
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArtemisia herba-alba sesquiterpenes: in silico inhibition in the ATP-binding pocket†
Tarik A. Mohamed *a,
Mohamed H. Abd El-Razekb,
Ibrahim A. Saleha,
Sherin K. Alia,
Abeer A. Abd El Atyc,
Paul W. Paréd and
Mohamed-Elamir F. Hegazy*a
*a,
Mohamed H. Abd El-Razekb,
Ibrahim A. Saleha,
Sherin K. Alia,
Abeer A. Abd El Atyc,
Paul W. Paréd and
Mohamed-Elamir F. Hegazy*a
aChemistry of Medicinal Plants Department, National Research Centre, 33 El-Bohouth St., Dokki, Giza, 12622, Egypt. E-mail: tarik.nrc83@yahoo.com; elamir77@live.com; Tel: +20-11-275-39-989 Tel: +20-33-371-635
bDepartment of Natural Compounds Chemistry, National Research Centre, 33 El-Bohouth St., Dokki, Giza, 12622, Egypt
cChemistry of Natural and Microbial Products Department, National Research Centre, 33 El-Bohouth St., Dokki, Giza, 12622, Egypt
dDepartment of Chemistry & Biochemistry, Texas Tech University, Lubbock, TX 79409, USA
First published on 28th June 2023
Abstract
To identify antimicrobial leads for medical applications, metabolites from the aerial part of Artemisia herba-alba were extracted and chromatographically purified. Two new sesquiterpenes, 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide (1) and 1β,6α,8α-trihydroxy, 11α-methyl-eudesma-4(15)-en-13-propanoate (2) along with a known eudesmanolide 11-epi-artapshin (3) were identified. Structures were determined by spectroscopic methods including 1D- and 2D-NMR as well as mass spectroscopy. Compound 3 inhibited Gram-positive bacteria Bacillus subtilis, Lactobacillus cereus and Staphylococcus aureus and exhibited antifungal activity against the pathogenic fungus F. solani. The mode-of-action of these antimicrobial sesquiterpenes as bacterial type II DNA topoisomerase and/or DNA gyrase B inhibitors were examined via in silico studies. Such molecular-docking studies were also employed to examine antifungal activity against an N-myristoyl transferase (NMT) target. Compound 3 had the greatest gyrase B binding affinity in the ATP-binding pocket and was found to possess an inhibitory action against non-invasive micro-test technology (NMT).
1. Introduction
Plant, microbial and marine natural products provide vital leads for drug discovery with more than 50% of currently authorized Western medications being derived from natural sources.1 Medicinal herbs also have a rich tradition in ancient and Eastern pharmacopeias.2 The family Asteraceae contains numerous plants rich in antioxidants and antimicrobials.3–7 The genus Artemisia (Asteraceae) is a widely-dispersed genera, with over 500 varieties spread over temperate areas including North America, Europe, and Asia.8 A number of recent phytochemical and pharmacological investigations utilizing species from Artemisia,9–12 can be contributed, as least in part to the recognition of the sesquiterpenoid lactone artemisinin as a highly active natural product that serves as an effective treatment for malaria. The elucidation of this natural product as an anti-malaria treatment resulted in Tu Youyou being awarded the Nobel Prize in 2015.13,14 Before the discovery of antibiotics, Artemisia was a common element in herbal tea blends, advised to TB patients to prevent sudation and later identified as the active component for chronic bronchitis.15 Along with treating mental and neurological illnesses, the plant is used as an insecticide in the treatment of sweating, fever, rheumatism, and rheumatoid arthritis.16,17 Artemisia is rich in terpenoids, flavonoids, coumarins, acetylenes, caffeoylquinic acids, and sterols. The plant has also been identified as having beneficial activity as an antimalarial, antiviral, antitumor, antipyretic, antihemorrhagic, anticoagulant, antianginal, antioxidant, anti-hepatitis agent.18–23Computer-Aided Drug Designing (CADD) has gained great acceptance among biologists and chemists as a part of an interdisciplinary drug discovery approach. It plays an essential role in drug discovery, design, and analysis in the pharmaceutical industry. It is significantly used to reduce cost and speed up early-stage development of biologically new active molecules.24 Recently, infections of resistant bacteria have become very common25–27 and many pathogens; have become resistant to different classes of antibiotics.27–29 A growing risk of antimicrobial resistance has provided a robust impetuous for identifying new antimicrobial agents with structural features different from existing antibiotics.27,30 DNA gyrases have become an attractive target for anticancer and antibacterial research as they are essential enzymes for cell survival in prokaryotes. Searching for new inhibitors of the ATP-binding pocket of gyrase B enzyme (PDB code 4GEE) is attracting the attention of the pharmaceutical industry.31,32
A. herba-alba, commonly known as white wormwood is a perennial dwarf shrub with greenish-silver leaves; it grows in dry and/or semi-arid areas.33 In is found in North Africa, Spain, the Sinai Peninsula, Middle East, Northwestern Himalayas, and India; it is also found along the Mediterranean Sea.34 Since ancient times, the plant has been utilized in folk medicine; Moroccan traditional medicine uses it to treat arterial hypertension and/or diabetes.34 The leaves can be prepared as an herbal tea as an analgesic, an antibiotic, an antispasmodic, and a hemostatic.35 An ethanol extract is used for neurological problems with demonstrated GABAA-benzodiazepine receptor action.36 Aqueous extracts have anti-initiation and anti-promotional action against tumors,37 and a methylene chloride![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol extract (1:1) exhibits antinociceptive activity.38
:methanol extract (1:1) exhibits antinociceptive activity.38
The plant is highly polymorphic with many documented chemotypes.39 This is significant as differing compound profiles will occur for specific chemotypes with differing medical effects depending on the chemotype. Based on the distinct sesquiterpene lactone profile observed in plant samples collected, we propose the emergence of a new A. herba-alba chemotype that is present in the south of the Sinai (Saint Catherine district), Egypt.
