
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDifferences in the catalytic properties of Fe isotopes
Songtao Xiao†
*,
Yubing Xue ,
Jing Zhao,
Xiechun Liu,
Haifeng Cong,
Tian Lan,
Xiaojuan Liu,
Dashu Pan,
Lingyu Wang,
Guoan Ye†* and
Yinggen Ouyang*
,
Jing Zhao,
Xiechun Liu,
Haifeng Cong,
Tian Lan,
Xiaojuan Liu,
Dashu Pan,
Lingyu Wang,
Guoan Ye†* and
Yinggen Ouyang*
China Institute of Atomic Energy, Beijing, 102413, China. E-mail: ouyang_yinggen@163.com; xiao_songtao@126.com; guo_anye_ciae@163.com
First published on 21st August 2023
Abstract
The significant differences in the catalytic properties caused by different ‘isotopic catalysts’ were discovered for the first time. The commonly purchased Fe2O3 is a ‘mixture’ of different Fe isotopic oxides which means the catalytic effect of Fe2O3 is theoretically a synthetical result of all isotopic compounds. In this work, the differences in catalytic properties of α-Fe2O3 with natural abundance ratio and separated isotopic α-Fe2O3 (α-54Fe2O3, α-56Fe2O3, and α-58Fe2O3) catalyzing thermal decomposition of ammonium perchlorate (AP) were investigated, and are mainly attributed to the difference in the charge distribution of the nuclei of different iron isotopes. The result suggests that isotope effects in different isotopes when utilized as catalysts are caused by nuclear morphology and the nuclear charge distribution. This study will serve as a base as well as an initiation for future studies of the isotopic catalyst.
Introduction
Isotopes are nuclides of the same element with the same charge number (Z) and electron shell structure but different neutron numbers. Since isotopes have a different mass number (A), isotopes can be classified into three groups viz. light isotopes (A ≤ 50), medium-weight isotopes (50 < A < 100), and heavy isotopes (A ≥ 100). Over 270 stable isotopes and over 2000 radioactive isotopes of 118 known elements have been found since the term ‘isotope’ was proposed by F. Soddy1 in 1910, in which only the elements with Z ≤ 83 have been found to have stable isotopes.Generally determined by the nuclear charge, the physical and chemical properties of isotopes are very similar, but not identical due to the distinct neutron number. Therefore, the differences in the properties of isotopes of an element or molecules substituted with sister isotopes (i.e., isotopomers) are called isotope effects (IE).2 According to the source of the isotope effect, it can be divided into the mass-dependent isotope effect (MDE) caused by the different masses and mass-independent isotope effect (MIE, also the anomalous isotope effect) caused by other nuclei properties like spin and volume.3
Isotope effect can also be categorized into equilibrium isotope effect (EIE) and kinetic isotope effect (KIE) according to the effect on the chemical behavior.4 EIE is the distribution difference of isotopes presented in different phases or chemical states at the equilibrium state and also the basis of chemical isotope separation. In addition, KIE is the kinetic difference when any atom of the reactant molecules is substituted with its sister isotopes, which is generally accepted as a consequence of the different minimum vibrational energy (i.e., zero-point energy) affected by different masses of isotopic isomers.
As a ubiquitous effect in chemical reactions, KIE is broadly utilized for studying mechanisms,4,5 preparing isotope reagents,6 controlling the reaction products,7,8 and so on. Most researchers in the field of KIE have focused on primary kinetic isotope effects (PKIE) and secondary kinetic isotope effects (SKIE) commonly expressed by the ratio of rate constants of light to heavy isotopomers, kL/kH.9 The value of PKIE is usually between 2 to 9, but in some special systems, it can be way more than 9 or less than 1 (inverse isotope effect). To date, two theories have been employed to explain the different values of KIE: one is the zero-point energy (ZPE), and the other is the quantum tunneling effect. Neither of them can explain all of the experiment results but only a part which means they all need to be further improved.9
Several studies have examined the difference of chemical properties for isotopes via the KIE method: (1) a large KIE effect (kH/kD = 40.4) was measured by U. Weisflog et al. at 157 °C with the compound without additional alkyl groups, C6H5–C2H2–O–C2H2–C6H5, as oxidation takes place also on the alkyl groups but not deuterated.10 (2) Huang Gang et al.11 from China Academy of Engineering Physics investigated the p-t curves of deuterium and tritium absorption by uranium at 150–300 °C and constant volume system through reaction rate analysis. They obtained the activation energy value of deuterium and tritium absorption as (−42.8 ± 0.3) and (−43.2 ± 1.2) kJ mol−1, respectively. (3) Eric M. Simmons and John F. Hartwig12 have reviewed the studies of deuterium KIE in C–H bond functionalization by transition-metal complexes and concluded factors affecting the KIE like multi-step reactions, the emergence, and structure of the transition state. Zhou Yu-jing et al.13 from Peking University also reviewed the studies as well as applications of KIE and calculated the difference of ZPE via symmetric stretching models obtaining the calculating result of the PKIE of 1H and D around 6.5, which is very close to the experiment result.
The present work has strongly confirmed the existence of differences in chemical characteristics of isotopes and some of the differences in certain systems are significant, although isotopes ‘behave similarly’ in most chemical systems due to the limitation of the precision of analysis instrument and the lacking of researches on isotope effects. Here, we first develop isotopes as catalysts and have found the significant differences at the chemical level, which can no longer be ignored.
