the particle size increased from 3.5 ± 1.57 nm to 8.1 ± 1.94 nm after HTT possibly due to nucleation and subsequent particle growth that took place during the HTT process leading to the formation of larger particles.43 VSM measurements (Fig. 1f) show that bare magnetite acquires a magnetism of about 35 emu/g which is similar to values reported in literature,44 while the hydrochar-coated magnetite exhibits a magnetism of about 12.4 emu/g which is less than that of bare magnetite due to the coating process, however higher than values reported in previous literature for magnetic hydrochar from orange residue, magnetic activated sawdust hydrochar and magnetized sesame oil cake hydrochar.45–47
O peak and has an increased intensity than the expired cellulose indicating a carbonyl or carboxyl formation during HTT which were possibly formed by dehydration involving the hydroxyl groups.32–34 The Raman spectrum (Fig. 1b) confirms a hydrochar structure as confirmed from the formation of D and G bands where G/D ratio was about 1.17 close to what was reported in literature indicating the formation of many defects or the formation of several functional groups on the structure of the hydrochar.35 In addition, the intensity of the Raman peak of expired cellulose is lower than that of fresh cellulose, possibly due to the degradation of cellulosic fibres.36 The TGA profiles (Fig. 1c) show that both Cel-HTT and the expired cellulose maintain high thermal stabilities up till 300 °C as they lose only about 10% of their weight, while Cel-HTT exhibits a slightly enhanced thermal stability over expired cellulose and fresh cellulose at the temperature range of 300–450 °C. This behavior is similar to that previously reported for raw cellulose and hydrothermally treated cellulose where the former underwent a major mass loss at 320 °C and 450 °C, which was encountered at higher temperatures in case of the hydrothermally treated cellulose.37 XRD patterns of expired cellulose and Cel-HTT (Fig. 1d) show the same characteristic peaks of fresh cellulose at about 14.5°, 16.5°, 22.5°, and 35.19° where the peak at about 22.5° indicates the presence of crystalline cellulose.38,39 The peak intensities for Cel-HTT are higher than their counterparts for the fresh and expired cellulose. This indicates an increase in the number of atoms/molecules due to phase change which could be the reason behind the higher thermal stability exhibited by Cel-HTT relative to the expired and fresh cellulose.40 The carbonization of fresh cellulose into expired cellulose is confirmed by the carbon content of expired cellulose which is comparable to the hydrochar Cel-HTT, as estimated by EDX analysis (Table 2). The porosity test for expired cellulose and Cel-HTT (Table 2) shows an increase in the BET surface area and pore volume of Cel-HTT over those of expired cellulose by 6.1 and 5.9 times, respectively, as a result of the HTT process which resulted in a mesoporous structure for Cel-HTT with pore sizes ranging between 2-50 nm as confirmed by the BET isotherm plots (Fig. 1e) which display a type V isotherm with H3 hysteresis type indicating slit like pores.41 The pore volume distribution curves (Fig. S2†) indicate that the majority of pores are in the mesoporous range. The increase in specific surface area of Cel-HTT is further confirmed by SEM analysis (Fig. 2e and f) which illustrates the rougher surface of Cel-HTT relative to expired cellulose. Regarding the fresh cellulose, it shows a comparable BET surface area to the expired one (Table 2), however double its BJH pore volume which implies a deformed porous structure for the expired cellulose relative to the fresh cellulose. Magnetization of Cel-HTT to form Cel-HTT/M enhanced the specific surface area by about 25 times and the pore volume by about 5 times, while the pore size decreased by about 4 times owing possibly to deformation in the pore shape and structure induced by the presence of magnetite. Similar findings were reported in literature where raw cellulose showed a surface with no cracks or trenches unlike that of the cellulose hydrochar,42 while the TEM histograms of expired cellulose and Cel-HTT (Fig. 2b and d, respectively) show that the particle size increased from 3.5 ± 1.57 nm to 8.1 ± 1.94 nm after HTT possibly due to nucleation and subsequent particle growth that took place during the HTT process leading to the formation of larger particles.43 VSM measurements (Fig. 1f) show that bare magnetite acquires a magnetism of about 35 emu/g which is similar to values reported in literature,44 while the hydrochar-coated magnetite exhibits a magnetism of about 12.4 emu/g which is less than that of bare magnetite due to the coating process, however higher than values reported in previous literature for magnetic hydrochar from orange residue, magnetic activated sawdust hydrochar and magnetized sesame oil cake hydrochar.45–47
