
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational insights into the mechanisms and origins of switchable selectivity in gold(I)-catalyzed annulation of ynamides with isoxazoles via 6π-electrocyclizations of azaheptatrienyl cations†
Abosede Adejoke Badeji *a,
Yuan Liub,
Segun D. Oladipoc and
Adejoke Deborah Osinubia
*a,
Yuan Liub,
Segun D. Oladipoc and
Adejoke Deborah Osinubia
aDepartment of Chemical Sciences, Tai Solarin University of Education, Ogun State, Nigeria. E-mail: ogunlanaaa@tasued.edu.ng
bSchool of Chemistry and Chemical Engineering, Nantong University, 9 Seyuan Road, Nantong 226019, China
cDepartment of Chemical Sciences, Olabisi Onabanjo University, 2002 Ago-Iwoye, Nigeria
First published on 14th June 2023
Abstract
Electrocyclizations of acyclic conjugated π-motifs have emerged as a versatile and effective strategy for accessing various ring systems with excellent functional group tolerability and controllable selectivity. Typically, the realization of 6π-electrocyclization of heptatrienyl cations to afford seven-membered motif has proven difficult due to the high-energy state of the cyclizing seven-membered intermediate. Instead, it undergoes the Nazarov cyclization, affording a five-membered pyrrole product. However, the incorporation of a Au(I)-catalyst, a nitrogen atom and tosylamide group in the heptatrienyl cations unexpectedly circumvented the aforementioned high energy state to afford a seven-membered azepine product via 6π-electrocyclization in the annulation of 3-en-1-ynamides with isoxazoles. Therefore, extensive computational studies were carried out to investigate the mechanism of Au(I)-catalyzed [4+3] annulation of 3-en-1-ynamides with dimethylisoxazoles to produce a seven-membered 4H-azepine via the 6π-electrocyclization of azaheptatrienyl cations. Computational results showed that after the formation of the key α-imino gold carbene intermediate, the annulation of 3-en-1-ynamides with dimethylisoxazole occurs via the unusual 6π-electrocyclization to afford a seven-membered 4H-azepine exclusively. However, the annulation of 3-cyclohexen-1-ynamides with dimethylisoxazole occurs via the commonly proposed aza-Nazarov cyclization pathway to majorly generate five-membered pyrrole derivatives. The results from the DFT predictive analysis revealed that the key factors responsible for the different chemo-, and regio-selectivities observed are the cooperating effect of the tosylamide group on C1, the uninterrupted π-conjugation pattern of the α-imino gold(I) carbene and the substitution pattern at the cyclization termini. The Au(I)-catalyst is believed to assist in the stabilization of the azaheptatrienyl cation.
Introduction
Recent advances in gold catalysis have inspired new annulations between alkynes and nucleophiles toward the synthesis of rings and ring systems with excellent functional group tolerability and controllable selectivity.1–8 Among the alkynes, significant attention has been paid to annulation involving ynamides due to the highly electrophilic keteniminium nature exhibited by their Au-π-alkyne complexes, as well as their higher reactivity compared to normal alkynes.2–8 In this regard, the annulation of ynamides with nucleophiles3–8 including isoxazoles5f,6-8 is one of the most powerful ring-forming strategies to assemble valuable azacyclic frameworks which are core structures found in myriads of natural products, bioactive molecules and pharmaceuticals.3–8 Since the seminal work reported by Ye and co-workers6a in 2015 to access various polysubstituted 2-aminopyrroles via the gold-catalyzed [3+2] annulation of ynamides with isoxazoles, several annulations involving ynamides and isoxazoles with its derivatives have been developed.6–8 For instance, Hashmi and co-workers6c elegantly revealed the Au(I)-catalyzed [3+2] annulation of ynamides with benzisoxazoles to deliver a facile, flexible, and atom-economical one-step route to unprotected 7-acylindoles. Furthermore, Gandon and Sahoo5f reported the combined experimental and DFT studies on the ring expansion and 1,2-migration cascade of benzisoxazoles with ynamides to produce benzo[e][1,3]oxazine of predominant E configuration. More recently, Hu et al.6g provided a comprehensive summary of the annulations involving ynamides and isoxazoles in an account of the reactivity of ynamides in catalytic intermolecular annulations.Moreover, the success of the annulations between ynamides and the isoxazoles has been attributed to the key α-imino gold carbene intermediates from which various regio-, chemo- and stereo-selective transformations could occur to afford diverse N-heterocyclic compounds.6–8 For example, the [3+2] annulation of ynamides with isoxazoles reported by Ye and coworkers to produce aminopyrroles, was made possible via the regioselective cyclization of the generated α-imino gold carbene intermediate. Just recently, Li et al.7a documented a series of annulations involving ynamides and isoxazoles in a review of the recent progress in the gold-catalyzed annulations of ynamides with isoxazoles derivatives. The annulations were reported to proceed via α-imino gold carbene intermediate which in turn afforded amino-heterocycles in a regioselective manner. In addition, Zhou et al.7b provided insights into the reaction mechanism and chemoselectivity in the cycloaddition of ynamides and isoxazoles with water. It was reported that the formation of the α-imino gold carbene complex as the key intermediate is very crucial for the formation of the product.
The synthetic prowess of pericyclic reactions has enormously increased with the development of catalytic methods.9–11 Of the known pericyclic processes, the electrocyclization of π-conjugated acyclic compounds has particularly attracted research attention because of their high level of reliability, stereo- and regiospecificity10,11 in constructing complex cyclic and polycyclic frameworks. Worthy of note is the Nazarov cyclization of pentadienyl cations12 and related 6π-electrocylization of trienes,11a,13 which are gainfully applied in synthetic organic chemistry.11–14 For instance, the electrocyclization of pentadienyl cations generally proceeds via a 4π electrocyclic ring closure to provide cyclopentadienyl cation.15a For heptatrienyl anion, they often undergoes facile 8π electrocyclization through rapid interconversions among various anion configurations to afford seven-membered motif.16 However, the possibility of the heptatrienyl cation15,17 A to exclusively