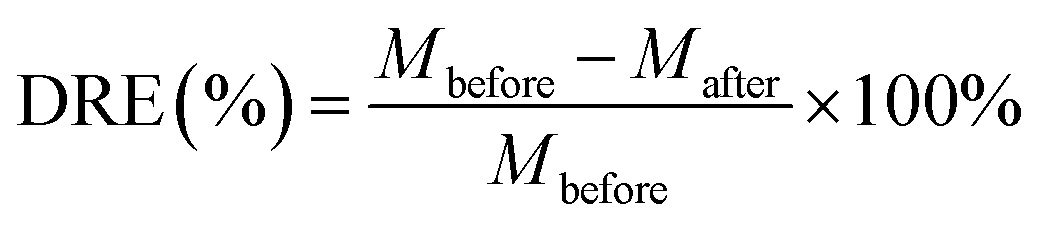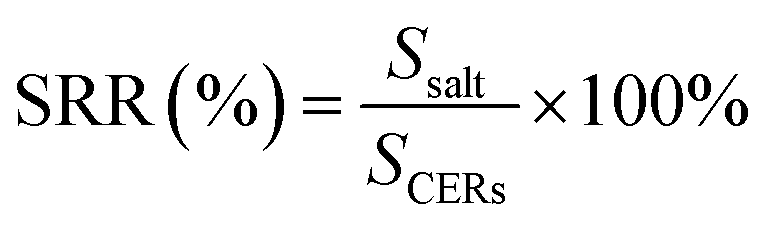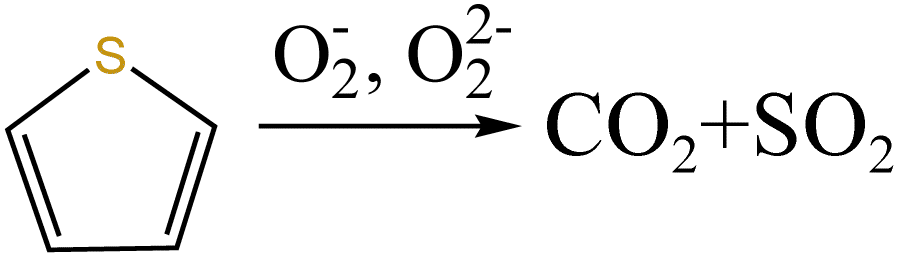DOI:
10.1039/D3RA02723F
(Paper)
RSC Adv., 2023,
13, 18347-18362
The effect of air on oxidation decomposition of uranium-containing cationic exchange resins in Li2CO3–Na2CO3–K2CO3 molten-salt system
Received
25th April 2023
, Accepted 9th June 2023
First published on 19th June 2023
Abstract
With the development of nuclear energy, spent cationic exchange resins after purification of radioactive wastewater must be treated. Molten-salt oxidation (MSO) can minimize the disposal content of resins and capture SO2. In this work, the decomposition of uranium-containing resins in carbonate molten salt in N2 and air atmospheres was investigated. Compared to N2 atmosphere, the content of SO2 released from the decomposition of resins was relatively low at 386–454 °C in an air atmosphere. The SEM morphology indicated that the presence of air facilitated the decomposition of the resin cross-linked structure. The decomposition efficiency of resins in an air atmosphere was 82.6% at 800 °C. The XRD analysis revealed that uranium compounds had the reaction paths of UO3 → UO2.92 → U3O8 and UO3 → K2U2O7 → K2UO4 in the carbonate melt, and sulfur elements in resins were fixed in the form of K3Na(SO4)2. The XPS result illustrated that peroxide and superoxide ions accelerated the conversion of sulfone sulfur to thiophene sulfur and further oxidized to CO2 and SO2. Besides, the ion bond formed by uranyl ions on the sulfonic acid group was decomposed at high temperature. Finally, the decomposition of uranium-containing resins in the carbonate melt in an air atmosphere was explained. This study provided more theoretical guidance and technical support for the industrial treatment of uranium-containing resins.
1. Introduction
Currently, ion exchange resins (IERs) are widely applied for the separation and purification of radioactive wastewater. When their capacity reaches saturation, waste resins are replaced by fresh resins. A large number of waste resins are produced in nuclear energy units.1 For example, the actual annual output of waste resins of two units of the Daya Bay nuclear power plant was 4–13 m3 per year, and the average annual output of waste resins of the two units of the Qinshan No. 2 nuclear power plant from 2002 to 2016 reached 46 m3 per year.2
After replacement, spent resins undergo chemical degradation and radiation decomposition, which can release hazardous substances into the environment. To reduce the risks of waste resins to the environment and humans, proper disposal of waste resins is required. The general treatment methods include curing method, incineration method, etc.3 Traditional curing methods are cement curing, asphalt curing and polymer curing.4–8 These are some advantages about these ways, such as a simple process, mature technology, low investment and operating costs, and stable properties of the obtained cured products. However, the volume reduction ratio after the curing of waste resins is low, and the degradation or swelling of waste resins is unfavorable for the long-term storage of cured products.9 The advantages of treating waste resins by incineration mainly contain a high weight loss rate and a volume reduction rate. The heat generated during the treatment process can be recovered. However, the treatment of waste resins with this technology makes radionuclides volatilize and releases large amounts of sulfur oxides, thereby polluting the environment.10–12 Therefore, it is critical to propose a safe and efficient method for the disposal of waste resins.
Molten-salt oxidation (MSO) is an efficient, flame-free thermal process that captures SO2 and minimizes the disposal content of wastes through oxidation destruction.13–16 Most importantly, this technology can be applied for the retention of radionuclides in molten salt. Our group investigated the destruction process of resins using the ternary eutectic carbonate (Li2CO3–Na2CO3–K2CO3), and demonstrated that this molten salt system was an effective medium to process resins at low temperatures, thereby reducing the volatilization of radionuclides.17 Antonetti et al.18 conducted pyrolysis experiments on cation exchange resins (CERs) doped with cobalt and cesium and demonstrated that the presence of Co and Cs on the resin inhibited the transfer of sulfur to the gas phase while accelerating the degradation of volatile carbon compounds. Yang et al.19 investigated the volatilization of Cd, Co, Ce, Cs, Pb, and Sr doped with different contents of resins in binary eutectic carbonate (K2CO3–Na2CO3) at 710 °C. The results showed that the retention rate of each metal reached 99.5 wt%. In addition, some experiments also explored certain metal impurities (such as unionized and ionized forms) can play a catalytic role in the oxidative cracking of resins.20–22 Juang et al.23 found that several metal salts containing CuO, CuSO4·5H2O and FeSO4·7H2O had a catalytic effect on the oxidative destruction of cationic resins. This was because the decomposition of functional groups and polymer matrices was enhanced by these salts. Matsuda et al.21 studied the effect of Fe impurities including unionized Fe (Fe, Fe2O3, Fe3O4) forms and ionized Fe (Fe3+) forms on resin oxidation. Ionized Fe (Fe3+) forms resulted in considerable increases in reaction rate constant, which indicated that Fe3+ functioned as a catalyst. However, unionized Fe (Fe, Fe2O3, Fe3O4) had no catalytic effect. In addition, the catalytic effect of other metal impurities (such as Pd2+, Cu2+, Fe2+, Al3+, Co2+ and Ni2+) on the resin oxidation reaction was also investigated using resins. The experimental analysis revealed that in the case of resins containing Pd2+, Cu2+, Fe2+ and Al3+, resins were catalysed and oxidized, and metallic sulfides in molten salt could be rapidly transferred to oxides. Sulfur compound formation was attributed to the functional sulfonic acid group (–SO3H) in the cation exchange resin.20 When metal compounds were present in sulfur-containing waste, sulfur, as a main environmental pollution source, was fixed in the form of metal sulfides and metal sulfates in the condensed phase during waste treatment, which decreased the output in sulfur compounds (such as H2S and SO2).24–28
Uranium element is a main radioactive nuclide in nuclear industry wastewater. Based on analysis of the above literature, resins after the purification of uranium-containing wastewater can be also disposed by MSO method and the content of spent resins can be reduced. However, the presence of uranium element could have a certain impact on the decomposition of resins in molten salt. Therefore, it is of great significance to study the oxidation decomposition of uranium-containing cationic exchange resins (CERs). In this study, due to its low melting point, the ternary carbonate (Li2CO3–Na2CO3–K2CO3) is chosen as molten salt for the oxidation decomposition of resins. Some experiments on the decomposition of uranium-containing resins and pure resins were conducted under air atmosphere. The characteristics of products after the decomposition of uranium-containing resins and pure resins were analysed, such as the release change of off gas, component of spent salt and structure variation of residues. The experiments focused on investigating the oxidation decomposition of uranium-containing resins in ternary carbonate (Li2CO3–Na2CO3–K2CO3) molten-salt system.
2. Materials and methods
2.1 Materials
Cationic exchange resins (CERs) used in our experiment were insoluble solid granules and had a strong acid form (–SO3–H+) based on the structure of a styrene-divinylbenzene (ST-DVB) matrix. Because fresh resins contain impurity ions, hydrochloric acid solution and sodium hydroxide solution were utilized to remove impurities in resins before the experiment. The resins after cleaning were dried to a constant weight in an oven at 105 °C. Through elemental analysis, the contents of C, O, S and H in resins were 38.6 wt%, 43.6 wt%, 12.2 wt% and 5.6 wt%, respectively. In the preparation process of uranium-containing resins, when resins were in contact with the solution containing a certain concentration of uranium element, uranyl ions in solution were adsorbed onto the resin using an ion exchange method. This uranium concentration approximately corresponded to that of spent resin generated from spent fuel reprocessing plant. In the experiment, uranium-containing resins and pure resins after drying were used as raw materials. The molten salt system contained 44 wt% Li2CO3, 30 wt% Na2CO3 and 26 wt% K2CO3.
2.2 Experimental operation process
Uranium-containing resins was decomposed in molten salt under nitrogen and air atmospheres in a tube furnace. First, carbonates (Li2CO3, Na2CO3 and K2CO3) were dried to remove moisture in an oven at 200 °C, and Li2CO3, Na2CO3 and K2CO3 were evenly mixed by 44 wt%, 30 wt% and 26 wt%, respectively. Second, 2 g of carbonates and 2 g of resins were weighed, respectively. 60 wt% of ternary eutectic carbonates was uniformly mixed with resins and the rest was covered on resins, which could absorb acid gas released from resin decomposition. This mixture was packed in the alundum crucible. The crucible was kept at a certain temperature for 30 min in a furnace and subsequently taken out. The products after resin decomposition were treated to collect waste salt and residues. Residues were washed with deionized water and then be dried to a constant weight in an oven at 105 °C. Waste salt was ground into a powder for further analysis. All experiments were repeated three times. The retention rate of sulfur in spent salt was analysed through the dichromic acid method. The degradation and removal efficiency (DRE) of resins and retention rate of sulfur (SRR) after the decomposition of resins were calculated by formulas (1) and (2).| |
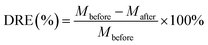 | (1) |
where Mbefore – the mass of resins before decomposition; and Mafter – the mass of resins after decomposition.| |
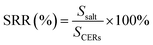 | (2) |
where SCERs – the amount of sulfur in resins before decomposition; and Ssalt – the amount of sulfur in spent salt after decomposition.
2.3 Analytical methods
The thermogravimetric analyzer (TGA) was utilized to explore the destruction laws of uranium-containing resins. The temperature was increased from room temperature to 800 °C at 10 °C min−1 in N2 or air flow rate of 20 mL min−1. On-line gas mass spectrometer (GM) was applied for the analysis of composition and release variation of gaseous products in the decomposition process of resins. The microscopic morphology of residue after resins destruction was observed by the scanning electron microscope (SEM). Energy dispersive spectrometry (EDS) was conducted to analyse the element composition of residues. The pictures of samples were taken at a magnification of ×50 and an excitation voltage of 5 kV. The chemical composition of spent salt was analyzed through X-ray diffraction (XRD). The scan degree range (2θ) of sample was from 5° to 85°. The Fourier transform infrared spectrometer (FT-IR) was used to determine the change of functional groups in residues. 2 mg of the sample was mixed with 40 mg of KBr and then pressed into round thin sheets. The thin sheets were measured in the 400–4000 cm−1 range. The binding energies of sulfur 2p in residues were determined by X-ray photoelectron spectroscopy (XPS). Al Kα radiation (hν = 1486.6 eV) from a monochromatic X-ray source and a 500 μm light spot size was utilized. Additionally, according to the experimental results, HSC Chemistry 6.0 thermodynamic software is used to analyse the relevant reactions, and study the Gibbs free energy of reaction between substances, which can verify the accuracy of the experimental results. In the calculation process of reaction Gibbs free energy, the analysis module of reaction equation in the software is mainly used. Before the calculation of Gibbs free energy, the reaction temperature range is set as 0–1000 °C, and the temperature step is 100 °C. The reaction equation between substances is balanced. Then, the Gibbs free energy of reaction is directly calculated through HSC Chemistry 6.0 thermodynamic software. Eventually, the relationship between Gibbs free energy of reaction and temperature can be obtained.