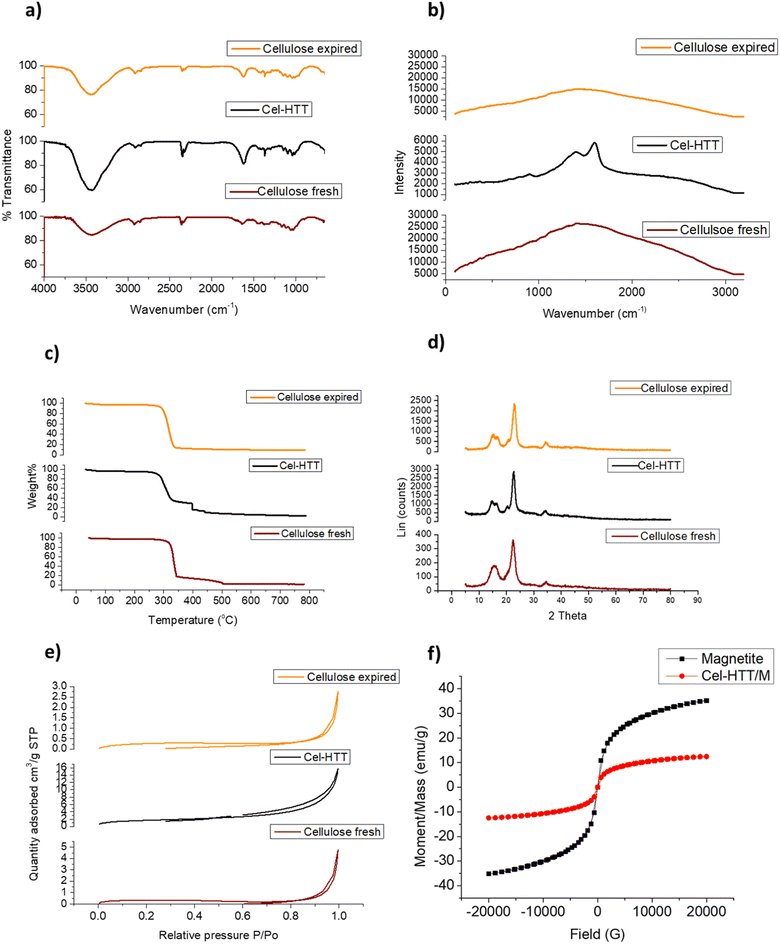 |
| | Fig. 1 FTIR spectra (a), Raman spectra (b) TGA profiles (c), XRD patterns (d), and BET isotherms (e) for expired cellulose and Cel-HTT as compared to fresh cellulose, together with VSM measurements of bare and hydrochar-coated magnetite Cel-HTT/M (f). | |
Table 2 Textural parameters and EDX analysis of the different cellulosic materials
| Porosity parameters |
Pore size (nm) |
BET surface area (m2 g−1) |
BJH pore volume (cm3 g−1) |
Langmuir surface area (m2 g−1) |
| Fresh cellulose |
20.9 |
1.05 |
0.0073 |
1.63 |
| Expired cellulose |
13.4 |
0.97 |
0.0039 |
1.59 |
| Cel-HTT |
14.4 |
5.94 |
0.0231 |
8.74 |
| Cel-HTT/M |
3.8 |
148.07 |
0.1166 |
216.71 |
| EDX analysis |
Carbon atom% |
Oxygen atom% |
| Expired cellulose |
87.04 |
12.96 |
| Cel-HTT |
92.96 |
7.04 |
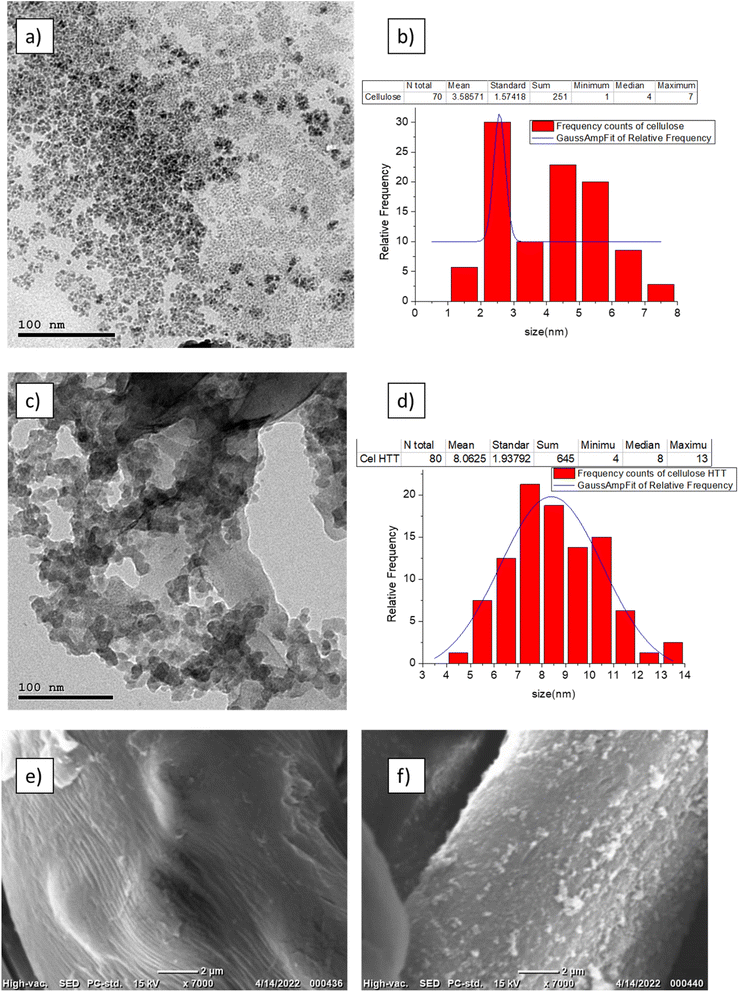 |
| | Fig. 2 TEM images of expired cellulose (a) and its particle size distribution (b), Cel-HTT (c) and its particle size distribution (d) and SEM images of expired cellulose (e) and Cel-HTT (f). | |
3.3. Adsorption performance of the hydrochar
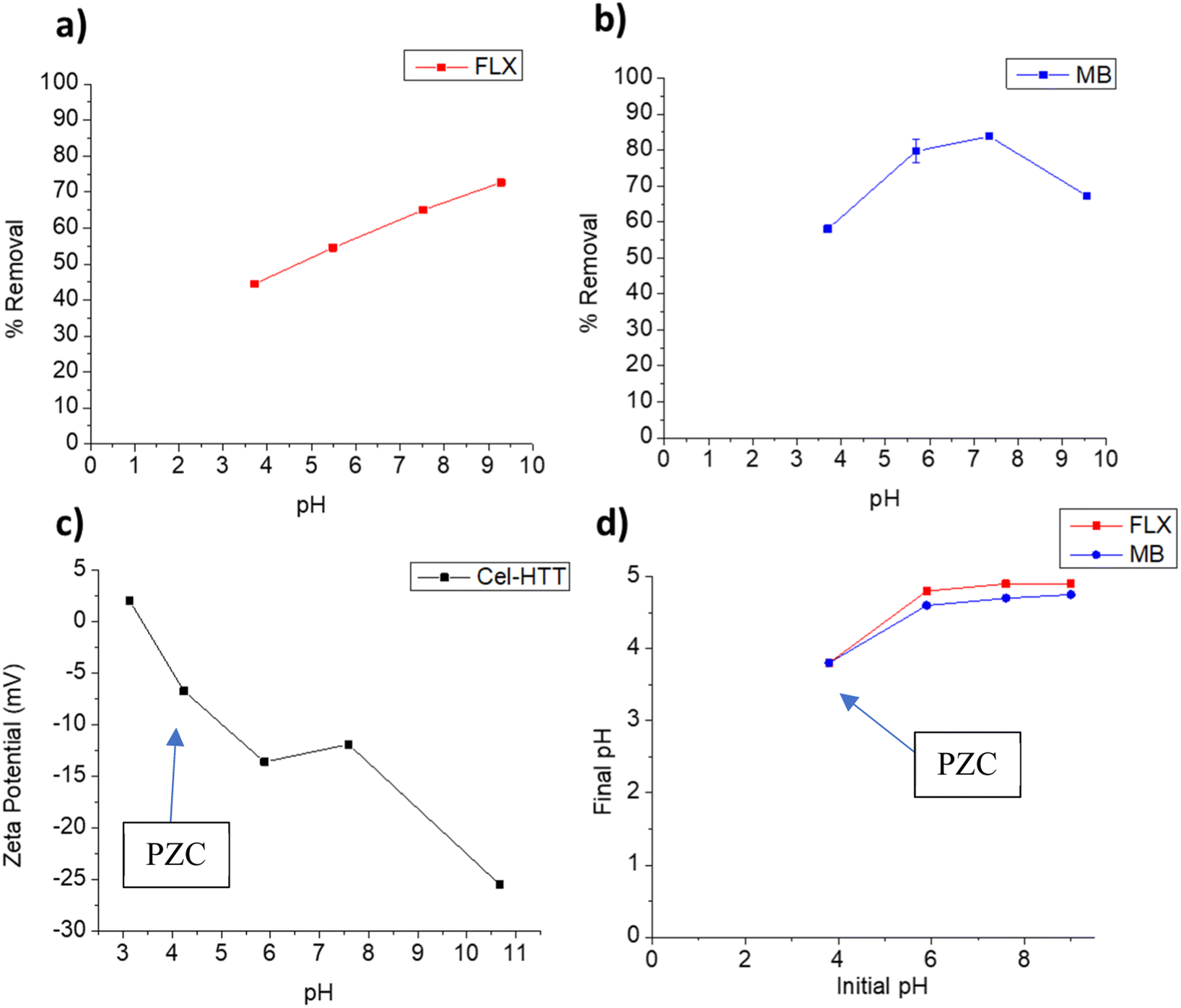
3.3.1. pH-controlled adsorption. pH is one of the main factors that control the adsorption behavior considering the protonation/deprotonation or surface charge changes of the adsorbent at the studied pHs. The effect of pH on the adsorption behavior of FLX and MB on Cel-HTT is illustrated in Fig. 3a and b, respectively. It is evident that increasing the pH increases the percent removal until it reaches its highest values of 72.7% and 83.8% at pH 9.3 and 7.5, respectively. According to the speciation diagrams of FLX (pKa 9.8) and MB (pKa 3.8), they are both positively charged in the studied pH range, while Cel-HTT is negatively charged as per the zeta potential measurements shown in Fig. 3c. Therefore, electrostatic attraction is likely to occur between each contaminant and the hydrochar. Similar behavior was previously observed for the adsorption of FLX onto low cost biosorbents such as pine cork and the best removal efficiencies were achieved at pH 7 and 9, due to electrostatic attraction.48 Electrostatic interactions were further confirmed when identifying the PZC of the adsorbent (Fig. 3d) which showed that below pH 3.8, the adsorbent is positively charged and above this pH, the adsorbent is negatively charged which concurs with the zeta potential measurements. Fig. 3b, however, shows that the removal of MB declined above pH 7.5 which could be attributed to other dominating adsorption mechanisms such as van der Waals forces or π–π interactions. Similar behavior was previously encountered when MB was adsorbed onto CO2-spherical activated carbon and a slight decrease in percent removal occurred above pH 7.49 Although the highest removal for FLX was obtained at pH 9, however the difference between FLX removal at pH 9 and pH 7 does not justify going for an alkaline pH especially that MB removal declines in the alkaline region. Therefore, we chose pH 7.5 as the working pH in which the removal of FLX and MB are about 65% and 83.8%, respectively, to avoid working under harsh alkaline conditions while maintaining a reasonably high percent removal. Comparing the adsorption performance of Cel-HTT to that of fresh and expired cellulose under the same operating conditions, it was found that fresh and expired cellulose exhibited almost zero to very low removal percentages (<20%) with both FLX and MB. The superior performance shown by Cel-HTT could be ascribed to its larger specific surface area and pore volume relative to fresh and expired cellulose, and its rougher defective surface that contains more functional groups as confirmed by SEM and Raman spectroscopy.