undergo 6π-electrocyclization through B to eventually generate a seven-membered motif D is difficult, but, cannot be ruled out (Scheme 1a, right).15 The reason for this difficulty is due to the high energy required for A through B to form all s-cis configured cations C than to generate E.15 Therefore, A exclusively undergoes typical Nazarov cyclization to afford a five-membered motif E (Scheme 1a, left).15a It has also been reported by Alickmann et al.15b that 1-aza- (Scheme 1b) and 1-oxaheptatrienyl cations (Scheme 1c) undergo various types of electrocyclizations to afford five-membered and seven-membered heterocyclic compounds, yet the path leading to the formation of I and I′, represent the kinetically preferred pathways among the pathways investigated (Scheme 1b and 1c).15b Moreover, Gir and Liu8 envisaged that the incorporation of Au(I)-catalyst and nitrogen atom into C might circumvent the aforementioned high energy state and successfully drive the reaction of the resulting C′ towards 6π-electrocyclization, thereby generating a seven-membered azacycle D′ (Scheme 2a, left). With respect to this, they developed a particularly interesting [4+3] annulation of 3-en-1-ynamides 1a, with 3,5-dimethylisoxazoles 2, to exclusively produce seven-membered 4H-azepine product 3 (Scheme 2a, right).8 It was also reported that when the substituent on the α-alkynyl carbon atom of the ynamide (1a) was replaced with a cyclohexenyl group, the annulation of the corresponding ynamide (1b) with 2 led to a switch in the product selectivity, majorly affording pyrrole 4 with traces of azepine product 5 (Scheme 2b).8
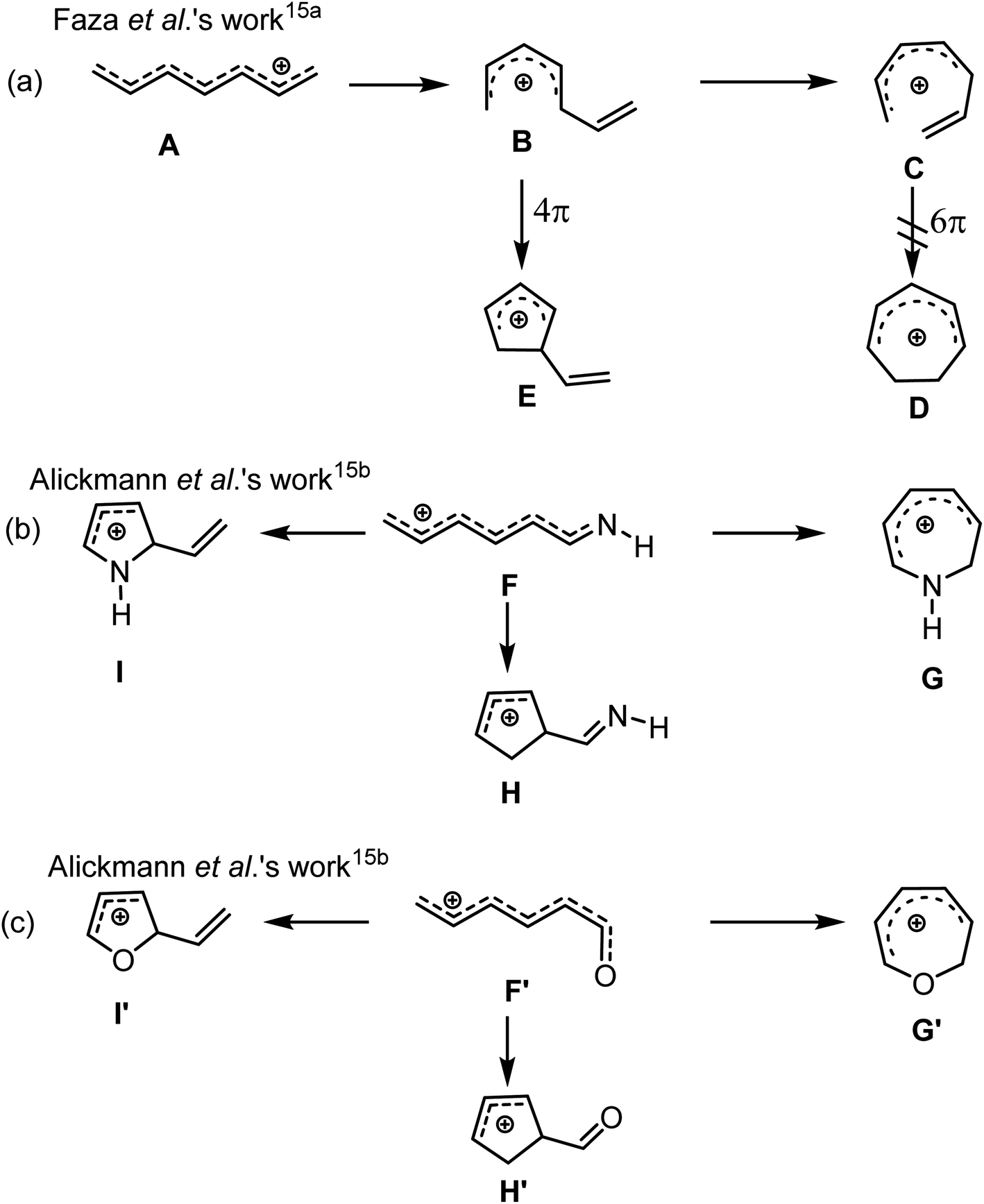 | ||
| Scheme 1 (a) The electrocyclization of heptatrienyl cations; (b) electrocyclization of 1-azaheptatrienyl cations; (c) electrocyclization of 1-oxaheptatrienyl cations. | ||
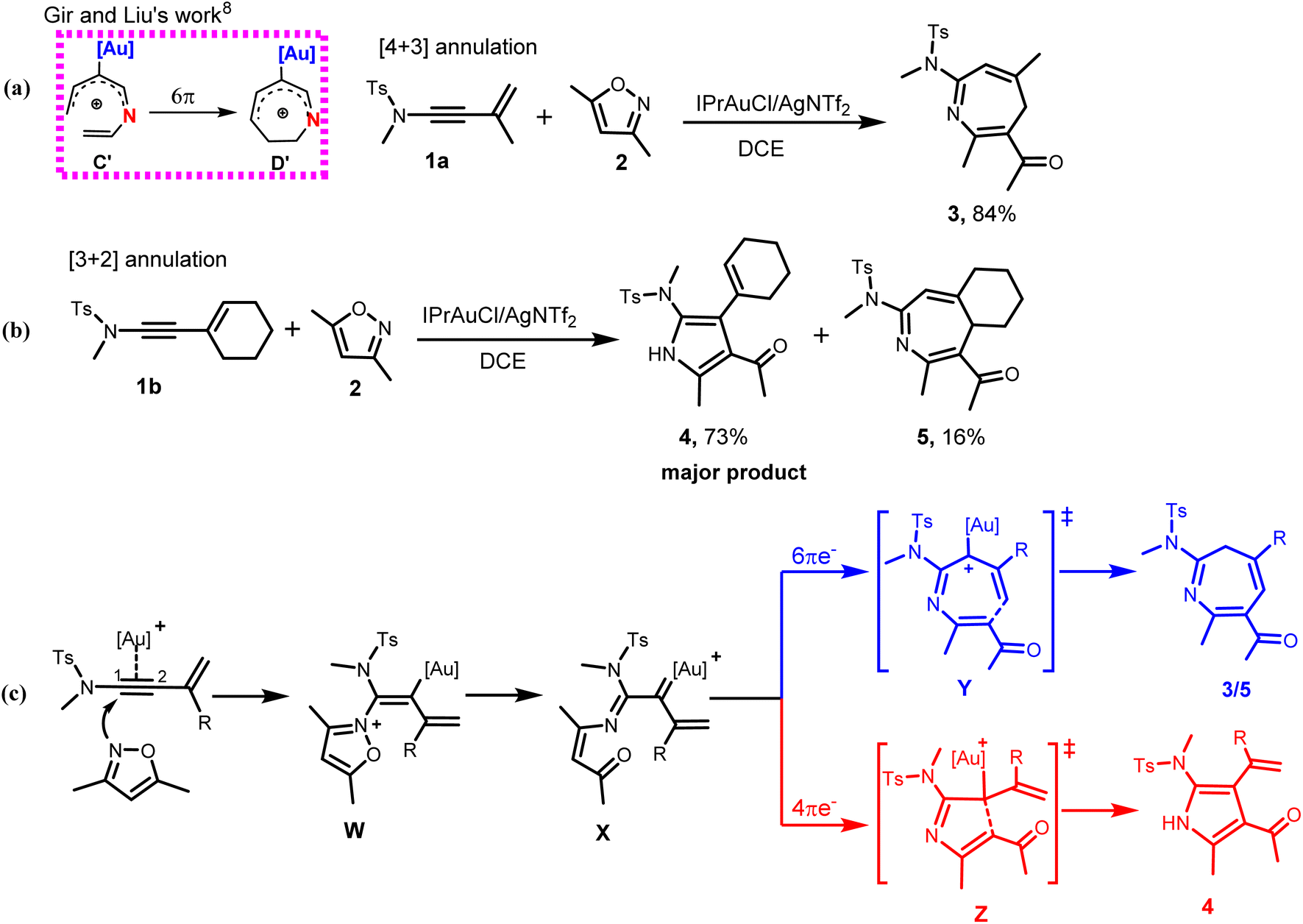 | ||
| Scheme 2 The Au(I)-catalyzed annulation of ynamides with isoxazoles. | ||
From previous studies,5f,6–8 the mechanism of the annulation of 1a/1b with 2 was postulated to involve an initial N-attack of 2 on the Au-π-alkyne complex of 1a/1b to afford an iminovinyl gold intermediate W. This then isomerizes to the key α-imino gold(I) carbenoid X via a ring-opening process.6–8 From this key intermediate, the unusual 6π-electron-7-atom electrocyclization could selectively take place to afford the seven-membered azepine derivatives 3/5 via the Au(I)-stabilized azaheptatrienyl cation Y (Scheme 2c). Alternatively, the reaction mechanism could follow the conventional reactivity of ynamides via 4π-electron-5-atom-electrocyclization (aza-Nazarov-type reaction) from X, to produce a five-membered pyrrole derivatives 4 via the Au(I)-stabilized pentadienyl cation Z (Scheme 2c).
Despite the recent advances in gold-catalyzed annulations, adequate mechanistic information is yet to be provided on how substrate modification influences the selectivity in the formation of ring systems. For instance, the successfully 6π-electrocyclization of all s-cis configured cations C, an isomer of heptatrienyl cation A, generated a seven-membered intermediate by the incorporation of Au(I)-catalyst and the nitrogen atom (Scheme 2)8 yet, Alickmann and co-workers15b reported that 1-aza- and 1-oxaheptatrienyl cations would prefer the Nazarov-type cyclization to afford a five-membered heterocycle, hence the need for mechanistic clarification. Intrigued by the switch in selectivity displayed in the above annulations due to substrate modification, some puzzling questions as to which factors control the selective formation of the seven-membered product as against the five-membered product are yet to be answered. For example, could it be that the Au(I)-catalyst is solely responsible for the successful 6π-electrocyclization to afford the seven-membered azacycle or the cooperating effect of the Au(I)-catalyst and the nitrogen atom? Could it also be that the incorporated nitrogen atom originated from the isoxalic substrate 2 or the tosylamide group? In addition, could there be other factors responsible for the preferred selectivity besides these two factors? To answer these questions, computational studies were employed. The origin of the chemoselectivity and the factors responsible for the selective formation of 3/5 as against 4 in the annulation of 1a/1b with 2 were unveiled. More so, the role of the incorporated Au(I)-catalyst, the nitrogen atom and the tosylamide group in facilitating the formation of the seven-membered product 3/5 were addressed.
Computational method
All the calculations were carried out by DFT method as implemented in the Gaussian 09 program package.18 For the geometry optimizations of all stationary points including the transition states and intermediates, the M06 density functional method19 was employed. For the geometry optimization, the 6-31G(d) basis set20 was utilized for the main group atoms (N, S, O, C, H, F) while the LANL2DZ basis set21 together with the LANL2DZ pseudopotential was employed for the Au atom. Vibrational frequency analyses at the same level of theory were performed on the optimized geometries to determine the nature of the stationary points as either minima or transition states. More so, intrinsic reaction coordinate calculations (IRC)22 were carried out to confirm that the optimized transition states connect to their appropriate reactants and products. In order to account for solvation effects, the optimized structures in the gas phase were subjected to single-point energy (SPE) calculations at the M06/SDD23(Au)-6-311++G(d,p)(N, S, O, C, H, F) level of theory. For these SPE calculations, the SMD solvation model as developed by Truhlar and Cramer24 was employed, and 1,2-dichloroethane (DCE) was used as the solvent.To account for the overestimation of entropy commonly known with DFT calculations, the translational entropy correction in solution as proposed by Whitesides et al.25 was incorporated. For discussion purposes, the solvation Gibbs free energy was used. These energy values were obtained by the addition of the solvation single-point energy and the thermal correction to the Gibbs free energy. The Gibbs free energies were evaluated at 298.15 K and 1 atm. The 3D structures of some key transition states are shown using the CYLview software.26 To reduce the computational cost, the isopropyl groups of the iPr ligand in the Au catalyst were replaced with methyl groups and the toluenesulfonyl group (Ts) were replaced with the methanesulfonyl group (Ms).