3. Results and discussion
3.1 Thermal destruction of uranium-containing resins
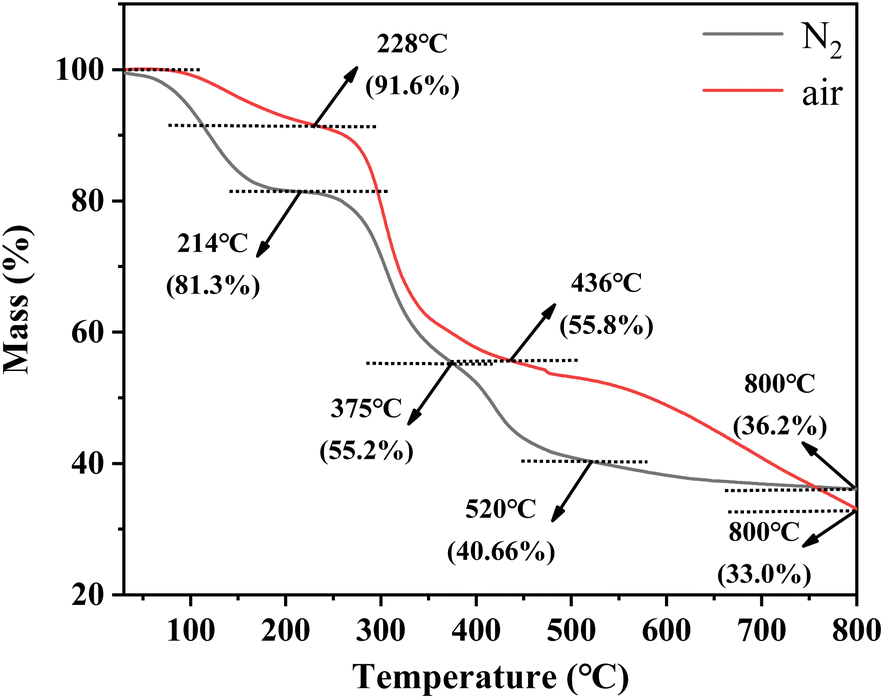
Fig. 1 describes the thermal destruction of uranium-containing resins in the temperature range of 30–800 °C in N2 and air atmospheres. According to the obtained thermogravimetric curve, the derivative thermogravimetric (DTG) curve was fitted and the main temperature destruction range was determined. It was determined that with increasing reaction temperature, the mass remaining efficiencies of uranium-containing resins progressively decreased, irrespective of the environment. Besides, there were three main variation stages in the thermal decomposition process of resins in N2 atmosphere, such as 30–214 °C, 214–375 °C and 375–800 °C, and in air atmosphere, such as 30–228 °C, 228–436 °C and 436–800 °C, i.e., the decomposition process of resins was different at every stage in N2 and air atmospheres.
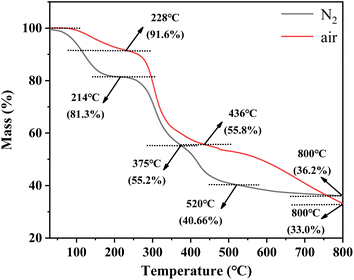 |
| | Fig. 1 The thermal characteristics of uranium-containing resins in the temperature range of 30–800 °C in N2 and air atmospheres. | |
From Fig. 1, in N2 atmosphere, the mass remaining efficiencies of uranium-containing resins gradually decreased with reaction temperature increasing from 30 °C to 214 °C and the weight loss of resins was 18.7% in this temperature range, which was due to the evaporation of water in resins. The maximum weight loss of resins (26.1%) occurs in 214–375 °C. This change of resin mass corresponded to the destruction of functional groups of sulfonic acid groups (–SO3H) in resins. When reaction temperature was increased to 800 °C, the mass remaining efficiencies of uranium-containing resins was decreased to 36.2%, and the weight loss of resins was 15.7%. The result was mainly ascribed to the decomposition of ST-DVB matrix and polymer chains. These results were similar to the conclusion in ref. 23. In addition, compared to N2 atmosphere, the mass remaining efficiencies of resins had an obvious decrease in air atmosphere above 436 °C, and reached 33.0% at 800 °C. It meant that the addition of air accelerated and prompted the decomposition of ST-DVB matrix and polymer chains in resins at the third stage.
To study the decomposition of uranium-containing resins in molten salt, some experiments were designed to analyse the content variation of exhaust gas during the decomposition of uranium-containing resins in N2 and air atmospheres with increasing temperature, and the results are shown in Fig. 2. From Fig. 2a and b, the release temperatures of CH4 and C2H4 during the decomposition of uranium-containing resins in N2 atmosphere were basically the same with those in air atmosphere, and were 423 °C and 386 °C, respectively. However, compared to N2 atmosphere, the production of CH4 at 527–755 °C and C2H4 at 386–452 °C in air atmosphere obviously decreased. Based on the TG analysis in Fig. 1, the generation of CH4 and C2H4 was due to the cleavage of hydrogen bonds (ST-DVB matrix and polymer chains) during the decomposition of resins in air atmosphere, and the hydrogen bonds in resins were immediately oxidized, resulting in the decrease in C2H4 release content and the increase in CO2 release content (Fig. 2b and c). Besides, the release content of CO2 above 368 °C in air atmosphere was significantly more than that in N2 atmosphere. Additionally, the release change of SO2 during resin decomposition in carbonates in N2 and air atmospheres was studied (Fig. 2d). The temperature range of SO2 release was 386–454 °C in N2 and air atmospheres. According to the TG analysis in Fig. 1, the production of SO2 in air atmosphere corresponded to the decomposition of sulfonic acid groups at 186–436 °C. The addition of carbonates absorbed the heat around resins and decreased the temperature around resins, which hindered the decomposition of sulfonic acid groups. It meant that the decomposition temperature of sulfonic acid groups was increased. Besides, compared to N2 atmosphere, the release content of SO2 in air atmosphere was comparatively low. It was concluded that more SO2 was absorbed by carbonates in air atmosphere.
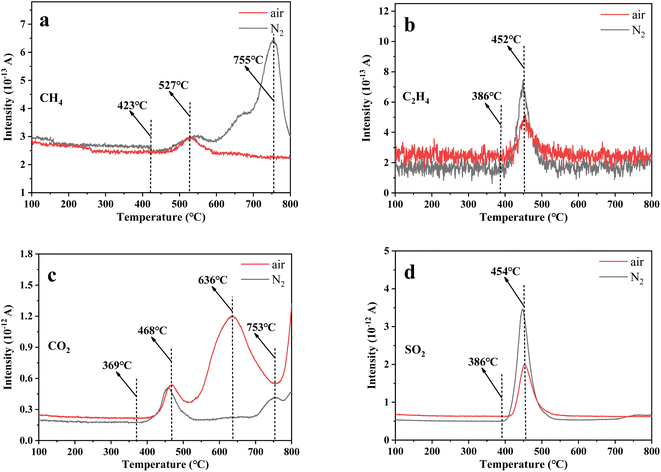 |
| | Fig. 2 The release variation of off gas (a) CH4, (b) C2H4, (c) CO2 and (d) SO2 during the decomposition of uranium-containing resins in carbonates with increasing temperature in N2 and air atmospheres. | |
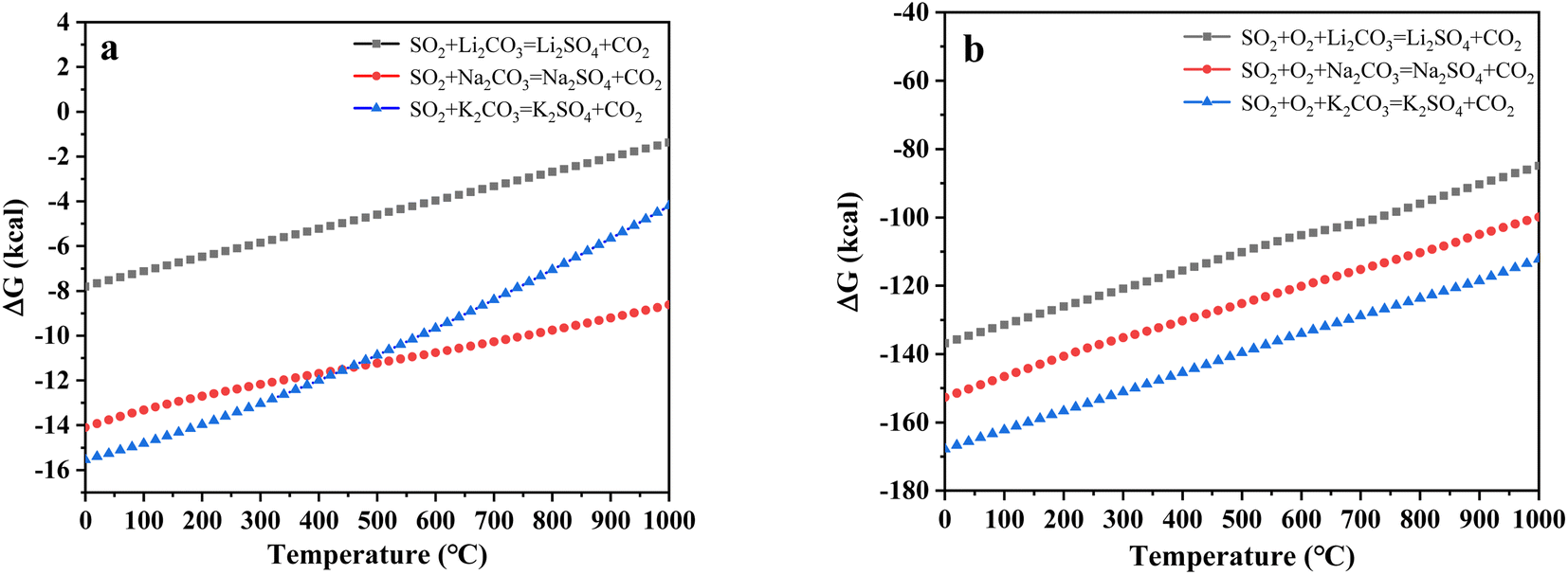
To further explore the content change of SO2 during uranium-containing resin decomposition in carbonates, the HSC Chemistry 6.0 thermodynamic software was applied for the analysis of the thermal characteristics of reaction between SO2 and carbonates in N2 and air atmospheres. Fig. 3a describes the relationship between reaction temperature and Gibbs free energy (ΔG) required for the reaction of SO2 and carbonates (Li2CO3, Na2CO3 and K2CO3) in N2 atmosphere. From Fig. 3a, it was seen that the Gibbs free energy required for the reaction of SO2 and carbonates gradually decreased with increasing reaction temperature and remained negative. The reaction between SO2 and carbonates was spontaneous to proceed. Compared to N2 atmosphere, the Gibbs free energy required for the reaction of SO2 and carbonates was relatively smaller in air atmosphere (Fig. 3b). It was determined that SO2 was easily absorbed by carbonates with the addition of air, which proved the decrease in SO2 release content at 386–454 °C in air atmosphere (Fig. 2d).