2. Results and discussion
2.1. Identification and structure elucidation
Chromatographic fractionation and purification of an organic extract of A. herba-alba afforded two new sesquiterpenes, 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide (1) and 1β,6α,8α-trihydroxy, 11α-methyl-eudesma-4(15)-en-13-propanoate (2) along with a known eudesmanolide 11-epi-artapshin (3) (Fig. 1).40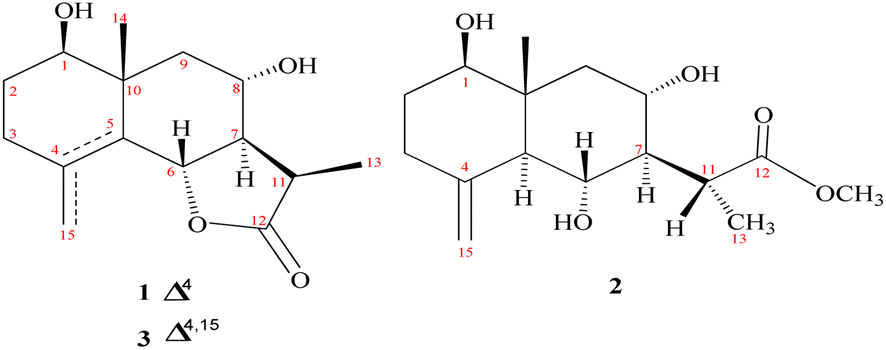 | ||
| Fig. 1 Identified metabolites (1–3). | ||
Compound 1 was purified as a clear gum with [α]25D = +46.5 (MeOH; c = 0.5); the chemical formula C15H22O4 was identified from the HRESIMS ion peak at m/z 267.1518 (calcd 267.1522). Twenty-two protons and fifteen carbon resonances were identified based on 1H and 13C NMR data (Table 1). The 13C NMR and DEPT spectra revealed three quartets, three triplets, five doublets, and four singlets. According to the IR spectrum, there is hydroxyl functionality at νmax 3300 and 3560 cm−1, as well as a γ-lactone carbonyl ring at νmax 1770 and 1785 cm−1. An HSQC experiment confirmed three methyls classified as: [one tertiary (δH/δC 1.03 s/19.6); one secondary (δH/δC 1.27 (d, J = 7.7 Hz)/9.4); and one vinylic (δH/δC 1.78 s/19.6)]; three saturated methylenes were [δH/δC 1.63 m, 1.66 dd (J = 14.0, 5.0 Hz)/26.4, δH/δC 2.15 td (11.7, 7.7), 1.95 brdd (J = 14.0, 3.5 Hz)/33.4 and δH/δC 1.13 t (J = 12.9 Hz), 2.26 dd (J = 12.9, 4.5 Hz)/47.8] and five methines three of them were oxygenated methines [δH/δC 3.44 dd (J = 12.4, 4.4 Hz)/77.1, δH/δC 4.73 brd (J = 11.7 Hz)/80.0 and δH/δC 3.89 td (J = 11.7, 4.1 Hz)/64.7] and the other two were aliphatic methines [δH/δC 2.10 dddd (14.0, 11.5, 3.5, 1.5)/54.6 and δH/δC 2.78 dq (J = 7.7, 7.7 Hz)/36.8]. The resonance frequencies for four non-protonated carbons were also observed in the 13C NMR spectra (Table 1); two of them olefinic at δC 126.9, a third was an aliphatic carbon at δC 41.0 and a fourth was a carbonyl unit at δC 180.0. In accordance with the above 1H and 13C NMR data (Table 1), compound 1 contains an eudesmane skeleton, with five degrees of unsaturation assigned to a double bond and a carbonyl while the other degrees of unsaturation are due to the tricyclic frame skeleton. Two spin systems, H-1-H-2-H-3 and H-6-H-7(H-11-H-13)-H-8-H-9, were identified in the 1H–1H COSY spectrum (Fig. 2). H2-2 (δH 1.66, 1.63) was correlated with a single oxygenated methine proton (δH 3.44, H-1) and vicinal protons (δH 2.15, 1.95, H2-3) leading to the initial spin system of C1(H)O–C2(H2)–C3(H2). In the second spin system, H-7 (δH 2.10) is coupled with H-6 (δH 4.73) and H-8 (δH 3.89), which in turn is coupled to H2-9 (δH 1.13, 2.26) (Fig. 2). This establishes connectivity between C-1 and C-3 as well as C-6 and C-9. The alpha methine proton of the γ-lactone ring (δH 2.10, H-7) correlates with a secondary methyl (δH 1.27, Me-13) and an aliphatic oxygenated methines located at the β-position adjacent to it in the 1H–1H COSY spectrum. The HMBC correlations (Fig. 2) of H-3/C-15, C-4; H-6/C-4, C-5; and H2-9/C-10, C-1, C-14 indicated a linkage between C-3 and C-5 via C-4 and between C-1 and C-9 via C-10 with a double bond between C-4 and C-5. The oxymethine group at δH 3.44 was assigned to C-1 based on HMBC long-range couplings between H3-14 (δH 1.03) and C-1 (δC 77.1), as well as between H-1 (δH 3.44) and C-3 (δC 33.4) and C-5 (δC 126.9). According a doublet coupling pattern (J = 11.7 Hz) and an HMBC cross-peak between H-7 (δH 2.10) and C-6, the second oxymethine group at δH 4.73 was assigned to C-6 (δC 80.0). Since C-8 is unique with a vicinal proton–proton couplings with three protons, the coupling pattern at δH 3.89 td (J = 11.7, 4.1 Hz) established an oxymethine unit. HMBC correlations between H-8 (δH 3.89) and C-7, C-9, C-10, C-6 and C-11, corroborated with a hydroxyl group at C-8. A HMBC coupling between C-5 and C-10 was identified via correlation of CH3-14 with C-5, H2-9 with C-5, and H-6 with C-10. The COSY couplings of CH3-13 and H-11, together with the agreement of H-11 HMBC correlations with C-12, C-7 and C-6, as well as de-shielding of C-12 at δC 179.94, indicated a γ-lactone ring. The NOESY data (Fig. 3) and coupling constants established the relative stereochemistry. Based on biogenetic precedent and in accordance with previously documented NMR chemical shifts for related sesquiterpene lactones, H-7 relative stereochemistry was assigned to an α-configuration.22 The large vicinal coupling (J = 11.7 Hz) indicated a trans H-6-H-7 conformation. The multiplicity on the chemical shift of the doublet characteristic for H-6 suggested a trans-eudesmane-12,6-olide. Based on COSY, HMBC, coupling constant and comparison with the previously eudesmanolides reported,22 the double doublets at δH 3.44 (J = 12.4, 4.4 Hz) originates from an axial proton at C-l, geminal to an OH group and a triplet doublet at δH 3.89 (J = 11.7, 4.1 Hz) indicating that the carbinol proton (H-8) is in an anti-periplanar orientation. These spectroscopic results supported an eudesmane-type sesquiterpene lactone. The NMR findings (Table 1) are comparable to those for 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide.41 Actually, the former compound was diagnosis from a mixture with other compounds and could be obtained from 8α-hydroxytaurin by reduction with sodium borohydride. The distinguishing signal for C-11 was discovered by a comparison between the biodiversity of such species and data from the literature.42
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 77.1 | 3.44 dd (12.4, 4.4) | 78.3 | 3.38 dd (4.1, 10.7) |
| 2axi | 26.4 | 1.63 m | 31.5 | 1.49 dddd (14.0, 11.0, 10.7, 4.3) |
| 2eq | 1.66 dd (14.0, 5.0) | 1.74 m | ||
| 3axi | 33.4 | 2.15 td (11.7, 7.7) | 34.9 | 2.02 m |
| 3eq | 1.95 brdd (14.0, 3.5) | 2.35 m | ||
| 4 | 126.9 | — | 145.1 | |
| 5 | 126.9 | — | 54.7 | 1.80 brd (10.6) |
| 6 | 80.0 | 4.73 brd (11.7) | 65.4 | 3.78 t (10.6) |
| 7 | 54.6 | 2.10 dddd (14.0, 11.5, 3.5, 1.5) | 54.4 | 2.00 t (11.0) |
| 8 | 64.7 | 3.89 td (11.7, 4.1) | 65.6 | 3.55 td (11.7, 4.1) |
| 9axi | 47.8 | 1.13 t (12.9) | 45.7 | 1.15 t (11.7) |
| 9eq | 2.26 dd (12.9, 4.5) | 2.33 dd (11.7, 4.3) | ||
| 10 | 41.0 | — | 40.8 | |
| 11 | 36.8 | 2.78 dq (7.7, 7.7) | 36.1 | 3.17 q (6.9) |
| 12 | 180.0 | — | 178.6 | |
| 13 | 9.4 | 1.27 d (7.7) | 10.5 | 1.12 d (6.9) |
| 14 | 19.6 | 1.03 s | 12.7 | 0.67 s |
| 15 | 19.6 | 1.78 s | 108.4 | 4.67 brd (1.5) |
| 4.97 brd (1.5) | ||||
| OCH3 | 52.0 | 3.64 s | ||
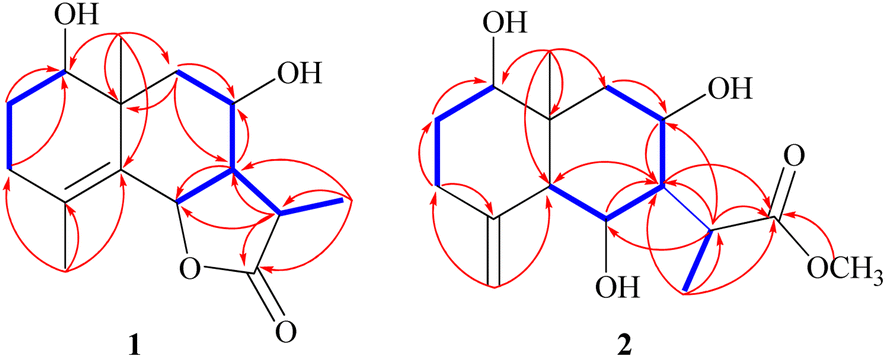 | ||
| Fig. 2 Observed 1H–1H COSY and HMBC correlations (shown in blue and red, respectively) for 1–2. | ||
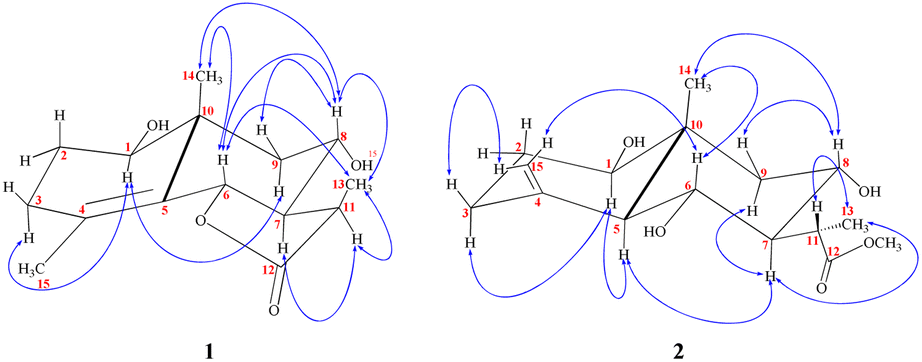 | ||
| Fig. 3 Observed NOESY correlations for 1–2. | ||