Thermal decomposition of NH4ClO4 (ammonium perchlorate, AP) catalyzed by Fe2O3 is a classical catalytic system14 that has been thoroughly studied and thus this system was selected to investigate the association between isotopes and catalytic ability. The band gap value and catalytic activity described by ΔTH of different catalytic materials from other public research are listed in Table 1.15 The data reveals an apparent law that the higher the Eg is, the better the catalytic performance is for Fe2O3 and other N-type semiconductor catalysts, while for the P-type the worse the catalytic performance is.
| Metal oxide semiconductor catalysts | Eg eV−1 | ΔTH/°C | |
|---|---|---|---|
| a ΔTH is the reduction of the peak temperature at high-temperature decomposition process of AP with addition of catalysts, compared with the pure AP. | |||
| P-type | NiO | 3.7 | 65 |
| La2O3 | 2.86 | 71 | |
| Cu2O | 2.02 | 90 | |
| Co3O4 | 0.9 | 117 | |
| N-type | MnO2 | 0.25 | 57 |
| Fe2O3 | 2.34 | 67 | |
| V2O5 | 2.49 | 88 | |
| ZnO | 3.35 | 132 | |
The commonly purchased Fe2O3 is actually a mixture of different Fe isotopic oxides consisted of 5.8% 54Fe2O3, 91.72% 56Fe2O3, 2.1% 57Fe2O3, and 0.28% 58Fe2O3, which means the catalytic performance of Fe2O3 on the thermal decomposition of AP is a synthetical result of all isotopic compounds. Here, nFe2O3 (abbreviation for Fe2O3 in natural abundance ratio), 54Fe2O3, 56Fe2O3 and 58Fe2O3 were transformed into the alpha-crystalline form through a same hydrothermal approach,16,17 and the differences in catalytic performance of these Fe-isotopomers were investigated by TG-DSC methods.
Results and discussion
Characterization results
The typical SEM images of the four kinds of α-Fe2O3 catalysts obtained via the hydrothermal procedure with different contents of Fe isotopes are shown in Fig. 1. It can be clearly seen from Fig. 1 that the particle shape of the catalysts are all irregular lump and the surface of the α-nFe2O3 is rougher with more pores and gullies on the surface compared with separated isotopic α-Fe2O3. | ||
| Fig. 1 SEM images of (a) α-nFe2O3, (b) α-54Fe2O3, (c) α-56Fe2O3 and (d) α-58Fe2O3. | ||
The adsorption and desorption isotherms of nitrogen at −196 °C obtained from four α-Fe2O3 catalysts are presented in Fig. 2. The four isotherms are identified as Type IV isotherms according to IUPAC (International Union of Pure and Applied Chemistry) classification because of the inflection point and the hysteresis loop, which indicating the existence of abundant mesopores in the catalysts. The hysteresis loops could be further divided into H2(b) as the α-nFe2O3 and H2(a) as other three separated isotopic α-Fe2O3 indicating that the α-nFe2O3 has much larger size distribution of neck widths of pore-blocking structure. The BET surface area and pore size data are listed in Table 2. The smaller average pore diameter and pore volume of α-nFe2O3 and the rougher surface support this result, which also explain why the surface area of α-nFe2O3 is about 14% smaller than that of separated isotopic α-Fe2O3.
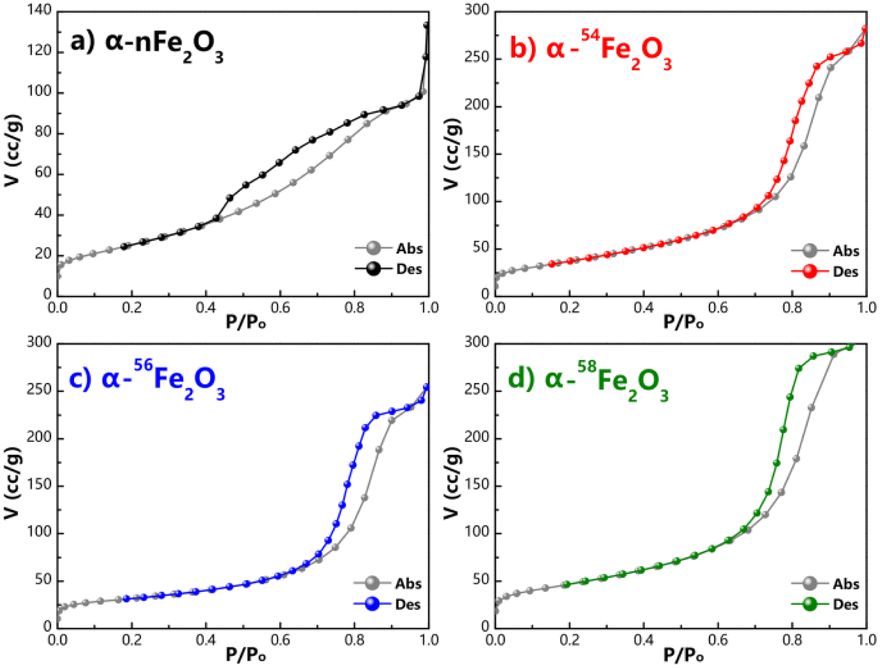 | ||
| Fig. 2 Adsorption and desorption isotherms of N2 of (a) α-nFe2O3, (b) α-54Fe2O3, (c) α-56Fe2O3, and (d) α-58Fe2O3. | ||
| Catalysts | Surface area/(m2 g−1) | Average pore diameter/nm | Pore volume /(cm3 g−1) |
|---|---|---|---|
| α-nFe2O3 | 120.29 | 3.91 | 0.211 |
| α-54Fe2O3 | 144.64 | 4.80 | 0.478 |
| α-56Fe2O3 | 144.01 | 5.16 | 0.432 |
| α-58Fe2O3 | 142.82 | 4.79 | 0.516 |
Based on the electron microscope images and the results of N2 adsorption and desorption isotherms, the morphology of four α-Fe2O3 catalysts is relatively similar that they are all bulk particle with abundant mesopores. The surface area of α-nFe2O3 (120.29 m2 g−1) is around 14% smaller than those of α-54Fe2O3 (144.64 m2 g−1), α-56Fe2O3 (144.01 m2 g−1), and α-58Fe2O3 (142.82 m2 g−1) because pore-blocking of α-nFe2O3 causing smaller pore diameter and volume than those of the separated isotopic α-Fe2O3 (around 5 nm and cm3 g−1).