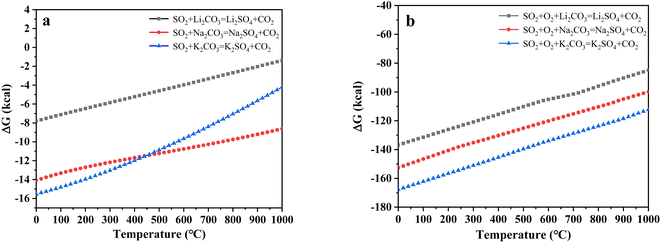 |
| | Fig. 3 The relationship between reaction temperature and Gibbs free energy (ΔG) required for the reaction of SO2 and carbonates in (a) N2 and (b) air atmospheres. | |
3.2 The characterization analysis of product
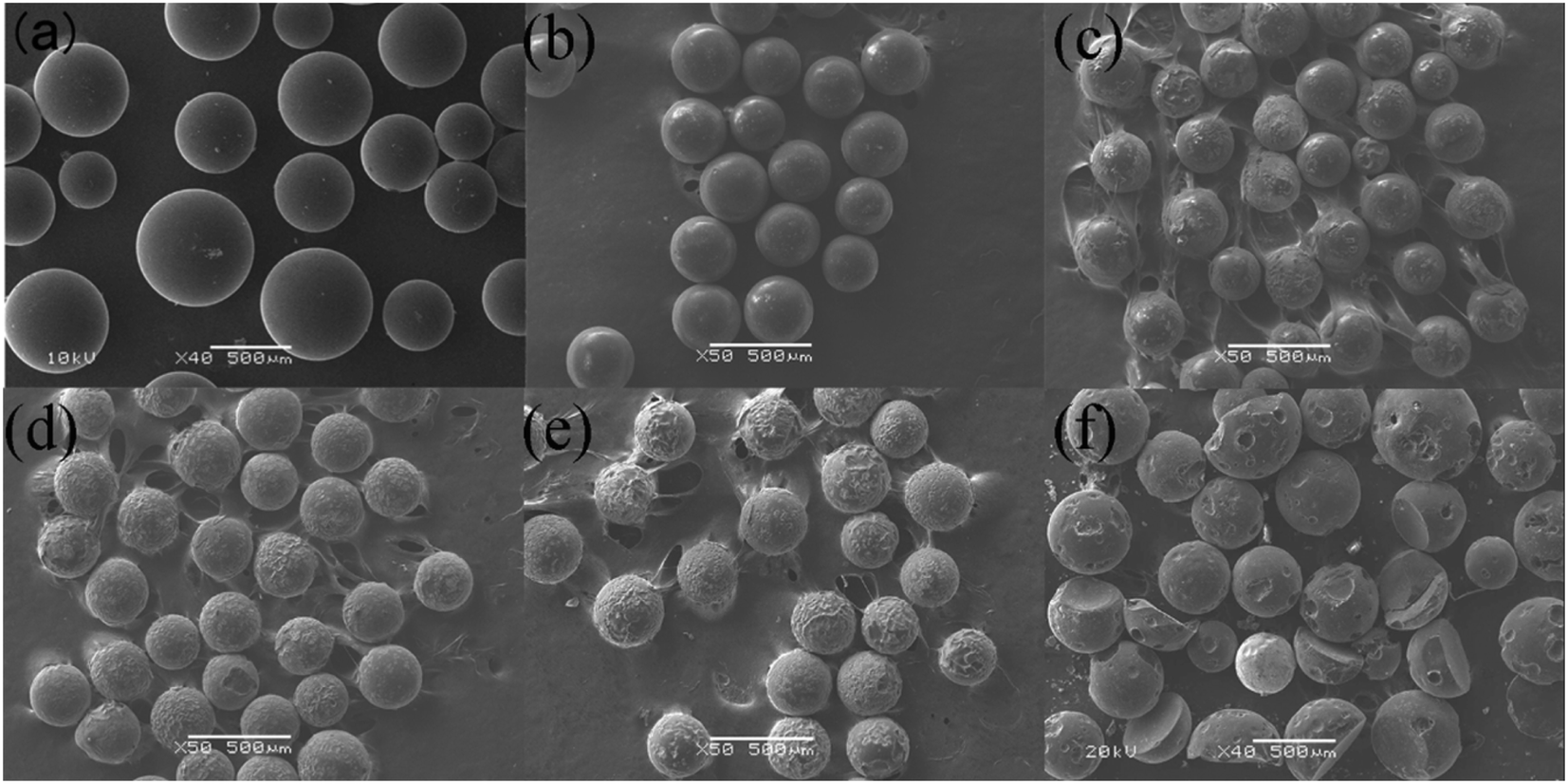
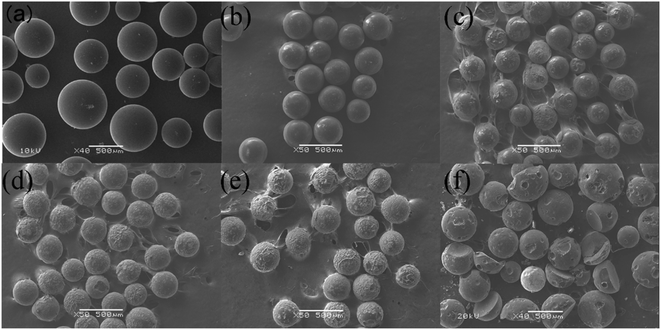
3.2.1 The morphology analysis of product. Fig. 4 shows the images of original resins and products after decomposition of uranium-containing resins in carbonate molten salt at different reaction temperatures in N2 and air atmospheres. The reaction temperature was chosen based on TG results of uranium-containing resins. From Fig. 4a, the color of original resins before destruction in N2 atmosphere was brown. With increasing reaction temperature, the resins changed from brown to black (Fig. 4b–d) and the remaining mass of residues gradually decreased, which confirmed that the amplify in temperature triggered the decomposition of resins.
 |
| | Fig. 4 The images of (a) original resins and product after the decomposition of uranium-containing resins in carbonate molten salt at (b) 460 °C, (c) 640 °C, (d) 720 °C in N2 atmosphere and at (e) 460 °C, (f) 640 °C, (g) 720 °C in air atmosphere. | |
From Fig. 4e–g, in contrast to N2 atmosphere, the remaining mass of residues after resin decomposition in carbonate molten salt in air atmosphere obviously decreased at the same temperature. It meant that the oxidation decomposition of resins was prompted due to the addition of air. Besides, from Fig. 4e and f, with reaction temperature increasing from 460 °C to 640 °C, the remaining mass of residues had a considerable decrease, which showed that majority of resins were oxidized to CO2 in air atmosphere. This result was validated by the conclusion in Fig. 2c.
3.2.2 The decomposition rate of resins and retention rate of sulfur. The decomposition rate of resins and retention rate of sulfur after the decomposition of uranium-containing resins in carbonate molten salt at different temperatures for 30 min in N2 and air atmospheres are presented in Fig. 5. From Fig. 5a, when temperature increased from 325 °C to 800 °C, the decomposition rate of resins increased from 37.3% to 54.3% in N2 atmosphere, and in air atmosphere, that of resins increased from 38.2% to 82.6%. During the decomposition of uranium-containing resins in carbonate molten salt in N2 and air atmospheres, the increase in temperature prompted the decomposition of resins. Associated with the TG analysis in Fig. 1, the decrease of resin decomposition rate at 325 °C was mainly ascribed to the dehydration of the remaining water and the decomposition of some sulfonic acid functional groups. Nevertheless, due to the stability of the ion bond formed by uranyl ions on the sulfonic acid group, the sulfonic acid group in uranium-containing resins was difficult to decompose at 325 °C, which resulted in a slightly low decomposition rate of resins. This conclusion was similar to the inhibition of resin pyrolysis with metal ion doping.29,30 With temperature increasing from 325 °C to 460 °C, the ion bonds on the sulfonic acid group of uranium-containing resins were gradually decomposed, which generated SO2 (Fig. 2d). Meanwhile, the hydrocarbon structure was destroyed, which released CH4, C2H4 and CO2 (Fig. 2a–c). The production of exhaust gas corresponded to the increase in resin decomposition rate at 325–460 °C. When temperature was increased to 550 °C, the decomposition rate of uranium-containing resins continuously greatly increased. This conclusion was because the ion bonds on the sulfonic acid group of uranium-containing resins were completely destroyed. Besides, the increase in decomposition rate of resins in air atmosphere was larger than that in N2 atmosphere at 460–550 °C, which meant that the presence of air prompted the oxidation decomposition of ST-DVB matrix and polymer chains in resins and resulted in the release increase in CO2. As temperature continuously increased, the decomposition rate of uranium-containing resins gradually increased, and the change curve of resin decomposition rate becomes steeper. Thus, the decomposition rate of resins in air atmosphere was higher than that in N2 atmosphere, which was consistent with the TG analysis in Fig. 1. Compared to the results of Fig. 1, the decomposition efficiency of resins in molten salt could be lower at the same temperature in Fig. 5. This was because in the thermal property analysis of resin in Fig. 1, resins without carbonate were directly decomposed in air atmosphere, and the cross-linked structure and carbon chain structure of the resin were destroyed, resulting in high weight loss of resins. However, in Fig. 5a, when samples contained carbonate, oxygen in air atmosphere could be first dissolved in molten salt and then, resins were oxidized and decomposed in molten carbonate. When the reaction temperature increased, more oxygen was dissolved in molten salt, and the oxidation decomposition of resins could be further improved. Therefore, the decomposition efficiency of resins in molten salt could be lower at the same temperature.
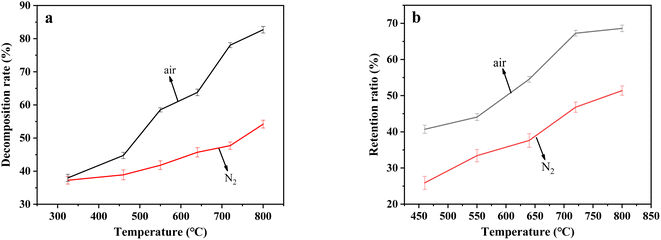 |
| | Fig. 5 (a) The decomposition rate of resins and (b) retention rate of sulfur after the decomposition of uranium-containing resins in carbonate molten salt at different temperatures for 30 min in N2 and air atmospheres. | |
From Fig. 5b, when temperature increased from 325 °C to 800 °C, the retention rate of sulfur after the decomposition of resins in carbonate molten salt in N2 atmosphere gradually increased from 17.7% to 51.4%, while that increased from 40.7% to 68.7% in air atmosphere. Compared to N2 atmosphere, organic sulfur in resins could be more easily retained by carbonate melt at high temperature in air atmosphere. According to the analytical results in Fig. 5a and b, it was determined that in air atmosphere, large quantities of resins were oxidized and decomposed to SO2, which was absorbed by carbonate molten salt and resulted in the substantial increase in sulfur retention rate.
In order to further investigate the decomposition of uranium-containing resins in (Li2CO3–Na2CO3–K2CO3) molten-salt system, the characteristics of products after the decomposition of resins at different reaction temperatures were studied using various analysis methods including SEM-EDS, XRD, FT-IR and XPS.
3.2.3 Scanning electron microscopy (SEM) analysis. Fig. 6 describes the SEM morphology of original resins and residues after the decomposition of uranium-containing resins in carbonate molten salt at different temperatures in N2 atmosphere. In Fig. 6a, resins after decomposition maintained a good spherical structure at 325 °C, since ion bonds on sulfonic acid groups and the cross-linked structure of resins have good thermal stability. At 460 °C, some holes were observed on the shell surface of resins in Fig. 6c, which was ascribed to resins being further destroyed at high temperature. With increasing temperature from 460 °C to 720 °C, more holes on the shell surface of resins were found. However, because the cross-linked structure of resins had good thermal stability and structural stability, parts of resins could maintain the spherical structure at 720 °C. Besides, some stable organic structures were generated in the resin structure, which made the resin structure difficult to decompose.
 |
| | Fig. 6 The SEM morphology of (a) original resins and residues after the decomposition of uranium-containing resins in carbonate molten salt at (b) 325 °C, (c) 460 °C, (d) 550 °C, (e) 640 °C and (f) 720 °C in N2 atmosphere. | |
Compared to N2 atmosphere, the SEM morphology of residues after decomposition of uranium-containing resins in carbonate molten salt in air atmosphere showed an obvious change at different temperatures (as shown in Fig. 7). When temperature was increased to 460 °C, the smooth surface of resins became uneven, and more holes appeared on the surface of resins in Fig. 7c, since the hydrocarbon structure of resins was decomposed. With increasing temperature, the spherical structure of parts of resins was destroyed and decomposed in Fig. 7d. Thus, the increase in temperature prompted the decomposition of ion bonds on sulfonic acid groups and resins, which resulted in the cleavage of resin morphology. The conclusion was confirmed by the increase in resins decomposition efficiency in Fig. 5a. Compared to N2 atmosphere (Fig. 6e), a majority of resins were destructed in air atmosphere at 640 °C (Fig. 7e), which implied that the cross-linked structure of resins was further destroyed by air. When temperature was 720 °C, the cross-linked structure of resins was basically completely destroyed in Fig. 7f. Based on the analytical result, the addition of air facilitated the oxidation decomposition of ion bonds on sulfonic acid groups and resin cross-linked structure at high temperature.