Results and discussion
Mechanistic studies on the Au(I)-catalyzed annulation of 1a with 2 to produce 3
The annulation of 1a with 2 commences with the coordination of the Au(I)-catalyst unto the alkyne moiety of 1a, affording an iminovinyl intermediate (INT1a) in an exergonic fashion.5f,6–8 Then, the various nucleophilic attacks of 2 on the two alkynyl carbons of INT1a to form vinyl gold intermediates that would eventually generate the key α-imino gold carbenoid were explored computationally. The transition state (TS) for the nucleophilic attack of the N-atom of 2 (N1) at the C1 of INT1a is optimized as TS1a, featuring the shortening of the N⋯C1 bond distance to 2.02 Å. The predicted energy barrier is only 6.2 kcal mol−1 relative to separated INT1a and 2 and the generated vinyl gold intermediate INT2a is thermodynamically stable by 11.1 kcal mol−1 (Path a). On the other hand, the O-atom of 2 could also compete with N1 in attacking the C1 of INT1a (Path b). For this possibility, the TS which is located as TS2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 is characterized by the O⋯C1 bond distance of 1.83 Å. This nucleophilic attack requires a barrier of 21.6 kcal mol−1 which is substantially higher than TS1a by 15.4 kcal mol−1, suggesting that Path “a” is more favourable than Path “b”. The greater nucleophilicity of the N-atom of 2 than the O-atom could be responsible for the preferred chemoselective N-attack at the C1 of INT1a.2f Conversely, N1 could chemoselectively attack the C2 of INT1a to generate the corresponding vinyl gold intermediate (Path c) with a (N1⋯C2) bond distance of 2.11 Å. The calculated energy barrier via TS3a is 17.5 kcal mol−1, which is also significantly higher than TS1a by 11.3 kcal mol−1. The great disparity between the energies of TS1a and TS3a could be attributed to the electron-donating ability of the N-atom of ynamides which might strongly polarize the triple bond, thus rendering C1 more electron deficient than C2. This greater electrophilicity of C1 than C2 as evident in their NPA charges (0.276e vs. −0.239e respectively) is beneficial for the N-nucleophilic attack of 2. Moreover, the O-attack at C2 of INT1a also represents another possibility to produce the vinyl gold complex, however, the high demanding energy barrier for this attack mode via TS4a (33.2 kcal mol−1 relative to separated INT1a and 2) ruled out its possibility in the mechanism of the reaction (Path d). Of all the nucleophilic attack modes examined, computational results showed that the N-atom of 2 would chemo-, and regio-selectively attack C1 of INT1a to produce INT2a, which is consistent with the experimental report (Fig. 1).8
27 is characterized by the O⋯C1 bond distance of 1.83 Å. This nucleophilic attack requires a barrier of 21.6 kcal mol−1 which is substantially higher than TS1a by 15.4 kcal mol−1, suggesting that Path “a” is more favourable than Path “b”. The greater nucleophilicity of the N-atom of 2 than the O-atom could be responsible for the preferred chemoselective N-attack at the C1 of INT1a.2f Conversely, N1 could chemoselectively attack the C2 of INT1a to generate the corresponding vinyl gold intermediate (Path c) with a (N1⋯C2) bond distance of 2.11 Å. The calculated energy barrier via TS3a is 17.5 kcal mol−1, which is also significantly higher than TS1a by 11.3 kcal mol−1. The great disparity between the energies of TS1a and TS3a could be attributed to the electron-donating ability of the N-atom of ynamides which might strongly polarize the triple bond, thus rendering C1 more electron deficient than C2. This greater electrophilicity of C1 than C2 as evident in their NPA charges (0.276e vs. −0.239e respectively) is beneficial for the N-nucleophilic attack of 2. Moreover, the O-attack at C2 of INT1a also represents another possibility to produce the vinyl gold complex, however, the high demanding energy barrier for this attack mode via TS4a (33.2 kcal mol−1 relative to separated INT1a and 2) ruled out its possibility in the mechanism of the reaction (Path d). Of all the nucleophilic attack modes examined, computational results showed that the N-atom of 2 would chemo-, and regio-selectively attack C1 of INT1a to produce INT2a, which is consistent with the experimental report (Fig. 1).8
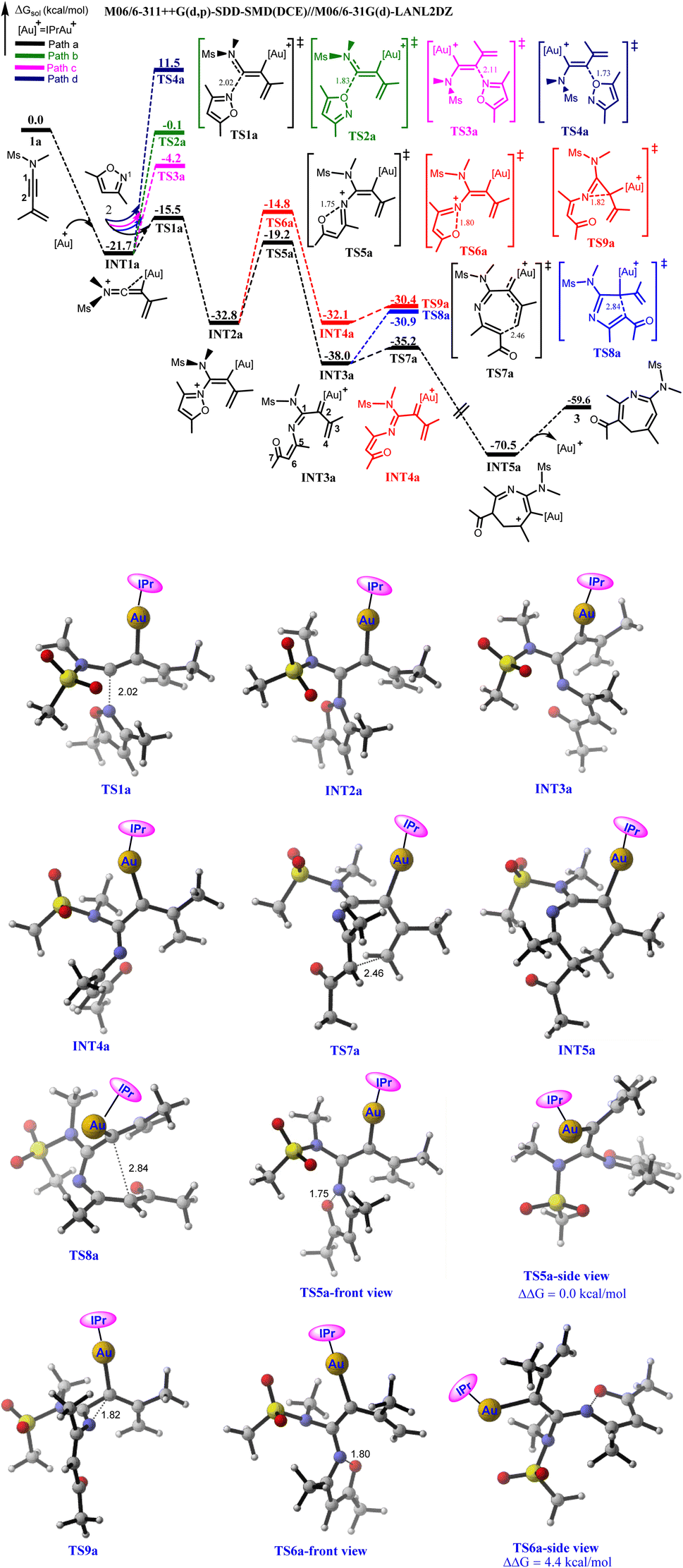 | ||
| Fig. 1 Energy profile showing the gold(I)-catalyzed annulation of 1a with 2 to afford 3. Bond distances are given in Å. | ||
Having addressed the formation of INT2a, the isomerization of INT2a to generate the key α-imino gold(I) carbenoid via the cleavage of the weak isoxalic N–O bond was examined next. Meanwhile, two possible structural conformers could result from the N–O bond cleavage to afford the α-imino gold(I) carbenoid. The first possible structural conformer features an increased N⋯O bond distance from 1.38 Å in INT2a to 1.75 Å in the optimized TS5a. With an energy barrier of 13.6 kcal mol−1 relative to INT2a, TS5a is expected to generate the α-imino gold carbenoid INT3a. The second conformer which is characterized by an N⋯O bond distance of 1.80 Å, has its TS located as TS6a. The predicted energy barrier to yield the corresponding α-imino gold(I) carbenoid INT4a is 18.0 kcal mol−1 relative to INT2a, implying that TS5a is 4.4 kcal mol−1 more stable than TS6a. The greater stability of TS5a than TS6a could be attributed to the geometrical conformation of these TSs. Examination of TS5a revealed that the lone pair of electrons on N1 is oriented towards the tosylamide nitrogen (N2) such that the π–π stacking interaction which could bring about stability to the transition state existed between the propenyl unit and the isoxazoic fragment (Fig. 1). However, for TS6a, the lone pair of electrons on N1 is inclined towards the propenyl unit and the aforementioned π–π stacking interaction is absent. The geometry orientation of TS5a is also advantageous for the generation of the corresponding α-imino gold(I) carbenoid as INT3a is thermodynamically more stable than INT4a by 5.9 kcal mol−1 (Fig. 1).
It is worth noting that the electrophilic and nucleophilic sites on the formed α-imino gold(I)carbenoid could allow various intramolecular cyclization modes to afford 3-, 5-, 6- and 7-membered heterocyclic products (Scheme 3).6–8
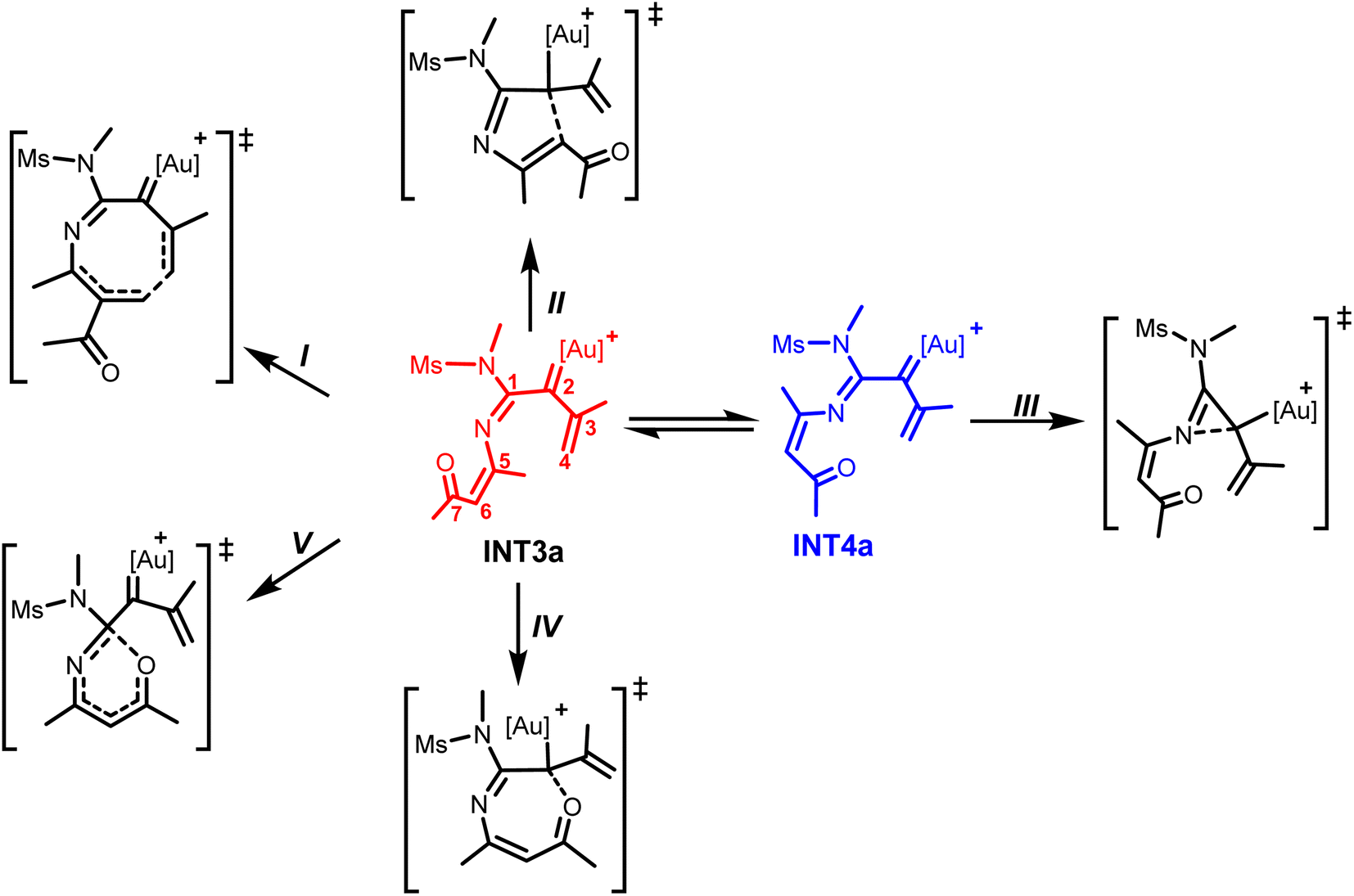 | ||
| Scheme 3 Possible reaction pathways from the α-imino gold(I)carbenoids INT3a and INT4a. | ||
The structural conformation of INT3a suggests that the intramolecular 6π-electrocyclization via the Au(I)-stabilized azaheptatrienyl would readily take place to afford a seven-membered heterocycle (Scheme 3, path I). This electrocyclization via the attack of the terminal alkene carbon atom (C4) at C6 is localized as TS7a with the C4⋯C6 bond distance shortened to 2.46 Å from 3.11 Å in INT3a. Interestingly, the calculated energy barrier which is only 2.8 kcal mol−1 relative to INT3a indicates that the formation of the seven-membered intermediate, INT5a is a very facile process.