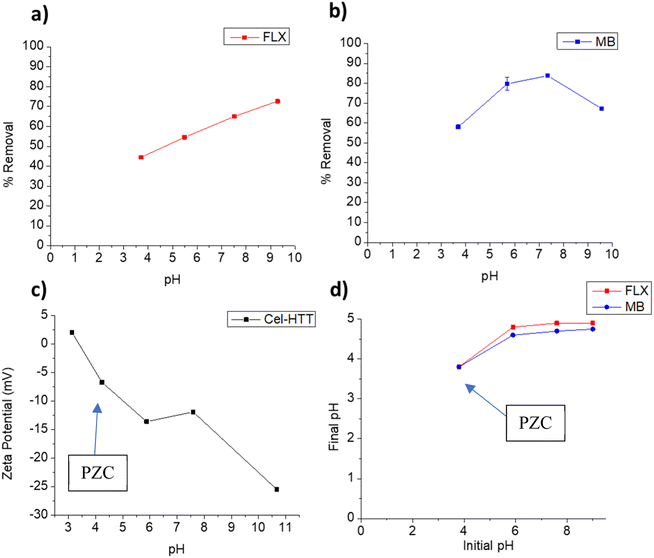 |
| | Fig. 3 Effect of solution pH on adsorption behavior of FLX (a) and MB (b) onto Cel-HTT, zeta potential profile of Cel-HTT (c), and determination of the Point-of-Zero Charge (PZC) for Cel-HTT (d). The arrows in panels (c) and (d) point towards the PZC. | |
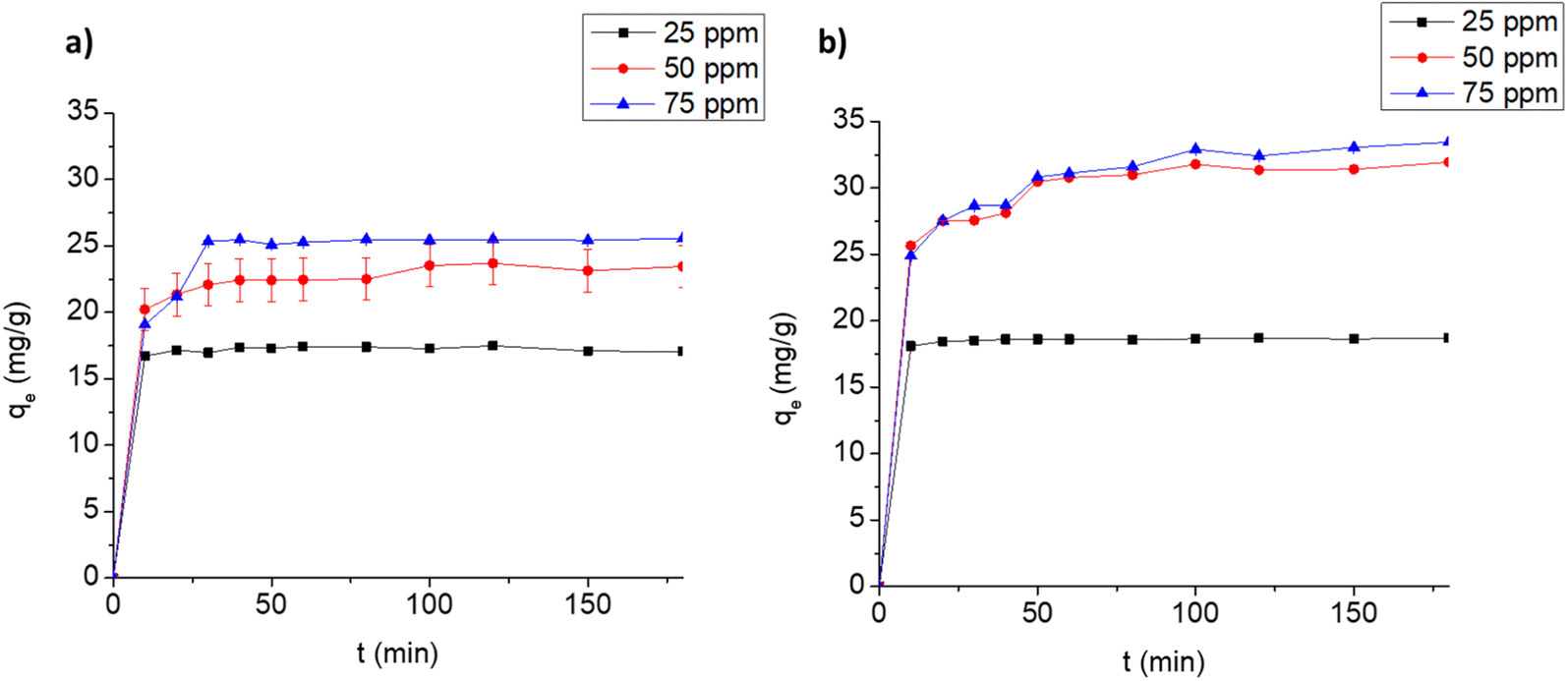
3.3.2. Adsorption kinetics. The time profiles for the adsorption of FLX and MB on Cel-HTT are depicted in Fig. 4a and b, respectively. The adsorption is carried out at three different concentrations at pH 7.5 and adsorbent dose of 1.33 g L−1. Equilibrium is reached at about 10, 20 and 30 min, and 10, 50 and 50 min for 25, 50 and 75 ppm of FLX and MB, respectively. This indicates that FLX uptake on Cel-HTT is faster than that of MB probably due its being more hydrophobic than MB since its water solubility (∼14 g L−1) is about one third that of MB (∼43 g L−1) and hence interacts better with the carbon-based Cel-HTT. The rate constants predicted by the pseudo-second-order model (k2) support this finding (Fig. S4 of ESI†) since the rate constant for FLX is higher than its MB counterpart implying faster rate of adsorption for FLX as presented in Table 3. The rate constants were predicted by the pseudo-second-order model since it better describes the uptake profiles as deduced from its higher R2 values relative to the pseudo-first-order model (Table 3, Fig. S3 and S4†). The pseudo-second-order model, thus, well predicted the qe values for the adsorption of both FLX and MB. This model assumes that adsorption occurs in three stages involving surface reaction, film diffusion, and pore diffusion. In various previous reports, adsorption of FLX on low-cost biochar followed pseudo-second-order model50 and the adsorption of dyes such as Malachite Green, MB and Rhodamine B also followed the same model.51–53 The plots for intra-particle diffusion of qt vs. t0.5 (Fig. S5 of ESI†) for FLX and MB are bi-linear. The boundary layer thickness coefficient (C) for MB adsorption exceeds that of FLX implying the bigger role of film diffusion in MB adsorption. The rate constant for intra-particle diffusion (kid) is greater for MB than FLX indicating a faster intra-particle diffusion process, however the overall diffusion of FLX was faster than MB as implied from the former's greater pseudo-second-order constant which signifies the overall surface diffusion, film diffusion and pore diffusion as stated earlier.