 |
| | Fig. 7 The SEM morphology of (a) original resins and residues after the decomposition of uranium-containing resins in carbonate molten salt at (b) 325 °C, (c) 460 °C, (d) 550 °C, (e) 640 °C and (f) 720 °C in air atmosphere. | |
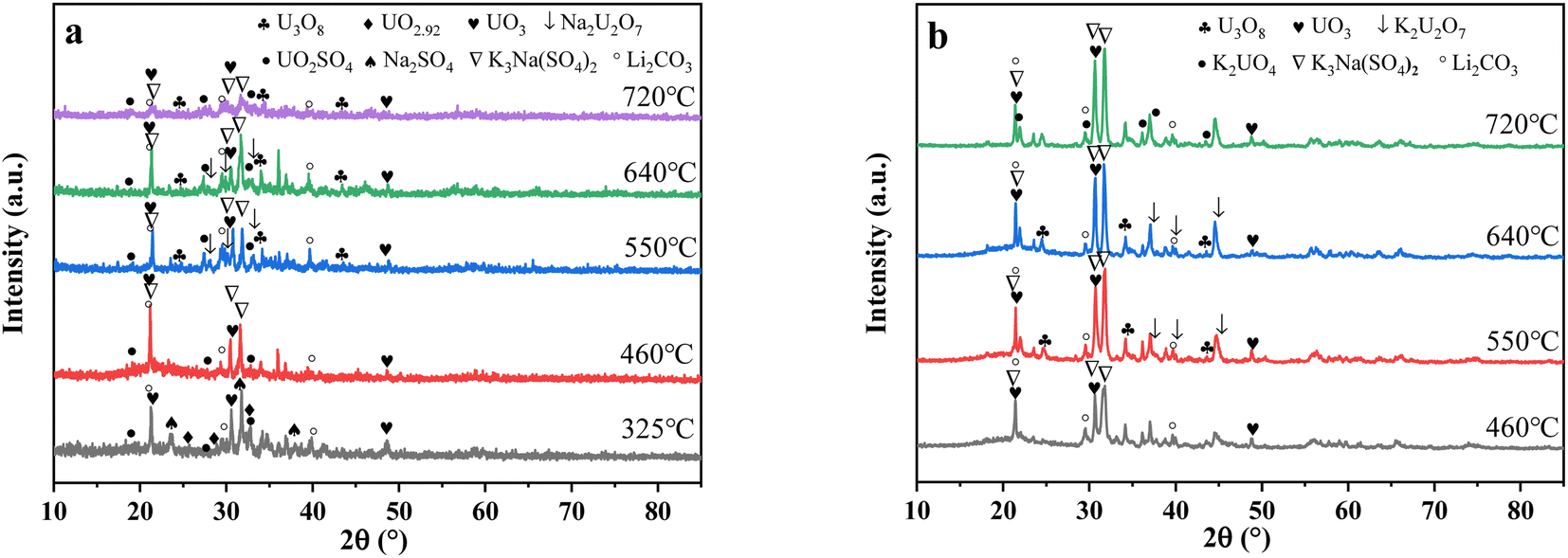
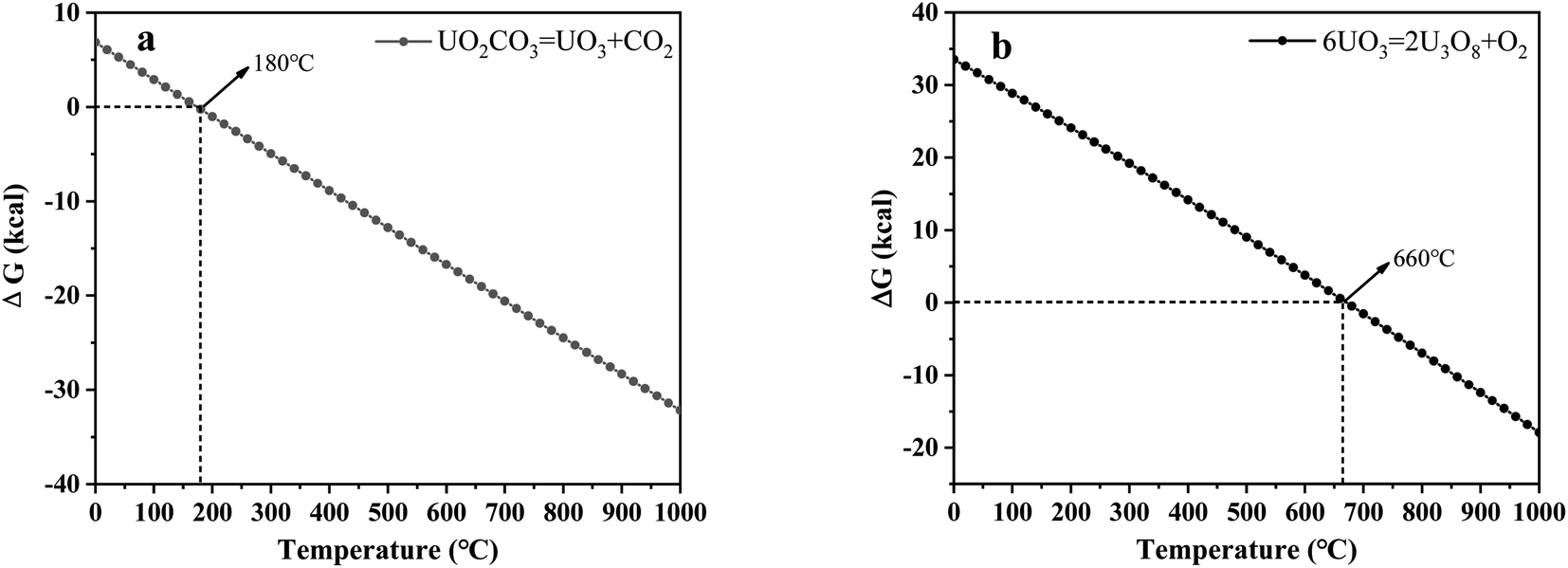
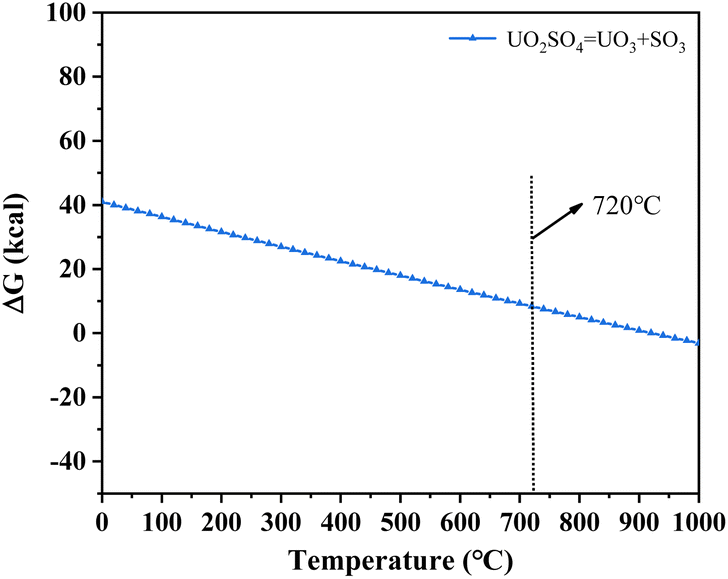
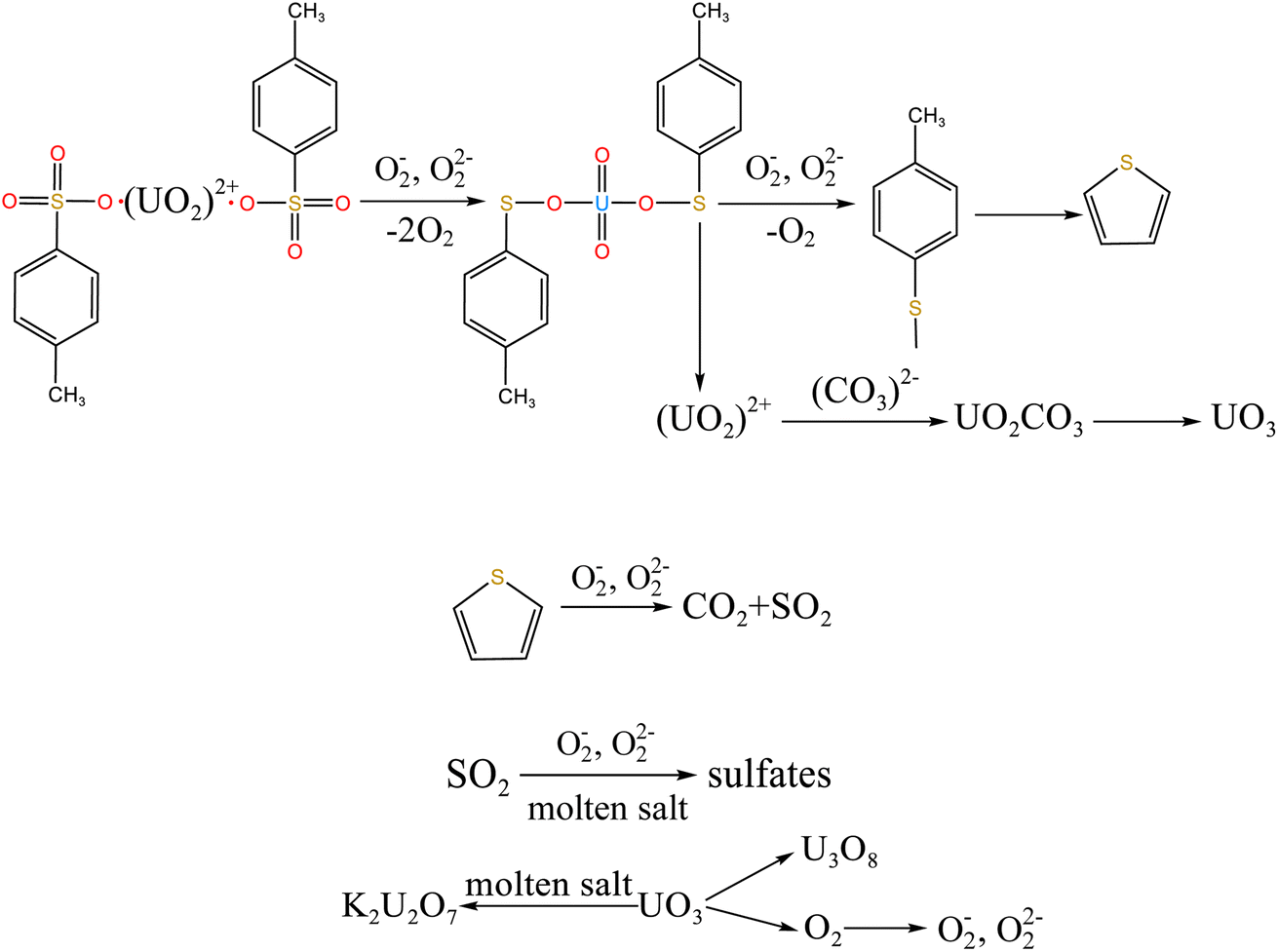
3.2.4 X-ray diffraction (XRD) analysis. XRD technology was applied for the analysis of chemical components in spent salt after uranium-containing resin decomposition in carbonate molten salt at different temperatures in N2 and air atmospheres and the results are shown in Fig. 8. It was seen from Fig. 8a that spent salt after the destruction of uranium-containing resins mainly contained sulfates, uranium oxides and alkali metal diuranate. The sulfur element in sulfates was from the sulfonic acid group functional group (–SO3H) in resins. When temperature was 325 °C, sulfates in the form of Na2SO4 (PDF card No. 24-1132) and UO2SO4 (PDF card No. 24-1378) and uranium oxides in the form of UO3 (PDF card No. 31-1419) and UO2.92 (PDF card No. 46-0948) appeared in spent salt. The formation of sulfates was ascribed to the absorption of SO2 by carbonates. The reaction between uranyl ions and sulfate ions in molten salt leaded to the formation of UO2SO4. UO3 could result from the decomposition of uranyl carbonate at high temperature.31 The thermal characteristics of the reaction between UO2CO3 and UO3 were studied using the HSC Chemistry 6.0 thermodynamic software, and the result is shown in Fig. 9a. When temperature gradually increased, the Gibbs free energy required for the transformation of UO2CO3 to UO3 gradually decreased and the transformation reaction easily proceeded. Parts of UO3 were converted to UO2.92. This conversion was used to establish the relationship between the final structures of UO3 to U3O8.32,33 With the increase in temperature, K3Na(SO4)2 (PDF card No. 74-1742) was present in spent salt at 460 °C. At 550 °C, U3O8 (PDF card No. 20-1345) and Na2U2O7 (PDF card No. 12-0106) were found in spent salt. The production of Na2U2O7 was ascribed to the reaction between Na2CO3 and U3O8.34 U3O8 was present due to the conversion of UO3.35 The thermal characteristics of the reaction between UO3 and U3O8 were analysed using the HSC Chemistry 6.0 thermodynamic software (as shown in Fig. 9b). Fig. 9b shows that with increasing temperature, the Gibbs free energy transforming UO3 to U3O8 gradually decreased and the transformation reaction easily proceeded. In Fig. 8a, as temperature was increased to 720 °C, Na2U2O7 phase essentially disappeared in spent salt, which was due to the decomposition of Na2U2O7.36 However, K3Na(SO4)2, UO2SO4 and U3O8 were still in spent salt. It meant that these compounds had a certain thermal stability at 720 °C. To further explain the stability of UO2SO4 in carbonate molten salt at 720 °C, the HSC Chemistry 6.0 thermodynamic software was utilized to analyse the thermal characteristics of UO2SO4, and the result is shown in Fig. 10. From Fig. 10, the Gibbs free energy transforming UO2SO4 to UO3 was relatively high at 720 °C, which illustrated that the transformation reaction did not easily proceed. In other words, UO2SO4 remained stable at 720 °C. In addition, the SEM morphology of product after the decomposition of resins in carbonate molten salt at 720 °C was also studied, and the result is shown in Fig. 11. From Fig. 11, it was found that uranium was evenly distributed in spent salt.