High-resolution transmission electron microscopy (HRTEM) images presented in Fig. 3a–d show the typical crystal plane spacing of the crystal plane like (110), (104) and (012). The particle distribution obtained from TEM images shown in Fig. 3e–h reveals the lumps in Fig. 1 are agglomeration of particles with around 5 nm average diameter.
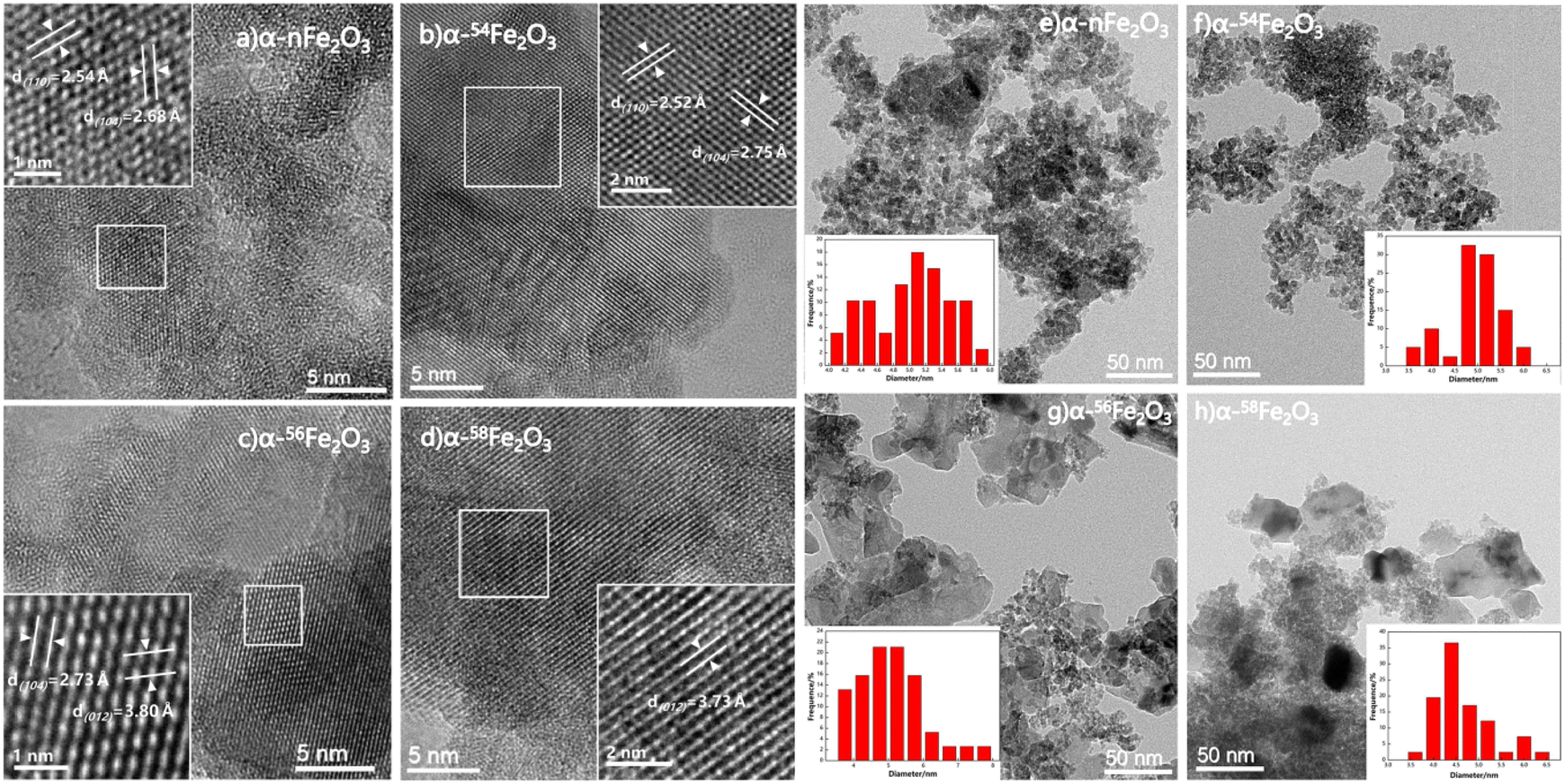 | ||
| Fig. 3 HRTEM images of synthesized catalysts (a) α-nFe2O3, (b) α-54Fe2O3, (c) α-56Fe2O3 and (d) α-58Fe2O3, and particle size distributions of synthesized catalysts (e) α-nFe2O3, (f) α-54Fe2O3, (g) α-56Fe2O3 and (h) α-58Fe2O3. | ||
It can be seen from Fig. 4a that the diffraction peaks in the XRD pattern of all four α-Fe2O3 catalysts are fit for the ICDD (International Center for Diffraction Data) No. 33-0664 of α-Fe2O3 with the a = b = 0.50356 nm and c = 1.37489 nm. The diffraction peaks indexed to the other phases are not observed in Fig. 4a. The diffraction peaks located in 2θ = 24.1°, 33.2°, 35.6°, 40.9°, 49.5°, 54.1°, 57.6°, 62.4°, and 64° are corresponding to the (012), (104), (110), (113), (024), (116), (018), (214), and (300) facets. HRTEM images also show the crystal plane spacing of the above crystal plane.