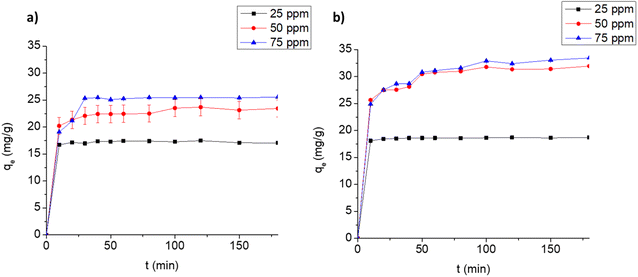 |
| | Fig. 4 Uptake profiles for the adsorption of FLX (a) and MB (b) on 1.33 g L−1 Cel-HTT at pH 7.5 and 25 ± 2 °C. | |
Table 3 Predicted kinetic parameters for the adsorption of FLX and MB at an initial concentration of 50 ppm, pH 7.5 and adsorbent dose of 1.33 g L−1
| |
Pseudo-first-order |
Pseudo-second-order |
Intra-particle diffusion |
| k1 min−1 |
qe mg g−1 |
R2 |
k2 g mg−1 min−1 |
qe mg g−1 |
R2 |
kid mg g−1 min−0.5 |
C mg g−1 |
R2 |
| FLX |
0.020 |
4.08 |
0.866 |
0.012 |
24.09 |
0.9995 |
0.585 |
18.6 |
0.9252 |
| MB |
0.029 |
6.94 |
0.939 |
0.009 |
32.26 |
0.9996 |
0.922 |
22.985 |
0.9234 |
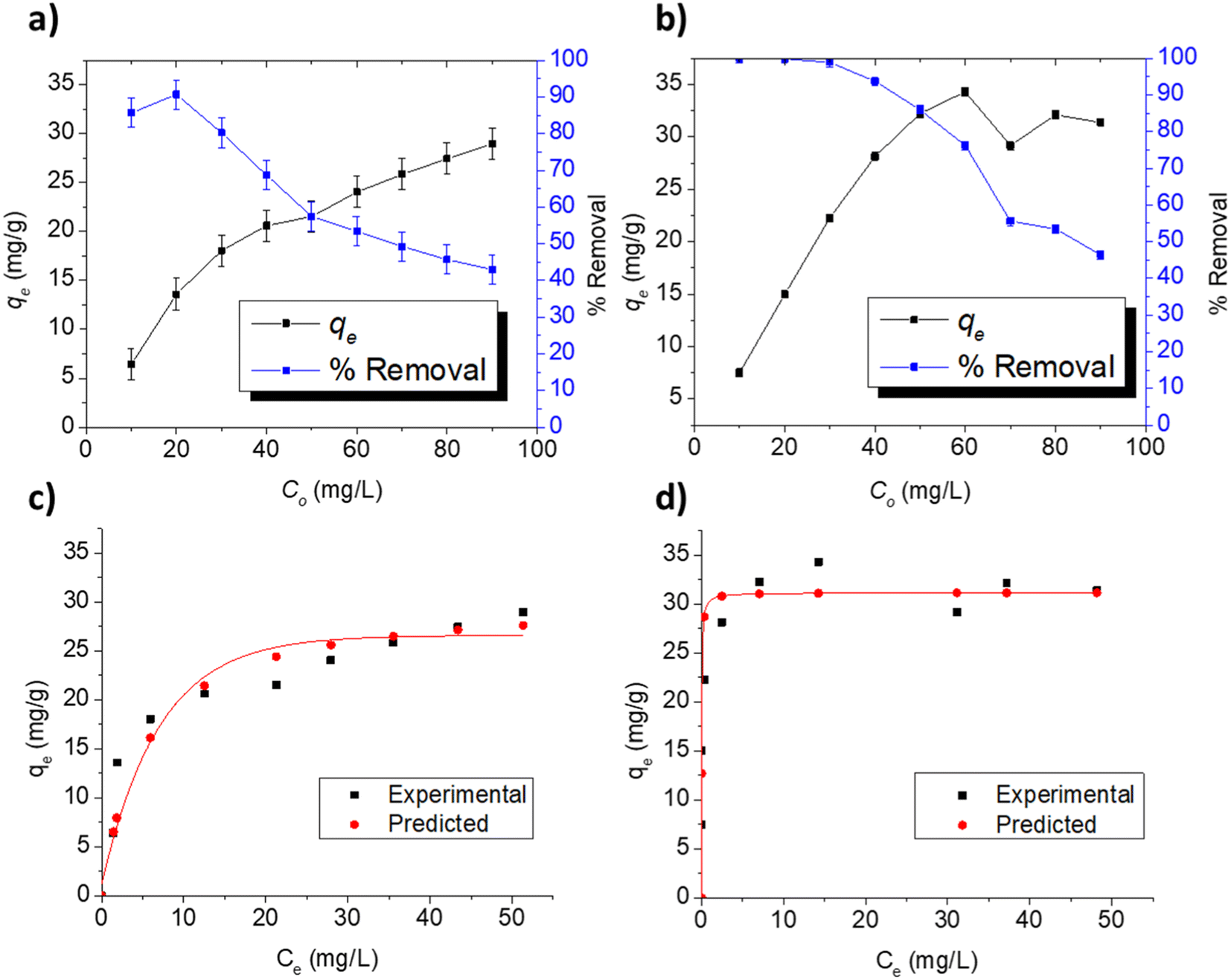
3.3.3. Adsorption at varying concentrations. The adsorption indicators of capacity (qe) and removal efficiency are presented in Fig. 5a and b for the respective adsorption of FLX and MB on Cel-HTT at the concentration range of 10–90 ppm using 1.33 g L−1 of Cel-HTT and pH 7.5. In the investigated concentration range, the percentage removal for FLX and MB decreased gradually from about 85 and 100% at 10 ppm to about 42 and 46% at 90 ppm for FLX and MB, respectively. This decrease can be ascribed to the occupation of available adsorption sites as the initial concentration increases.54 On the other hand, qe increases from about 6 and 7.5 mg g−1 at 10 ppm to about 28 and 31 mg g−1 at 90 ppm for FLX and MB, respectively, due to the increase in the concentration gradient driving force of the adsorbate molecules at higher initial concentrations.55,56
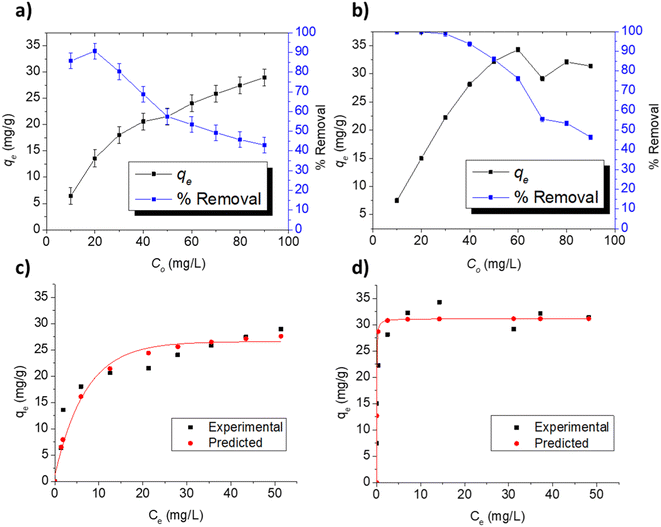 |
| | Fig. 5 Adsorption indicators for the adsorption of FLX and MB on 1.33 g L−1 of Cel-HTT at pH 7.5, 25 ± 2 °C and under different initial concentrations (a and b), experimental and predicted isotherm profiles for FLX (c) and MB (d) adsorption on Cel-HTT at pH 7.5 and 25 ± 2 °C. | |
To predict the adsorption behavior, the experimental equilibrium isotherms of FLX and MB adsorption on Cel-HTT (Fig. 5c and d, respectively) were fitted to the Langmuir and Freundlich models (Fig. S6 and S7 of ESI,† respectively). Langmuir model assumes a monolayer adsorption on energetically equivalent and homogenous sites, while Freundlich assumes a multi-layer adsorption on energetically non-equivalent