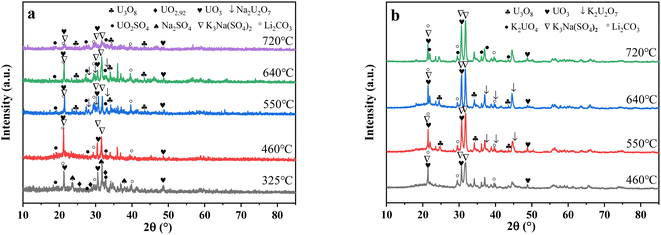 |
| | Fig. 8 The XRD patterns of spent salt after the decomposition of uranium-containing resins at different temperatures in carbonate molten salt in (a) N2 and (b) air atmospheres. | |
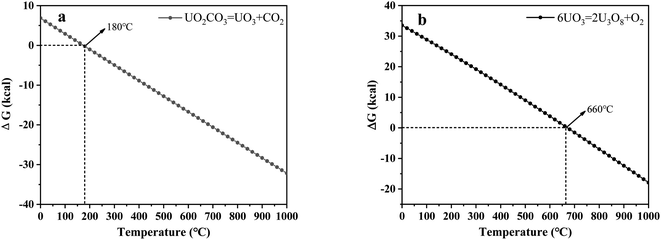 |
| | Fig. 9 Analysis of the thermal characteristics of reaction for (a) UO2CO3–UO3 and (b) UO3–U3O8 using the HSC Chemistry 6.0 thermodynamic software. | |
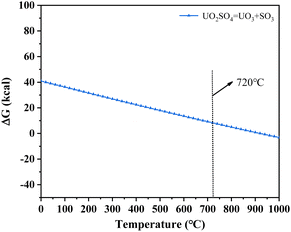 |
| | Fig. 10 The thermal characteristics of UO2SO4 analysed using the HSC Chemistry 6.0 thermodynamic software. | |
 |
| | Fig. 11 Images of product at 720 °C: (a) SEM image, (b) uranium element distribution image. | |
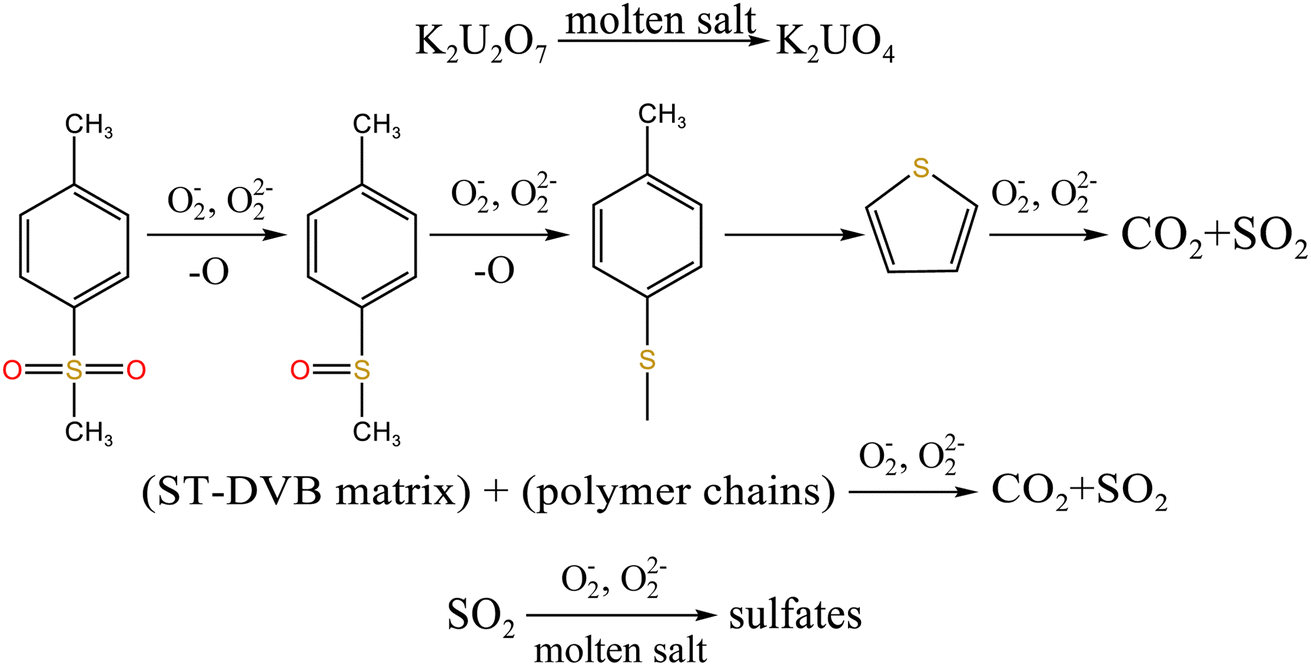
From Fig. 8b, K2U2O7 (PDF card No. 13-0081) occurred in spent salt in air atmosphere at 460 °C. The formation of K2U2O7 resulted from the reaction between UO3 and K2CO3 and the formation process is showed in reaction (3).37 When temperature was 720 °C, a new peak of K2UO4 was observed in spent salt. Because carbonate was in excess, K2U2O7 reacted with an excess of K2CO3 to form K2UO4 and this process is showed in reaction (4).37 Compared to N2 atmosphere (Fig. 8a), the peak intensities of K3Na(SO4)2 obviously strengthened in air atmosphere (Fig. 8b), which meant that more sulfate remained in spent salt. The result was consistent with the high retention rate of sulfur in air atmosphere at 720 °C in Fig. 5b.
| | |
K2CO3 + 2UO3 → K2U2O7 + CO2
| (3) |
| | |
K2CO3 + K2U2O7 → K2UO4 + CO2
| (4) |
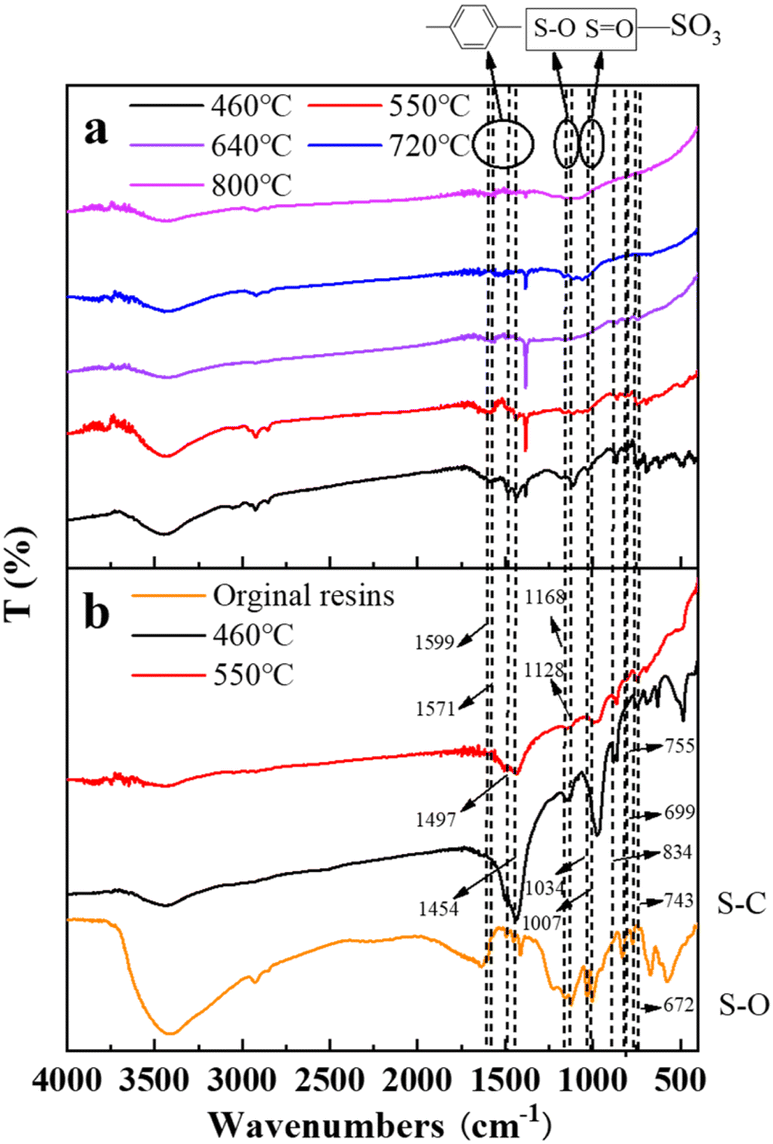
3.2.5 Fourier transform infrared spectrometer (FT-IR) analysis. Fig. 12 describes the FT-IR spectrograms of residue after the decomposition of uranium-containing resins at different temperatures in carbonate molten salt in N2 and air atmospheres. The peak (743 cm−1) was the characteristic absorption of the S–C bond. The peaks (699 and 755 cm−1) were ascribed to the twisting and wobbling of the benzene ring.38,39 The peak (834 cm−1) was due to outside surface bending of C–H in the benzene ring. The peaks (1007 and 1034 cm−1) were attributed to the characteristic absorption of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in the sulfonic acid group functional group (–SO3H), and the peaks (672, 1128 and 1168 cm−1) were the characteristic absorption of the S–O bond.40,41 The peaks (1454, 1497, 1571 and 1599 cm−1) corresponded to the CC stretching of the aromatic benzene ring.42,43
O bond in the sulfonic acid group functional group (–SO3H), and the peaks (672, 1128 and 1168 cm−1) were the characteristic absorption of the S–O bond.40,41 The peaks (1454, 1497, 1571 and 1599 cm−1) corresponded to the CC stretching of the aromatic benzene ring.42,43
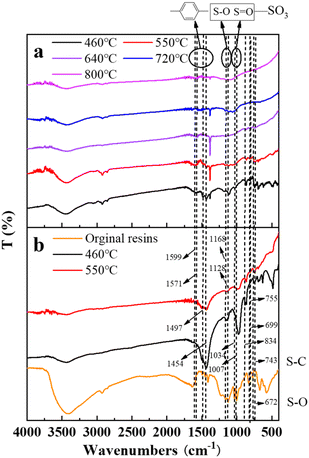 |
| | Fig. 12 The FT-IR spectrograms of residue after the decomposition of uranium-containing resins at different temperatures in carbonate molten salt in (a) air and (b) N2 atmospheres. | |
In Fig. 12, compared to N2 atmosphere (Fig. 12b), the peak intensities of the benzene ring and sulfonic acid group functional group (–SO3H) after the decomposition of resins in carbonate molten salt in air atmosphere were relatively weaker at 460 °C in air atmosphere (Fig. 12a). It was determined that the addition of air prompted the destruction of the benzene ring and sulfonic acid group functional group (–SO3H). Associated with the analysis of Fig. 5a, the decomposition efficiency of resins (37.3%) in N2 atmosphere was slightly higher than that (44.7%) in air atmosphere, which meant that the benzene ring was destructed and could be further converted to other structures. These structures remained good thermal stability. In addition, the ion bond formed by uranyl ions on the sulfonic acid group had a certain thermal stability, which inhibited the decomposition of the sulfonic acid group in residues. When temperature increased from 550 °C to 720 °C, the intensity of characteristic peaks of the S–O bond and S–C bond in residues gradually weakened and basically disappeared at 720 °C, which was ascribed to the oxidation decomposition of the S–O bond and S–C bond in air atmosphere.