Next, the intramolecular cyclizations via N-, C-, and O-attack at the carbenoid moiety (C2) to generate various heterocycles were analyzed. For instance, the Nazarov-type cyclization which is commonly proposed to access five-membered heterocycles6a–d could occur via the intramolecular attack of C6 at the C2 of INT3a (Scheme 3, path II). Relative to INT3a, the optimized TS8a for this possibility, with a C2⋯C6 bond distance of 2.84 Å would require a barrier of 7.1 kcal mol; an energy barrier higher than TS7a by 4.3 kcal mol−1. This computational result implies that the formation of the seven-membered intermediate via the C4⋯C6 attack is more favorable than the formation of the five-membered intermediate.
Moreover, the structural orientation of INT4a suggests that the intramolecular cyclization leading to the formation of a three-membered intermediate would readily take place via N-attack at C2 (Scheme 3, path III). The TS for the intramolecular cyclization is located as TS9a with its N1⋯C2 bond distance stabilized to 1.82 Å. Although the calculated energy barrier is only 1.7 kcal mol−1 relative to INT4a, the previous N–O bond cleavage resulting in INT4a is less favorable compared to that leading to INT3a.28 Thus, the formation of seven-membered intermediate via the C6-induced cyclization at the terminal propenyl carbon atom is more favorable than the formation of the three-membered intermediate (Fig. 1). Another possible pathway explored from INT3a which involves an O-atom induced intramolecular cyclization at the C2 of INT3a (Scheme 3, path IV) via TS10a gave a higher energy barrier than TS7a by 5.7 kcal mol; suggesting that this pathway is also unlikely (the O⋯C2 attack pathway is less favourable than the C4⋯C6 attack pathway in the formation of a seven-membered intermediate, Fig. S1†). Similarly, the possibility of constructing a six-membered heterocycle via a 6π-electrocyclization facilitated by O-attack at C1 of INT3a along TS11a (Scheme 3, path V) would also require an energy barrier higher than TS7a by 13.3 kcal mol−1 (Fig. S1†). Overall, these computational results indicate an overwhelming preference for the exclusive formation of a seven-membered intermediate (INT5a) over other possibilities explored. Therefore the reaction of 1a with 2 would proceed through an initial N-attack of the isoxazole at the C1 of the iminovinyl functionality of INT1a, after the π-alkyne activation by the Au(I) catalyst had taken place, to subsequently produce the key α-imino Au(I)-carbenoid INT3a via the N–O bond cleavage. C6-induced intramolecular cyclization of the Au(I)-stabilized azaheptatrienyl cation at the carbenoid carbon would follow to readily produce the seven-membered intermediate INT5a (Fig. 1).
Subsequently, the bis(trifluoromethane)sulfonimide ion (NTf2 counter-ion) would act as a proton shuttle in assisting the proton transfer from C6 of INT5a to C2. The TS for the deprotonation of C6 is optimized as TS12a, requiring an energy barrier of 25.6 kcal mol−1 relative to INT5a. Then, the protonation of C2 in the resulting INT6a would readily occur via TS13a to produce the product complex INT7a with a barrier of 4.8 kcal mol−1 relative to INT6a. Finally, the dissociation of the Au(I)-catalyst and the counter-ion would release the expected 4H-azepine product 3. (Fig. S2†).
Mechanistic studies on the Au(I)-catalyzed annulation of 1b with 2 to produce 4 and 5
It was experimentally reported that the annulation of 1b with 2 would generate pyrrole 4 as the major product with traces of azepine 5.8 After the formed iminovinyl intermediate (INT1b), the aforementioned nucleophilic attacks of 2 on INT1b were investigated. The located TS1b for the N-attack of 2 at C1 of INT1b having its N⋯C1 bond distance shortened to 2.01 Å, demands a barrier of 5.2 kcal mol−1 relative to separated INT1b and 2. The resulting vinyl gold(I) intermediate INT2b is produced in an exergonic manner. For the competitive O-attack at C1, the computed energy barrier via TS2b27 is 24.6 kcal mol−1 relative to INT1b and 2, which is substantially higher than TS1b by 19.4 kcal mol−1. The N-and O-attack at C2 of INT1b via TS3b and TS4b were disregarded due to their higher energy barriers than TS1b by 11.7 kcal mol−1 and 26.4 kcal mol−1 respectively (Fig. 2). Thus, the reaction mechanism for the annulation of 1b with 2 would chemo-, and regio-selectively proceed via the N-attack of 2 at C1 of INT1b to produce INT2b, which is in excellent agreement with that for the annulation of 1a and 2.
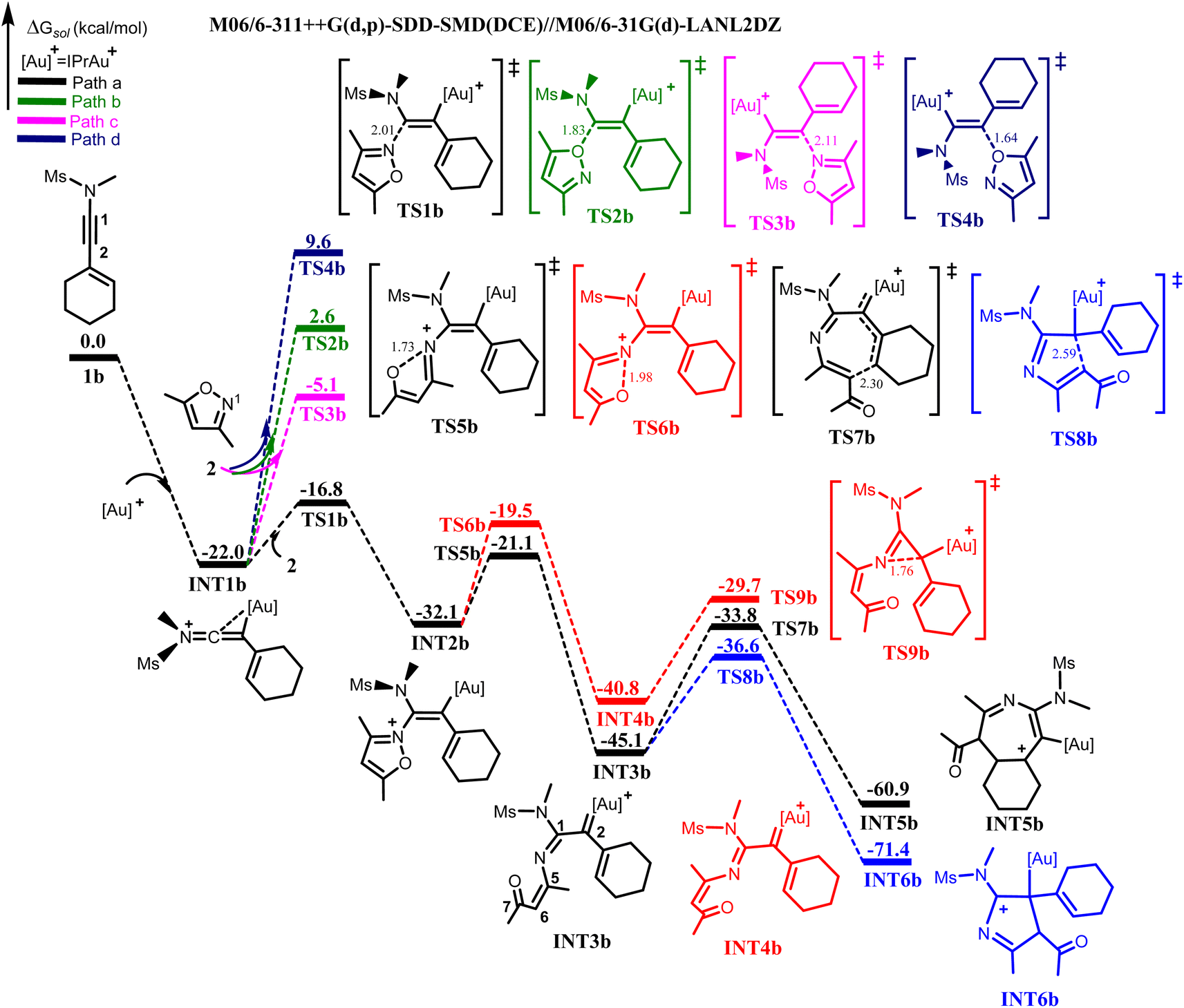 | ||
| Fig. 2 Energy profile showing the gold(I)-catalyzed annulation of 1b with 2 to generate INT5b and INT6b respectively. | ||
The formed vinyl gold intermediate INT2b will subsequently undergo N⋯O bond cleavage to generate the key α-imino gold(I) carbene intermediate. For the bond cleavage via TS5b to produce α-imino gold(I) carbene intermediate INT3b, the N⋯O bond distance of the TS is increased to 1.73 Å and with an energy barrier of 11.0 kcal mol−1 relative to INT2b. On the other hand, the N–O bond cleavage of INT2b via TS6b is characterized by an increased N⋯O bond distance of 1.98 Å. The required energy barrier for this bond cleavage to afford the α-imino gold(I) carbenoid INT4b is 12.6 kcal mol−1, which is slightly higher than TS5b by 1.6 kcal mol−1. However, the INT4b formed is less stable than INT3b from both the kinetic and thermodynamic standpoint (Fig. 2). This result suggests that the geometrical orientation and the π–π stacking interaction between the cyclohexenyl group and the isoxazoic fragment present in TS5b are more beneficial for the generation of the α-imino gold carbenoid than that in TS6b, a finding consistent with that for the annulation of 1a and 2.