heterogenous adsorption sites. Langmuir gave the best fit for both FLX and MB adsorption isotherms as it yielded higher R2 and lower Root Mean Square Error (RMSE) values than those of Freundlich plots (Table 4). Also, the experimental qm values of 30.39 and 31.15 mg g−1 were better predicted by Langmuir model for FLX and MB adsorption, respectively. The predicted Langmuir profiles are also shown in Fig. 5c and d for FLX and MB adsorption, respectively. On the other hand, the dissociation or desorption constant for MB is much lower than that for FLX indicating that MB is more strongly bound to the hydrochar. In previous literature, the adsorption of FLX on activated carbon from almond tree under different temperature conditions was studied where crushed almond tree was subjected to pyrolysis at 600 °C for 1 h, then was carbon dioxide activated at 700–950 °C for 1 h and subsequently steam activated at 650–850 °C for 1 h. Adsorption onto the formed activated carbon also followed Langmuir isotherm with maximum adsorption capacities between 110 and 224 mg g−1 at a pH of about 6–7 and a temperature of 25 °C.57 In addition, the adsorption of Malachite Green on activated carbon modified by aminobenzoic acid followed Langmuir model with a maximum adsorption capacity of 66.87 mg g−1 at pH 7 and 25 °C.58
Table 4 Isotherm parameters for the adsorption of FLX and MB at pH 7.5 and 25 ± 2 °C
| |
Langmuir isotherm |
Freundlich isotherm |
| qm mg g−1 |
Kd L mg−1 |
R2 |
RMSE |
1/n |
Kf (mg g−1) (L mg−1)n |
R2 |
RMSE |
| FLX |
30.39 |
5.24 |
0.9868 |
0.066 |
0.32 |
8.41 |
0.8450 |
0.175 |
| MB |
31.15 |
0.028 |
0.9972 |
0.029 |
0.09 |
23.84 |
0.8721 |
0.092 |
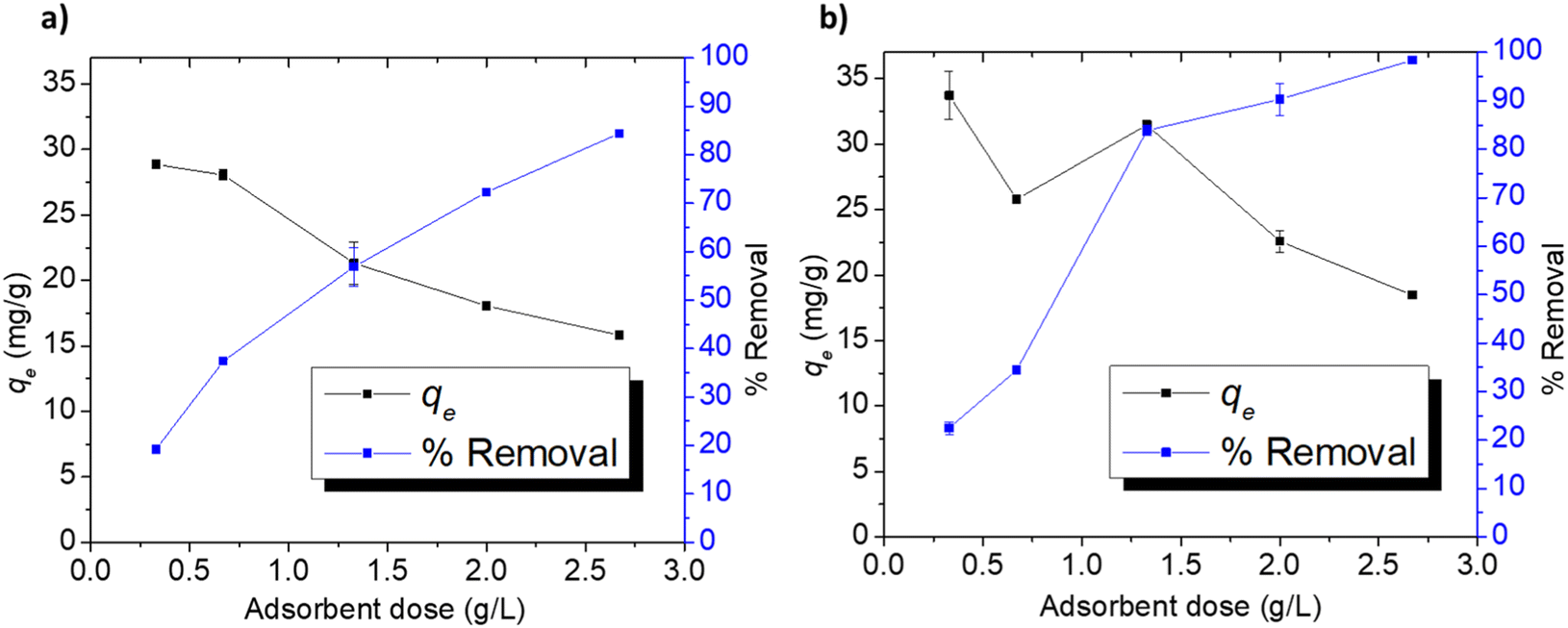
3.3.4. Adsorption at different doses. The effect of Cel-HTT dose on qe and % removal of 50 ppm FLX and MB at pH 7.5 is depicted in Fig. 6a and b. The removal of FLX increases from 19 to 84%, while that of MB rises from 22 to 98% with increasing the adsorbent dose from 0.33 to 2.67 g L−1. As for qe, it decreases correspondingly from about 28 to 15 mg g−1 and from 33 to 18 mg g−1, respectively. The increase in %removal might be attributed to the increase in available binding sites and surface areas, while the decrease in qe might be attributed to the aggregation of the adsorbent particles leading to a limitation in the active surfaces available for adsorption.59,60