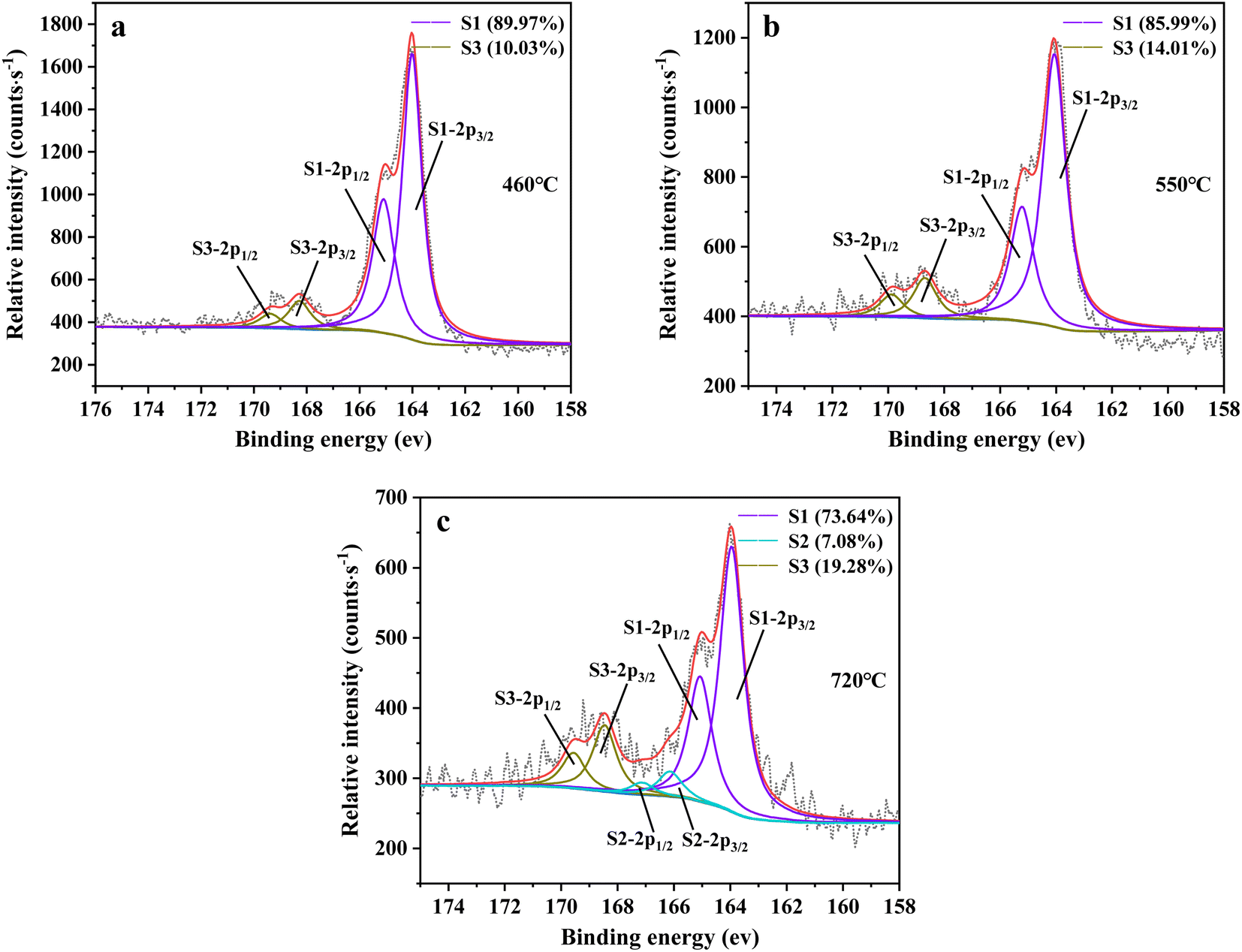
3.2.6 X-ray photoelectron spectrometer (XPS) analysis. XPS spectroscopy can be further utilized to investigate the change in sulfur speciation and component percentage of sulfur species in residue. According to the binding energy of sulfur compounds, sulfur (2p) spectra was determined using components at the peaks of 2p1/2 and 2p3/2 with separated energy of 1.18 eV.44,45 The fitting result is presented in Fig. 13. From Fig. 13, there were three sulfur compounds existed in residues, such as thiophene sulfur (164.1 ± 0.2 eV), sulfoxide sulfur (165.7 ± 0.3 eV) and sulfone sulfur (168.0 ± 0.5 eV).46–49 The binding energy gradually increased with the oxidation state of sulfur rising, which was consistent with the experimental result of Urban et al.50
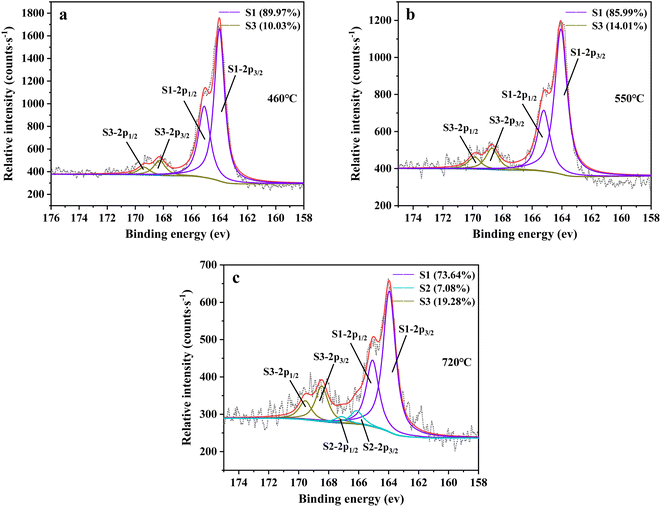 |
| | Fig. 13 Sulfur speciation in residue after the decomposition of uranium-containing resins and the spectra of S 2p conducted through the XPS spectrograph analysis (S1: thiophene sulfur, S2: sulfoxide sulfur, S3: sulfone sulfur) at (a) 460 °C, (b) 550 °C and (c) 720 °C in carbonate molten salt in N2 atmosphere. | |
From Fig. 13a, there were two sulfur species existing in residues at 460 °C, including thiophene sulfur (89.97%) and sulfone sulfur (10.03%). The formation of thiophene sulfur was ascribed to the condensation reaction of organic sulfide.51 Whereas, organic sulfide was relatively unstable and easily destroyed at high temperatures,52 so organic sulfide was not observed in Fig. 13a. When resins were destroyed at 550 °C, the relative proportion of thiophene sulfur decreased from 89.97% to 85.99% and sulfone sulfur increased from 10.03% to 14.01%. The change in sulfur compounds in residues was due to the transformation of sulfur species.53 The increase in temperature accelerated the decomposition of parts of thiophene sulfur. This decomposition of thiophene sulfur is shown in reaction (5).
| |
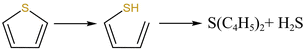 | (5) |
In addition, combined with the component analysis of spent salt (Fig. 8a), parts of UO3 were converted to U3O8 at 550 °C and this conversion was accompanied by the production of oxygen (as exhibited in Fig. 9b).31 When temperature was increased to 720 °C, sulfoxide sulfur was found in residues and the relative proportion was 7.08% in Fig. 13c. The relative proportion of sulfone sulfur increased from 14.01% to 19.28%, while that of thiophene sulfur significantly decreased from 85.99% to 73.64%. The increase in relative proportion of sulfone sulfur and sulfoxide sulfur corresponded to the decrease in relative proportion of thiophene sulfur, which was due to the decomposition of thiophene sulfur and the conversion of sulfone sulfur to sulfoxide sulfur. Meanwhile, the release of oxygen gradually increased with increasing temperature and a small amount of oxygen was chemically dissolved in molten salt.54,55 The solubility of oxygen in carbonate molten salt also resulted in the oxidation of fractional thiophene sulfur and the conversion of some sulfone sulfur to sulfoxide sulfur. This transformation can be expressed in reactions (6)–(8), which was confirmed by the decrease in thiophene sulfur and the increase in sulfoxide sulfur during the molten salt decomposition of resins.56 Additionally, the production of CO2 and SO2 in these reactions corresponded to the experimental results in Fig. 2c and d.
| | |
S(C4H5)2 + 11.5O2 → 8CO2 + SO2 + 5H2O
| (7) |
| |
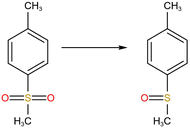 | (8) |
Fig. 14 shows sulfur speciation in residue after the decomposition of uranium-containing resins and the spectra of S 2p conducted through the XPS spectrograph at different temperatures in carbonate molten salt in air atmosphere. In Fig. 14a, there were three sulfur compounds existed in residues at 325 °C, such as thiophene sulfur (25.61%), sulfone sulfur (47.53%) and sulfonic acid sulfur (26.86%). When temperature was 460 °C, the relative proportion of thiophene sulfur after the decomposition of resins was increased to 85.09% in Fig. 14b. The increase in the relative proportion for thiophene sulfur was ascribed to the transformation of sulfone sulfur and sulfonic acid sulfur. When temperature was increased to 550 °C, the relative proportion of sulfone sulfur and thiophene sulfur increased in Fig. 14c. Thus, the relative proportion in formation of sulfone sulfur and thiophene sulfur were more than that in consumption. When temperature was 720 °C, the decrease in relative proportion of thiophene sulfur corresponded to the increase in relative proportion of sulfate sulfur in Fig. 14d. It was determined that the increase in relative proportion of sulfate sulfur resulted from the oxidation decomposition of thiophene sulfur in residues. The presence of sulfate sulfur could be because alkali metal sulfates in molten salt came into the resin structure, which corresponded to the uniform distribution of elements (Na, K, S and O) in residues in Fig. 15 and was confirmed by the analytical result in Fig. 8b.