Next, the previously described cyclization pathways were investigated from INT3b. For the 6π-electrocyclization via the Au(I)-stabilized azaheptatrienyl cation to afford the seven-membered heterocyclic intermediate INT5b, the optimized TS7b for the electrocyclization has its C4⋯C6 bond distance shortened to 2.30 Å, with an energy barrier of 11.3 kcal mol−1 relative to INT3b. On the other hand, the TS for the formation of the five-membered intermediate, INT6b is localized as TS8b. This intramolecular cyclization has its C2⋯C6 bond distance stabilized to 2.59 Å. Surprisingly, the computed energy barrier for TS8b is 8.5 kcal mol−1, which is 2.8 kcal mol−1 lower than that of TS7b. In addition, the generated INT6b is also thermodynamically more stable than INT5b by 10.5 kcal mol−1, indicating that the formation of the five-membered intermediate is more favourable than that of the seven-membered from INT3b (Fig. 2).
Moreover, the cyclization leading to the formation of the three-membered intermediate (TS9b) via the N-attack at C2 of INT4b would require a higher energy barrier than TS8b by 6.9 kcal mol−1. Similar to 1a, other possible O-atom-induced intramolecular cyclization modes including the 7-, and 6-membered intramolecular cyclization were investigated from INT3b via TS10b and TS11b respectively. The results showed that the energy barriers of both transition states were higher than that of TS8b by 3.4 kcal mol−1 and 15.4 kcal mol−1 respectively (Fig. S3†).
With these computational results, it is therefore evident that a dramatic switch in the selectivity for the annulation of 1b with 2 had occurred, giving preference to the formation of the five-membered intermediate over that of the seven-membered. This is against what was operational for the annulation between 1a and 2. It should be noted that the ΔΔG‡ between TS7b and TS8b in the annulation of 1b with 2 (2.8 kcal mol−1) is smaller in comparison with that between TS7a and TS8a (4.3 kcal mol−1). This suggests that the formation of the seven-membered INT5b cannot be completely ruled out in the annulation of 1b with 2. Thus, from INT6b, proton transfer is expected to take place to deliver the desired five-membered pyrrole product 4 as the major product, and from INT5b, the release of the seven-membered azepine product 5 as the side product is also expected. The computational results further validate the experimental results8 from both kinetic and thermodynamic standpoints.
Origin of the chemo- and regio-selectivities in the annulation of the ynamides (1a/1b) with 2
It was previously stated above that clarification is needed as to why the heptatrienyl cation A would proceed through the high-energy intermediate C to generate a seven-membered heterocycle D by the incorporation of Au(I)-catalyst and nitrogen atom instead of the known Nazarov-type cyclization via B to produce a five-membered heterocycle E (Scheme 1a & 2). To address the factor(s) controlling the selective formation of the seven-membered and the five-membered product, and the role played by the incorporated Au(I) catalyst and nitrogen atom in the annulation of 1a/1b with 2, predictive analysis on some optimized α-imino gold(I) carbenes were carried out by computational means.It is worth noting that Gir and Liu8 only illustrated that the incorporation of nitrogen atom assists in the selective formation of the seven-membered azepines, however, it was not specified where the incorporated nitrogen atom originated from; whether from the isoxazole substrate 2 or the tosylamide group.
To address this, the intramolecular cyclization of the vinyl gold(I)carbenes with and without the nitrogen atom, to generate a five-membered and a seven-membered intermediate were investigated respectively to examine the role of the nitrogen atom in controlling the selectivity. The predictive analysis of the cyclization of the vinyl gold(I) carbene A, an intermediate having no nitrogen atom, suggests that the Nazarov-type cyclization leading to the formation of a five-membered heterocycle via 6a-TS would be more favoured than the cyclization of the Au(I)-stabilized heptatrienyl motif resulting in the formation of the seven-membered heterocycle (7a-TS), by 13.5 kcal mol−1 (Schemes 4a and S1a†). When the nitrogen atom was incorporated at the position adjacent to C1 of carbene A, the predictive analysis of the resulting α-imino gold(I) carbene (subsequently referred to as carbene) B revealed that the incorporated nitrogen atom plays no significant role in altering the selectivity; as the formation of the five-membered heterocycle via C6 attack at C2 (6b-TS) is still more favourable than the cyclization of the Au(I)-stabilized azaheptatrienyl cation (7b-TS) to form the seven-membered heterocycle, by 4.2 kcal mol−1 (Schemes 4a and S1a†). This finding, therefore, showed that the incorporated nitrogen atom which might stem from the isoxazole substrate is not responsible for the formation of the seven-membered azepines.
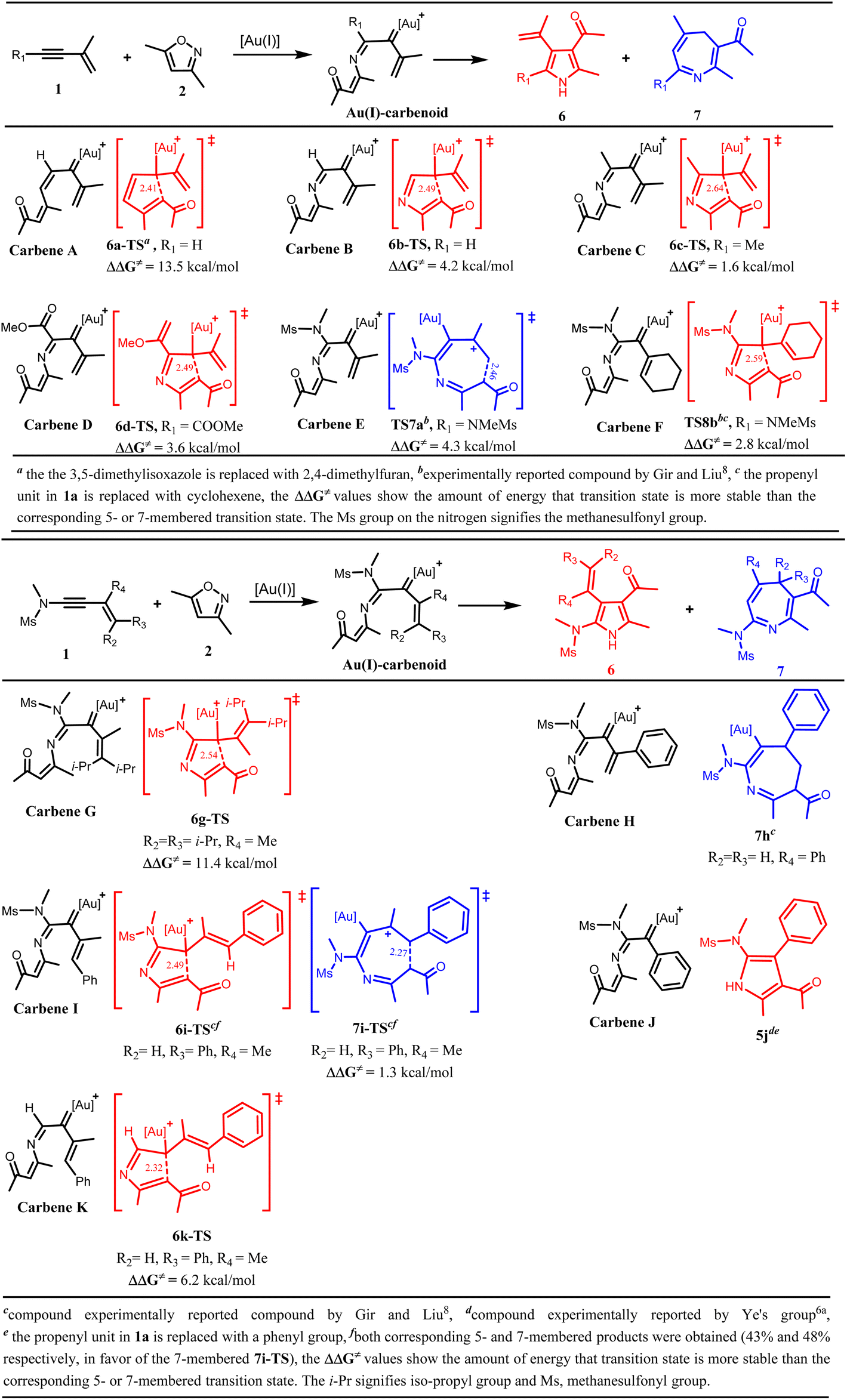 | ||
| Scheme 4 (a)Predictive analysis of some α-imino gold(I)-carbene intermediates. Bond distances are given in Å. (b) Predictive analysis of some α-imino gold(I)-carbene intermediates. Bond distances are given in Å. | ||
Next, the electronic effect on the substituent at C1 of carbene B was examined. Carbene C and D corresponding to the methyl group and ester group as substituents at C1 of carbene B were optimized respectively. The results also showed that neither the electron-donating methyl group substituent in carbene C nor the electron-withdrawing ester group substituent in carbene D alters the selectivity from the five-membered heterocycle (6c-TS and 6d-TS respectively) to the seven-membered heterocycle (7c-TS and 7d-TS respectively); suggesting that electronic factor at this position have no profound effect on the selectivity. Surprisingly, with the tosylamide group as a substituent at C1 of carbene B to generate carbene E (INT3a), the result of the cyclization showed a switch in the product selectivity; favouring the formation of the seven-membered heterocycle via TS7a (the cyclized azaheptatrienyl cation) more than that of the five-membered (TS8a) by 4.3 kcal mol−1 (Schemes 4a and S1a†). This result clearly indicates that the tosylamide group has a profound effect on the selectivity. The reason for the observed dramatic switch in selectivity in the cyclization of carbene E and not in carbene C or D could be due to the interaction of the lone pair of electrons on the nitrogen atom with the π-conjugated system in carbene E which further stabilizes the cyclizing transition state towards 6π-electrocyclization. However such interaction was absent in carbene C or D. From these findings, it is therefore believed that the incorporated nitrogen atom reported by Gir and Liu8 which aids the selective formation of the seven-membered azepine stemmed from the nitrogen atom of the tosylamide group.