The stereochemistry at C-11 was established using the value of the NMR coupling between H-7 and H-11.42 The C-11 proton of 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide,41 appears as a double quartet at δH 2.34. The JH7–11 = 11.7 Hz and JH11–13 = 7.0 Hz frequencies represent the X of the A3MX pattern. In accordance with the Newman projection, the C-11 proton is positioned in a pseudo-axial orientation (Fig. 4A). As seen in Fig. 4B, the C-11 proton moves into a pseudo-equatorial orientation and the pattern changes to be located at δH 2.78 with JH7–11 = 7.7 Hz. Based on spectral data for 1 in comparison with 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide,41 the two compounds are epimeric eudesmanolides. However, significant downfield changes in the signals of H-6, H-7, and H-11, along with a significant upfield shift of the Me-13 are seen for 1, together with a decline in the coupling constant from 11.7 Hz in 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide,41 to 7.7 Hz in 1. The inverted order of the Me-13 absorption observed in the NMR spectra, where the resonance of Me-13 equatorial (sharing a co-planarity with C-12, Fig. 4A) is found further downfield (ΔδH = +0.12 ppm) than that of 1, is attribute to the anisotropic deshielding effect of the carbonyl group that is a component of the γ-lactone ring (Fig. 4B). 13C NMR spectral comparisons between 1 and 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide41 shows significant differences in some chemical shifts (Table 1). These changes include an up-field shift for C-7 (ΔδC = −4.0 ppm), C-8 (ΔδC = −6.3 ppm) and C-11 (ΔδC = −4.0 ppm), as well as C-13 (ΔδC = −5.0 ppm). The methyl equatorial nature (Fig. 4A) results in a decrease in the β-effect on C-7 and γ-effect on C-8, as predicted, causing downfield shifts C-7 and C-8 in comparison with 1. NOESY data (Fig. 3) validated the relative stereochemistry for 1 with the opposite configuration for the C-12 stereogenic center, as well as changes in the coupling constant for H-12. Thus, 1 is indeed a 12-epimer of 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide.
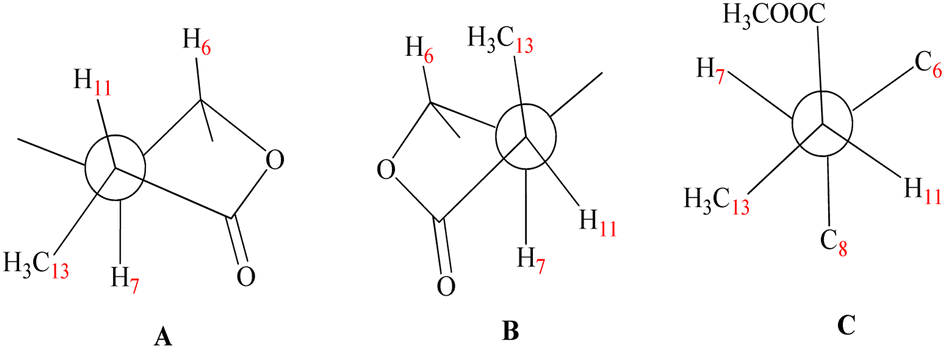 | ||
| Fig. 4 Newman projections [A] H-11 displays data for the coupling constants (JH7–11 = 11.7 Hz) and (JH11–13 = 7.0 Hz) in a pseudo-axial orientation. [B] (JH7–11 = 7.7 Hz) and (JH11–13 = 7.0 Hz) are values of the coupling constants which might be seen in the pseudo-equatorial configuration of H-11. [C] Confirmed that compound 2 virtually completely dismisses the coupling between H-7 and H-11. | ||
Compound 2 is a colorless oil with a positive optical rotation [α]25D = +55.2 (MeOH; c = 0.5). A prominent IR band at 1740 cm−1 indicated a carbonyl ester group, while broad band's at νmax 3300 and 3560 cm−1 suggested free hydroxyl functionality. Other bands at νmax 3090, 1650, and 900 cm−1 were attributed to the di-substituted carbon–carbon double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2). HRESIMS (m/z 299.1782) analysis indicated a molecular formula of C16H26O5 and 13C NMR data (Table 1) was consistent with an additional carbon deviating from the common sesquiterpene carbon number and four degrees of unsaturation. One methyl ester was detected based on the 13C and DEPT NMR [δC 178.6 (s), 52.0 (q)]. Additional signals at δC 145.1 (s), 108.4 (t), were attributed to an exocyclic double bond accounting for two degrees of unsaturation with a bicyclic frame accounting for the other degrees of unsaturation. 13C NMR and DEPT data (Table 1), indicated two methyls (one of which was angular δC 12.7); and the other was secondary (δC 10.5), three sp3 methylenes (δC 31.5, 34.9, and 45.7), six sp3 methines, including three oxygenated carbons (δC 78.3, 65.4 and 65.6), as well as the other three aliphatics (δC 54.7, 54.3 and 36.1) and one sp3 quaternary carbon at δC 40.8. These NMR findings complemented those of 1, assuming that 2 contain an eudesmane skeleton. NMR signal similarity from C-1–C-10 suggests shared rings of eudesmane derivatives with the respect of the different position of a double bond. The exocyclic double bond Δ4,15 in 2 was replaced by a methyl group (Me-15) and relocated to be Δ4,5 as an olefinic bond in 1. This was demonstrated by the lack of sp2 methylene [δC 145.1(C-4); δH/δC 4.67 br d (J = 1.5 Hz, H-15a), 4.97 br d (J = 1.5 Hz, H-15b)/δC 108.4 (C-15)] in 1 and the presence of one vinylic methyl (CH3-15) [δH/δC 1.78 s/19.6] and two sp2 quaternary carbons (δC 126.9). Also in 2, the γ-lactone ring was missing with instead an ester side chain. HMBC and 1H–1H COSY data (Fig. 2) indicated connectivities between these units. Three isolated proton spin systems that correspond to the C-1/C-2/C-3, C-5/C-6/C-7/C-8/C-9, and C-11/C-13 subunits of structure 2 were observed. An eudesmane moiety containing a Δ4,15 double bond was created by the HMBC correlations of H-1 with C-10 and C-9, H-14 with C-1, C-5 and C-9, H-3 with C-5, H-15 with C-3 and C-5, and H-7 with C-11 and C-12. A 7-propanoate moiety was ascribed as a result of correlations between H-13, C-11, C-7, and the carbonyl ketone C-12, as well as between H-11, C-7, C-6, C-8, and the methoxy group. And furthermore, it was proposed based on HMBC correlations of H-7 with C-11, C-13, and carbonyl ester C-12 that the propanoate moiety and the eudesmane skeleton are linked at C-7. The HMBC connection between the methoxy group at δH 3.64 and C-13 (δC 178.6) established that it is a component of a methyl ester group. H-7 relative stereochemistry was ascribed to the α-configuration based on biogenetic precedence and in agreement with previously recorded NMR chemical shifts for associated sesquiterpene lactones.22 The trans-5,6/trans-6,7 disposition in 2, was consistent with the well-defined triplet at configuration δH 3.78 (J = 10.6 Hz) that was produced by the carbinol proton at C-6. The key distinctions between 1 and 2 was the emergence of a propanoate moiety coupled with C-7 in 2 and the elimination of the γ-lactone ring that is part of the skeleton in 1. The up-field shifted of H-6, H-7, and H-8 combined with a down-field H-11 for 2 served as confirmation of this assignment. The aspect of the signal from H-11 that was supposed to have arisen as a quintuplet (J = 7.5, 7.5 Hz) at coupled with C-7 in 2 and the elimination coupled with C-7 in 2 and the elimination δH 2.21, or rather, as a double quartet at 1.67 (J = 12.0, 7.0 Hz), in many eudesman skeletons a downfield shift to δH 3.17 is observed with a reduction of the coupling constant equal to 6.9 Hz. Additionally, the multiplicity of the H-7 signal that appears as a triplet at δH 2.00 (J = 11.0 Hz) and a quartet signal at δH 3.17 (J = 6.9 Hz) characteristic for H-11, that confirmed the equatorial orientation of the propanoate moiety at C-7 with a dihedral angle between the two vicinal protons (H-7 & H-11) being close to 90° (Fig. 4C). As a result, it is possible to reveal the Me-13 as having an α-orientation. A stereoisomer of 2 has been previously isolated from A. illicola43 with the trivial name of sericin methyl ester A. The stereochemistry of sericin methyl ester A was determined by X-ray with C-11 being pro-S. By 13C NMR comparisons, C-11 of 2 was assigned to an R-configuration with a shift changes of +4.0 ppm along with a change in multiplicity from doublet quartet to be quartet. The NOSY correlation (Fig. 3) between CH3-13 and H-7, also was consistent with a H-11 β-orientation. The NOESY spectrum showed distinct interactions between H-1 (δH 3.38)/H-5 (δH 1.80)/H-3axi (δH 2.02), which showed that H-1 is in an α-orientation. Significant evidence for the β-orientation of H-6 and H-8 were derived by correlations between H-6 (δH 3.78) and CH3-14 (δH 0.67), and between H-8 (significant evidence δH 3.55) with CH3-14 (δH 0.67) and H-9eq (δH 2.33). Based on the aforementioned data, 2 was identified as 1β,6α,8α-trihydroxy,11α-methyl-eudesma-4(15)-en-13-propanoate. The 11,12-hydrogenation pattern for an un-lactonized sesquiterpene is rare, particularly with hydroxylation at C-8.