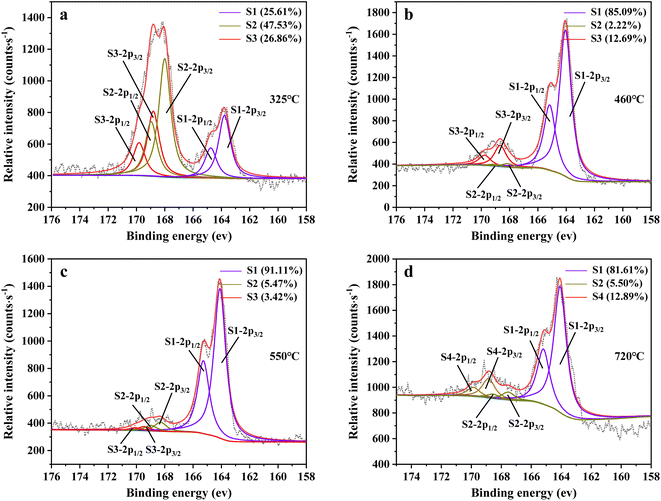 |
| | Fig. 14 Sulfur speciation in residue after the decomposition of uranium-containing resins and the spectra of S 2p conducted through the XPS spectrograph analysis (S1: thiophene sulfur, S2: sulfone sulfur, S3: sulfonic acid sulfur, S4: sulfate sulfur) at (a) 325 °C, (b) 460 °C, (c) 550 °C and (d) 720 °C in carbonate molten salt in air atmosphere. | |
 |
| | Fig. 15 EDS of residues after resin decomposition at 720 °C for 30 min. | |
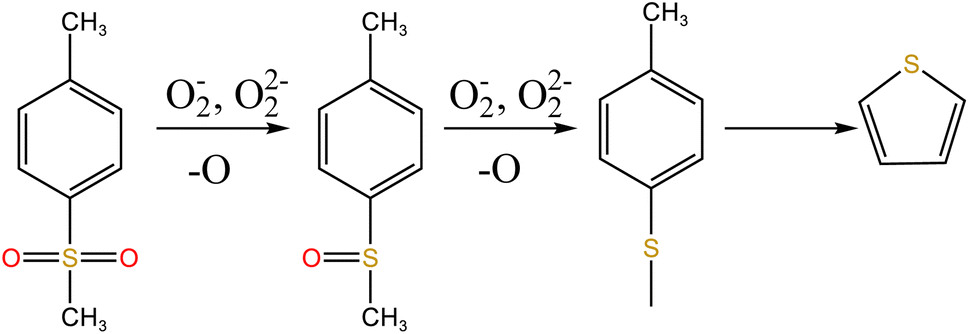
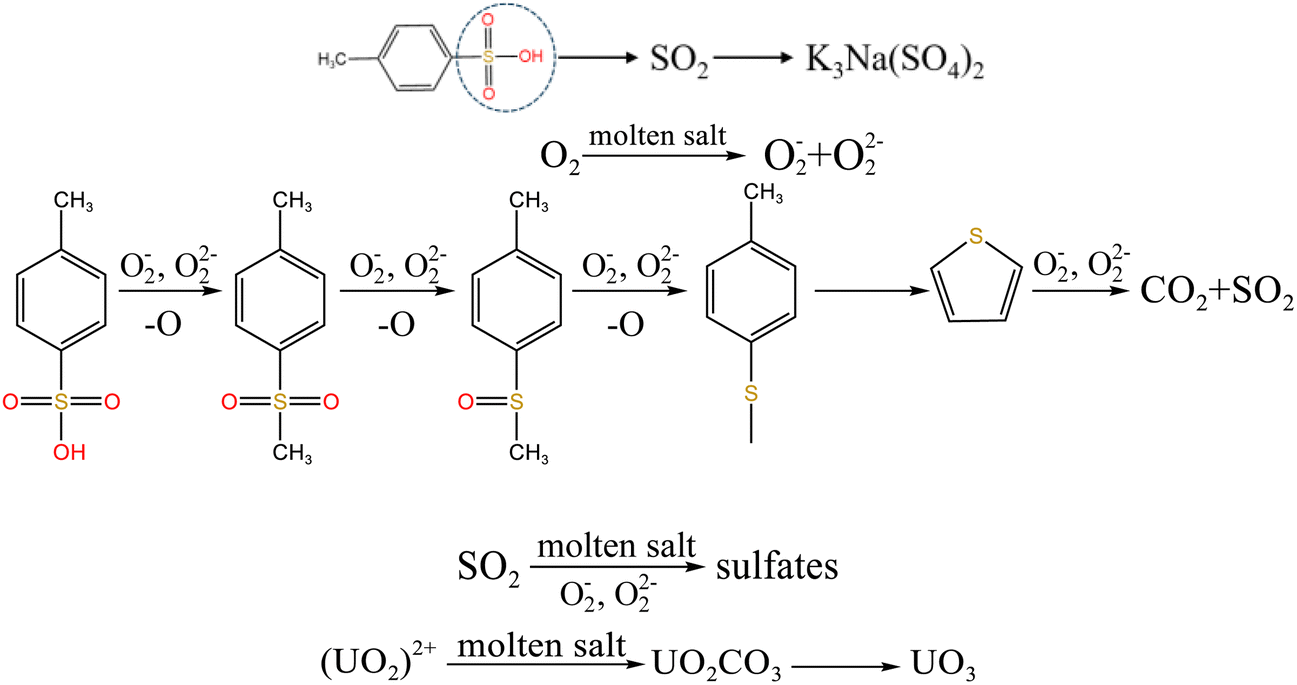
To explore the oxidation decomposition of uranium-containing resins in carbonate molten salt in air atmosphere, the characteristics of products after resin decomposition were studied. During the decomposition of resins, SO2 released from the cleavage of functional sulfonic acid groups was absorbed by carbonates to form sulfate at 325 °C. When temperature was 460 °C, K3Na(SO4)2 and UO3 were observed in spent salt. Compared to N2 atmosphere, the production temperatures of CH4, C2H4, CO2 and SO2 released from the decomposition of resins were essentially unchanged (Fig. 2). The relative proportions of sulfone sulfur (2.22%) in residues in air atmosphere greatly decreased at 460 °C (Fig. 14b). Due to the existence of oxygen in air atmosphere, oxygen was chemically dissolved in molten salt and formed peroxide and superoxide ions, which could accelerate the conversion of sulfone sulfur to thiophene sulfur and generate CO2 and SO2 (Fig. 2c and d). This conversion is showed in reactions (9)–(12). However, sulfonic acid sulfur (12.69%) still existed in residues. This could be because parts of uranyl ions in spent salt formed some stable ion bonds with sulfonic acid group in residue structure. These ion bonds were attached to the benzene ring of ST-DVB matrix, which could keep resin structure stable in Fig. 7c.
| | |
3O2 + 2CO2−3 → 4O−2 + 2CO2
| (9) |
| | |
O2 + 2CO2−3 → 2O2−2 + 2CO2
| (10) |
| |
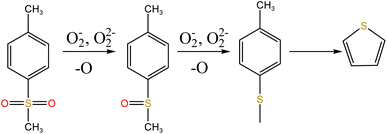 | (11) |
| |
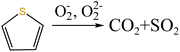 | (12) |
When temperature was 550 °C, U3O8, UO3 and K2U2O7 as the main uranium compounds existed in spent salt. The conversion of UO3 to U3O8 was accompanied by the production of a small amount of oxygen. Besides, these stable ion bonds between uranates and sulfonic acid group were destroyed. Fractional sulfonic acid groups were decomposed, which leaded to the increase in relative proportion of sulfone sulfur and thiophene sulfur. Associated with the TG analysis of Fig. 1, ST-DVB matrix and polymer chains in resins was oxidized by peroxide and superoxide ions in molten salt at 550 °C, which resulted in the increase in CO2 release content (Fig. 2c) and the cleavage of resin structure (Fig. 7d). With the increase of temperature, the release content of CO2 gradually increased. The uranium compounds essentially unchanged in spent salt at 640 °C. As temperature was 720 °C, UO3 and K2UO4 was the main uranium compounds in spent salt. A majority of reins were oxidized and decomposed, and ST-DVB matrix and polymer chains were basically completely destroyed, which corresponded to the increase in the decomposition efficiency of resins (Fig. 5a) and the morphology change of residues (Fig. 7f). Meanwhile, thiophene sulfur in residues was further oxidized and decomposed (Fig. 14d). This inference was confirmed by the increase in retention rate of sulfur in molten salt with increasing temperature from 550 °C to 720 °C (Fig. 5b). Because thiophene sulfur had excellent stability, thiophene sulfur still existed in spent salt at 720 °C. Based on these analytical results, the decomposition of uranium-containing resins in molten carbonates in air atmosphere is presented in Table 1.
Table 1 The decomposition of uranium-containing resins in carbonate molten salt in air atmosphere
| Temperature range |
Decomposition process |
| 325–460 °C |
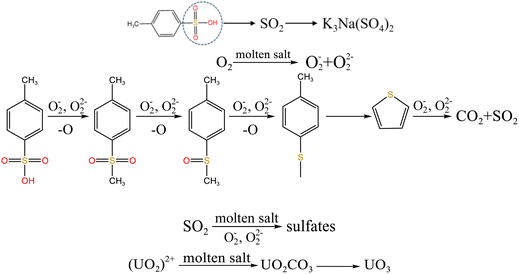 |
| 460–550 °C |
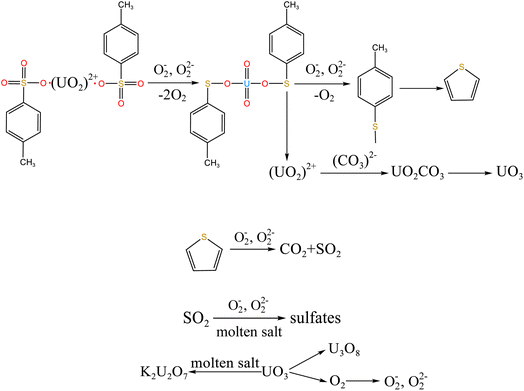 |
| 550–720 °C |
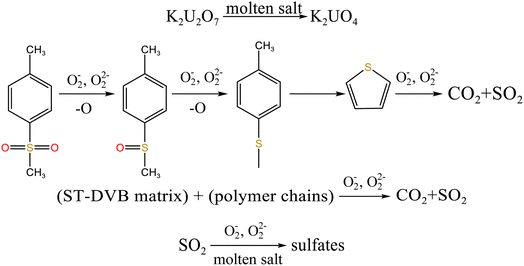 |
4. Conclusions
In this work, the oxidation decomposition of uranium-containing resins in Li2CO3–Na2CO3–K2CO3 molten salt system was investigated at different temperatures in air atmosphere. Based on the experimental analysis, the following conclusions were obtained:
(1) Compared to N2 atmosphere, the release content of exhaust gas CO2 released from the destruction of uranium-containing resins in air atmosphere was relatively high above 368 °C, which was because oxygen in the form of peroxide and superoxide ions in molten salt oxidized and decomposed ST-DVB matrix and polymer chains. The decrease in release content of SO2 at 454 °C was assigned to the absorption of SO2 by molten salt. The decomposition rate of resins in air atmosphere reached 82.6% at 800 °C in 30 min.
(2) Spent salt after resin decomposition was composed of sulfates and uranium compounds, and the retention rate of organic sulfur in spent salt at 800 °C was 68.7%. Besides, there were the transformation of UO3 to U3O8 and K2U2O7 to K2UO4 occurring in carbonate melt at 325–720 °C.
(3) These sulfur species showed a conversion law during the decomposition of uranium-containing resins in carbonate molten salt in air atmosphere. Peroxide and superoxide ions accelerated the conversion of sulfone sulfur to thiophene sulfur and generated CO2 and SO2 at 460 °C. The ion bond formed by uranyl ions on the sulfonic acid group inhibited the decomposition of the sulfonic acid group in residues. With the increase in temperature, sulfonic acid group was converted to sulfone sulfur (–SO2–), sulfoxide sulfur (–SO–) and further to thiophene sulfur. Finally, the oxidation decomposition of uranium-containing resins in molten carbonates in air atmosphere was explained.
(4) This work provided more theoretical guidance for the industrial treatment of uranium-containing resins and technical support for the sustainable development of the nuclear industry.
Author contributions
Zhi Zhang: data curation, supervision, conceptualization, writing-original draft, writing-reviewing & editing. Yun Xue: methodology, formal analysis, writing-reviewing & editing. Yong-De Yan: conceptualization, resources, investigation, writing-reviewing & editing. Guo-Qiang Li: data curation, supervision, conceptualization. Wen-Da Xu: project advisor, validation, formal analysis, writing-reviewing & editing. Fu-Qiu Ma: supervision, writing-reviewing & editing. Xin Liu: conceptualization, formal analysis, writing-reviewing & editing. Qing-Guo Zhang: conceptualization, formal analysis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was financially supported by Key R&D Program of Shandong Province, China, the National Natural Science Foundation of China (22176045 and 21976047), and University and Local Integration Development Project of Yantai, China (2020XDRHXMPT36), and the Sino-Russian Cooperation Fund of Harbin Engineering University (2021HEUCRF004).
References
- J. L. Wang and Z. Wan, Treatment and disposal of spent radioactive ion-exchange resins produced in the nuclear industry, Prog. Nucl. Energy, 2015, 78, 47–55 CrossRef CAS.
- L. X. Huang, W. X. He and D. G. Chen, Solid radioactive waste management in Daya Bay nuclear power station, Radiat. Prot., 2004, 24, 211–226 CAS.
- K. Kinoshita, M. Hirata and T. Yahata, Treatment of ion-exchange resins by fluidized bed incinerator equipped with copper oxide catalyst fundamental studies, J. Nucl. Sci. Technol., 1991, 28, 228–238 CrossRef CAS.
- C. M. Jantzen, W. E. Lee and M. I. Ojovan, Radioactive waste conditioning, immobilization, and encapsulation process and technologies: overview and advances, Radioactive Waste Management and Contaminated Site Clean-Up: Process, Technologies and International Waste Experience, 2013, pp. 171–272 Search PubMed.
- J. F. Li and J. L. Wang, Advances in cement solidification technology for waste radioactive ion exchange resins: A review, J. Hazard. Mater., 2006, 135, 443–448 CrossRef CAS PubMed.
- N. Moriyama, S. Dojiri, S. Emura, T. Sugo and S. Machi, Incorporation of radioactive spent ion exchange resins in plastics, J. Nucl. Sci. Technol., 1975, 12, 362–369 CrossRef CAS.
- L. Gong, D. H. Du, Li Cheng and Q. X. Zhao, Feasibility study of immobilization of simulated radioactive wastes using unsaturated polyester, Radiat. Prot., 1991, 11, 352–357 CAS.