As previously stated, the experimental report for the annulation of 1b and 2 via carbene F (INT3b) majorly afforded a five-membered product.8 The observed experimental result was validated by DFT calculations in this study (the preferred formation of the five-membered intermediate via TS8b over that of the seven-membered intermediate via TS7b by 2.8 kcal mol−1). It should be noted that 1b differs from 1a in that the pendent propenyl group in 1a is substituted with cyclohexenyl group in 1b. Comparing the computational result of the cyclization of carbene E with that of F, we hypothesized that the bulkiness of the cyclohexenyl group being at the cyclization termini in carbene F as against the flexible pendent propenyl group in carbene E might allow steric hindrance during the cyclization process, hence altering the selectivity from the 6π-electrocyclization which could have afforded the seven-membered heterocycle, to the formation of the five-membered heterocycle.
To verify this hypothesis, carbene G, having the terminal alkenyl hydrogen atoms in carbene E substituted with isopropyl groups was optimized. The result of the cyclization step demonstrated that the bulkiness of the substituent indeed hindered the 6π-electrocyclization; as the formation of the five-membered heterocycle via 6g-TS is more favourable than that of the seven-membered (7g-TS) by 11.4 kcal mol−1, thus validating our hypothesis (Schemes 4b and S1b†). In support of this finding, Faza et al.15a also reported that for heptatrienyl cation having a bulky group at the cyclization termini, the 4π-electrocyclization leading to the five-membered product is more favoured than the 6π-electrocyclization leading to the seven-membered product. It can therefore be summed that the presence of the bulky cyclohexenyl group at the terminus of 1b as against the more flexible tethered propenyl unit in 1a causes substantial steric hindrance during the cyclization process thereby obstructing the cyclization of the azaheptatrienyl cation that could have produced the seven-membered product 5 as the major product.
Besides the steric effect being a factor in controlling the selectivity of the cyclization process of carbenes, one might wonder if the π-conjugation pattern in the carbenes can also influence such selectivity. With the predictive results of the cyclization of carbenes E–G, it can be presumed that the uninterrupted π-conjugation pattern in carbene E which is not in carbene F and G might allow the delocalization of π-electrons which stabilizes the cyclizing transition state, and such is beneficial for the 6π-electrocyclization leading to the formation of the seven-membered product. However, for carbene F and G, such extended π-conjugation pattern was interrupted, hence, the preference for the aza-Nazarov-type cyclization to afford the five-membered product. Interestingly, the experimental report documented by Gir and Liu8 further validates our presumption. The experimental report8 showed that when the methyl group of the propenyl unit in 1a was replaced by a phenyl group to allow an uninterrupted π-conjugated system, the cyclization of the resulting carbene H prefers the formation of a seven-membered product (Scheme 4b).8
Again, the role of the conjugation pattern in controlling the selectivity was further validated in the cyclization of carbene I; an intermediate in which one of the terminal hydrogen atoms of the propenyl unit in carbene E is substituted with a phenyl group. The experimental results8 showed that carbene I marginally favoured the formation of the seven-membered product with almost equal proportion with that of the five-membered product. In agreement, DFT calculations also revealed that the cyclization of carbene I gave a slight preference for the formation of the seven-membered heterocycle via 7i-TS over that of the five-membered (6i-TS) by about 1.3 kcal mol−1, which is consistent with the experimental findings8 (Scheme 4b). The reason for the marginal difference in the formation of the two heterocycles was rationalized based on the competition between the aforementioned stabilizing π-orbital interaction brought about by the uninterrupted π-conjugation and the destabilizing steric hindrance caused by the presence of the bulky phenyl group at the cyclization terminus.
Meanwhile, the combined experimental and DFT studies documented by Wu and Ye's group6a on the cyclization of carbene J resulting from the Au(I) annulation of 2 with 1j (the cyclohexenyl group in 1b being replaced with a phenyl group to allow an uninterrupted π-conjugated system) demonstrated that carbene J prefers the formation of a five-membered product 5j exclusively (Scheme 4b).6a This finding coupled with that on the cyclization of carbenes E–I clearly reveals that while the π-conjugation pattern of the carbenes influences selectivity during cyclization, the absence of steric hindrance at the cyclization terminus of the cyclizing carbenes is crucial for the exclusive formation of seven-membered products. Otherwise, the Au(I) annulation of 1 with 2 will result in the exclusive formation of five-membered products. These findings are in consonant with that reported by Faza et al.15a in the computational study on the regio-, peri-, and torquoselectivity in hydroxy heptatrienyl cation electrocyclizations.
Interestingly, the attempt to replace the tosylamide group in carbene I with a hydrogen atom reveals that the tosylamide group still plays a crucial role in controlling the selectivity as the cyclization of the resulting carbene K reverts to favouring the five-membered heterocycle over the seven-membered heterocycle (Schemes 4b and 1b). Therefore, it can be summarized that the cooperating effect of the tosylamide group on C1, the uninterrupted π-conjugation pattern of the α-imino gold(I) carbenes and the substitution pattern at the cyclization termini to avoid steric hindrance are the key factors responsible for the different chemo-, and regio-selectivities observed in the Au(I)-catalyzed annulation of 1a/1b with 2 to afford either 3/5 or 4. The role of the Au(I)-catalyst is believed to assist in the stabilization of the azaheptatrienyl cation.
Conclusion
In summary, a comparative study has been carried out by DFT calculations to gain insights into the mechanisms and the origin of switchable selectivity in Au(I)-catalyzed annulations of ynamides (1a/1b) with 3,5-dimethylisoxazoles (2) via 6π-electrocyclizations of azaheptatrienyl cations. For both annulations, computational results support the overwhelming preference for the initial N-attack of the isoxazole at the electron-deficient carbon atom of the Au-π-ynamide complex to eventually form the key α-imino gold(I) carbene intermediate after N–O bond cleavage had occurred.For the annulation of 3-en-1-ynamides (1a) with the 2, the unusual 6π-electrocyclization of the Au(I)-stabilized azaheptatrienyl cations to exclusively produce a seven-membered 4H-azepine product 3, after the formation of the key cationic α-imino gold(I) carbene intermediate had taken place, is preferred over the commonly proposed aza-Nazarov-type-cyclization which would afford a five-membered product. However, upon substitution of the α-alkynyl carbon atom of 1a with a cyclohexenyl group, computational results reveal that the annulation of the corresponding ynamide 1b with the isoxazole leads to a dramatic switch in the product selectivity; majorly affording the five-membered pyrrole 4 as the predominant product over the seven-membered azepine product 5. The reason for the selective formation of the seven-membered azepine in the annulation of 1a and 2 was not due to the incorporated Au(I) catalyst nor the nitrogen atom in the azaheptatrienyl cation, however, the selectivity was rationalized based on the alkene substitution pattern at the cyclization termini of the ynamides; as substituent flexibility in the cyclizing step to avoid significant steric hindrance is beneficial for the 6π-electrocyclization to afford the seven-membered product. This is against the bulky substituents at the cyclizing terminus in the annulation of 1b and 2. More so, the uninterrupted π-conjugation pattern in the α-imino gold(I) carbene intermediate for the annulation of 1a with 2 allows the delocalization of π-electrons which stabilizes the transition structure, hence promoting the 6π-electrocyclization to produce the seven-membered azepine over the aza-Nazarov-type cyclization which would afford the five-membered pyrrole.
Finally, the tosylamide group as a substituent on C1 of the Au(I)-stabilized azaheptatrienyl cation was also discovered to play a crucial role in facilitating the 6π-electrocyclization process toward the formation of the seven-membered azepine product.
It is believed that the Au(I)-catalyst helps to stabilize the azaheptatrienyl cation. It is hoped that the insight gleaned from this study will contribute significantly to the success of challenging electrocyclic reactions and facilitate the development of novel annulations that involve ynamides and other nucleophiles for the synthesis of valuable polycyclic compounds.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful to the Chinese Scholarship Council, Soochow University, Suzhou, China, and Tai Solarin University of Education, Ijebu-Ode, Ogun State, Nigeria. Also, a sincere appreciation goes to Prof. Xiaoguang Bao for his supervision when the work was carried out in his laboratory.References
- For reviews, see (a) C. M. Friend and A. S. K. Hashmi, Gold catalysis, Acc. Chem. Res., 2014, 47, 729–730 CrossRef CAS PubMed; (b) D. J. Gorin and F. D. Toste, Relativistic effects in homogeneous gold catalysis, Nature, 2007, 446, 395–403 CrossRef CAS PubMed; (c) M. E. Muratore, A. Homs, C. Obradors and A. M. Echavarren, Meeting the Challenge of Intermolecular Gold (I)-Catalyzed Cycloadditions of Alkynes and Allenes, Chem.–Asian J., 2014, 9, 3066–3082 CrossRef CAS PubMed; (d) D. Pflästerer and A. S. K. Hashmi, Gold catalysis in total synthesis–recent achievements, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC; (e) W. Debrouwer, T. S. Heugebaert, B. I. Roman and C. V. Stevens, Homogeneous Gold-Catalyzed Cyclization Reactions of Alkynes with N-and S-Nucleophiles, Adv. Synth. Catal., 2015, 357, 2975–3006 CrossRef CAS; (f) R. Dorel and A. M. Echavarren, Gold (I)-catalyzed activation of alkynes for the construction of molecular complexity, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (g) A. Fürstner, Gold and platinum catalysis—a convenient tool for generating molecular complexity, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC; (h) F.-L. Hong and L.-W. Ye, Transition metal-catalyzed tandem reactions of ynamides for divergent N-heterocycle synthesis, Acc. Chem. Res., 2020, 53, 2003–2019 CrossRef CAS PubMed; (i) F. Hu and M. Szostak, Recent developments in the synthesis and reactivity of isoxazoles: metal catalysis and beyond, Adv. Synth. Catal., 2015, 357, 2583–2614 CrossRef CAS; (j) D. B. Huple, S. Ghorpade and R. S. Liu, Recent advances in gold-catalyzed N-and O-functionalizations of alkynes with nitrones, nitroso, nitro and nitroxy species, Adv. Synth. Catal., 2016, 358, 1348–1367 CrossRef CAS; (k) E. Jimenez-Nunez and A. M. Echavarren, Gold-catalyzed cycloisomerizations of enynes: a mechanistic perspective, Chem. Rev., 2008, 108, 3326–3350 CrossRef CAS PubMed; (l) W. Zi and F. D. Toste, Recent advances in enantioselective gold catalysis, Chem. Soc. Rev., 2016, 45, 4567–4589 RSC.