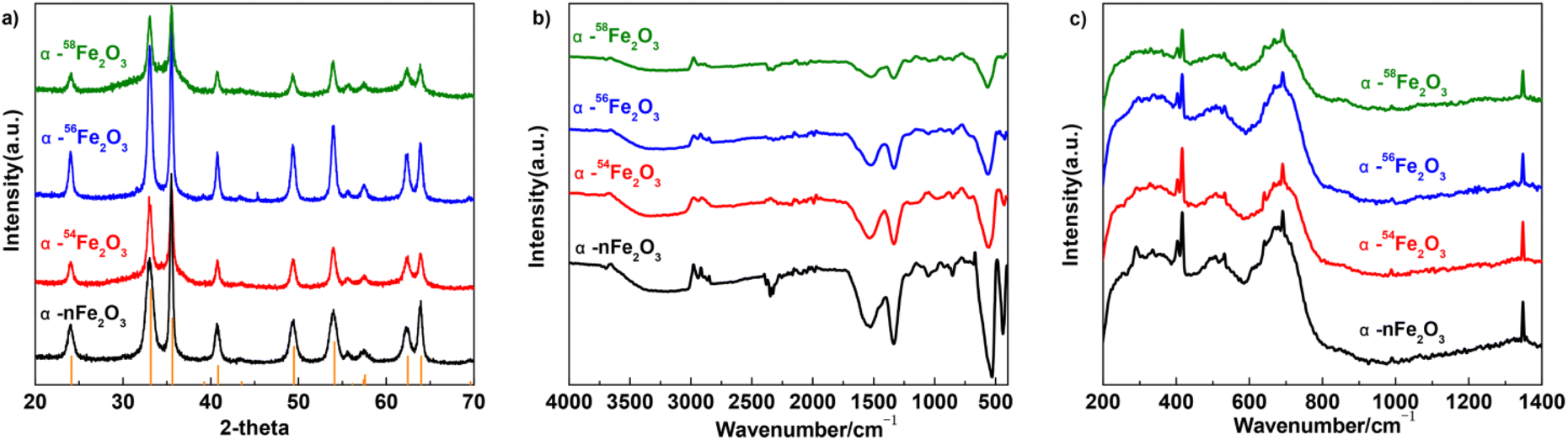 | ||
| Fig. 4 The (a) XRD patterns, (b) FTIR spectra, and (c) Raman spectra of four α-Fe2O3 catalysts. | ||
The FTIR spectra of prepared four α-Fe2O3 catalysts basically shows no difference in the peaks as presented in Fig. 4b. The peaks around 560 and 430 cm−1 are distinct for the vibrations of Fe–O from α-Fe2O3 which means the samples are all in the high-purity of α-Fe2O3 phase. The peaks around 3000 and 1530 cm−1 originate from the absorbed water molecules caused by the potassium bromide pellet technique, and peaks around 1340 cm−1 originate from residual impurities.18
From Fig. 4c, the four kinds of α-Fe2O3 catalysts have similar peaks in the Raman spectra while the peaks located at 403 cm−1, 669 cm−1, and 1348 cm−1 are related to the A1g peak of α-Fe2O3, and the peaks at around 416 cm−1, 532 cm−1, 691 cm−1 are related to the E1g of α-Fe2O3. There's no obvious difference between the four α-Fe2O3 catalysts with different Fe isotopes.19
To determine the surface chemical states of four α-Fe2O3 catalysts, the XPS spectra was measured and displayed in Fig. 5. Fig. 5a is the survey spectrum that confirms the presence of Fe and O elements located at around 720 eV and 530 eV, respectively. For the high-resolution O 1s spectra in Fig. 5b, the peak at 531.6 eV is attributed to the absorbed water molecules, and the peak at 530.0 eV is the lattice oxygen of α-Fe2O3. All the catalysts show two main peaks of Fe 2p3/2 at 710.9 eV and Fe 2p1/2 at 724.4 eV from Fig. 5c. The peak positions of the XPS O 1s and Fe 2p peak are listed in Table 3. The major compound α-Fe2O3 in the prepared catalysts was further demonstrated by all the characteristic peaks mentioned above.20
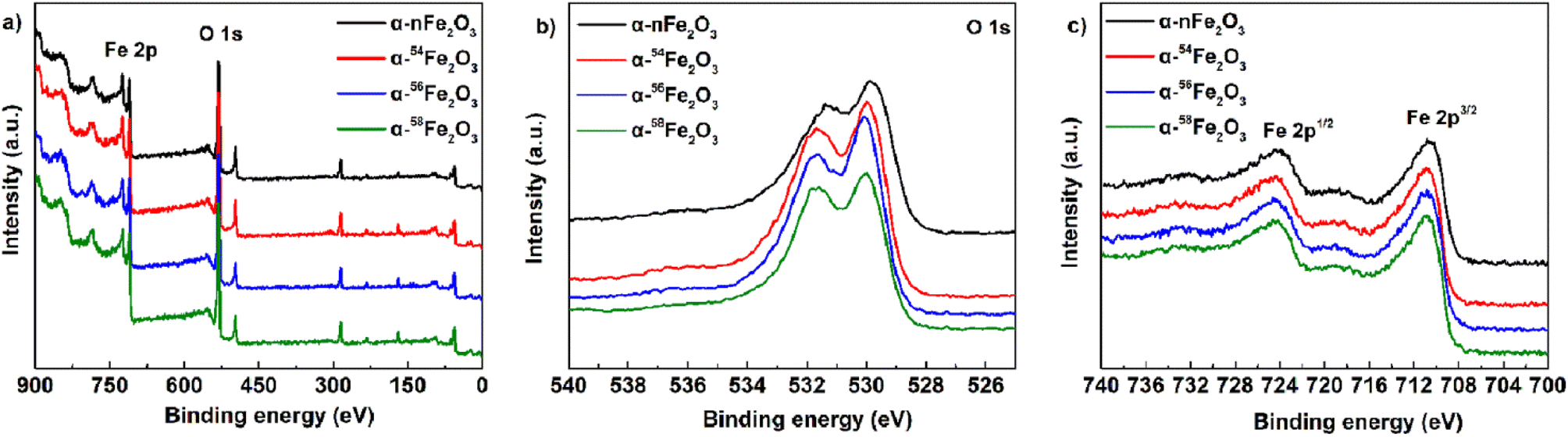 | ||
| Fig. 5 XPS spectra of four α-Fe2O3 catalysts: (a) survey spectrum, (b) O 1s, and (c) Fe 2p. | ||
| Compound | Peak position of O 1s/eV | Peak position of Fe 2p/eV | |||