CH2). HRESIMS (m/z 299.1782) analysis indicated a molecular formula of C16H26O5 and 13C NMR data (Table 1) was consistent with an additional carbon deviating from the common sesquiterpene carbon number and four degrees of unsaturation. One methyl ester was detected based on the 13C and DEPT NMR [δC 178.6 (s), 52.0 (q)]. Additional signals at δC 145.1 (s), 108.4 (t), were attributed to an exocyclic double bond accounting for two degrees of unsaturation with a bicyclic frame accounting for the other degrees of unsaturation. 13C NMR and DEPT data (Table 1), indicated two methyls (one of which was angular δC 12.7); and the other was secondary (δC 10.5), three sp3 methylenes (δC 31.5, 34.9, and 45.7), six sp3 methines, including three oxygenated carbons (δC 78.3, 65.4 and 65.6), as well as the other three aliphatics (δC 54.7, 54.3 and 36.1) and one sp3 quaternary carbon at δC 40.8. These NMR findings complemented those of 1, assuming that 2 contain an eudesmane skeleton. NMR signal similarity from C-1–C-10 suggests shared rings of eudesmane derivatives with the respect of the different position of a double bond. The exocyclic double bond Δ4,15 in 2 was replaced by a methyl group (Me-15) and relocated to be Δ4,5 as an olefinic bond in 1. This was demonstrated by the lack of sp2 methylene [δC 145.1(C-4); δH/δC 4.67 br d (J = 1.5 Hz, H-15a), 4.97 br d (J = 1.5 Hz, H-15b)/δC 108.4 (C-15)] in 1 and the presence of one vinylic methyl (CH3-15) [δH/δC 1.78 s/19.6] and two sp2 quaternary carbons (δC 126.9). Also in 2, the γ-lactone ring was missing with instead an ester side chain. HMBC and 1H–1H COSY data (Fig. 2) indicated connectivities between these units. Three isolated proton spin systems that correspond to the C-1/C-2/C-3, C-5/C-6/C-7/C-8/C-9, and C-11/C-13 subunits of structure 2 were observed. An eudesmane moiety containing a Δ4,15 double bond was created by the HMBC correlations of H-1 with C-10 and C-9, H-14 with C-1, C-5 and C-9, H-3 with C-5, H-15 with C-3 and C-5, and H-7 with C-11 and C-12. A 7-propanoate moiety was ascribed as a result of correlations between H-13, C-11, C-7, and the carbonyl ketone C-12, as well as between H-11, C-7, C-6, C-8, and the methoxy group. And furthermore, it was proposed based on HMBC correlations of H-7 with C-11, C-13, and carbonyl ester C-12 that the propanoate moiety and the eudesmane skeleton are linked at C-7. The HMBC connection between the methoxy group at δH 3.64 and C-13 (δC 178.6) established that it is a component of a methyl ester group. H-7 relative stereochemistry was ascribed to the α-configuration based on biogenetic precedence and in agreement with previously recorded NMR chemical shifts for associated sesquiterpene lactones.22 The trans-5,6/trans-6,7 disposition in 2, was consistent with the well-defined triplet at configuration δH 3.78 (J = 10.6 Hz) that was produced by the carbinol proton at C-6. The key distinctions between 1 and 2 was the emergence of a propanoate moiety coupled with C-7 in 2 and the elimination of the γ-lactone ring that is part of the skeleton in 1. The up-field shifted of H-6, H-7, and H-8 combined with a down-field H-11 for 2 served as confirmation of this assignment. The aspect of the signal from H-11 that was supposed to have arisen as a quintuplet (J = 7.5, 7.5 Hz) at coupled with C-7 in 2 and the elimination coupled with C-7 in 2 and the elimination δH 2.21, or rather, as a double quartet at 1.67 (J = 12.0, 7.0 Hz), in many eudesman skeletons a downfield shift to δH 3.17 is observed with a reduction of the coupling constant equal to 6.9 Hz. Additionally, the multiplicity of the H-7 signal that appears as a triplet at δH 2.00 (J = 11.0 Hz) and a quartet signal at δH 3.17 (J = 6.9 Hz) characteristic for H-11, that confirmed the equatorial orientation of the propanoate moiety at C-7 with a dihedral angle between the two vicinal protons (H-7 & H-11) being close to 90° (Fig. 4C). As a result, it is possible to reveal the Me-13 as having an α-orientation. A stereoisomer of 2 has been previously isolated from A. illicola43 with the trivial name of sericin methyl ester A. The stereochemistry of sericin methyl ester A was determined by X-ray with C-11 being pro-S. By 13C NMR comparisons, C-11 of 2 was assigned to an R-configuration with a shift changes of +4.0 ppm along with a change in multiplicity from doublet quartet to be quartet. The NOSY correlation (Fig. 3) between CH3-13 and H-7, also was consistent with a H-11 β-orientation. The NOESY spectrum showed distinct interactions between H-1 (δH 3.38)/H-5 (δH 1.80)/H-3axi (δH 2.02), which showed that H-1 is in an α-orientation. Significant evidence for the β-orientation of H-6 and H-8 were derived by correlations between H-6 (δH 3.78) and CH3-14 (δH 0.67), and between H-8 (significant evidence δH 3.55) with CH3-14 (δH 0.67) and H-9eq (δH 2.33). Based on the aforementioned data, 2 was identified as 1β,6α,8α-trihydroxy,11α-methyl-eudesma-4(15)-en-13-propanoate. The 11,12-hydrogenation pattern for an un-lactonized sesquiterpene is rare, particularly with hydroxylation at C-8.