- For reactions, see (a) H. Jin, B. Tian, X. Song, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold-Catalyzed Synthesis of Quinolines from Propargyl Silyl Ethers and Anthranils through the Umpolung of a Gold Carbene Carbon, Angew. Chem., Int. Ed., 2016, 55, 12688–12692 CrossRef CAS PubMed; (b) S. N. Karad, W.-K. Chung and R.-S. Liu, Gold-catalyzed formal [4π+2π]-cycloadditions of propiolate derivatives with unactivated nitriles, Chem. Sci., 2015, 6, 5964–5968 RSC; (c) S. N. Karad, W.-K. Chung and R.-S. Liu, Gold-catalyzed formal [4π+2π]-cycloadditions of tert-butyl propiolates with aldehydes and ketones to form 4H-1,3-dioxine derivatives, Chem. Commun., 2015, 51, 13004–13007 RSC; (d) R. L. Sahani and R.-S. Liu, Gold-catalyzed [4+3] and [4+4]-annulation reactions of t-butyl propiolate derivatives with epoxides and oxetanes for the construction of 1,4-dioxepane and 1,5-dioxocane cores, Chem. Commun., 2016, 52, 7482–7485 RSC; (e) R. R. Singh, M. Skaria, L.-Y. Chen, M.-J. Cheng and R.-S. Liu, Gold-catalyzed (4+3)-annulations of 2-alkenyl-1-alkynylbenzenes with anthranils with alkyne-dependent chemoselectivity: skeletal rearrangement versus non-rearrangement, Chem. Sci., 2019, 10, 1201–1206 RSC; (f) A. A. Ogunlana and X. Bao, Mechanistic insights into the gold (I)-catalyzed annulation of propiolates with isoxazoles: a DFT study, Chem. Commun., 2019, 55, 11127–11130 RSC; (g) E. M. Arce, S. G. Lamont and P. W. Davies, Sulfenyl ynamides in gold catalysis: synthesis of oxo-functionalised 4-aminoimidazolyl fused compounds by intermolecular annulation reactions, Adv. Synth. Catal., 2020, 362, 2503–2509 CrossRef CAS; (h) H.-C. Hsieh, K.-C. Tan, A. S. K. Raj and R.-S. Liu, Gold-catalyzed [4+1]-annulation reactions between anthranils and 4-methoxy-1,2-dienyl-5-ynes involving a 1,2-allene shift, Chem. Commun., 2019, 55, 1979–1982 RSC; (i) X. Tian, L. Song, C. Han, C. Zhang, Y. Wu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold (III)-catalyzed formal [3+2] annulations of N-acyl sulfilimines with ynamides for the synthesis of 4-aminooxazoles, Org. Lett., 2019, 21, 2937–2940 CrossRef CAS PubMed; (j) L. Ye, L. Cui, G. Zhang and L. Zhang, Alkynes as equivalents of α-diazo ketones in generating α-oxo metal carbenes: a gold-catalyzed expedient synthesis of dihydrofuran-3-ones, J. Am. Chem. Soc., 2010, 132, 3258–3259 CrossRef CAS PubMed.
- (a) S. N. Karad and R. S. Liu, Regiocontrolled Gold-Catalyzed [2+2+2] Cycloadditions of Ynamides with Two Discrete Nitriles to Construct 4-Aminopyrimidine Cores, Angew. Chem., Int. Ed., 2014, 53, 9072–9076 CrossRef CAS PubMed; (b) S. K. Pawar, R. L. Sahani and R. S. Liu, Diversity in Gold-Catalyzed Formal Cycloadditions of Ynamides with Azidoalkenes or 2H-Azirines:[3+2] versus [4+3] Cycloadditions, Chem.–Eur. J., 2015, 21, 10843–10850 CrossRef CAS PubMed; (c) S. K. Pawar, D. Vasu and R. S. Liu, Gold-and Silver-Catalyzed [4+2] Cycloadditions of Ynamides with Oxetanes and Azetidines, Adv. Synth. Catal., 2014, 356, 2411–2416 CrossRef CAS; (d) L. Zhu, Y. Yu, Z. Mao and X. Huang, Gold-catalyzed intermolecular nitrene transfer from 2H-azirines to ynamides: a direct approach to polysubstituted pyrroles, Org. Lett., 2015, 17, 30–33 CrossRef CAS PubMed; (e) R. B. Dateer, K. Pati and R.-S. Liu, Gold-catalyzed synthesis of substituted 2-aminofurans via formal [4+1]-cycloadditions on 3-en-1-ynamides, Chem. Commun., 2012, 48, 7200–7202 RSC; (f) P. W. Davies, A. Cremonesi and L. Dumitrescu, Intermolecular and Selective Synthesis of 2,4,5-Trisubstituted Oxazoles by a Gold-Catalyzed Formal [3+2] Cycloaddition, Angew. Chem., 2011, 38, 9093–9097 CrossRef.
- (a) M. Chen, N. Sun, H. Chen and Y. Liu, Dioxazoles, a new mild nitrene transfer reagent in gold catalysis: highly efficient synthesis of functionalized oxazoles, Chem. Commun., 2016, 52, 6324–6327 RSC; (b) S. S. Giri and R. S. Liu, Gold-Catalyzed [4+2]- and [3+3]-Annulations of Ynamides with 1-Yn-3-ols to Access Six-Membered Carbocycles and Oxacycles via Three Distinct Cyclizations, Adv. Synth. Catal., 2017, 359, 3311–3318 CrossRef CAS; (c) W. Xu, G. Wang, N. Sun and Y. Liu, Gold-catalyzed formal [3+2] cycloaddition of ynamides with 4,5-dihydro-1,2,4-oxadiazoles: Synthesis of functionalized 4-aminoimidazoles, Org. Lett., 2017, 19, 3307–3310 CrossRef CAS PubMed; (d) Z. Zeng, H. Jin, J. Xie, B. Tian, M. Rudolph, F. Rominger and A. S. K. Hashmi, α-Imino gold carbenes from 1,2,4-oxadiazoles: Atom-economical access to fully substituted 4-aminoimidazoles, Org. Lett., 2017, 19, 1020–1023 CrossRef CAS PubMed; (e) D. P. Zimin, D. V. Dar’in, V. Y. Kukushkin and A. Y. Dubovtsev, Oxygen atom transfer as key to reverse regioselectivity in the gold (I)-catalyzed generation of aminooxazoles from ynamides, J. Org. Chem., 2020, 86, 1748–1757 CrossRef PubMed.
- (a) R. R. Singh, S. K. Pawar, M.-J. Huang and R.-S. Liu, Gold-catalyzed [3+2]-annulations of α-aryl diazonitriles with ynamides and allenamides to yield 1-amino-1 H-indenes, Chem. Commun., 2016, 52, 11434–11437 RSC; (b) L. Song, X. Tian, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold (III)-catalyzed chemoselective annulations of anthranils with N-allylynamides for the synthesis of 3-azabicyclo [3.1.0] hexan-2-imines, Chem. Commun., 2019, 55, 9007–9010 RSC; (c) X. Zhao, X. Song, H. Jin, Z. Zeng, Q. Wang, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold-Catalyzed Intermolecular [4+2] Annulation of 2-Ethynylanilines with Ynamides: An Access to Substituted 2-Aminoquinolines, Adv. Synth. Catal., 2018, 360, 2720–2726 CrossRef CAS; (d) C. Zhu, L. Kou and X. Bao, Au (I)-Catalyzed Annulation of Benzofurazan N-oxides with Ynamides: From Predicting the Chemo-Selectivity to the Synthesis of 7-Nitroindole Derivatives, Chin. J. Chem., 2020, 38, 57–62 CrossRef CAS; (e) Y. Yu, G. Chen, L. Zhu, Y. Liao, Y. Wu and X. Huang, Gold-Catalyzed β-Regioselective Formal [3+2] Cycloaddition of Ynamides with Pyrido[1,2-b]indazoles: Reaction Development and Mechanistic Insights, J. Org. Chem., 2016, 81, 8142–8154 CrossRef CAS PubMed; (f) R. Vanjari, S. Dutta, B. Prabagar, V. Gandon and A. K. Sahoo, Ring Expansion and 1, 2-Migration Cascade of Benzisoxazoles with Ynamides: Experimental and Theoretical Studies, Chem.–Asian J., 2019, 14, 4828–4836 CrossRef CAS PubMed.
- (a) A.-H. Zhou, Q. He, C. Shu, Y.-F. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L.-W. Ye, Atom-economic generation of gold carbenes: gold-catalyzed formal [3+2] cycloaddition between ynamides and isoxazoles, Chem. Sci., 2015, 6, 1265–1271 RSC; (b) X. Y. Xiao, A. H. Zhou, C. Shu, F. Pan, T. Li and L. W. Ye, Atom-Economic Synthesis of Fully Substituted 2-Aminopyrroles via Gold-Catalyzed Formal [3+2] Cycloaddition between Ynamides and Isoxazoles, Chem.–Asian J., 2015, 10, 1854–1858 CrossRef CAS PubMed; (c) H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold-Catalyzed C-H Annulation of Anthranils with Alkynes: A Facile, Flexible, and Atom-Economical Synthesis of Unprotected 7-Acylindoles, Angew. Chem., Int. Ed., 2016, 55, 794–797 CrossRef CAS PubMed; (d) L. Li, T.-D. Tan, Y.-Q. Zhang, X. Liu and L.-W. Ye, Recent advances in transition-metal-catalyzed reactions of alkynes with isoxazoles, Org. Biomol. Chem., 2017, 15, 8483–8492 RSC; (e) M.-H. Tsai, C.-Y. Wang, A. S. K. Raj and R.-S. Liu, Gold-catalyzed annulations of N-aryl ynamides with benzisoxazoles to construct 6H-indolo[2,3-b] quinoline cores, Chem. Commun., 2018, 54, 10866–10869 RSC; (f) Y. C. Hsu, S. A. Hsieh and R. S. Liu, Gold-Catalyzed Annulations of N-Propargyl Ynamides with Anthranils with Two Distinct Chemoselectivities, Chem.–Eur. J., 2019, 25, 5288–5297 CrossRef CAS PubMed; (g) Y.-C. Hu, Y. Zhao, B. Wan and Q.-A. Chen, Reactivity of ynamides in catalytic intermolecular annulations, Chem. Soc. Rev., 2021, 50, 2582–2625 RSC.