|---|---|---|---|---|---|
| O–H | Fe–O | Fe 2p1/2 | Satellite | Fe 2p3/2 | |
| α-nFe2O3 | 531.4 | 529.9 | 724.4 | 720.0 | 710.8 |
| α-54Fe2O3 | 531.7 | 530.0 | 724.4 | 720.4 | 710.9 |
| α-56Fe2O3 | 531.6 | 530.1 | 724.4 | 720.0 | 710.9 |
| α-58Fe2O3 | 531.6 | 530.0 | 724.4 | 719.9 | 710.9 |
As presented in Fig. 6a the UV-Vis diffuse reflectance spectra of four separated isotopic α-Fe2O3 catalysts showing the variation of absorption coefficient A, as a function of photon energy. The A values for all catalysts decreased with the wavelength increasing around 570 nm. The optical band gap was extracted from the UV-Vis diffuse reflectance spectra according to the Tauc plot method,21,22
| (Ahν)2 = B(hν − Eg) | (1) |
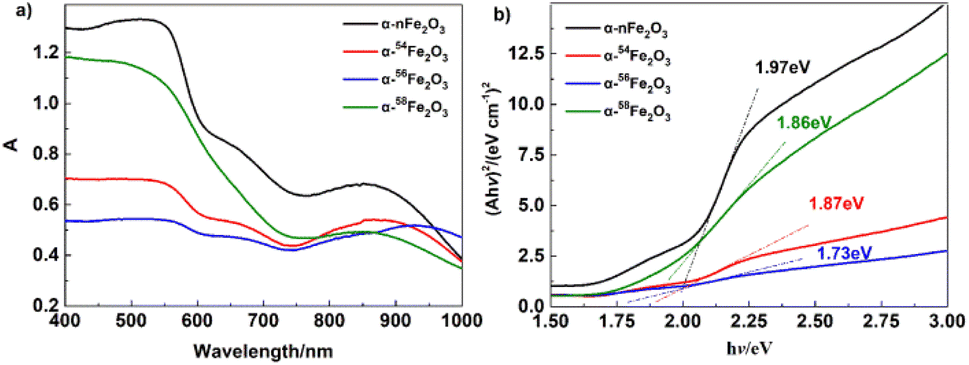 | ||
| Fig. 6 (a) UV-Vis diffuse reflectance spectra, and (b) (Ahν)2 − hν curves of α-Fe2O3 catalysts. | ||
| Compound | Endothermal peak | Exothermal peak | Eg/eV | ||
|---|---|---|---|---|---|
| Tendo/°C | RSD | Texo/°C | RSD | ||
| AP | 242.50 | 0.28% | 439.76 | 0.21% | — |
| α-nFe2O3/AP | 242.59 | 0.08% | 341.34 | 0.47% | 1.97 |
| α-54Fe2O3/AP | 243.36 | 0.11% | 341.98 | 0.32% | 1.87 |
| α-56Fe2O3/AP | 243.20 | 0.15% | 346.25 | 0.32% | 1.73 |
| α-58Fe2O3/AP | 243.95 | 0.11% | 344.34 | 0.16% | 1.86 |
Catalytic performance
The TG-DSC results of pure AP and AP in presence of α-Fe2O3 are listed in Table 4. Fig. 7a shows the three processes of thermal decomposition of pure AP: (1) phase transition corresponding to the negative peak in DSC curves. AP is transforming into the cubic crystal from the orthorhombic crystal without weight loss. (2) The low-temperature decomposition (LTD) corresponding to the first exothermic peak (310 °C) is related to the partial decomposition of AP to form NH3 and HClO4 intermediates by dissociation and sublimation. And (3) the high-temperature decomposition (HTD) corresponding to the last peak (440 °C) related to the complete decomposition of the intermediates to N2O, NO, H2O, HCl, Cl2, etc. products.23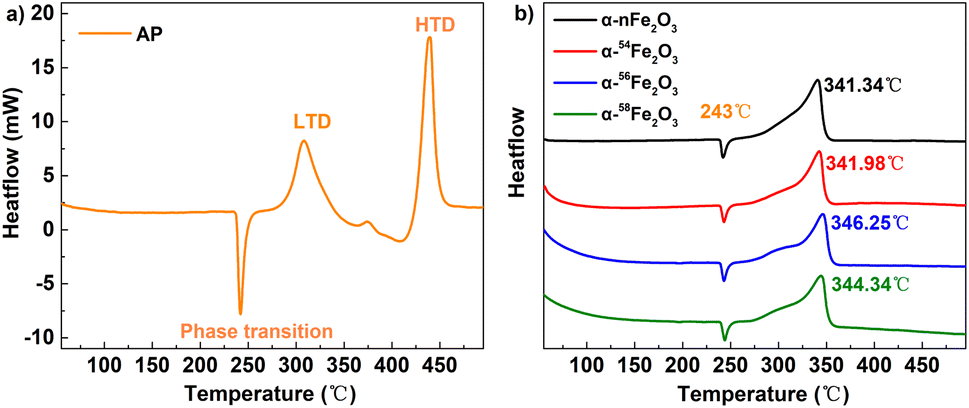 | ||