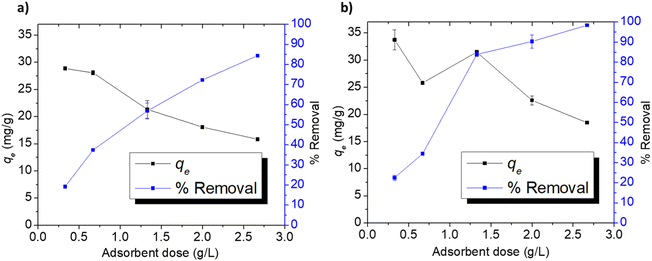 |
| | Fig. 6 Adsorption indicators for FLX (a) and MB (b) on Cel-HTT at an initial concentration of 50 ppm, pH 7.5, and 25 ± 2 °C. | |
3.4. Regeneration of Cel-HTT
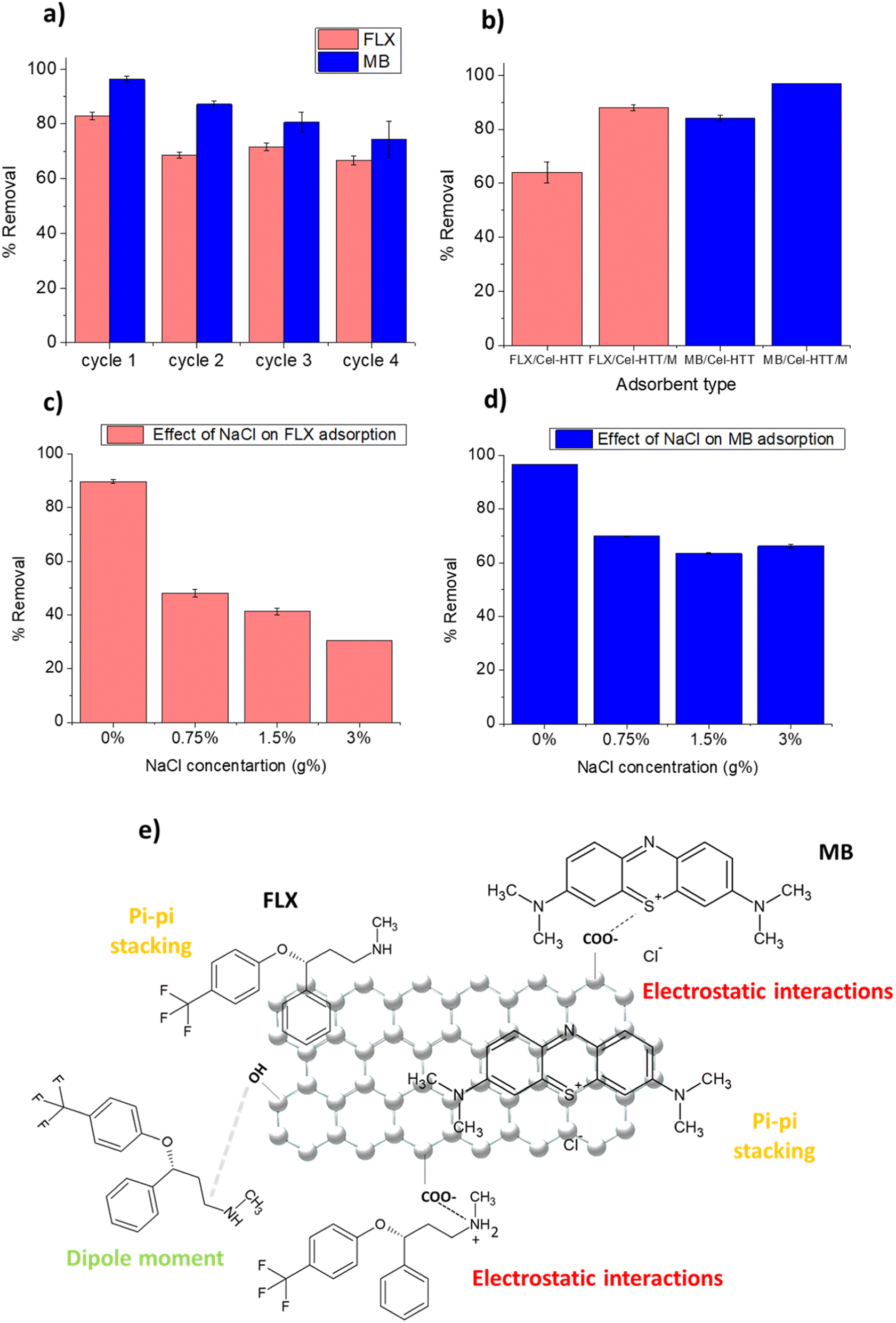
Regeneration is an important criterion in selecting cost-effective adsorbents. The regeneration of Cel-HTT after adsorbing 25 ppm of FLX or MB as depicted in Fig. 7a was successful for four cycles of regeneration using ethanol absolute. The % removal decreased by about 15–20% from 83 to 67% and from 96 to 74% for FLX and MB, respectively, after the four cycles. MB shows more resistance for desorption than FLX due to its stronger binding to the hydrochar as corroborated by its lower Kd value (Table 4). Activated carbon from waste edible fungus was also successfully regenerated using ethanol.61 It is thus evident that Cel-HTT is efficiently recycled using the applied desorbing agent.
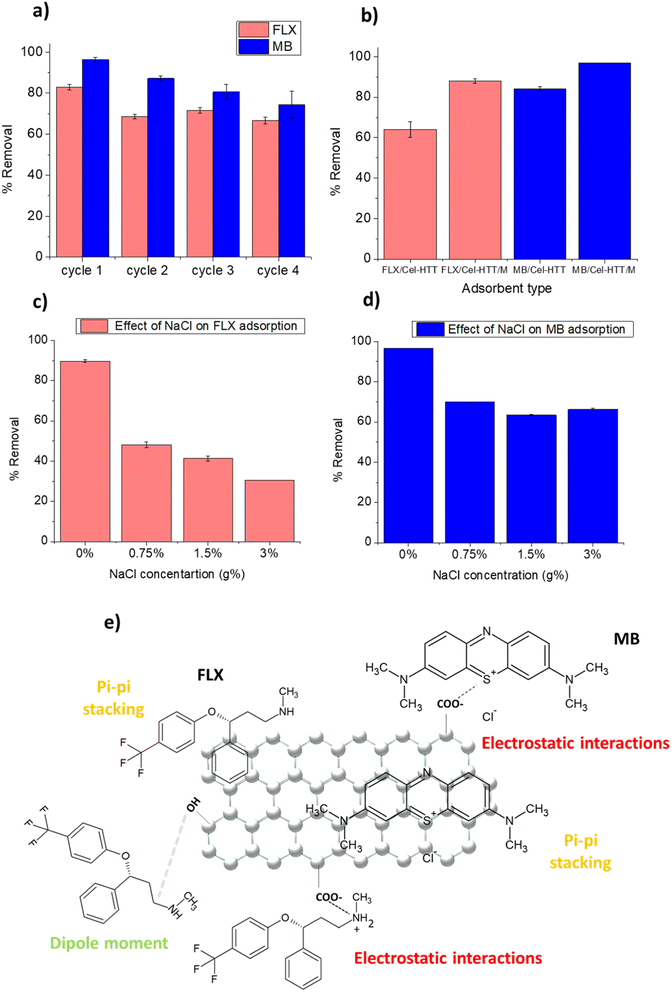 |
| | Fig. 7 Regeneration study of Cel-HTT using ethanol absolute after adsorption of 25 ppm FLX or MB at pH 7.5 and adsorbent dose of 1.33 g L−1 (a), comparison of % removal of FLX and MB on Cel-HTT/M and Cel-HTT at initial concentration of 50 ppm, adsorbent dose of 1.33 g L−1 and pH 7.5 (b), effect of the addition of NaCl on the adsorption of FLX (c) and MB (d) at initial concentration of 50 ppm, adsorbent dose of 1.33 g L−1 and pH 7.5, schematic diagram illustrating some possible adsorption mechanisms for FLX and MB on Cel-HTT (e). | |
4. Adsorption using magnetic hydrochar
To facilitate the collection of the hydrochar from water in real life applications, Cel-HTT was magnetized to form the magnetic hydrochar Cel-HTT/M then it was tested for adsorbing FLX and MB. The removal efficiencies of FLX and MB on Cel-HTT are compared to their counterparts on Cel-HTT/M under the same conditions of 1.33 g L−1 dose, 50 ppm and pH 7.5 (Fig. 7b). It can be deduced that the percent removal of FLX and MB on Cel-HTT/M exceeded their removal on Cel-HTT by about 27.2 and 13.1%, respectively. This could be owed to the enhanced surface area and pore volume of Cel-HTT/M relative to Cel-HTT.