- (a) L. Li, W.-F. Luo and L.-W. Ye, Recent progress in the gold-catalyzed annulations of ynamides with isoxazole derivatives via α-Imino gold carbenes, Synlett, 2021, 32, 1303–1308 CrossRef CAS; (b) L. Zhou, L. Yang, S. Dai, Y. Gao, R. Fang, A. M. Kirillov and L. Yang, Insight into the reaction mechanism and chemoselectivity in the cycloaddition of ynamides and isoxazoles with H2O, Catal. Sci. Technol., 2020, 10, 240–251 RSC; (c) P. D. Jadhav, X. Lu and R.-S. Liu, Gold-catalyzed [5+2]-and [5+1]-annulations between ynamides and 1,2-benzisoxazoles with ligand-controlled chemoselectivity, ACS Catal., 2018, 8, 9697–9701 CrossRef CAS.
- S. S. Giri and R.-S. Liu, Gold-catalyzed [4+3]-and [4+2]-annulations of 3-en-1-ynamides with isoxazoles via novel 6π-electrocyclizations of 3-azahepta trienyl cations, Chem. Sci., 2018, 9, 2991–2995 RSC.
- (a) P. von Ragué Schleyer, J. I. Wu, F. P. Cossío and I. Fernández, Aromaticity in transition structures, Chem. Soc. Rev., 2014, 43, 4909–4921 RSC; (b) N. Jana and T. G. Driver, Assembly of functionalized carbocycles or N-heterocycles through a domino electrocyclization–[1,2]migration reaction sequence, Org. Biomol. Chem., 2015, 13, 9720–9741 RSC; (c) E. C. Taylor and I. J. Turchi, 1,5-Dipolar cyclizations, Chem. Rev., 1979, 79, 181–231 CrossRef CAS; (d) N. Li and Y. Huang, Phosphine-Catalyzed Sequential [3+3]/Aza-6π-Electrocyclization Reaction of Cross-Conjugated Azatrienes and δ-Sulfonamido-Allenoates, Org. Lett., 2020, 22, 9392–9397 CrossRef CAS PubMed.
- (a) M. Bian, L. Li and H. Ding, Recent Advances on the Application of Electrocyclic Reactions in Complex Natural Product Synthesis, Synthesis, 2017, 49, 4383–4413 CrossRef CAS; (b) S. Raja, M. Nakajima and M. Rueping, Experimental and Computational Study of the Catalytic Asymmetric 4π-Electrocyclization of N-Heterocycles, Angew. Chem., Int. Ed., 2015, 54, 2762–2765 CrossRef CAS PubMed; (c) B. Iglesias, A. R. de Lera, J. Rodríguez-Otero and S. López, Torquoselectivity in the cationic cyclopentannelation of (2Z)-hexa-2,4,5-trienal acetals, Chem.–Eur. J., 2000, 6, 4021–4033 CrossRef CAS PubMed.
- (a) V. A. Guner, K. Houk and I. W. Davies, Computational Studies on the Electrocyclizations of 1-Amino-1,3,5-hexatrienes, J. Org. Chem., 2004, 69, 8024–8028 CrossRef CAS PubMed; (b) J. Rodríguez-Otero, Study of the electrocyclization of (Z)-hexa-1,3,5-triene and its heterosubstituted analogues based on ab initio and DFT calculations, J. Org. Chem., 1999, 64, 6842–6848 CrossRef PubMed; (c) S. A. Bonderoff, T. N. Grant, F. West and M. Tremblay, Nazarov reactions of vinyl cyclopropylamines: an approach to the imino-Nazarov problem, Org. Lett., 2013, 15, 2888–2891 CrossRef CAS PubMed.
- (a) A. J. Frontier and J. J. Hernandez, New twists in Nazarov cyclization chemistry, Acc. Chem. Res., 2020, 53, 1822–1832 CrossRef CAS PubMed; (b) W. T. Spencer III, T. Vaidya and A. J. Frontier, Beyond the divinyl ketone: innovations in the generation and Nazarov cyclization of pentadienyl cation intermediates, Eur. J. Org. Chem., 2013, 2013, 3621–3633 CrossRef PubMed; (c) M. J. Riveira, L. A. Marsili and M. P. Mischne, The iso-Nazarov reaction, Org. Biomol. Chem., 2017, 15, 9255–9274 RSC; (d) R. L. Davis and D. J. Tantillo, Theoretical studies on pentadienyl cation electrocyclizations, Curr. Org. Chem., 2010, 14, 1561–1577 CrossRef CAS; (e) T. N. Grant, C. J. Rieder and F. G. West, Interrupting the Nazarov reaction: domino and cascade processes utilizing cyclopentenyl cations, Chem. Commun., 2009, 5676–5688 RSC; (f) M. J. Di Grandi, Nazarov-like cyclization reactions, Org. Biomol. Chem., 2014, 12, 5331–5345 RSC; (g) N. Shimada, C. Stewart and M. A. Tius, Asymmetric Nazarov cyclizations, Tetrahedron, 2011, 67, 5851 CrossRef CAS PubMed.
- (a) R. von Essen, D. Frank, H. W. Sünnemann, D. Vidović, J. Magull and A. de Meijere, Domino 6π-Electrocyclization/Diels–Alder Reactions on 1,6-Disubstituted (E,Z,E)-1,3,5-Hexatrienes: Versatile Access to Highly Substituted Tri-and Tetracyclic Systems, Chem.–Eur. J., 2005, 11, 6583–6592 CrossRef CAS PubMed; (b) N. A. Magomedov, P. L. Ruggiero and Y. Tang, Remarkably facile hexatriene electrocyclizations as a route to functionalized cyclohexenones via ring expansion of cyclobutenones, J. Am. Chem. Soc., 2004, 126, 1624–1625 CrossRef CAS PubMed; (c) E. Marvell, G. Caple, B. Schatz and W. Pippin, Electrocyclic reactions: stereochemistry of the triene electrocyclization, Tetrahedron, 1973, 29, 3781–3789 CrossRef CAS; (d) P. Qin, L. A. Wang, J. M. O'Connor, K. K. Baldridge, Y. Li, B. Tufekci, J. Chen and A. L. Rheingold, Transition-Metal Catalysis of Triene 6π Electrocyclization: The π-Complexation Strategy Realized, Angew. Chem., Int. Ed., 2020, 59, 17958–17965 CrossRef CAS PubMed; (e) C. L. Benson and F. West, Highly stereoselective 6π electrocyclization of bridged bicyclic 1,3,5-trienes, Org. Lett., 2007, 9, 2545–2548 CrossRef CAS PubMed; (f) P. E. Tessier, N. Nguyen, M. D. Clay and A. G. Fallis, Aryl Annulation of Cyclic Ketones via a Magnesium Carbometalation−6-π-Electrocyclization Protocol, Org. Lett., 2005, 7, 767–770 CrossRef CAS PubMed.
- G. A. Barcan, A. Patel, K. Houk and O. Kwon, A torquoselective 6π electrocyclization approach to reserpine alkaloids, Org. Lett., 2012, 14, 5388–5391 CrossRef CAS PubMed.
- (a) O. N. Faza, C. S. López, R. Álvarez and Á. R. de Lera, Regio-, Peri-, and Torquoselectivity in Hydroxy Heptatrienyl Cation Electrocyclizations: The Iso/Homo-Nazarov Reaction, Chem.–Eur. J., 2009, 15, 1944–1956 CrossRef CAS PubMed; (b) D. Alickmann, R. Fröhlich, A. H. Maulitz and E. U. Würthwein, Electrocyclization Reactions of 1-Aza-and 1-Oxapentadienyl and-heptatrienyl Cations: Synthesis of Pyrrole and Furan Derivatives, Eur. J. Org. Chem., 2002, 2002, 1523–1537 CrossRef.
- (a) R. B. Bates, W. Deines, D. McCombs and D. Potter, Preparation of heptatrienyl anions. Heptatrienyl-cycloheptadienyl anion rearrangement, J. Am. Chem. Soc., 1969, 91, 4608 CrossRef CAS; (b) K. Marx and W. Eberbach, 8-Phenyl-10,10a-dihydropyrido [1,2-a] azepines by 1,7-Electrocyclization of Conjugated Pyridinium Ylides—Rationalization of the Periselectivity, Chem.–Eur. J., 2000, 6, 2063–2068 CrossRef CAS PubMed; (c) M. Reisser and G. Maas, Synthesis of pyrroles from 1-dialkylamino-3-phosphoryl (or phosphanyl) allenes through 1,5-cyclization of conjugated azomethine ylide intermediates, J. Org. Chem., 2004, 69, 4913–4924 CrossRef CAS PubMed.
- N. Deno, C. U. Pittman Jr and J. O. Turner, Carbonium Ions. XVIII. Cyclizations of Pentadienyl and Heptatrienyl Cations, J. Am. Chem. Soc., 1965, 87, 2153–2157 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) P. C. Hariharan and J. A. Pople, The influence of polarization functions on molecular orbital hydrogenation energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS; (b) W. J. Hehre, R. Ditchfield and J. A. Pople, Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- K. Fukui, The path of chemical reactions-the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- P. Fuentealba, H. Preuss, H. Stoll and L. Von Szentpály, A proper account of core-polarization with pseudopotentials: single valence-electron alkali compounds, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Mammen, E. I. Shakhnovich, J. M. Deutch and G. M. Whitesides, Estimating the Entropic Cost of Self-Assembly of Multiparticle Hydrogen-Bonded Aggregates Based on the Cyanuric Acid.Melamine Lattice, J. Org. Chem., 1998, 63, 3821–3830 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Montreal, 2009, http://www.Cylview.org Search PubMed.
- Partial optimization of TS2a and TS2b were carried out since the full optimization of the transition state could not be located Search PubMed.
- All attempts made to locate other transition states to generate the corresponding 5-, 6-, and 7-membered intermediates from INT4a failed Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02839a |
| This journal is © The Royal Society of Chemistry 2023 |