| Fig. 7 The DSC curves of (a) pure AP and (b) AP catalyzed by α-Fe2O3. | ||
It's obvious from Fig. 7b that the HTD peaks of the separated isotopic α-nFe2O3 shift to the lower temperature direction much more than that of α-Fe2O3 shifts, indicating the existence of remarkable differences in catalytic performance between separated isotopic α-Fe2O3 and α-nFe2O3. From Table 4 we can further see that the HTD peak exothermal temperatures of four kinds of α-Fe2O3/AP show significant differences which reveals that the catalytic performance is affected by the Fe isotopes in α-Fe2O3. The catalytic performance order from the best to the least of the four α-Fe2O3 catalysts is α-nFe2O3, α-54Fe2O3 α-58Fe2O3, and α-56Fe2O3. The catalytic performance positively correlates with the optical band gap, which follows the rules found in experiment results of thermal decomposition of AP catalyzed by N-type metal oxide semiconductor materials.22
According to the electron-transfer theory,24 the partially filled 3d orbit in α-Fe2O3 catalysts promotes the LTD process by providing more pathways for electrons and intermediate products. Then the gases arising from LTD process would be absorbed on the surface of catalysts enhancing the HTD process. The mechanism of thermal decomposition process of AP/α-Fe2O3 is illustrated in Fig. 8.
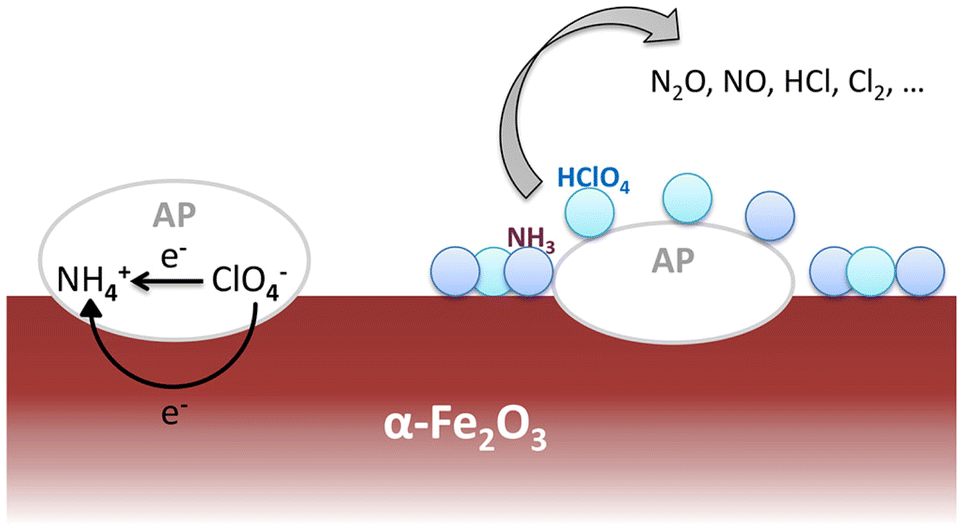 | ||
| Fig. 8 Schematic of the thermal decomposition process of AP on the α-Fe2O3 catalytic surface. | ||
Although the surface area of α-nFe2O3 is the smallest, it still shows the best catalytic performance which is not in accordance with the rule that larger surface area leads to better catalytic performance. We infer that the difference of catalytic activity in α-nFe2O3, α-54Fe2O3, α-56Fe2O3 and α-58Fe2O3 is due to the different Fe isotope content.