- X. Fan, M. Q. H Lin, H. Z. Li, J. L. Tan, Y. H. Chang and L. Liu, A study on immobilization of spent resins with polyester, At. Energy Sci. Technol., 1994, 28, 348–355 CAS.
- Y. J. Huang, H. P. Wang, S. H. Liu and M. C. Hsiao, Pyrolysis kinetics of spent low-level radioactive resin, Nucl. Technol., 2002, 138, 206–210 CrossRef CAS.
- A. Nezu, K. Moro and T. Watanabe, Thermal plasma treatment of waste ion exchange resins by CO2 injection, Thin Solid Films, 2006, 506, 432–435 CrossRef.
- M. J. Quina, J. C. M. Bordado and R. M. Quinta-Ferreira, Chemical stabilization of air pollution control residues from municipal solid waste incineration, J. Hazard. Mater., 2010, 179, 382–392 CrossRef CAS PubMed.
- H. C. Yang, Y. J. Cho, H. C. Eun, J. H. Yoo and J. H. Kim, Molten salt oxidation of ion-exchange resins doped with toxic metals and radioactive metal surrogates, J. Nucl. Sci. Technol., 2005, 42, 123–129 CrossRef CAS.
- P. C. Hsu, K. G. Foster, T. D. Ford, P. H. Wallman, B. E. Watkins, C. O. Pruneda and M. G. Adamson, Treatment of solid wastes with molten salt oxidation, Waste Manage., 2000, 20, 363–368 CrossRef CAS.
- H. Nasir, P. Kassandra, L. Tristan and A. Tn, Thermal analysis and immobilization of spent ion exchange resin in borosilicate glass, New J. Glass Ceram., 2012, 2, 111–120 Search PubMed.
- H. C. Yang, M. W. Lee, I. H. Yoon, D. Y. Chung and J. K. Moon, Scale-up and optimization of a two-stage molten salt oxidation reactor system for the treatment of cation exchange resins, Chem. Eng. Res. Des., 2013, 91, 703–712 CrossRef CAS.
- N. Sathi Sasidharan, D. S. Deshingkar and P. K. Wattal, Vitrification of spent organic ion exchange resins-137Cesium volatility during oxidation, Bhabha Atomic Research Centre, 2006 Search PubMed.
- X. Z. Wang, Y. H. Zheng, Y. Xue, Y. D. Yan, F. Q. Ma, M. L. Zhang, H. Y. Bai, Z. Q. Kou and J. P. Liu, Study on the destruction process of cationic exchange resins treated by Li2CO3–Na2CO3–K2CO3 molten salt, J. Environ. Chem. Eng., 2021, 9, 105948 CrossRef CAS.
- P. Antonetti, Y. Claire, H. Massit, P. Lessart, C. Pham Van Cang and A. Perichaud, Pyrolysis of cobalt and cesium doped cationic ion-exchange resin, J. Anal. Appl. Pyrolysis, 2000, 55, 81–92 CrossRef CAS.
- H. C. Yang, J. S. Yun, M. J. Kang, J. H. Kim and Y. Kang, Mechanisms and kinetics of cadmium and lead capture by calcined kaolin at high temperatures, Korean J. Chem. Eng., 2001, 18, 499–505 CrossRef CAS.
- M. Matsuda, K. Funabashi and H. Yusa, Effect of metallic impurities on ion exchange resin oxidation rate, J. Nucl. Sci. Technol., 1985, 22, 241–243 CrossRef CAS.
- M. Matsuda, K. Funabashi and H. Yusa, Effect of Metallic Impurities on Oxidation Reaction of Ion Exchange Resin, (I) Catalytic Effect of Ionized and Unionized Irons, J. Nucl. Sci. Technol., 1986, 23, 244–252 CrossRef CAS.
- M. Matsuda, K. Funabashi and H. Yusa, Effect of Metallic Impurities on Oxidation Reaction of Ion Exchange Resin, (II) Comparison of Catalytic Activity between Six Metals, J. Nucl. Sci. Technol., 1986, 23, 813–818 CrossRef CAS.
- R. S. Juang and T. S. Lee, Oxidative pyrolysis of organic ion exchange resins in the presence of metal oxide catalysts, J. Hazard. Mater., 2002, 92, 301–314 CrossRef CAS PubMed.
- S. H. Yu, C. Zhang, X. P. Zhang, X. Liu, B. Wei, P. Tan, Q. Y. Fang, G. Chen and J. Xia, Release and transformation characteristics of Na/Ca/S compounds of Zhundong coal during combustion/CO2 gasification, J. Energy Inst., 2020, 93, 752–765 CrossRef CAS.
- C. Y. Cao, Y. Ren, H. Wang, H. Y. Hu, B. J. Yi, X. Li, L. L. Wang and H. Yao, Insights into the role of CaO addition on the products distribution and sulfur transformation during simulated solar-powered pyrolysis of waste tires, Fuel, 2022, 314, 122795 CrossRef CAS.
- J. R. Lanteigne, J. P. Laviolette and J. Chaouki, Behavior of sulfur during the pyrolysis of tires, Energy Fuels, 2015, 29, 763–774 CrossRef CAS.
- H. Darmstadt, C. Roy and S. Kaliaguine, Inorganic components and sulfur compounds in carbon blacks from vacuum pyrolysis of used tires, Kautsch. Gummi Kunstst., 1994, 47, 891–895 CAS.
- P. R. Westmoreland, J. B. Gibson and D. P. Harrison, Comparative kinetics of high temperature reaction between H2S and selected metal oxides, Environ. Sci. Technol., 1977, 11, 488–491 CrossRef CAS.
- J. Luo, W. R. Hu, Z. L. Suo, Y. B. Wang and Y. K. Zhang, Co-pyrolysis of spent radioactive ion exchange resin and manganese dioxide: Decrease the decomposition temperatures of functional groups, J. Hazard. Mater., 2021, 418, 126275 CrossRef CAS PubMed.
- H. C. Eun, H. C. Yang, Y. Z. Cho and H. S. Lee, Study on a stable destruction method of radioactive waste ion exchange resins, J. Radioanal. Nucl. Chem., 2009, 281, 585–590 CrossRef CAS.
- T. R. Griffiths and V. A. Volkovich, A review of the high temperature oxidation of uranium oxides in molten salts and in the solid state to form alkali metal uranates, and their composition and properties, J. Nucl. Mater., 1999, 274, 229–251 CrossRef CAS.
- A. F. Andresen, The structure of U3O8 determined by neutron diffraction, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 211, 612–614 Search PubMed.
- H. R. Hoekstra, S. Siegel, L. H. Fuchs and J. J. Katz, The Uranium-Oxygen System: UO2.5 to U3O8, J. Phys. Chem., 1955, 59, 136–138 CrossRef CAS.
- K. M. Efremova, E. A. Ippolitova and Y. P. Simanov, et al., An investigation of the composition of alkali element uranates obtained by a dry procedure, Dokl. Akad. Nauk SSSR, 1959, 124, 29 Search PubMed.
- A. R. Beketov, V. N. Strekalovskii and V. G. Vlasov, A study of the structure of solid solutions of uranium oxides in the range α-UO3-U3O8, J. Struct. Chem., 1965, 6, 64–67 CrossRef.
- E. H. P. Cordfunke and B. O. Loopstra, Sodium uranates: Preparation and thermochemical properties, J. Inorg. Nucl. Chem., 1971, 33, 2427–2436 CrossRef CAS.
- V. A. Volkovich, T. R. Griffiths, D. J. Fray and R. C. Thied, Solubilities
and solubilisation enthalpies of alkali metal uranates(vi) in carbonate melts, Phys. Chem. Chem. Phys., 1999, 1, 3297–3302 RSC.
- P. J. Wibawa, M. A. Agam, H. Nur and H. Saim, Changes in physical properties and molecular structure of polystyrene nanospheres exposed with solar flux, 2010 International Conference on Enabling Science & Nanotechnology, AIP Conf. Proc., 2011, 1341, 54–61 CrossRef CAS.
- J. Sheng and Y. Z. Wan, Preparation and characterization of TiO2/polystyrene core-shell nanospheres via microwave-assisted emulsion polymerization, Mater. Lett., 2008, 62, 37–40 CrossRef.
- L. J. Koenig, Application of fourier transform infrared spectroscopy to chemical systems, Appl. Spectrosc., 1975, 29, 293–308 CrossRef.
- R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric identification of organic compounds, J. Chem. Educ., 2014, 826–827 Search PubMed.
- A. Refik and U. Nurseli, Study of the morphological and thermal properties of polystyrene nanocomposites based on modified halloysite nanotubes with styrene-maleic anhydride copolymers, Mater. Today Commun., 2017, 13, 255–262 CrossRef.
- S. Luciano, M. S. Mariana, L. S. Arxel, A. S. C. Adriana, M. A. Diana, F. M. Rachel and B. Roberto, Synthesis and characterization of styrene-vinyl tetrazole copolymers for their application as a solid electrolyte, React. Funct. Polym., 2021, 167, 105007 CrossRef.
- L. Wang, P. Z. Zhang and M. Zheng, Study on structural characterization of three Chinese coals of high organic sulphur content using XPS and solid-state NMR spectroscopy, J. Fuel Chem. Technol., 1996, 24, 539–543 CAS.
- M. A. Olivella, J. M. Palacios, A. Vairavamurthy, J. C. Delrio and F. X. C. Delasheras, A study of sulfur functionalities in fossil fuels using destructive-(ASTM and Py-GC-MS) and non-destructive-(SEM-EDX, XANES) and (XPS) techniques, Fuel, 2002, 81, 405–411 CrossRef CAS.
- L. Flandinet, F. Tedjar, V. Ghetta and J. Fouletier, Metals recovering from waste printed circuit boards (WPCBs) using molten salts, J. Hazard. Mater., 2012, 213, 485–490 CrossRef PubMed.
- C. Q. Lin, Y. Chi, Y. Q. Jin, X. G. Jiang, A. Buekens, Q. Zhang and J. Chen, Molten salt oxidation of organic hazardous waste with high salt content, Waste Manage. Res., 2018, 36, 140–148 CrossRef CAS PubMed.
- S. Kelemen, G. George and M. Gorbaty, Direct determination and quantification of sulphur forms in heavy petroleum and coals: 1. The X-ray photoelectron spectroscopy (XPS) approach, Fuel, 1990, 69, 939–944 CrossRef CAS.
- S. R. Kelemen, M. Afeworki, M. L. Gorbaty, M. Sansone, P. J. Kwiatek, C. Walters, H. Freund, M. Siskin, A. E. Bence, D. Curry, M. S. Solum and R. Pugmire, Direct characterization of kerogen kerogen by X-ray and solid-state, Energy Fuels, 2007, 21, 1548–1561 CrossRef CAS.
- N. R. Urban, K. Ernst and S. Bernasconi, Addition of sulfur to organic matter during early diagenesis of lake sediments, Geochim. Cosmochim. Acta, 1999, 63, 837–853 CrossRef CAS.
- J. L. Hou, Y. Ma and S. Y. Li, Transformation of sulfur and nitrogen during Shenmu coal pyrolysis, Fuel, 2018, 231, 134–144 CrossRef CAS.
- H. Y. Hu, Y. Fang and H. Liu, The fate of sulfur during rapid pyrolysis of scrap tires, Chemosphere, 2014, 97, 102–107 CrossRef CAS PubMed.
- S. Cheng, Y. Qiao, J. Huang, L. Cao, H. Yang, H. Liu, Y. Yu and M. Xu, Effect of alkali addition on sulfur transformation during low temperature pyrolysis of sewage sludge, Proc. Combust. Inst., 2017, 36, 2253–2261 CrossRef CAS.
- J. A. Appleby, Solubilities of Oxygen and Carbon Monoxide in Carbonate Melts, J. Electrochem. Soc., 1980, 8, 1655–1659 CrossRef.
- T. Nishina, Y. Masuda and I. Uchida, Gas solubility and diffusivity of H2, CO2 and
O2 in molten alkali carbonates, Proc. Int. Symp. Molten Salt Chemistry and Technology, 1993, 424–435 CAS.
- T. Kamo, K. Takaoka, J. Otomo and H. Takahashi, Effect of steam and sodium hydroxide for the production of hydrogen on gasification of dehydrochlorinated poly(vinyl chloride), Fuel, 2006, 85, 1052–1059 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Guo-Qiang Lib,
Wen-Da Xuc,
Fu-Qiu Maa,
Xin Liua and
Qing-Guo Zhanga
*a,
Guo-Qiang Lib,
Wen-Da Xuc,
Fu-Qiu Maa,
Xin Liua and
Qing-Guo Zhanga