On studying the effect of adding varying concentrations of NaCl salt on the adsorption of FLX and MB (Fig. 7c and d), it was found that 0.75 g% NaCl addition reduced the percent removal of FLX and MB by about 46% and 27%, respectively. The presence of salt decreases the removal owing to the salt shielding effect which decreases the electrostatic interaction between the contaminant and the hydrochar. Similar results were previously obtained for the adsorption of MB on coal-based activated carbon.62 Percent removal of FLX further declined upon adding higher salt concentrations while the removal of MB almost remained steady. This shows that the magnetic hydrochar is more efficient in removing MB than FLX under saline conditions, likely due to the stronger electrostatic interactions taking place between MB and the hydrochar as was previously discussed.
5. Adsorption mechanism
The FTIR measurements of Cel-HTT before and after adsorption with FLX and MB (Fig. S8†) shows that the peak at about 3400 cm−1 assigned for OH group exhibited a decreased intensity after adsorption with FLX and MB. Also, the peak at 1650 cm−1 assigned for CO group exhibited a slight shifting to 1656 and 1655 cm−1 for FLX and MB, respectively. Thus, it is concluded that Cel-HTT could possibly adsorb FLX and MB via a carboxylic group as illustrated in Fig. 7e. As per the zeta potential measurements of Cel-HTT and the speciation diagrams of FLX and MB, Cel-HTT is negatively charged at the applied pH while FLX and MB are positively charged and hence can interact via electrostatic attraction. This was further confirmed by the pH control test for determination of PZC which showed the pH changes after adsorption thus indicating the role of electrostatic interactions in adsorption. In addition, ionic strength studies manifested a clear salt shielding effect which further supports the role of electrostatic interactions. Surface specific reactions can possibly occur via electron transfer from the electron-rich oxygen containing groups of the hydrochar and the electron-deficient nitrogen atom on FLX or MB or the electron-deficient sulfur atom on MB.63 This was also confirmed by the best kinetic fitting model which is pseudo-second-order model indicating chemisorption. Also, dispersive interactions or pi–pi stacking could take place between the delocalized carbon electrons on Cel-HTT rings and the aromatic rings of the adsorbates FLX and MB.64,65
6. Conclusion
Expired cellulose was successfully converted to a functional hydrochar by means of hydrothermal treatment and compared to the expired and fresh cellulose. Bulk density of expired cellulose was less than that of fresh cellulose by about 14%. Cel-HTT exhibited larger porosity and surface area as well as higher thermal stability than the expired cellulose. When tested for adsorption of FLX and MB, Cel-HTT yielded a maximum adsorption capacity of about 30 mg g−1 for both contaminants at pH 7.5 and removal efficiencies that exceeded 90%, while the underlying interactions were suggested to be electrostatic and pi–pi stacking. However, FLX was adsorbed faster than MB and kinetics followed the pseudo-second-order model. Cel-HTT was successfully regenerated after FLX or MB adsorption using ethanol absolute for four cycles. When Cel-HTT was magnetized, better percent removal was attained owing to the larger surface area and pore volume of the magnetic nanocomposite relative to the bare one. Exploiting expired chemicals and converting them to hydrochar adsorbents for water treatment promises to be a sustainable approach that supports circular economy through integrating the concepts of solid waste management, pollution reduction, and reuse/regeneration of resources that are outlined by the UN SDGs.
Author contributions
H. H. F., conceptual design, performing all the experiments, data analysis, and preparing the original draft with figures; M. N. writing and supervision; M. M. H. E. conceptual design, data analysis and curation, writing, and supervision.
Conflicts of interest
Authors declare no competing interests.
Acknowledgements
This work was funded by the American University in Cairo through a PhD research grant for purchasing chemicals and consumables, and by USAID ASHA grant number: AID-ASHA-G-17-00010 for capacity building, which provided the lab instruments.
References
- S. Sivakanthan, S. Rajendran, A. Gamage, T. Madhujith and S. Mani, Food Res. Int., 2020, 136, 109327 CrossRef CAS PubMed.
- Y. Luo, Z. Lin and G. Guo, Nanoscale Res. Lett., 2019, 14, 1–10 CrossRef PubMed.
- M. Niaounakis, Eur. Polym. J., 2019, 114, 464–475 CrossRef CAS.
- Y. Gao, J. Remón and A. S. Matharu, Green Chem., 2021, 23, 3502–3525 RSC.
- H. Mumtaz, S. Sobek, S. Werle, M. Sajdak and R. Muzyka, Sustainable Chem. Pharm., 2023, 32, 100991 CrossRef CAS.
- A. Sharma, M. Thakur, M. Bhattacharya, T. Mandal and S. Goswami, Biotechnol. Rep., 2019, 21, e00316 CrossRef PubMed.
- G. Gadaleta, C. Ferrara, S. De Gisi, M. Notarnicola and G. De Feo, Sci. Total Environ., 2023, 871, 161958 CrossRef CAS PubMed.
- N. M. van der Velden, M. K. Patel and J. G. Vogtländer, Int. J. Life Cycle Assess., 2014, 19, 331–356 CrossRef.
- C. Pan, F. Zhu, M. Wu, L. Jiang, X. Zhao and M. Yang, Chemosphere, 2022, 287, 132434 CrossRef CAS PubMed.
- V. H. O. Silva, A. P. dos Santos Batista, A. C. Silva Costa Teixeira and S. I. Borrely, Environ. Sci. Pollut. Res., 2016, 23, 11927–11936 CrossRef CAS PubMed.
- T. L. Palma and M. C. Costa, Anaerobe, 2021, 68, 102356 CrossRef CAS PubMed.
- H.-y. Shao, M.-h. Wu, F. Deng, G. Xu, N. Liu, X. Li and L. Tang, Chemosphere, 2018, 190, 184–190 CrossRef CAS PubMed.
- D. A. G. Sumalinog, S. C. Capareda and M. D. G. de Luna, J. Environ. Manage., 2018, 210, 255–262 CrossRef CAS PubMed.
- F. Mashkoor and A. Nasar, J. Magn. Magn. Mater., 2020, 500, 166408 CrossRef CAS.
- M. R. Khan, M. A. Khan, Z. A. Alothman, I. H. Alsohaimi, M. Naushad and N. H. Al-Shaalan, RSC Adv., 2014, 4, 34037–34044 RSC.
- B. Sun, Y. Yuan, H. Li, X. Li, C. Zhang, F. Guo, X. Liu, K. Wang and X. Zhao, Chem. Eng. J., 2019, 371, 55–63 CrossRef CAS.
- Z. Feng, K. Odelius, G. K. Rajarao and M. Hakkarainen, Chem. Eng. J., 2018, 346, 557–566 CrossRef CAS.
- T. M. A. Babeker and Q. Chen, Curr. Pollut. Rep., 2021, 7, 54–71 CrossRef CAS.
- D. S. Tong, C. W. Wu, M. O. Adebajo, G. C. Jin, W. H. Yu, S. F. Ji and C. H. Zhou, Appl. Clay Sci., 2018, 161, 256–264 CrossRef CAS.
- B. Li, J. Guo, K. Lv and J. Fan, Environ. Pollut., 2019, 254, 113014 CrossRef CAS PubMed.
- F. Zhou, K. Li, F. Hang, Z. Zhang, P. Chen, L. Wei and C. Xie, RSC Adv., 2022, 12, 1885–1896 RSC.