Experimental
Materials
54Fe2O3 (≥99.88%) 56Fe2O3 (≥99.7%) and 58Fe2O3 (≥93.13%) were purchased from Trace Sciences International. Fe2O3 (≥99.95%), NaOH solid, concentrated hydrochloric acid, concentrated nitric acid and NH₄ClO₄ (≥99.5%) were purchased from Aladdin Industrial Corporation. All the above reagents are analytically pure and used directly as received without further purification.Preparation and characterization
Four kinds of α-Fe2O3 catalysts, i.e., α-nFe2O3, α-54Fe2O3, α-56Fe2O3, and α-58Fe2O3 were all prepared through the typical hydrothermal procedure from the four kinds of purchased Fe2O3, respectively. The mixture of 0.2 g Fe2O3 and 8 mL aqua regia was sealed into a Teflon-lined stainless-steel autoclave maintained at 100 °C for 3 hours and then cooled to room temperature naturally. Adding 8 mL deionized water into the solution and then adding 5 M NaOH solutions drop by drop with magnetic stirring until the pH of the supernatant arrived at 6–7. The obtained precipitate was collected by centrifugation and washed 3 times with deionized water as well as absolute ethanol. The precipitate after being dried through a vacuum dryer for 8 h at 60 °C was roasted at 400 for 2 h in the Muffle furnace to get the α-Fe2O3 catalysts.The form of the catalysts was investigated by scanning electron microscopy (SEM) using a GeminiSEM300. The specific surface area was measured by the Brunner–Emmett–Teller (BET) measurements on an SSA-7000 analyzer. Powder X-ray diffraction (XRD) analyses were characterized by Bruker D8 Advance equipped with Cu Kα radiation at 14° min−1, and the high-resolution transmission electron microscopy (HRTEM) measurements were obtained by FEI Tecnai G2 F30 for a comprehensive understanding of the crystal structure. Fourier transform infrared spectra (FTIR, Nicolet Is 10) were measured from 4000 to 400 cm−1 using the potassium bromide pellet method. X-ray photoelectron spectroscopy (XPS) was carried out on Escalab 250Xi.
Catalytic activity
AP and four kinds of α-Fe2O3 catalysts were grounded for about 20 min in an agate mortar with absolute ethanol and further dispersed by ultrasonic wave for 10 min. The mixtures were dried through a vacuum dryer for 12 h at 60 °C and then ground to get the evenly dispersed α-Fe2O3/AP sample with the stable 5% mole fraction of the catalyst.Under the condition of an N2 gas flow rate of 10 mL min−1, thermal decomposition processes were investigated by the TG-DSC method (TGA/DSC 3+ Simultaneous Thermal Analyzer from METTLER TOLEDO) at a heating rate of 10 °C min−1 in the temperature range from 50–500 °C.
Conclusions
Four kinds of α-Fe2O3 catalysts (α-nFe2O3, α-54Fe2O3, α-56Fe2O3, and α-58Fe2O3) were prepared through same hydrothermal procedure to investigate the existence of the difference of catalytic performances of the isotopic catalysts. The characterization results of α-Fe2O3 present the uniformity of physical morphology among the α-Fe2O3 in natural abundance ratio and the separated isotopic α-Fe2O3 with the similar SEM and HRTEM images, adsorption and desorption isotherms of N2, XRD patterns, FTIR and Raman spectra. While the surface area of α-nFe2O3 is about 14% smaller than those of other three separated isotopic α-Fe2O3 because of much larger size distribution of neck widths of pore-blocking for the α-nFe2O3.The TG-DSC thermal analysis results of thermal decomposition of AP in presence of α-Fe2O3 confirm the existence of differences in the catalytic performance of the isotopic catalysts. The catalytic performance is affected by the contents of Fe isotopes in α-Fe2O3 and the order of it is: α-nFe2O3 < α-54Fe2O3 < α-58Fe2O3 < α-56Fe2O3 in line with the order of band gap obtained by UV-Vis DRS, which suggests that the catalytic performance of α-Fe2O3 catalysts with different contents of Fe isotopes positively correlated with the band gap value but they are not strictly proportional.
Although the surface area of α-nFe2O3 is the smallest, it still shows the best catalytic performance, so we infer that the difference in their catalytic activity in AP decomposition is mainly attributed to the difference in the charge distribution of the nuclei of different iron isotopes. The s-orbital is spherically symmetric when the nuclear charge is regarded as a point charge, but it's not a point charge under the real condition. The difference in the number of neutrons in isotopes leads to the difference in the nuclear forms of different atoms of the same element, and the distribution of the nuclear charge also varies.25 The distribution difference of nuclear charge with the change of nuclear form makes the nuclear field an equipotential surface consistent with the distribution of nuclear charge. Then for the s-orbital electrons, for example, instead of having a spherical symmetry distribution, they have a probability distribution along the equipotential surface of the nuclear field. For the p- and d-orbitals, there exist the same distortion to a certain degree for the same isotopes, but different for different isotopes. When separated isotopes are utilized as materials, the distortion of the same isotope is completely coincident, resulting in a significant cluster effect.26 Based on the theories of isotope effect and the data obtained in this work, we infer that the natural α-Fe2O3 is weakened by the overlapping of distorted orbitals, while the separated isotopic α-Fe2O3 shows a remarkable cluster effect because of the overlapping of distorted orbitals, which would reflect in the band gap energy, that is, the band gap energy of oxides of natural elements is larger than that of oxides of separated isotopes as the characterization results. The band gap energies of different separated isotopes are apparently distinct due to the different structures of orbital distortion. In this work, band gap energy is a clue to detect the catalytic difference between isotopic catalysts. We also expect to find out other parameters or methods to more directly as well as precisely reflect the difference.