2.2. Chemosystematic significance
The taxonomy of the genus Artemisia had already sparked debate. Several researchers have divided the genus into different taxa below the genus level.44,45 For particular, Artemisia was split into three subgenera:46 Artemisia, Seriphidium Besser, and Dracunculus Besser. Tridentatae (Rydb.) McArthur, a novel group that is solely found in North America, was introduced by McArthur et al.47 Others like,46 have traditionally classified the genus Artemisia into three subgenera: (1) A. subg., all of the blooms on Artemisia are fertile, the marginal ones are female, and the center ones are hermaphrodite. It has heterogamous capitula; (2) A. subg. Dracunculus has heterogamous capitula, a glabrous receptacle, marginal female flowers, centre hermaphrodite blooms, and all or some of the inner flowers are fertile; and (3) A. subg. Seriphidium has hermaphrodite blooms throughout and a homogamous capitula with a glabrous receptacle. Valles and McArthur (2001)45 categorized Artemisia into the five groupings, mostly based on capitula type and floret fertility: Tridentatae, Absinthium DC., Artemisia, Dracunculus, Seriphidium, and (Rydb.) Seriphidium and Artemisia were separated to form a new genus by Ling48–50, acknowledged this division; nevertheless, Seriphidium and Artemisia were once more joined by Watson et al. in 2002.51 Internal transcribed spacers (ITS) of nuclear ribosomal DNA and chloroplast DNA (cpDNA) restriction site variation have both been used in molecular research to explore this separation.52 However, there is still considerable debate over how to classify Artemisia and the interactions between its many components.52The aerial parts of A. herba-alba stored many structural varieties of sesquiterpene lactones. The most common lactone types in this species are eudesmanolides and germacranolides, respectively. In all, there are 64 sesquiterpene lactones, of which 41 are eudesmanolides and around 23 are germacranolides.22,34 Investigation of A. herba-alba plants in Israel's Negev and Judean desert led to the identification of five distinct chemotypes based on variations in their sesquiterpene lactone composition.34 Segal and colleagues studied the chemotypes of A. herba-alba gathered in various places in Egypt and Israel and discovered many novel germacranolides and eudesmanolides, known as herbolides A–J.53,54 There were confirmed to be 10 sesquiterpene lactones, of which 3 were eudesmanolide skeleton.34 Sesquiterpene lactones (37) from A. herba-alba, (31 of which were eudesmanolide) collected in various geographic locations, have been the subject of certain phytochemical research in Spain.34 The presence of oxygen functions at C-9 (germacrane and eudesmane numbering) in lactones from the African specimens, markedly different compounds from Spain and those obtained by Segal et al.53,54 These sort of lactones were not uncovered from plant material gathered in the Sinai desert.55 However, a study on an Egyptian specimen produced a few novel sesquiterpene lactones, with two of them having oxygen functionalities at C-9.56 The findings reported from Spain suggest that lactones isolated from North African A. herba-alba may in fact have a unique chemical property called oxygenation at C-9. A. herba-alba growing in Egypt has been the subject of several studies on its chemistry. Sesquiterpene lactones have been the subject of the majority of investigations. All lactones identified (of which 4 eudesmanolides and 5 germacranolides) are different from those previously found in Israel-grown A. herba-alba. All collections, apart from one, were found to be 1,3-dihydroeudesmanolides and 1,3-dihydrogermacranolides containing at least an oxygen function at C-9, a rare characteristic in the genus Artemisia. However, the instance under research totally lacks the eudesmanolides oxygenated at C-9, which are among the most distinctive chemical characteristics of the sesquiterpene lactones isolated from North African A. herba-alba.34 Additionally, a range of sesquiterpene lactones with at least a C-8 oxygen function have been found in Egyptian A. herba-alba specimens. A. herba-alba species from Morocco (of which 2 eudesmanolides out of 6) and Algeria (of which 2 eudesmanolides) were examined chemically in a small number of literature studies, demonstrating the abundance of sesquiterpenes from this genus.34 Eudesmanolides makes up over 60% of A. herba-alba, whereas germacranolides makes up around 35%. Seriphidium is a section of the genus Artemisia that contains A. herba-alba. These chemical characteristics of A. herba-alba are generally in accordance with what is anticipated for members of the section Seriphidium.57,58 The pronounced propensity for the manufacture of 11,13-dihydroeudesmanolides is a characteristic of plants in this section.34,57–59 However, the large prevalence of lactones with a 11αH stereochemistry is far less typical. It's interesting to note that the majority of the lactones identified in the Spanish subspecies of A. herba-alba match this structural characteristic. However, over the past several years, a variety of sesquiterpene lactones have been identified in A. herba-alba specimens from Egypt and Spain reflected that the percentage of eudesmanolides with a 11βH stereochemistry about 66% in contrast to 34% with 11αH stereochemistry. This reflects that the majority of the isolated eudesmanolides belong to 11αH stereochemistry. The published 13C-NMR spectrum data, suggests that structures need to be updated. The chemical shift values for the atom C-13 in several the lactones are in the 9–10 ppm range. This indicates that these compounds are truly 11α,13-dihydrolactones since they feature a β-Me group at C-11.60 We looked into A. herba-alba, which was gathered in Spain and North Africa, since we were intrigued by these variations in the chemical makeup, which may indicate taxonomic changes at the subspecies or even species level. We thus feel that the chemical information that is currently accessible supports our earlier findings that were based on morphological criteria.61
2.3. Antimicrobial activity
Results in Tables 2 and 3 indicated that 3 is the most effective against all tested bacteria with IZDs of 7–8 mm and MIC values 25–100 mg. Compounds 1 and 2 showed moderate antibacterial activity with IZDs around 7 mm against two bacterial strains. In addition, antifungal activity was observed (IZD 8 mm) and an MIC value of 25 for 3 (Tables 2 and 3). Moderate antifungal effects also observed with 1–2 (IZD 7 mm). In contrast, each compound was inactive against the E. coli and the yeast strain C. albicans. Compound 3 showed strong binding affinity (−7.08 kcal mol−1) with 4GEE, while compounds 1–2 showed more moderate binding (−6.7 and −6.8 kcal mol−1, respectively). Additionally, the inhibition constant (pKi) for the tested metabolites were: 11.6 μM, 9.9 μM and 6.4 μM, 1–3, respectively (Fig. 5).| Compound | Inhibition zone diameter (IZD) (mm) | |||||
|---|---|---|---|---|---|---|
| Bacterium | Yeast | Fungus | ||||
| B. subtilis ATCC6633 | L. cereus ATCC14579 | S. aureus ATCC29213 | E. coli ATCC 25922 | C. albicans ATCC 10321 | F. solani NRC15 | |
| a Values of inhibition are the average of three different points around the discs ± SD. N.A. No activity. | ||||||
| 1 | N.A. | 7 ± 0.6 | 7 ± 0.7 | N.A. | N.A. | 7 ± 1.4 |
| 2 | 7 ± 0.6 | 7 ± 0.7 | N.A. | N.A. | N.A. | 7 ± 0.6 |
| 3 | 7 ± 0.8 | 8 ± 0.2 | 8 ± 0.4 | N.A. | N.A. | 8 ± 0.00 |
| Thiophenicol | 20 ± 0.7 | 23 ± 0.0 | 18 ± 1.4 | 15 ± 0.7 | N.A. | N.A. |
| Treflucan | N.A. | N.A. | N.A. | N.A. | 22 ± 0.4 | 12 ± 0.7 |
| Compound | Minimum inhibitory concentration (μg per disc) | |||
|---|---|---|---|---|
| Bacterium | Fungus | |||
| B. subtilis ATCC6633 | L. cereus ATCC14579 | S. aureus ATCC29213 | F. solani NRC15 | |
| 1 | — | 100 | 50 | 100 |
| 2 | 50 | 50 | — | 50 |
| 3 | 100 | 25 | 50 | 25 |
| Thiophenicol | 3.13 | 3.13 | 3.13 | — |
| Treflucan | — | — | — | 50 |
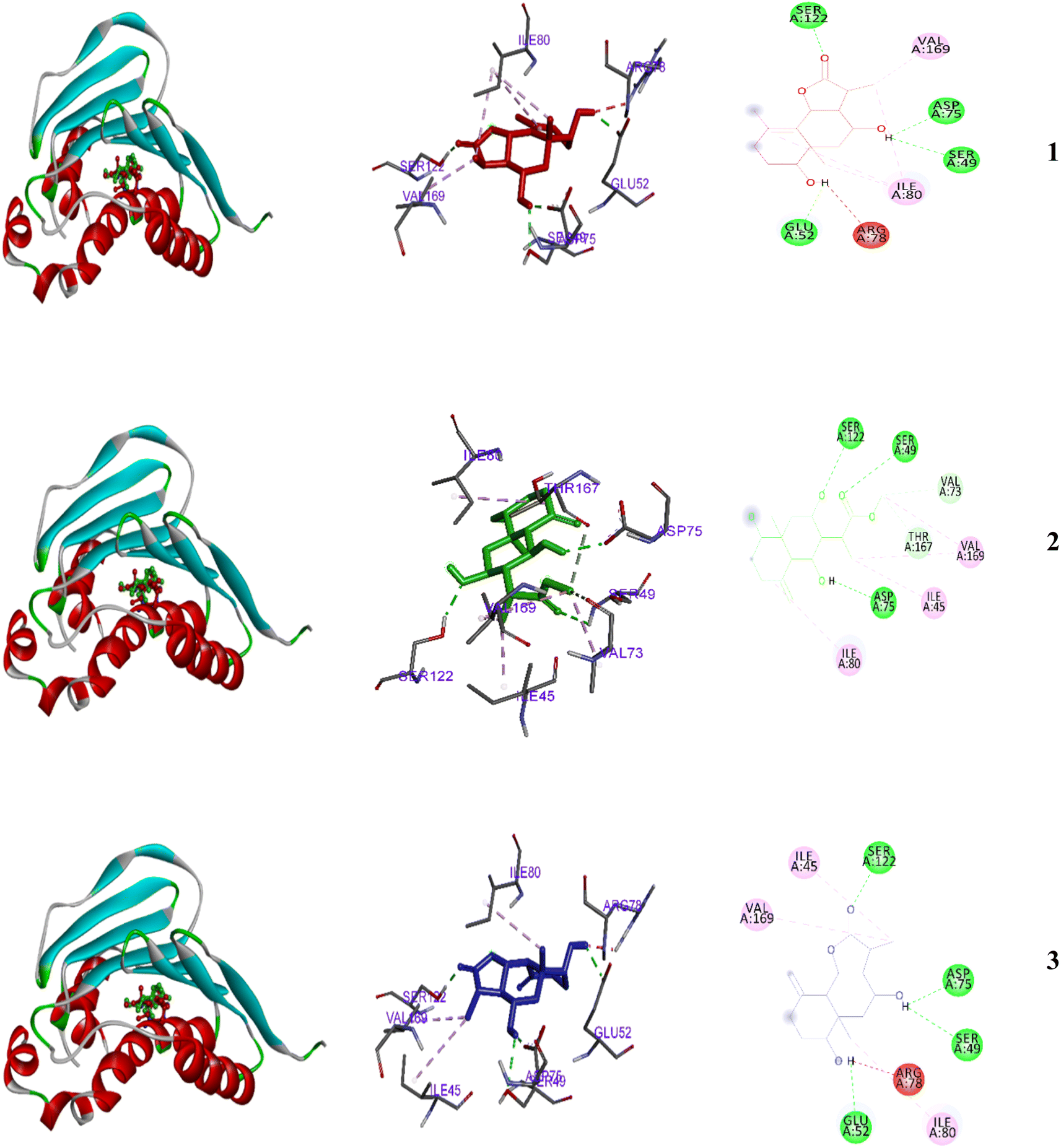 | ||
| Fig. 5 2D and 3D representations of the predicted binding modes inside the active site of the bacterial 4GEE for compounds 1–3. | ||
A single N-myristoyltransferase present in fungi, protozoa, and insects is essential for survival.62,63 The binding score of 3 with the enzyme was the highest (−6.99 kcal mol−1) and moderate with 1–2 (−6.8 and −6.1 kcal mol−1 respectively). The inhibition constants (pKi) were 10.1, 34.6, and 7.5 μM, for 1–3, respectively. With growing occurrence of antimicrobial resistance25–29 a need to identify new classes of antimicrobial agents is essential.27,30 DNA gyrases have become an attractive target for antibacterial research as they are essential enzymes for cell survival in prokaryotes. Today, searching for new inhibitors of the ATP-binding pocket of gyrase B enzyme (PDB code 4GEE) is attracting the attention of pharmaceutical industries.31,32 The N-myristoyltransferase appears to be present in eukaryotes. Fungi, protozoa, and insects possess a single N-myristoyltransferase, essential for their survival, while mammals possess various N-myristoyltransferases (Fig. 6).62,63
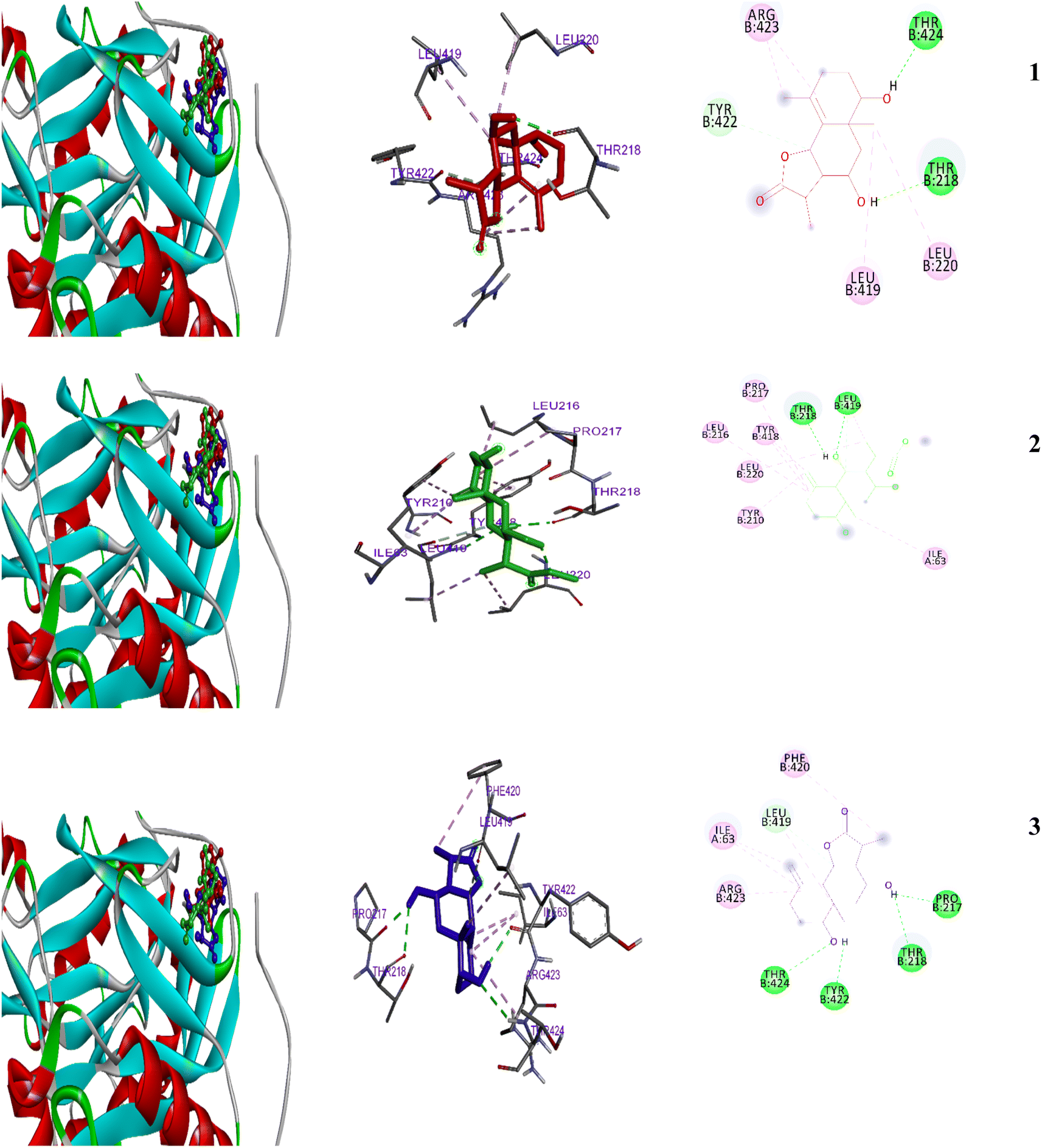 | ||
| Fig. 6 2D and 3D representations of the predicted binding modes inside the fungal active site of 1IYK for compounds 1–3. | ||
3. Experimental
3.1. General experimental procedures
For specific rotation, a Horiba SEPA-300 digital polarimeter (5 cm) was used and IR data was collected from a Shimadzu FTIR-8100 spectrometer. For mass spectrometry measurements, IMS was performed on a Finnegan LCQ ion trap mass spectrometer and HR-EI-MS experiments were performed on Fourier transform ion cyclotron mass spectrometer. EI-MS experiments were performed a JEOL JMS-GCMATE mass spectrometer. Using tetramethylsilane as an internal standard, the 1H (600 MHz) and 13C (150 MHz) NMR spectra were achieved on a JEOL JNM-ECA 600 spectrometer. Compound separation for analytical and preparative separation was carried out using YMC-Pack ODS-A (250 × 4.6 mm i.d.) and (250 × 20 mm i.d.) columns, respectively. Purification was performed using a Shimadzu HPLC system furnished with a RID-10A refractive index detector. Fuji Silysia Chemical, Ltd.'s normal-phase silica BW-200 (Fuji Silysia Chemical, Ltd., 150–350 mesh) and Chromatorex's ODS reverse phase DM1020T (Fuji Silysia Chemical, Ltd., 100–200 mesh) were used for chromatography separations (Fuji Silysia Chemical, Ltd., 100–200 mesh). Additionally, silica gel 60 F254 (Merck, 0.25 mm) and RP-18 WF254 were used (Merck, 0.25 mm) for TLC analysis with compound visualization by with H2SO4–MeOH (1:9) spray followed by heating.
3.2. Plant material
A. herba-alba was harvested in June 2017 in the South Sinai (Saint Catherine) region of Egypt; a voucher specimen (SH-1109) was deposited in the herbarium of the protectorate of St. Katherine, Egypt.3.3. Extraction and isolation
Aerial stems and leaves (2.5 kg) were crushed and extracted for 2 days at room temperature with CH2Cl2–MeOH (1:1). The extract was concentrated in vacuo at 45 °C to produce a dark brown residue (200 g). The material was purified on a silica gel column (6 × 120 cm) with a gradient of n-hexane up to 100% CHCl3 and CHCl3–MeOH up to 50% MeOH (3 L of each solvent combination). To create two sub-fractions, the n-hexane:CH2Cl2 (1:1) fraction (8.3 g) was run over a second silica gel column (3 × 120 cm). Sub-fraction 1 A (2.3 g) underwent further HPLC purification and MeOH:H2O elution (20:80). The flow rate was adjusted to 1.5 mL min−1 and was at 0–70 min to produce 1 (30 mg, purity > 97% by HPLC) (eluent CH2Cl2/MeOH/H2O 80:3:1, Rf = 0.30) and 3 (55 mg, purity > 97% by HPLC) (eluent CH2Cl2/MeOH/H2O 80:3:1, Rf = 0.20), using an eluent ratio of 80:3:1. HPLC was used to further purify sub-fraction 2 A (1.2 g), which was eluted with MeOH:H2O. (20:80). HPLC was further used to increase purity for 2 (40 mg, purity > 97% by HPLC) (eluent CH2Cl2/MeOH/H2O 70:3:1, Rf = 0.20), the flow rate was adjusted to 2.0 mL min−1 and was at 0–60 min.
3.4. Spectral data
O), 3090, 1650, 900 (CC).3.5. Antibacterial and antifungal assay
The antimicrobial activities were assayed in vitro against Gram positive (Bacillus subtilis ATCC6633, Lactobacillus cereus ATCC14579 and Staphylococcus aureus ATCC29213), Gram negative (Escherichia coli ATCC25922), yeast (Candida albicans ATCC10321), and fungi (Fusarium solani NRC15), using an agar diffusion technique.22 Filter paper discs (5 mm in diameter) saturated with (100 μg/5 μL DMSO per disc) for the test compounds. Thiophenicol and treflucan are used as positive controls for antibacterial and antifungal activity, respectively (100 μg per disc). The DMSO solvent negative control showed no inhibition zone. Prepared discs were placed on the surface of each plate, at a concentration of 1 × 106/mL of fungi (potato dextrose agar medium) and 1 × 108/mL of bacteria (nutrient agar medium). The plates left for 30 min at 4 °C and then incubated for 24 h at 30 °C for bacteria and 72 h at 28 °C for the fungus. The zone of inhibition zone was recorded at three different points and an average value are reported as mean ± SD.3.6. Molecular docking
The chemical structures of 1–3 were downloaded as SDF files from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/(accessed on 29 January 2021)), and then converted to a PDB format via Avogadro software (https://avogadro.cc/). The protein crystal structures for 4GEE (antimicrobial) and 1Iyk (anti-fungal) were downloaded from the protein databank (https://www.rcsb.org/) using a previously published molecular docking protocol (Mohamed et al., 2020).4. Conclusions
Artemisia herba-alba afforded two new sesquiterpenes, 1β,8α-dihydroxyeudesm-4-en-6β,7α,11βH-12,6-olide (1) and 1β,6α,8α-trihydroxy, 11α-methyl-eudesma-4(15)-en-13-propanoate (2) along with a known eudesmanolide 11-epi-artapshin (3). Molecular docking simulations indicate that the natural-product antibiotic, eudesmanolide 11-epi-artapshin (3) can bind in the ATP-binding pocket to function as a gyrase B inhibitor; compound 3 also is able to bind as a fungal inhibitor to N-myristoyltransferase.Author contributions
Conceptualization, T. A. M., M. H. A., P. W. P. and M.-E. F. H.; formal analysis, T. A. M., M. H. A., I. A. S., A. A. A. and M.-E. F. H.; investigation, T. A. M., M. H. A., S. K. A., A. A. A., P. W. P. and M.-E. F. H.; writing—original draft preparation, T. A. M., M. H. A., I. A. S., S. K. A., A. A. A. and M.-E. F. H.; writing—review and editing, T. A. M., M. H. A., I. A. S., S. K. A., A. A. A. and M.-E. F. H.; funding acquisition, T. A. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- S.-Y. Pan, G. Litscher, S.-H. Gao, S.-F. Zhou, Z.-L. Yu, H.-Q. Chen, S.-F. Zhang, M.-K. Tang, J.-N. Sun and K.-M. Ko, Evid.-Based Complementary Altern. Med., 2014, 2014, 1–20 Search PubMed.
- U. Anand, N. Jacobo-Herrera, A. Altemimi and N. Lakhssassi, Metabolites, 2019, 9, 258 CrossRef CAS PubMed.
- U. Anand, S. Nandy, A. Mundhra, N. Das, D. K. Pandey and A. Dey, Drug Resistance Updates, 2020, 51, 100695 CrossRef PubMed.
- C. L. Gorlenko, H. Y. Kiselev, E. V. Budanova, A. A. Zamyatnin Jr and L. N. Ikryannikova, Antibiotics, 2020, 9, 170 CrossRef CAS PubMed.
- Y. Zu, H. Yu, L. Liang, Y. Fu, T. Efferth, X. Liu and N. Wu, Molecules, 2010, 15, 3200–3210 CrossRef CAS PubMed.
- D.-c. Hao and P.-g. Xiao, Chin. Herb. Med., 2020, 12, 104–117 CrossRef PubMed.
- K. S. Bora and A. Sharma, Pharmaceut. Biol., 2011, 49, 101–109 CrossRef PubMed.
- R. X. Tan, W. Zheng and H. Tang, Planta Med., 1998, 64, 295–302 CrossRef CAS PubMed.
- M. Willcox, J. Altern. Complementary Med., 2009, 15, 101–109 CrossRef PubMed.
- B. Koul, P. Taak, A. Kumar, T. Khatri and I. Sanyal, J. Glycomics Lipidomics, 2017, 7, 142 Search PubMed.
- H. Ekiert, J. Świątkowska, E. Knut, P. Klin, A. Rzepiela, M. Tomczyk and A. Szopa, Front. Pharmacol., 2021, 12, 653993 CrossRef CAS PubMed.
- H. Dlugónska, Ann. Parasitol., 2015, 61, 299–301 Search PubMed.
- T. Efferth, S. Zacchino, M. I. Georgiev, L. Liu, H. Wagner and A. Panossian, Phytomedicine, 2015, 22, A1–A3 CrossRef PubMed.
- A. Alamgir, in Therapeutic Use of Medicinal Plants and Their Extracts, Springer, 2017, vol. 1, pp. 177–293 Search PubMed.
- T. A. Oliveira, M. B. Santiago, V. H. Santos, E. O. Silva, C. H. Martins and A. E. Crotti, Chem. Biodiversity, 2022, 19, e202200097 CAS.
- R. D. Bidgoli, J. Food Sci. Technol., 2021, 58, 1313–1318 CrossRef PubMed.
- Y.-r. Zhao, G.-a. Zou and H. A. Aisa, Phytochemistry, 2022, 197, 113108 CrossRef CAS PubMed.
- J.-H. Ahn, E.-J. Song, D.-H. Jung, Y.-J. Kim, I.-S. Seo, S.-C. Park, Y.-S. Jung, E.-S. Cho, S. H. Mo and J. J. Hong, Phytomedicine, 2022, 99, 153934 CrossRef CAS PubMed.
- D. Bisht, D. Kumar, D. Kumar, K. Dua and D. K. Chellappan, Arch. Pharmacal Res., 2021, 44, 439–474 CrossRef CAS PubMed.
- T. A. Mohamed, H. A. Albadry, A. I. Elshamy, S. H. Younes, A. A. Shahat, M. T. El-wassimy, M. F. Moustafa and M. E. F. Hegazy, J. Chin. Chem. Soc., 2021, 68, 338–342 CrossRef CAS.
- T. A. Mohamed, A. A. Abd El Aty, A. A. Shahat, N. S. Abdel-Azim, K. A. Shams, A. A. Elshamy, M. M. Ahmed, S. H. Youns, T. M. El-Wassimy and S. A. El-Toumy, Nat. Prod. Res., 2021, 35, 1959–1967 CrossRef CAS PubMed.
- T. A. Mohamed, M.-E. F. Hegazy, A. A. Abd El Aty, H. A. Ghabbour, M. S. Alsaid, A. A. Shahat and P. W. Paré, J. Asian Nat. Prod. Res., 2017, 19, 1093–1101 CrossRef CAS PubMed.
- T. Usha, D. Shanmugarajan, A. K. Goyal, C. S. Kumar and S. K. Middha, Curr. Top. Med. Chem., 2017, 17, 3296–3307 CrossRef CAS PubMed.
- G. V. De Socio, P. Rubbioni, D. Botta, E. Cenci, A. Belati, R. Paggi, M. B. Pasticci and A. Mencacci, J. Global Antimicrob. Resist., 2019, 19, 154–160 CrossRef PubMed.
- M. Frieri, K. Kumar and A. Boutin, J. Infect. Public Health, 2017, 10, 369–378 CrossRef PubMed.
- E. M. Mohi El-Deen, E. A. Abd El-Meguid, S. Hasabelnaby, E. A. Karam and E. S. Nossier, Molecules, 2019, 24, 3650 CrossRef PubMed.
- B. Li and T. J. Webster, J. Orthop. Res., 2018, 36, 22–32 Search PubMed.
- D. C. Kaur and S. S. Chate, J. Glob. Infect. Dis., 2015, 7, 78 CrossRef CAS PubMed.
- G. Teitzel, Trends Microbiol., 2019, 27, 285–286 CrossRef CAS PubMed.
- A. H. Al-Nadaf, S. A. Salah and M. O. Taha, Comput. Biol. Chem., 2018, 74, 263–272 CrossRef CAS PubMed.
- E. H. Reda, Z. T. A. Shakour, A. M. El-Halawany, E.-S. A. El-Kashoury, K. A. Shams, T. A. Mohamed, I. Saleh, A. I. Elshamy, M. A. Atia and A. A. El-Beih, Antibiotics, 2021, 10, 252 CrossRef CAS PubMed.
- H. Mohsen and F. Ali, Molecules, 2009, 14, 1585–1594 CrossRef CAS PubMed.
- A. E.-H. H. Mohamed, M. El-Sayed, M. E. Hegazy, S. E. Helaly, A. M. Esmail and N. S. Mohamed, Rec. Nat. Prod., 2010, 4 CAS.
- M. Laid, M.-E. F. Hegazy, A. A. Ahmed, K. Ali, D. Belkacemi and S. Ohta, Phytochem. Lett., 2008, 1, 85–88 CrossRef CAS.
- S. M. Salah and A. K. Jäger, J. Ethnopharmacol., 2005, 97, 145–149 CrossRef PubMed.
- M. M. Mokhtar, H. M. Shaban, M. E.-a. F. Hegazy and S. S. Ali, Bull. Fac. Pharm. Cairo Univ., 2017, 55, 195–201 Search PubMed.
- I. Kadi, M. Ouinten, N. Gourine and M. Yousfi, Nat. Prod. Res., 2019, 33, 875–878 CrossRef CAS PubMed.
- S. Amkiss, A. Dalouh and M. Idaomar, Arab. J. Chem., 2021, 14, 102976 CrossRef CAS.
- J. A. Marco, J. F. Sanz-Cervera, V. Garcia-Lliso, M. Guara and J. Vallès-Xirau, Phytochemistry, 1997, 45, 751–754 CrossRef CAS.
- J. A. Marco, Phytochemistry, 1989, 28, 3121–3126 CrossRef CAS.
- C. Narayanan and N. Venkatasubramanian, J. Org. Chem., 1968, 33, 3156–3162 CrossRef CAS.
- A. Ajmal, J. Wu and A. Tulak, CN114292253A, 2022.
- E. D. McArthur, Proc. RMRS, 1998, 67 Search PubMed.
- J. Valles and E. D. McArthur, USDA Forest Service Proceedings RMRS-P-21, 2001, pp. 67–74 Search PubMed.
- D. Podlech, Compositae ‘VI-’Anthemideae, Akademische Druck-und Verlagsanstalt, 1986 Search PubMed.
- E. D. McArthur, C. L. Pope and D. C. Freeman, Am. J. Bot., 1981, 68, 589–605 CrossRef.
- Y. R. Ling, Bull. Bot. Res., 1991, 11, 1–40 Search PubMed.
- Y. R. Ling, Advances in Compositae Systematics, 1995, pp. 283–291 Search PubMed.
- K. Bremer and A. A. Anderberg, Asteraceae: cladistics and classification, 1994 Search PubMed.
- L. E. Watson, P. L. Bates, T. M. Evans, M. M. Unwin and J. R. Estes, BMC Evol. Biol., 2002, 2, 1–12 CrossRef PubMed.
- S. Zahra, M. Iraj, A. Mostafa and N. Taher, J. Biodivers. Environ. Sci., 2014, 5, 608–624 Search PubMed.
- R. Segal, L. Eden, A. Danin, M. Kaiser and H. Duddeck, Phytochemistry, 1985, 24, 1381–1382 CrossRef CAS.
- R. Segal, I. Feuerstein and A. Danin, Biochem. Systemat. Ecol., 1987, 15, 411–416 CrossRef CAS.
- M. M. Gordon, D. Van Derveer and L. H. Zalkow, J. Nat. Prod., 1981, 44, 432–440 CrossRef CAS.
- A. Ahmed, M. Abou-El-Ela, J. Jakupovic, A. S. El-Din and N. Sabri, Phytochemistry, 1990, 29, 3661–3663 CrossRef CAS.
- A.-u. Rahman, Studies in natural products chemistry, Elsevier, 2015 Search PubMed.
- J. Marco and O. Barbera, Stud. Nat. Prod. Chem., 1990, 7, 201–264 CAS.
- T. Tutin, K. Persson and W. Gutermann, Flora europaea, 1976, 4, pp. 178–186 Search PubMed.
- J. F. Sanz, G. Castellano and J. A. Marco, Phytochemistry, 1990, 29, 541–545 CrossRef CAS.
- J. Vallès and M. Torrell, Fontqueria, 1996, 44, pp. 17–24 Search PubMed.
- T. A. Farazi, J. K. Manchester, G. Waksman and J. I. Gordon, Biochemistry, 2001, 40, 9177–9186 CrossRef CAS PubMed.
- R. Khalil, S. Ashraf, A. Khalid and Z. Ul-Haq, ACS Omega, 2019, 4, 13658–13670 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02690f |
| This journal is © The Royal Society of Chemistry 2023 |