- I. Kurnia, S. Karnjanakom, I. Irkham, H. Haryono, Y. A. Situmorang, A. Indarto, A. R. Noviyanti, Y. W. Hartati and G. Guan, RSC Adv., 2023, 13, 220–227 RSC.
- H. Ouachtak, A. El Guerdaoui, R. El Haouti, R. Haounati, H. Ighnih, Y. Toubi, F. Alakhras, R. Rehman, N. Hafid and A. A. Addi, RSC Adv., 2023, 13, 5027–5044 RSC.
- X. Huang, N.-y. Gao and Q.-l. Zhang, J. Environ. Sci., 2007, 19, 1287–1292 CrossRef CAS PubMed.
- R. H. Althomali, K. A. Alamry, M. A. Hussein and R. Guedes, RSC Adv., 2023, 13, 4303–4313 RSC.
- A. E. Ofomaja, Bioresour. Technol., 2010, 101, 5868–5876 CrossRef CAS PubMed.
- A. Idris and K. Saed, Environmentalist, 2003, 23, 329–334 CrossRef.
- M. A. Ahmed, M. A. Ahmed and A. A. Mohamed, RSC Adv., 2023, 13, 5337–5352 RSC.
- N. Kruer-Zerhusen, B. Cantero-Tubilla and D. B. Wilson, Cellulose, 2018, 25, 37–48 CrossRef CAS.
- V. Hospodarova, E. Singovszka and N. Stevulova, Am. J. Anal. Chem., 2018, 9, 303–310 CrossRef CAS.
- N. Abidi and M. Manike, Text. Res. J., 2018, 88, 719–730 CrossRef CAS.
- K. Sheng, S. Zhang, J. Liu, E. Shuang, C. Jin, Z. Xu and X. Zhang, J. Cleaner Prod., 2019, 237, 117831 CrossRef CAS.
- N. Saha, A. Saba and M. T. Reza, J. Anal. Appl. Pyrolysis, 2019, 137, 138–145 CrossRef CAS.
- S. Kang, X. Li, J. Fan and J. Chang, Ind. Eng. Chem. Res., 2012, 51, 9023–9031 CrossRef CAS.
- A. Brown, B. McKeogh, G. Tompsett, R. Lewis, N. Deskins and M. Timko, Carbon, 2017, 125, 614–629 CrossRef CAS.
- K. Kavkler and A. Demšar, Spectrochim. Acta, Part A, 2011, 78, 740–746 CrossRef PubMed.
- H. Düdder, A. Wütscher, R. Stoll and M. Muhler, Fuel, 2016, 171, 54–58 CrossRef.
- J. Gong, J. Li, J. Xu, Z. Xiang and L. Mo, RSC Adv., 2017, 7, 33486–33493 RSC.
- C. Trilokesh and K. B. Uppuluri, Sci. Rep., 2019, 9, 1–8 CrossRef CAS PubMed.
- K. Balani, V. Verma, A. Agarwal and R. Narayan, Biosurfaces: a materials science and engineering perspective, John Wiley & Sons, 2015 Search PubMed.
- Z. A. ALOthman, Materials, 2012, 5, 2874–2902 CrossRef CAS.
- Y. Gao, X.-H. Wang, H.-P. Yang and H.-P. Chen, Energy, 2012, 42, 457–465 CrossRef CAS.
- N. Saha, K. McGaughy and M. T. Reza, J. Anal. Appl. Pyrolysis, 2020, 152, 104965 CrossRef CAS.
- C. Okoli, M. Sanchez-Dominguez, M. Boutonnet, S. Jaras, C. Civera, C. Solans and G. R. Kuttuva, Langmuir, 2012, 28, 8479–8485 CrossRef CAS PubMed.
- A. Burbano, G. Medina, F. Sánchez, V. Lassalle, M. Horst, G. Gascó and A. Méndez, Biomass Convers. Biorefin., 2022, 1–14 Search PubMed.
- S.-Q. Chen, Y.-L. Chen and H. Jiang, Ind. Eng. Chem. Res., 2017, 56, 3059–3066 CrossRef CAS.
- M. A. Khan, A. A. Alqadami, S. M. Wabaidur, M. R. Siddiqui, B.-H. Jeon, S. A. Alshareef, Z. A. Alothman and A. E. Hamedelniel, J. Hazard. Mater., 2020, 400, 123247 CrossRef CAS PubMed.
- B. Silva, M. Martins, M. Rosca, V. Rocha, A. Lago, I. C. Neves and T. Tavares, Sep. Purif. Technol., 2020, 235, 116139 CrossRef CAS.
- K. C. Bedin, I. P. Souza, A. L. Cazetta, L. Spessato, A. Ronix and V. C. Almeida, J. Mol. Liq., 2018, 269, 132–139 CrossRef CAS.
- S. Escudero-Curiel, U. Penelas, M. Á. Sanromán and M. Pazos, Chemosphere, 2021, 268, 129318 CrossRef CAS PubMed.
- W. Qu, T. Yuan, G. Yin, S. Xu, Q. Zhang and H. Su, Fuel, 2019, 249, 45–53 CrossRef CAS.
- A. H. Jawad, M. Bardhan, M. A. Islam, M. A. Islam, S. S. A. Syed-Hassan, S. Surip, Z. A. ALOthman and M. R. Khan, Surf. Interfaces, 2020, 21, 100688 CrossRef CAS.
- W. Xiao, Z. N. Garba, S. Sun, I. Lawan, L. Wang, M. Lin and Z. Yuan, J. Cleaner Prod., 2020, 253, 119989 CrossRef CAS.
- N. Kannan and M. M. Sundaram, Dyes Pigm., 2001, 51, 25–40 CrossRef CAS.
- Y. Kuang, X. Zhang and S. Zhou, Water, 2020, 12, 587 CrossRef.
- S. Afshin, Y. Rashtbari, B. Ramavandi, M. Fazlzadeh, M. Vosoughi, S. A. Mokhtari, M. Shirmardi and R. Rehman, Korean J. Chem. Eng., 2020, 37, 80–92 CrossRef CAS.
- S. Román, J. M. V. Nabais, J. F. González, C. M. González-García and A. L. Ortiz, Clean: Soil, Air, Water, 2012, 40, 698–705 Search PubMed.
- M. Naushad, A. A. Alqadami, A. A. Al-Kahtani, T. Ahamad, M. R. Awual and T. Tatarchuk, J. Mol. Liq., 2019, 296, 112075 CrossRef CAS.
- H. A. Ahsaine, M. Zbair, Z. Anfar, Y. Naciri, N. El Alem and M. Ezahri, Mater. Today Chem., 2018, 8, 121–132 CrossRef.
- F. Keyvani, S. Rahpeima and V. Javanbakht, Solid State Sci., 2018, 83, 31–42 CrossRef CAS.
- H. Li, L. Liu, J. Cui, J. Cui, F. Wang and F. Zhang, RSC Adv., 2020, 10, 14262–14273 RSC.
- J. Wang, J. Ma and Y. Sun, Water, 2022, 14, 3576 CrossRef CAS.
- S. Román, J. M. Valente Nabais, B. Ledesma, C. Laginhas and M.-M. Titirici, Molecules, 2020, 25, 2264 CrossRef PubMed.
- J. V. Nabais, A. Mouquinho, C. Galacho, P. Carrott and M. R. Carrott, Fuel Process. Technol., 2008, 89, 549–555 CrossRef CAS.
- A. M. Vargas, A. L. Cazetta, M. H. Kunita, T. L. Silva and V. C. Almeida, Chem. Eng. J., 2011, 168, 722–730 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Marianne Nebsenb and
Mayyada M. H. El-Sayed
*a,
Marianne Nebsenb and
Mayyada M. H. El-Sayed