All the experimental results indicate that the difference of the energy state of isotopomers when utilizing as catalysts is big enough to be taken into consideration at the chemical level. The law of it is not clear at present but the discovery of the catalytic isotope effect in this system is meaningful. Further studies on the difference of catalytic activity between separated isotopic and natural abundance oxides would not only explore the influence of nuclear charge distribution on the molecular states of extra nuclear electrons but also be possible to obtain a new kind of catalyst, namely isotopic catalyst.
Author contributions
Songtao Xiao, the secondary corresponding author (Songtao Xiao and Guoan Ye contributed equally to this work), has made substantial contributions to the design of the work and been visualizing during the whole work. He also has drafted the work. Yubing Xue has also drafted the work, made contributions to the acquisition and interpretation of data, and substantively revised the manuscript. Jing Zhao contributed the project administration. Xiechun Liu contributed the formal analysis of data. Haifeng Cong contributed the data curation and acquisition. Tian Lan has been providing the necessary resources. Dashu Pan contributed the analysis of the data. Lingyu Wang contributed resources and data curation. Xiaojuan Liu contributed the review and editing work. Guoan Ye, the secondary corresponding author (Songtao Xiao and Guoan Ye contributed equally to this work), contributed the funding acquisition and methodology. Yinggen Ouyang, the first corresponding author, has made contributions to the funding acquisition, conceptualization, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support for this study provided by the Continuous-Support Basic Scientific Research Project [grant number BJ22003103].Notes and references
- F. Soddy, J. Chem. Soc. Trans., 1911, 99, 72–83 RSC.
- P. Krumbiegel, Isotopes Environ. Health Stud., 2011, 47, 1–17 CrossRef CAS.
- A. L. Buchachenko, J. Phys. Chem. B, 2013, 117, 2231–2238 CrossRef CAS PubMed.
- W. W. Cleland, Arch. Biochem. Biophys., 2005, 433, 2–12 CrossRef CAS PubMed.
- C. Gunnemann, D. W. Bahnemann and P. K. J. Robertson, ACS Omega, 2021, 6, 11113–11121 CrossRef PubMed.
- K. Fujiki, Y. Kanayama and S. Yano, et al., Chem. Sci., 2019, 10, 1936–1944 RSC.
- P. E. Yankwich and M. Calvin, J. Chem. Phys., 1949, 17, 109–110 CrossRef CAS.
- J. Bigeleisen and M. Wolfsberg, J. Chem. Phys., 1955, 23, 1535–1539 CrossRef CAS.
- J. F. Marlier, Accounts Chem. Res., 2001, 34, 283–290 CrossRef CAS.
- U. Weisflog, P. Krumbiegel and H. Hübner, Isot. Environ. Health Stud., 1970, 6, 285–287 CrossRef CAS.
- H. Gang, L. Xinggui and L. Jianhua, et al., Rare Met. Mater. Eng., 2011, 40, 2010–2013 Search PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- Y. Zhou and J. Wang, Sci. China Chem., 2016, 46, 573–578 Search PubMed.
- S. Chaturvedi and P. N. Dave, J. Saudi Chem. Soc., 2013, 17, 135–149 CrossRef CAS.
- B. Shu-hong, C. Si-yu and F. Lu, Chin. J. Energ. Mater., 2021, 29, 460–470 Search PubMed.
- H. Li, Y. Chen, X. Zhao and S. Deng, Chin. J. Inorg. Chem., 2018, 34, 1670–1676 CAS.
- F. Wang, X. F. Qin and Y. F. Meng, et al., Mater. Sci. Semicond. Process., 2013, 16, 802–806 CrossRef CAS.
- X. Li, S. Li, H. Hu, T. Sun and S. Yang, Catal. Lett., 2022, 152, 3479–3488 CrossRef CAS.
- D. T. M. Phan, T. Häger and W. Hofmeister, J. Raman Spectrosc., 2017, 48, 453–457 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- T. J. Tauc, R. Grigorovici and A. Vancu, Phys. Status Solidi, 1966, 15, 627–637 CrossRef.
- C. Laberty and A. Navrotsky, Geochim. Cosmochim. Acta, 1988, 62, 2905–2913 CrossRef.
- V. V. Boldyrev, Thermochim. Acta, 2006, 443, 1–36 CrossRef CAS.
- K. Gao, G. Li and Y. Luo, et al., J. Therm. Anal. Calorim., 2014, 118, 43–49 CrossRef CAS.
- J. Bigeleisen, J. Am. Chem. Soc., 1966, 118, 3676–3680 CrossRef.
- S. Zhang, Q. Liu, M. Tang and Y. Liu, ACS Earth Space Chem., 2020, 4, 420–433 CrossRef CAS.
Footnote |
| † Songtao Xiao and Guoan Ye contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |