
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of novel β-lactam-metallo β-lactamase inhibitors†
Mbongeni Shungube a,
Ayanda K. Hlophea,
Letisha Girdharia,
Victor T. Sabea,
Byron B. Petersa,
Nakita Reddya,
Kehinde F. Omolabia,
Lloyd Chettya,
Thilona Arumugamb,
Anil Chuturgoonb,
Hendrik G. Krugera,
Per I. Arvidssonac,
Hua-Li Qind,
Tricia Naicker*a and
Thavendran Govender*e
a,
Ayanda K. Hlophea,
Letisha Girdharia,
Victor T. Sabea,
Byron B. Petersa,
Nakita Reddya,
Kehinde F. Omolabia,
Lloyd Chettya,
Thilona Arumugamb,
Anil Chuturgoonb,
Hendrik G. Krugera,
Per I. Arvidssonac,
Hua-Li Qind,
Tricia Naicker*a and
Thavendran Govender*e
aCatalysis and Peptide Research Unit, University of KwaZulu Natal, Durban, 4001, South Africa. E-mail: naickert1@ukzn.ac.za; Tel: +27 312601845
bSchool of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban, South Africa
cScience for Life Laboratory, Drug Discovery & Development Platform & Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden
dSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, 205 Luoshi Road, Wuhan, 430070, P. R. China
eDepartment of Chemistry, University of Zululand, Private Bag X1001, KwaDlangezwa 3886, South Africa. E-mail: govendert@unizulu.ac.za
First published on 22nd June 2023
Abstract
β-lactamases are enzymes that deactivate β-lactam antibiotics through a hydrolysis mechanism. There are two known types of β-lactamases: serine β-lactamases (SBLs) and metallo β-lactamases (MBLs). The two existing strategies to overcome β-lactamase-mediated resistance are (a) to develop novel β-lactam antibiotics that are not susceptible to hydrolysis by these enzymes; or (b) to develop β-lactamase inhibitors that deactivate the enzyme and thereby restore the efficacy of the co-administered antibiotics. Many commercially available SBL inhibitors are used in combination therapy with antibiotics to treat antimicrobial resistant infections; however, there are only a handful of MBL inhibitors undergoing clinical trials. In this study, we present 11 novel potential MBL inhibitors (via multi-step chemical synthesis), that have shown to completely restore the efficacy of meropenem (≤2 mg L−1) against New Delhi metallo-β-lactamase (NDM) producing Klebsiella pneumoniae in vitro. These compounds contain a cyclic amino acid zinc chelator conjugated to various commercially available β-lactam antibiotic scaffolds with the aim to improve the overall drug transport, lipophilicity, and pharmacokinetic/pharmacodynamic properties as compared to the chelator alone. Biological evaluation of compounds 24b and 24c has further highlighted the downstream application of these MBLs, since they are non-toxic at the selected doses. Time-kill assays indicate that compounds 24b and 24c exhibit sterilizing activity towards NDM producing Klebsiella pneumoniae in vitro using minimal concentrations of meropenem. Furthermore, 24b and 24c proved to be promising inhibitors of VIM-2 (Ki = 0.85 and 1.87, respectively). This study has revealed a novel series of β-lactam MBLIs that are potent, efficacious, and safe leads with the potential to develop into therapeutic MBLIs.
Introduction
The World Health Organisation (WHO) has predicted that drug resistance will cause the deaths of 10 million people worldwide every year by 2050.1,2 Incorrect prescription, dispensing and usage of commercialised antibiotics have contributed significantly to antibiotic resistance.1,3,4 The Antimicrobial Resistance (AMR) National Strategy Framework (2018–2024), as well as the Global Research and Development priority setting for AMR, and the Global Antibiotic Research and Development Partnership, prioritise research regarding antimicrobial treatment to help prevent obsolete antibiotics emanating as a result of mutations and bacterial evolution.5–7 Antibiotic resistance has worsened, due to the empirical treatment of hospitalised COVID-19 patients.3,8,9 There are several initiatives in place to either raise awareness, help reduce, or keep track of resistance, such as: the Global Action Plan on Antimicrobial Resistance (GAP); World Antimicrobial Awareness Week (WAAW); The Global Antimicrobial Resistance and Use Surveillance System (GLASS); Global Research and Development priority setting for AMR; Access Watch Reserve (AWaRe); and Global Antibiotic Research and Development Partnership (GARDP).7 Murray et al. estimated that 4.95 million deaths occurred in the year 2019 as a result of antibiotic resistance.6 Identified Klebsiella pneumoniae, as a common pathogen accounting for 29% of all reported bacterial infections.7 Klebsiella pneumoniae has recently been identified as a bacteria of concern, as mentioned in several reports, studies, and reviews, and is further substantiated by the resistance map (Fig. 1) generated from the CDDEP (Centre for Disease Dynamics, Economics and Policy).10 Unfortunately, most countries, especially the lower-middle-income countries, either do not supply their data to surveillance systems such as GLASS, or do not track and identify the bacterial infections; therefore, the resistance percentages indicated in this map are an underestimation of the actual prevalence. These alarming trends call for urgent intervention to treat AMR infections.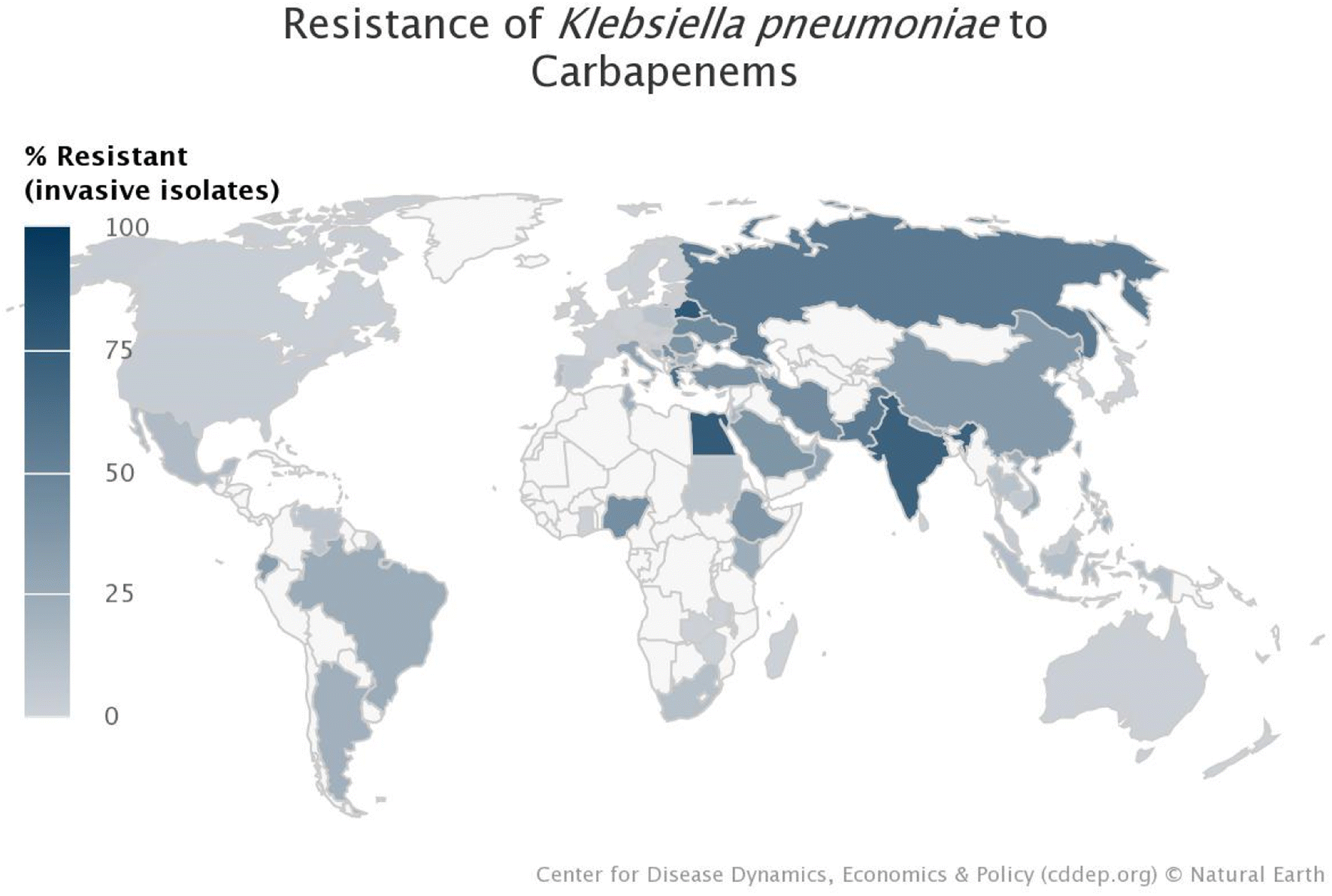 | ||
| Fig. 1 World map indicating the incidence of carbapenem-resistant Klebsiella pneumoniae in each country.10 | ||
β-lactamases are enzymes produced by the bacteria as a defence mechanism, which irreversibly deactivates β-lactam antibiotics by hydrolysing/cleaving the amide bond in the β-lactam ring before it has had the opportunity to bind to the penicillin-binding proteins (PBPs).3,11–17 These enzymes have been characterised by Ambler classes A, B, C and D, based on their amino acid homology and molecular properties.11–14 Classes A, C and D (SBLs) utilise a serine residue at the active site to effect the hydrolyses of the β-lactams while class B enzymes utilize either one or two zinc(II) ions at their active sites. The latter are referred to as MBLs and use a hydroxide ion from an activated water molecule as a nucleophile in the ring-opening process.11,14,18–21
Several SBL inhibitors are used in combination therapy with β-lactam antibiotics e.g., ampicillin with sulbactam, piperacillin with tazobactam, and a host of other combinations.12 Unlike the SBLs, there are no MBLs with clinically approved inhibitors8,12,13 or enhancers.22 MBLs have been shown to deactivate all β-lactam-containing antibiotics, except for monobactams (specifically aztreonam).23,24 Aztreonam is used in combination with ceftazidime/avibactam as a treatment since most MBL-producing Enterobacterales co-produce SBLs, which can hydrolyse aztreonam.11,23–29 A 2003 study found that metal chelators inhibit the Verona Integron-encoded metallo-β-lactamase 2 (VIM-2) enzyme, presumably through complexation of its zinc ion.12 This led to the hypothesis that metal chelators could be developed to quell resistance caused by MBLs.12
Metal chelating agents, such as 2,6-dipicolinic acid (DPA) and ethylenediaminetetraacetic acid (EDTA), have proven to inhibit the MBL's catalytic activity by isolating or removing the metal ions from its active site.17,30–32 One of the drawbacks of using these “metal stripping” agents for clinical use is the lack of selectivity and specificity towards MBLs as their use could result in the removal of essential metals and the deactivation of physiological metalloproteins.17,33
Our group recently investigated the antibacterial effect of cyclic amino acidic zinc chelators when co-administered with the clinically-used carbapenem antibiotics, meropenem and imipenem (the most widely prescribed carbapenem antibiotics worldwide).12,34,35 We found that conjugation to existing β-lactam antibiotic scaffolds improved the transport of the chelators to the MBL active site, as well as their pharmacokinetics/pharmacodynamics (PK/PD) profile.36,37 Our rationale was based on the premise that the chelating moiety would be responsible for MBL inhibition whilst the β-lactam scaffold would provide for increased lipophilicity and selective transport to the bacteria.
Results and discussion
Following from our studies conducted for NOTA (1)12,34 and the reported BP series,36,37 we decided to investigate the effect of (i) NOTA and (ii) β-lactam scaffolds as potential MBLIs. We envisaged that these changes could offer alternate PK/PD profiles of such MBLIs. We first investigated the effect of varying the chelator to having an additional donor atom for metal bonding and an additional carbon spacer for steric effects (Fig. 2).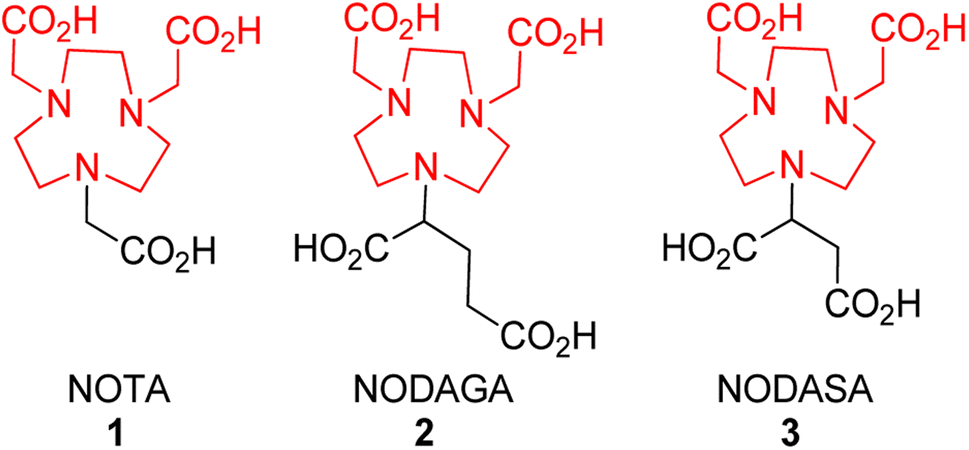 | ||
| Fig. 2 1,4,7-Triazocyclononane chelators: NODASA derivative, NOTA and NODAGA. | ||
For this to be achieved, one of the carboxylic acids from these zinc chelators was to be employed in an amide bond linkage to the lactam scaffold. The protected form of NOTA (7) or NODAGA (4) chelators were purchased and used for coupling to the commercially available 7-aminocephalosporanic acid (7-ACA) (Scheme 1). 7-ACA is a cephalosporin core which has been used to generate a number of cephalosporin antibiotics. Its solubility has proved to be very poor in solvents such as dimethylformamide (DMF) and acetonitrile (ACN), which are commonly employed for amide couplings. 7-ACA had to be dissolved in a mixture of acetone and water with base, then coupled to 4 or 7 using the peptide coupling agent 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) at −10 °C, to furnish compounds 5 and 8 in yields of 40% and 25%, respectively, after purification with preparative supercritical fluid chromatography (prep-SFC). Since compound 4 formed an asymmetric centre, compound 5 and its subsequent products were obtained as an epimeric mixture. Attempts to improve the yield with higher temperatures resulted in the formation of undesired by-products. Compounds 5 and 8 were deprotected with trifluoroacetic acid (TFA), using thioanisole as a scavenger to afford the target compounds 6 and 9 in 60% and 70% yields respectively.
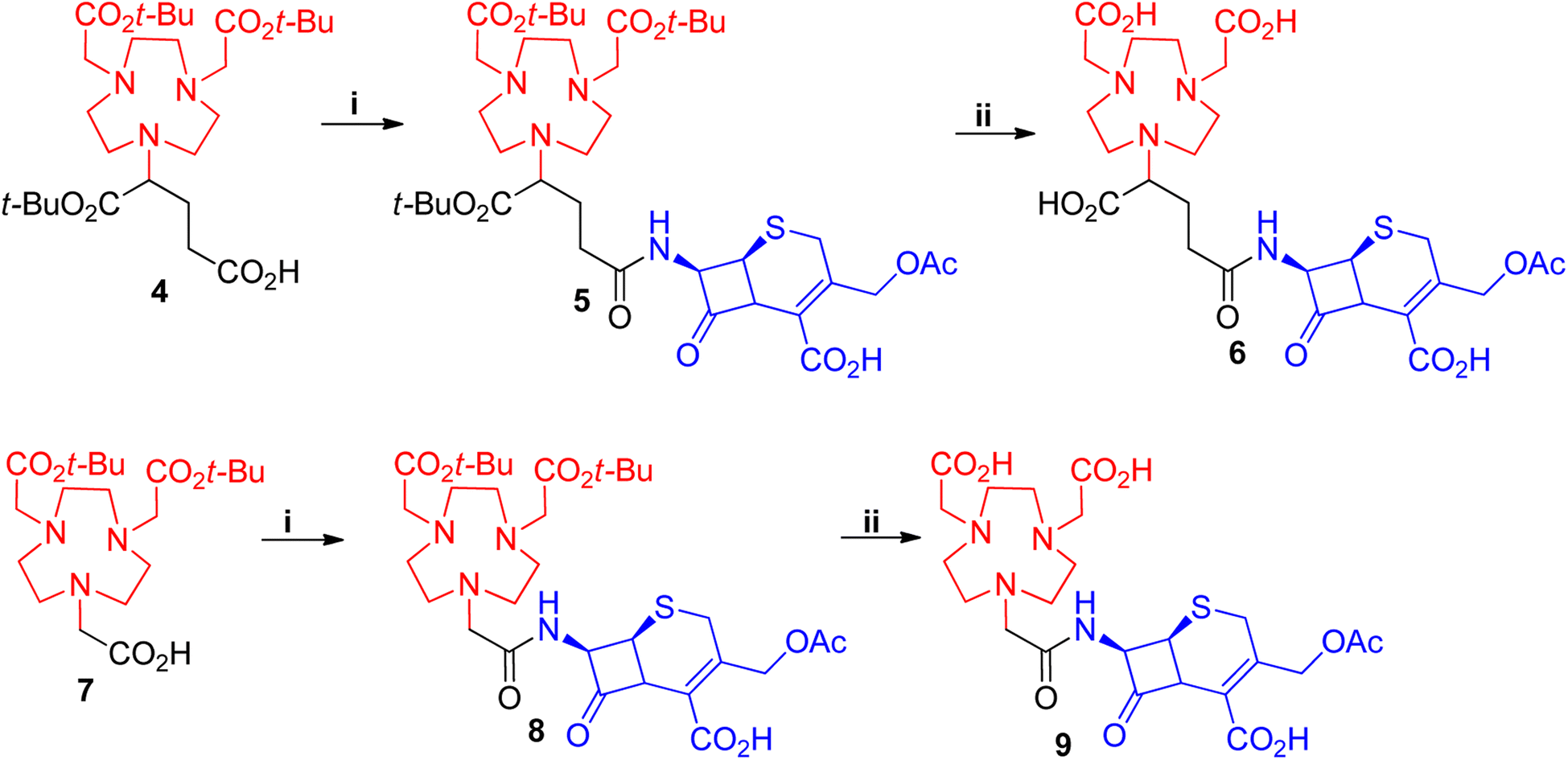 | ||
| Scheme 1 Synthesis of NOTA-7-ACA and NODAGA-7-ACA. i = HATU, DIEA, ACN, 7-ACA (dissolved in a mixture of NaHCO3, H2O, acetone, ACN), −20 °C, 1 h ii = thioansole, TFA, rt, 3 h. | ||
Unlike NOTA and NODAGA, protected NODASA could not be obtained commercially or synthetically. Challenges with its synthesis are discussed in the ESI.† An alternative route was envisaged whereby we first attach the β-lactam to the chelator, followed by the alkylation in order to provide enough steric hindrance to prevent the over-alkylation step. We synthesised 11 using a procedure reported by Dutta et al.,38 1,4,7-triazanonane (10) underwent a Michael-addition reaction with mono-methyl fumarate to afford 11 in excellent yields (>95%) (Scheme 2). Compound 11 was Boc-protected to afford 12 in yields of 85–96% after purification on a silica gel column. Compound 12 was coupled to 7-ACA to afford 13 with yields of 30–50%, following similar protocols adopted for the coupling of NOTA and NODAGA. Similar to compound 4 all compounds obtained after the coupling reaction of 11 with enantiomerically pure β-lactams were also obtained as epimeric mixtures. The Boc-groups were removed at room temperature using TFA and anisole as the scavenger in dichloroethane (DCE) and washed in ether to afford the TFA salt of 14 in excellent yields (>90%), without the need for further purification. Compound 14 was alkylated with tert-butyl bromoacetate to afford 15, without any observable over-alkylation and was purified via prep-SFC to provide the pure product in 46–50% yields. Compound 15 was subjected to a hydrolysis reaction of the tert-butyl groups with TFA to afford the target compound 16 in 65% yield. All reactions were monitored with LC-MS and characterized using standard techniques.
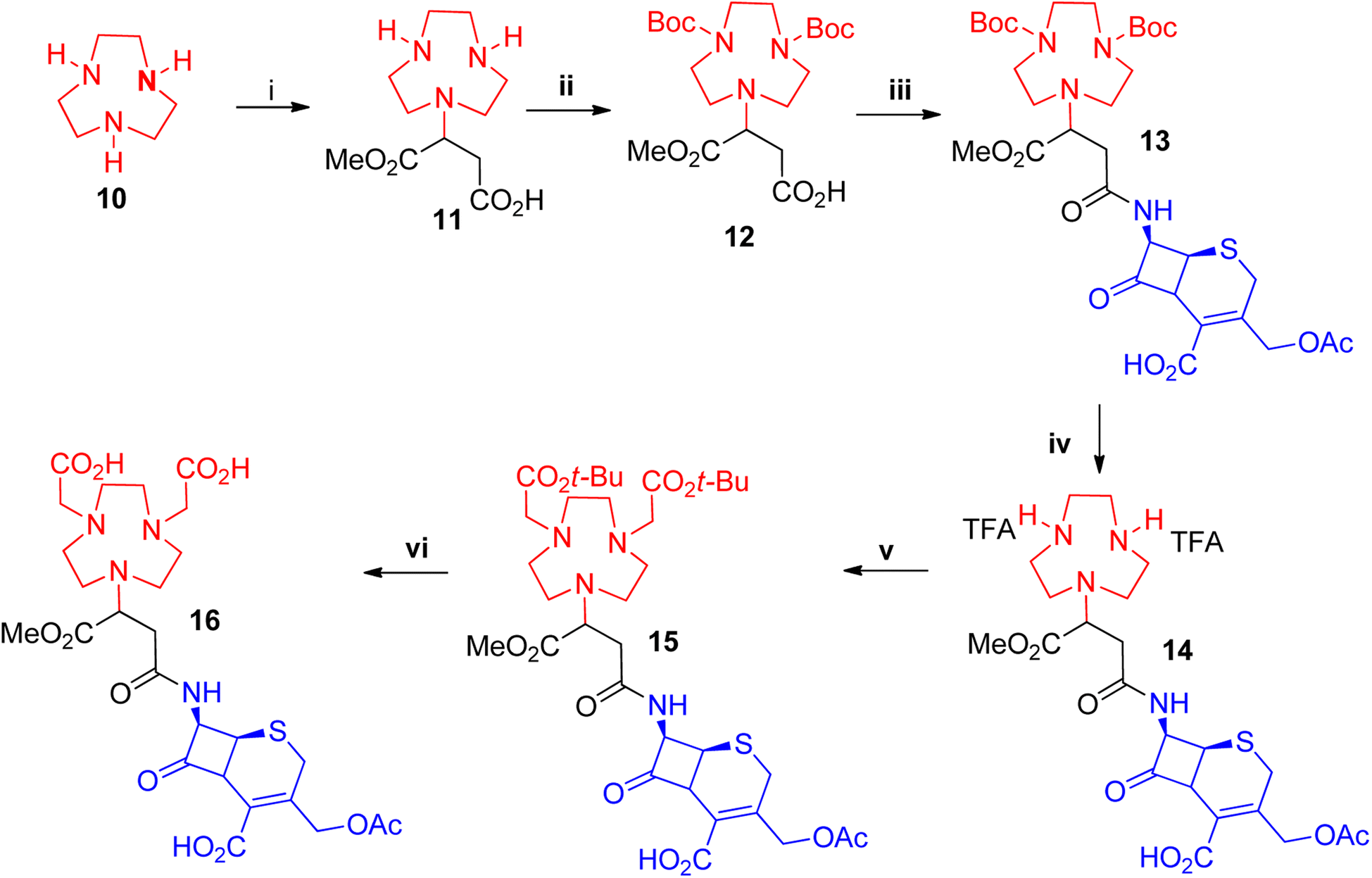 | ||
| Scheme 2 Synthesis of NODASA-7-ACA. i = mono-methyl fumarate, DIEA, DCM, rt, 4 h ii = Boc2O, DIEA, DCM, rt, 4 h iii = HATU, DIEA, ACN, 7-ACA (dissolved in a mixture of NaHCO3, H2O, acetone, ACN), −20 °C, 1 h iv = anisole, TFA, DCE, rt, 20 min. v = tert-butyl bromoacetate, DIEA, DCE, 0–25 °C, 1 h vi = thioansole, TFA, rt, 3 h. | ||
Analogues 6, 9 and 16 were evaluated, in combination with meropenem, for their ability to inhibit NDM harbouring strains of Escherichia coli39 and Klebsiella pneumoniae.10 Meropenem is a carbapenem antibiotic in clinical use and currently displays little-to-no activity against carbapenemase-producing bacteria. Hence, it was chosen to assess the synergistic effect of these new MBLIs (Table 1, entry 1).40 The compounds were evaluated using the checkerboard method and the results are shown in Table 1. Unless otherwise stated, the minimum inhibitory concentrations (MIC) were obtained by co-administration of meropenem with an inhibitor and all experiments were performed in triplicate.
| Entry | Inhibitor | Minimum inhibitory concentrationa-mg L−1 | |||
|---|---|---|---|---|---|
| Escherichia coli NDM-1 | Klebsiella pneumoniae NDM | ||||
| Meropenem | MBLI | Meropenem | MBLI | ||
| a The checkerboard assays were conducted in triplicate p < 0.01. | |||||
| 1 | None | 128 | 0 | 128 | 0 |
| 2 | 7-ACA | 2 | 64 | 2 | 32 |
| 3 | NOTA (1) | 0.06 | 4 | 0.125 | 4 |
| 4 | NODAGA (2) | 0.06 | 4 | 0.06 | 16 |
| 5 | 6 | 0 | >128 | 0 | >128 |
| 6 | 9 | 0 | >128 | 0 | >128 |
| 7 | 16 | 0 | >128 | 0 | >128 |
| 8 | 6 | 0.25 | 32 | 0.25 | 16 |
| 9 | 9 | 0.125 | 16 | 0.125 | 16 |
| 10 | 16 | 1 | 16 | 2 | 8 |
| 11 | NOTA-Zn | >128 | >128 | >128 | >128 |
Table 1 shows that meropenem alone did not inhibit the growth of neither E. coli NDM-1 nor Klebsiella pneumoniae NDM at a concentration of 128 mg L−1. Co-administration with high concentrations of 7-ACA resulted in only a modest regain of meropenem activity against both strains. However, co-administration with chelator compounds 1 and 2 drastically enhanced the activity of meropenem to achieve MICs between 0.06–0.125 mg L−1 with low chelator concentrations of 4–16 mg L−1. Although the results for chelators 1 and 2 were excellent, they are not viable options for therapy due to the low bioavailability, non-specificity towards metal chelators, and off-target activities that often accompanies these types of compounds. The synthesized compounds 6, 9, and 16 were found to lack any antibacterial activities on their own, even with concentrations greater than 128 mg L−1. Combination of these chelators with meropenem restored its MIC efficacies which is evidence for their MBL inhibitory activities. Importantly, zinc pre-complexed NOTA was also evaluated as a potential MBL inhibitor and as expected, produced no bacterial inhibition, confirming that zinc chelation is essential to restore the activity of meropenem. Additionally, the compounds in Table 1 displayed an absence of activity towards SBL expressing bacteria Serratia marcescens (KPC-2) and E. coli (OXA-28), indicating specific activity towards MBLs; NDM (Tables 1–3), IMP and VIM (Table 3).
| Entry | Inhibitor | Minimum inhibitory concentration (mg L−1)a | |||
|---|---|---|---|---|---|
| Escherichia coli NDM-1 | Klebsiella pneumoniae NDM | ||||
| Meropenem | MBLI | Meropenem | MBLI | ||
| a All assays were conducted in triplicate p < 0.001. | |||||
| 1 | 9 | 0.125 | 16 | 0.125 | 16 |
| 2 | 20a | 0.125 | 16 | 0.25 | 16 |
| 3 | 20b | 0.5 | 16 | 0.5 | 16 |
| 4 | 24a | 0.06 | 16 | 0.125 | 8 |
| 5 | 24b | 0.125 | 8 | 0.25 | 8 |
| 6 | 24c | 0.06 | 8 | 0.25 | 8 |
| 7 | 24d | 0.125 | 16 | 0.125 | 16 |
| MBL-producing bacteria | Minimum inhibitory concentration (mg L−1)a | |||
|---|---|---|---|---|
| Meropenem | 24b | Meropenem | 24c | |
| a All assays were conducted in triplicate p < 0.001. | ||||
| E. coli NDM-4 | 0.06 | 8 | 0.25 | 8 |
| E. coli IMP-1 | 0.03 | 8 | 0.5 | 8 |
| E. coli IMP-8 | 0.125 | 8 | 0.25 | 8 |
| Enterobacter cloacae NDM-1 | 0.25 | 8 | 0.25 | 8 |
| E. cloacae VIM-1 | 0.5 | 8 | 0.25 | 8 |
The biological activity confirmed that 7-ACA, coupled with various zinc chelators, could restore the activity of meropenem towards MBL-expressing pathogens. The high cost of commercial starting materials 7 and 4, prompted us to pursue the structural optimisation with the more affordable NODASA (3) derivatives.
The synthesis of further derivatives was based on related and commercially available cephalosporins: cefadroxil; cefaclor; ceftizoxime; ceftibuten; ceftiofur and ceftazidime. Structurally similar β-lactams cefadroxil and cefaclor were used to synthesise compounds 20a and 20b (Scheme 3). Unlike 7-ACA, cefadroxil had excellent solubility in DMF was therefore the first choice solvent for its HATU facilitated coupling to 12. However, poor yields were observed due to incompletion of the reaction. We anticipated that the reaction could benefit from longer reaction times and employed the more stable coupling reagent combination of EDC.HCl with HOBt to get yields of 40–55% for compound 17a after prep-SFC purification. Cefaclor displayed better solubility and gave optimum couplings to 12 in acetonitrile with COMU in just two hours to afford 17b in 50–70% yields.
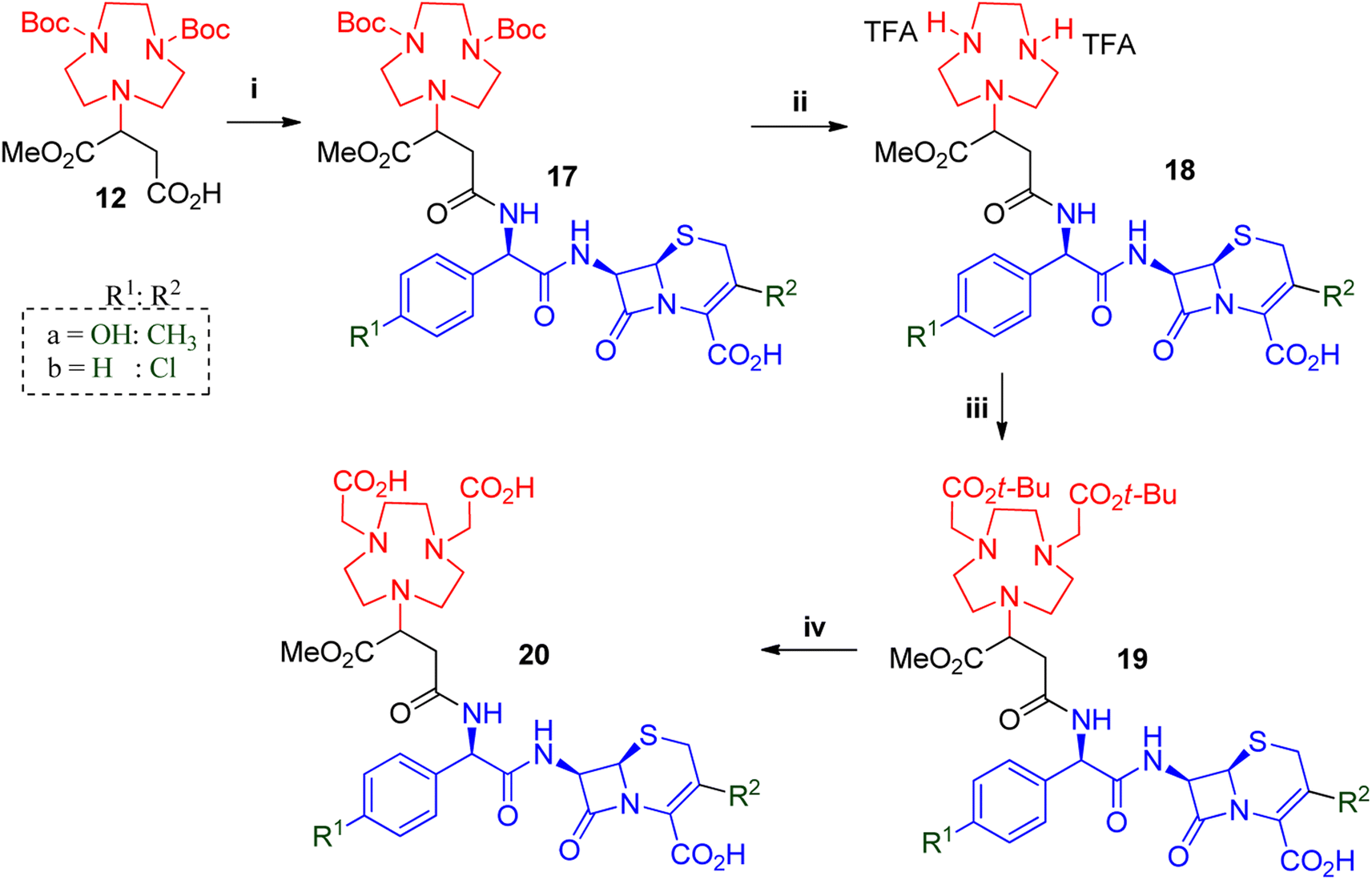 | ||
| Scheme 3 General synthesis of NODASA-β-lactam antibiotic used in studies A. i = β-lactam, HOBt, EDC.HCl, DMF/DMSO, rt, 6–12 h or COMU, DIEA, ACN, rt, 2 h ii = anisole, TFA, DCE, rt, 2–6 h iii = tert-butyl bromoacetate, DIEA, DCE, 0–25 °C, 2–6 h iv = thioansole, TFA, rt, 3–6 h. | ||
Once the β-lactams were coupled to the chelator to afford 17a and 17b, their solubility properties became similar. Therefore, the amine deprotection to afford 18a and 18b was similar to that of 13, with the only difference being the reaction times. The reactions with 17a took between two and three hours while 17b required just one hour to complete, before affording compounds 18a and 18b in quantitative yields. Thereafter, compounds 18a and 18b were washed with diethyl ether, and alkylated similarly to 14, with varying reaction times. Compounds 19a and 19b were purified using prep-SFC to give products with yields of 30–45%. Thereafter, 19a and 19b underwent hydrolysis with TFA to afford the desired compounds 20a and 20b respectively.
Another set of structurally related β-lactams was also explored, namely ceftizoxime, ceftibuten, ceftiofur and ceftazidime. These were used to synthesise compounds 24a–d, respectively (Scheme 4). Solubility tests showed that all of the starting β-lactams were only applicable in DMF and DMSO. Compound 12 was used to couple to each of these β-lactams with EDC.HCl and HOBt in dry DMF/DMSO. Pure compounds 21a–d were obtained after prep-SFC purification with yields ranging from 50–65%. Compounds 21a–d were Boc-deprotected, using TFA with anisole as a scavenger in DCE. All of the reactions were completed in 1–2 hours with quantitative yields obtained after the TFA was removed over a stream of nitrogen gas and washed with diethyl ether. The crude salts of compounds 22a–d were alkylated with tert-butyl bromoacetate to afford 23a–d. The reactions were performed in DCM in the presence of DIEA at 0 °C to room temperature and the times varied from 4–7 hours. The crude reactions were purified using SFC to afford 23a–d with 30–40% yields. Subsequently, the tert-butyl groups of 23a–d were cleaved using TFA to afford the desired compounds 24a–d in 89–95% yields.
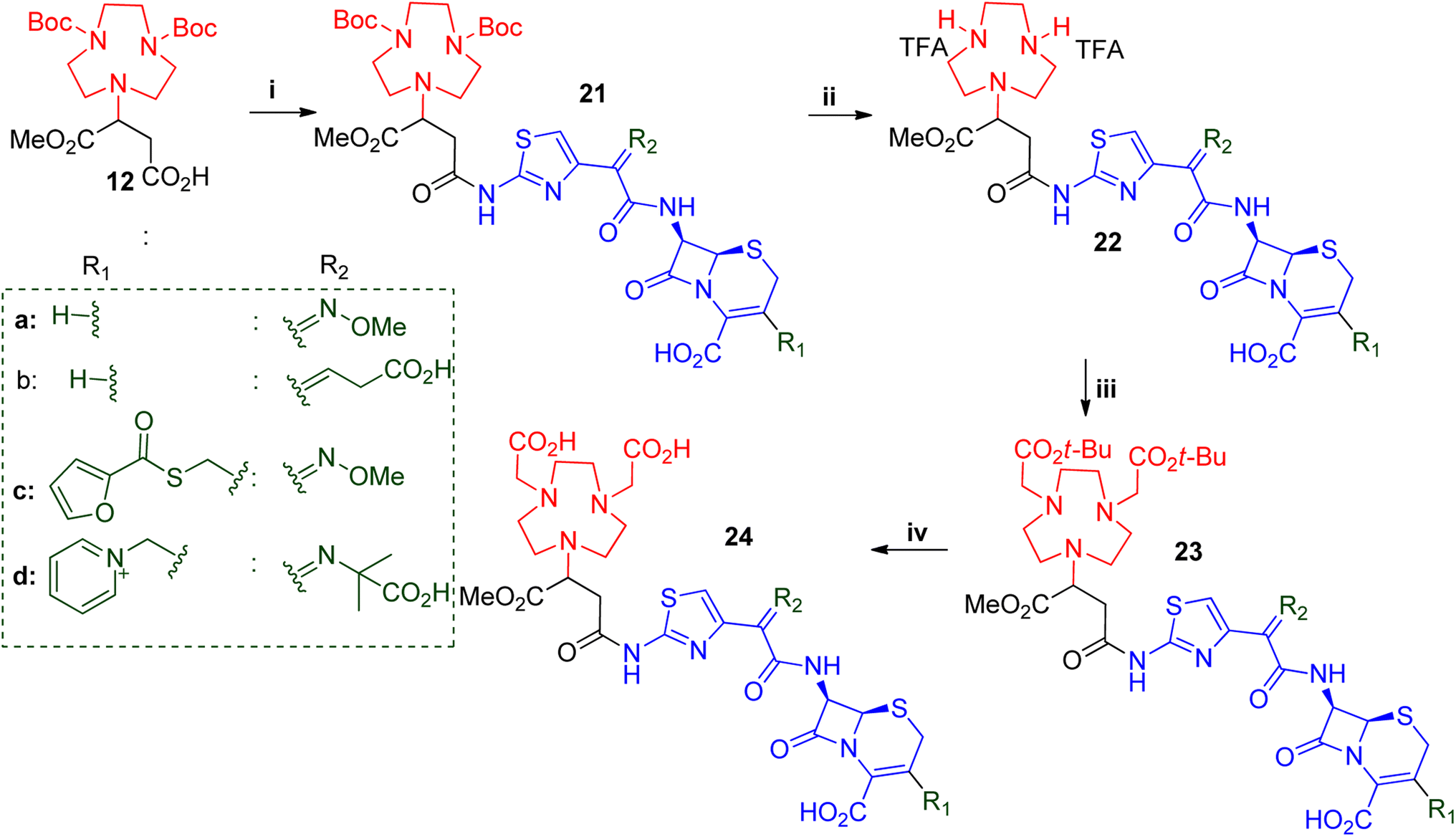 | ||
| Scheme 4 General synthesis of NODASA-β-lactam antibiotic used in studies B. i = β-lactam, HOBt, EDC.HCl, DMF or DMSO, rt, 6–12 h ii = anisole, TFA, DCE, rt, 2–6 h iii = tert-butyl bromoacetate, DIEA, DCE, 0–25 °C, 2–6 h iv = thioansole, TFA, rt, 3–8 h. | ||
All six synthesised derivatives 20a–b and 24a–d (Fig. 3), were evaluated as MBLIs against Escherichia coli NDM-1 (ref. 39) and Klebsiella pneumoniae NDM,10 in combination with meropenem.
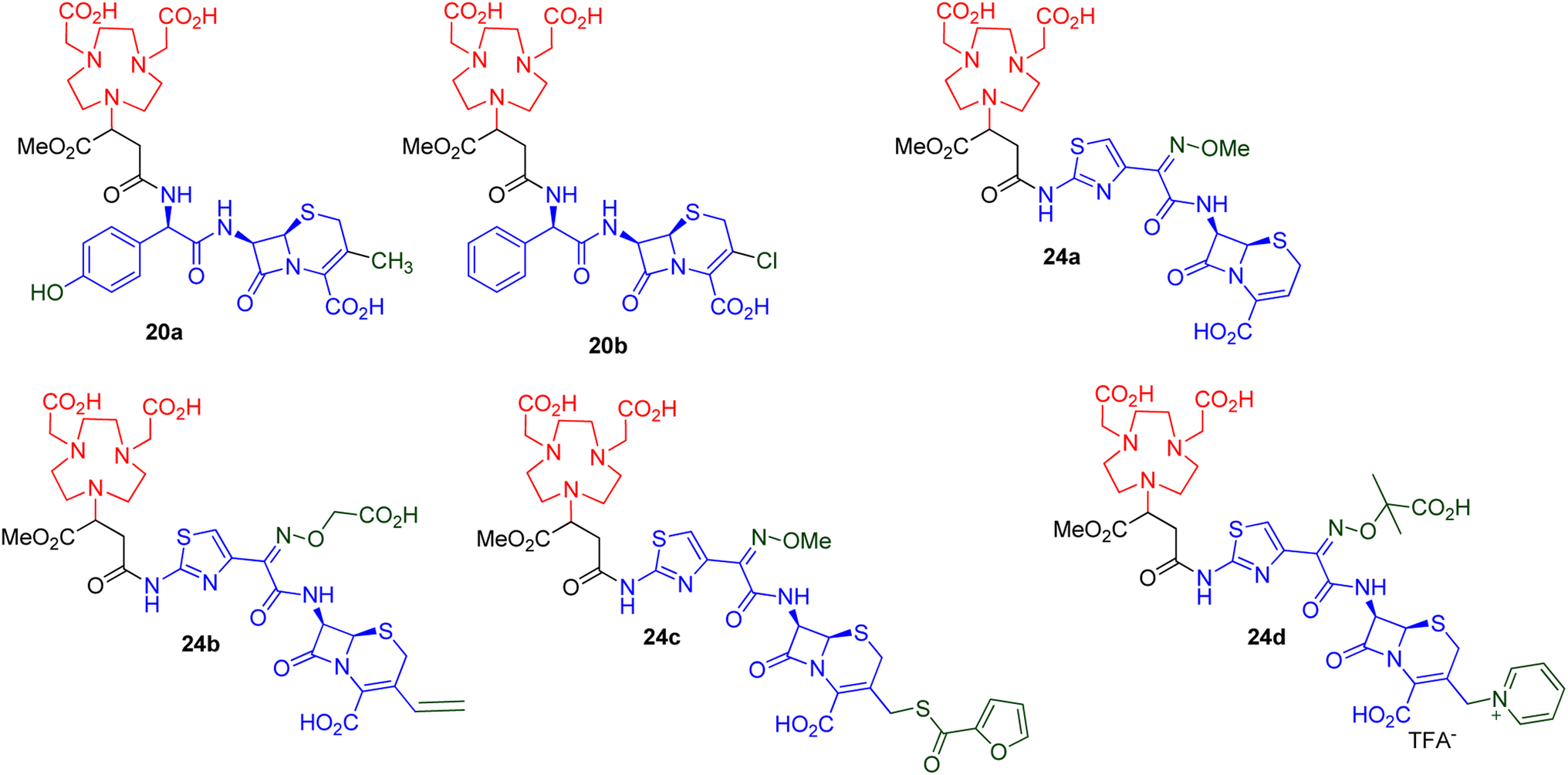 | ||
| Fig. 3 Synthesized NODASA derivatives as MBLIs.41 | ||
The data in Table 2 demonstrates that all of the evaluated compounds, 20a–b and 24a–d, successfully restored the activity of meropenem to MICs ranging from 0.06–0.5 mg L−1 with inhibitor concentrations between 8 and 16 mg L−1. When comparing the activity between the synthesized MBLIs, 24b and 24c showed the best activities.
All six derivatives, 20a–b and 24a–d, fully restored meropenem's efficacy (≤2 mg L−1) according to the EUCAST breakpoints.42 These derivatives demonstrated similar activities when compared to the lead inhibitor in Table 1, i.e., compound 9, while the thiazole-containing compounds' (24a–d) MICs produced better results than the non-thiazole-containing compounds (20a–b). It is also noted that compounds 24a–d have a longer distance between the chelator moiety and the lactam moiety than 20a and 20b. The binding free energies of the two families of compounds (20 versus 24) were previously calculated by our group using advanced molecular dynamics studies. In general, the system with the longer chain distance between the chelator and the lactam moieties was superior.36,37 It is also possible that a thiazole ring, which is present in compounds 24, is necessary to enhance the MBL inhibition. This could be investigated further by trying different approaches to insert unsubstituted carbon chain linkers with varying chain lengths between compounds 20 or a structurally similar compound; a linker containing a phenyl group; or a linker containing a thiazole group, and comparing the activities of these compounds (beyond the scope of this manuscript).
Based on the synthetic viability from Table 2, compounds 24b and 24c were further screened for activity against pathogens harbouring VIM and IMP MBLs. Table 3 indicates that 24b and 24c restored the efficacy of meropenem against pathogens harbouring blaNDM-4, blaVIM-1, blaIMP-1 and blaIMP-8 genes, in addition to bacteria expressing blaNDM-1 genes (Tables 1 and 2). Compounds 24b and 24c are therefore effective inhibitors of the most clinically relevant subclass, B1 MBLs.
The cytotoxicity using a human liver (HepG2) cell line was evaluated for 24b and 24c. From the cytotoxicity data, IC50 values of 42.34 mg mL−1 and 51.83 mg mL−1 were obtained for 24b and 24c, respectively (Fig. 4). In general, compound 24b reduced cell viability compared to the control, with 10 μg mL−1 being significant; compound 24c increased cell viability compared to the control, with 200 μg mL−1 being significant (Fig. 4A). The methyl thiazol tetrazolium (MTT) assay measures cellular metabolic output and compound 24c increases metabolic output. It is known that cancer cells utilise anaerobic glycolysis for ATP generation and minimises mitochondrial output as this will activate apoptosis. The lactate dehydrogenase (LDH) assay further confirmed the non-toxic properties of both compounds 24b and 24c with the former significantly decreasing LDH membrane leakage at 8 μg mL−1 (Fig. 4B). Compound 24c significantly decreased LDH membrane leakage at all evaluated concentrations (Fig. 4B). The chelator concentrations selected for drug susceptibility screenings (≤ 32 mg L−1) were below the IC50's, which allowed us to proceed safely with biological evaluation.
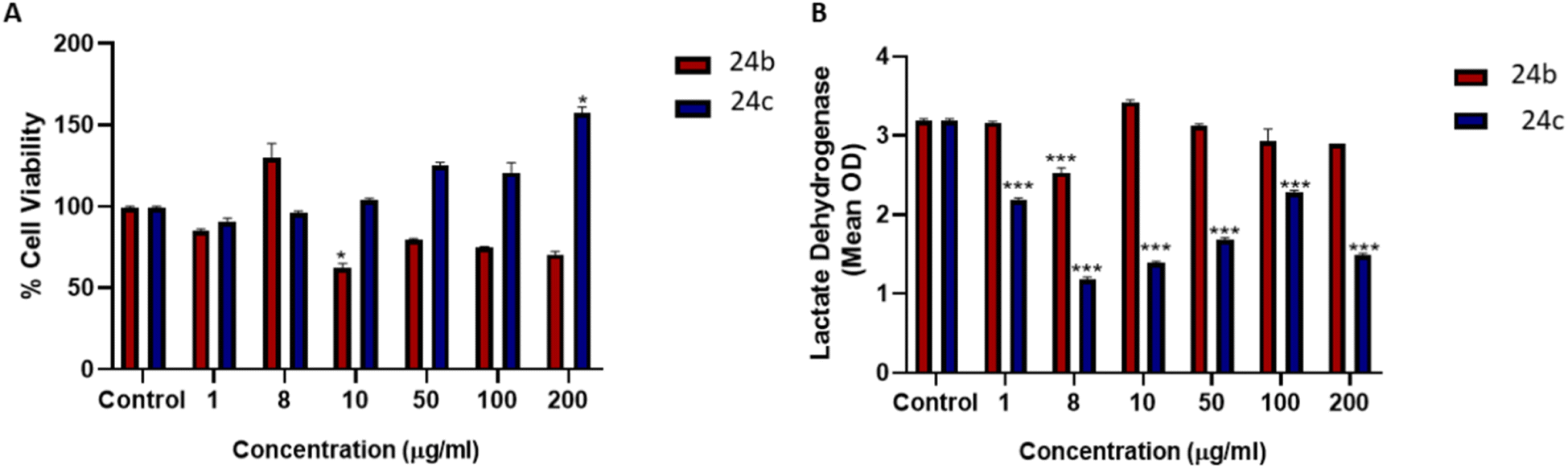 | ||
| Fig. 4 Cell viability studies conducted on HepG2 cells using varying concentrations of 24b and 24c. (A) Cell viability was reduced at 1 μg L−1 and 10–200 μg L−1 with proliferation observed at 8 μg L−1 for 24b. *p < 0.5 and **p < 0.01, relative to the control. 24c exhibited a dose-dependent increase in the cell viability of HepG2 cells. However, cell viability was only significantly altered at 200 μg L−1 *p < 0.05 relative to control. (B) LDH levels were significantly reduced at 8 μg L−1 and remained unaffected at 1 μg L−1 and 10–200 μg L−1, indicating 24b does not induce necrosis in HepG2 cells after exposure. Similarly, 24c also did not induce necrosis, as LDH was significantly reduced at all concentrations. ***p < 0.001 relative to control. All assays were conducted in triplicate. | ||
Fig. 5 represents the effect exhibited by chelator 24b/24c + meropenem over 24 hours against the virulent K. pneumoniae NDM-expressing bacteria, respectively. Both chelators displayed excellent bactericidal activity against this strain, with meropenem concentrations of 0.5, 1 and 2 mg L−1. A > 3 log10 decrease in the bacterial load was observed 6 hours post-inoculation for combination therapy, relative to meropenem monotherapy. Although a sharp decrease of bacteria was observed at 4 hours post-inoculation with meropenem monotherapy, this effect could not be sustained and K. pneumoniae NDM continued to proliferate. This was expected, as the chelator is essential to potentiate the activity of meropenem to susceptible levels. 32 mg L−1 of compounds 24b and 24c were needed to achieve complete bactericidal activity with 0.5 mg L−1 meropenem. Increasing the meropenem dose to 2 mg L−1 resulted in faster sterilizing activity from 8 hours post-inoculation, without any regrowth in a 24 hour period.
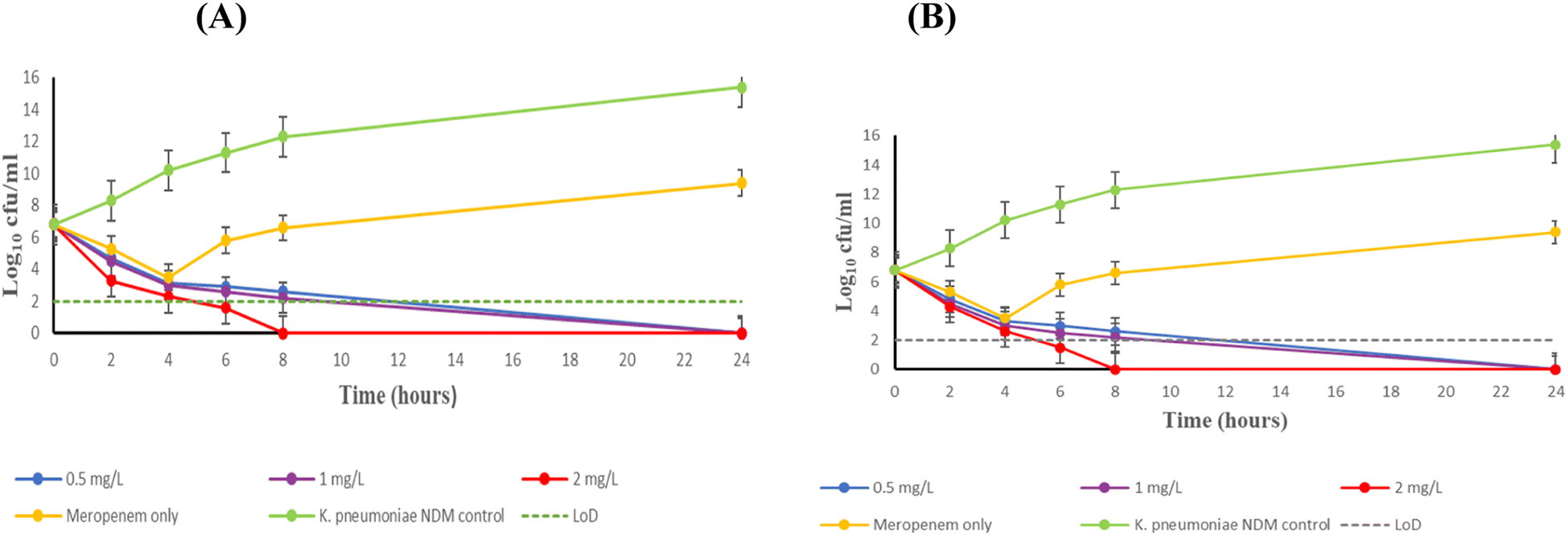 | ||
| Fig. 5 Time-kill kinetic study of K. pneumoniae NDM. (A) The combination of 24b + meropenem. (B) The combination of 24c + meropenem. All assays were conducted in triplicate. The combination of 32 mg L−1 24c + 0.5 mg L−1 meropenem produced p < 0.0441 when compared to monotherapy and p < 0.0013 for no therapy. | ||
To understand the kinetics of MBLs; VIM-2 and IMP-1, enzyme analyses were undertaken utilizing Michaelis–Menten conditions and Lineweaver–Burk plots (Fig. S1†). The kinetics parameters of both MBLs (Table 4), shared a similarity with regards to the Vmax and catalytic turn over numbers. The resultant Km values, however, were not similar, demonstrating that VIM-2 was a more catalytically efficient enzyme between the two MBLs. This infers that pathogens expressing VIM-2 can neutralize antibiotics even at low concentrations, much faster as compared to IMP-1, that requires a larger antibiotic concentration to reach Vmax and produce optimal hydrolysis. Therefore, much higher doses of the antibiotic may need to be administered in treatment regimens to eradicate infectious disease caused by VIM expressing bacteria. Conversely these required doses are not safe to administer and have therefore contributed to the soaring resistance rates.
| Kinetics parameter | IMP-1 | VIM-2 |
|---|---|---|
| Vmax (μmol min−1) | 2.41 ± 0.19 | 2.62 ± 0.11 |
| Km (μM) | 60.42 ± 0.54 | 4.08 ± 0.04 |
| kcat (s−1) | 0.40 | 0.44 |
| kcat/Km (μM−1 s−1) | 6.64 × 10−3 | 10.70 × 10−2 |
To further assess the potential of 24b/24c, inhibition of enzyme kinetics was performed, to generate the compounds' inhibitory constant (Ki). The activity plots of 24b/24c followed a graphical pattern characteristic of non-competitive inhibitors (Fig. S2 and S3†). The ln (% residual activity) values of each enzyme decreased over time, with the highest inhibitor concentration having the lowest residual activity values. The Kobs values were observed to increase as the inhibitor concentration increased. The Kobs values were deduced from the residual activity plot and the Ki values (Table 5) were determined by interpolation of the Kobs versus inhibitor concentration plot.
| Enzymes (MBLs) | 24b | 24c |
|---|---|---|
| Ki (μM) | Ki (μM) | |
| a All the data is shown as means ± SD, n = 3. | ||
| IMP-1 | 23.87 ± 0.00 | 74.44 ± 0.00 |
| VIM-2 | 0.85 ± 0.00 | 1.87 ± 0.00 |
Table 5 conveys that stronger inhibition was exhibited by 24b against both IMP-1 and VIM-2, as compared to 24c. This finding concurs with the results of the antimicrobial susceptibility tests in which 24b achieved a 1–4 2-fold dilution decrease in the meropenem MIC, whilst utilizing the same inhibitor concentration for both 24b and 24c (Table 3). Furthermore, compound 24b and 24c displayed stronger inhibition towards VIM-2 as compared to IMP-1. Compounds 24b and 24c were superior in potency to predecessor compound BP1 (Kiapp = 24.8 μM).36
Conclusion
The study has shown that β-lactam antibiotics covalently linked to cyclic amino acidic zinc chelators can potentially re-sensitize meropenem and target metallo-β-lactamase resistance in E. coli NDM-1 and K. pneumoniae NDM. Between 8–32 mg L−1 of compounds, 6, 9, 16, 20a–b and 24a–d were able to restore E. coli NDM-1 and K. pneumoniae NDM sensitivity towards meropenem at MICs ranging from 0.06–2 mg L−1. Subsequent biological evaluation of the potent 24b and 24c revealed the compounds to be safe to administer, as neither cell death nor reduced cell viability was observed at the concentrations investigated herein. An added advantage of compound 24c (increases metabolic output and decreases LDH leakage) will aid eukaryotic cells to increase ATP production and help fight bacterial insult. Time-kill studies demonstrated that compounds 24b and 24c produced sterilizing activity at 24 hours post-inoculation, utilizing a minimal meropenem dose of 0.5 mg L−1. Bactericidal activity was achieved faster (from 8 hours post-inoculation) by increasing the meropenem dose to 2 mg L−1. Enzyme inhibition studies indicated 24b and 24c are non-competitive, potent inhibitors of VIM-2. The overall findings of this study are that the novel series of β-lactam MBLIs reported herein, are potent, viable, and non-toxic candidates, with promising potential. Our research group is further exploring the scope of this class of MBLIs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The South African National Research Foundation grant no. 120419, 137979, 145774, 105236, 105216, 105303, BRICAMR/202109/002, and the Technology Innovation Agency of South Africa (UKZN_17-18_1). The authors wish to thank Patrice Nordmann and David P. Nicolau for the CRE and NDM strain, respectively.References
- B. C. N. Torres, Antibiotic Use and Resistance in South Africa: The Need for Better Data, available from: https://www.hsrc.ac.za/en/review/hsrc-review-june-2019/antibiotic-use-and-resistance-in-sa, [cited 2021 14 September 2021] Search PubMed.
- L. J. Shallcross, S. J. Howard and T. Fowler, et al., Tackling the threat of antimicrobial resistance: from policy to sustainable action, Philos. Trans. R. Soc., B, 2015, 370(1670), 20140082, DOI:10.1098/rstb.2014.0082.
- G. Bahr, L. J. González and A. J. Vila, Metallo-β-lactamases in the Age of Multidrug Resistance: from Structure and Mechanism to Evolution, Dissemination, and Inhibitor Design, Chem. Rev., 2021, 121(13), 7957–8094, DOI:10.1021/acs.chemrev.1c00138.
- G. A. Durand, D. Raoult and G. Dubourg, Antibiotic discovery: history, methods and perspectives, Int. J. Antimicrob. Agents, 2019, 53(4), 371–382, DOI:10.1016/j.ijantimicag.2018.11.010.
- J. Ruiz, Enhanced antibiotic resistance as a collateral COVID-19 pandemic effect?, J. Hosp. Infect., 2021, 107, 114–115, DOI:10.1016/j.jhin.2020.11.010.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, A. G. Robles, G. Authia, C. Han, C. Bisignano, P. Rao, E. Wool and S. C. Johnson, Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis, Lancet, 2022, 399(10325), 629–655, DOI:10.1016/s0140-6736(21)02724-0.
- R. M. Kariyawasam, D. A. Julien and D. C. Jelinski, et al., Antimicrobial resistance (AMR) in COVID-19 patients: a systematic review and meta-analysis (November 2019–June 2021), Antimicrob. Resist. Infect. Control, 2022, 11(1), 45, DOI:10.1186/s13756-022-01085-z.
- M. F. Mojica, M. A. Rossi and A. J. Vila, et al., The urgent need for metallo-β-lactamase inhibitors: an unattended global threat, Lancet Infect. Dis., 2022, 22(1), e28–e34, DOI:10.1016/s1473-3099(20)30868-9.
- B. J. Langford, M. So and S. Raybardhan, et al., Antibiotic prescribing in patients with COVID-19: rapid review and meta-analysis, Clin. Microbiol. Infect., 2021, 27(4), 520–531, DOI:10.1016/j.cmi.2020.12.018.
- S. H. MacVane, J. L. Crandon and W. W. Nichols, et al., Unexpected in vivo activity of ceftazidime alone and in combination with avibactam against New Delhi metallo-β-lactamase-producing Enterobacteriaceae in a murine thigh infection model, Antimicrob. Agents Chemother., 2014, 58(11), 7007–7009, DOI:10.1128/aac.02662-14.
- X. Tan, H. S. Kim and K. Baugh, et al., Therapeutic Options for Metallo-β-Lactamase-Producing Enterobacterales, Infect. Drug Resist., 2021, 14, 125–142, DOI:10.2147/idr.S246174.
- A. M. Somboro, D. Tiwari and L. A. Bester, et al., NOTA: a potent metallo-β-lactamase inhibitor, J. Antimicrob. Chemother., 2015, 70(5), 1594–1596, DOI:10.1093/jac/dku538.
- N. Reddy, M. Shungube and P. I. Arvidsson, et al., A 2018–2019 patent review of metallo beta-lactamase inhibitors, Expert Opin. Ther. Pat., 2020, 30(7), 541–555, DOI:10.1080/13543776.2020.1767070.
- M. W. Crowder, J. Spencer and A. J. Vila, Metallo-β-lactamases: novel Weaponry for Antibiotic Resistance in Bacteria, Acc. Chem. Res., 2006, 39(10), 721–728, DOI:10.1021/ar0400241.
- M. Aitha, T. K. Richmond and Z. Hu, et al., Dilution of dipolar interactions in a spin-labeled, multimeric metalloenzyme for DEER studies, J. Inorg. Biochem., 2014, 136, 40–46, DOI:10.1016/j.jinorgbio.2014.03.010.
- R. Tsivkovski, M. Totrov and O. Lomovskaya, Biochemical Characterization of QPX7728, a New Ultrabroad-Spectrum Beta-Lactamase Inhibitor of Serine and Metallo-Beta-Lactamases, Antimicrob. Agents Chemother., 2020, 64(6) DOI:10.1128/aac.00130-20.
- C. A. Thomas, Z. Cheng and K. Yang, et al., Probing the mechanisms of inhibition for various inhibitors of metallo-β-lactamases VIM-2 and NDM-1, J. Inorg. Biochem., 2020, 210, 111123, DOI:10.1016/j.jinorgbio.2020.111123.
- M. R. Meini, L. I. Llarrull and A. J. Vila, Overcoming differences: the catalytic mechanism of metallo-β-lactamases, FEBS Lett., 2015, 589(22), 3419–3432, DOI:10.1016/j.febslet.2015.08.015.
- L. C. Ju, Z. Cheng and W. Fast, et al., The Continuing Challenge of Metallo-β-Lactamase Inhibition: Mechanism Matters, Trends Pharmacol. Sci., 2018, 39(7), 635–647, DOI:10.1016/j.tips.2018.03.007.
- T. Palzkill, Metallo-β-lactamase structure and function, Ann. N. Y. Acad. Sci., 2013, 1277, 91–104, DOI:10.1111/j.1749-6632.2012.06796.x.
- K. H. M. E. Tehrani, N. Wade and V. Mashayekhi, et al., Novel Cephalosporin Conjugates Display Potent and Selective Inhibition of Imipenemase-Type Metallo-β-Lactamases, J. Med. Chem., 2021, 64(13), 9141–9151, DOI:10.1021/acs.jmedchem.1c00362.
- S. Butler Mark, V. Gigante and H. Sati, et al., Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed, Antimicrob. Agents Chemother., 2022, 66(3), e01991, DOI:10.1128/aac.01991-21.
- J. A. Karlowsky, K. M. Kazmierczak and B. L. M. de Jonge, et al., In Vitro Activity of Aztreonam-Avibactam against Enterobacteriaceae and Pseudomonas aeruginosa Isolated by Clinical Laboratories in 40 Countries from 2012 to 2015, Antimicrob. Agents Chemother., 2017, 61(9) DOI:10.1128/aac.00472-17.
- M. Falcone and D. Paterson, Spotlight on ceftazidime/avibactam: a new option for MDR Gram-negative infections, J. Antimicrob. Chemother., 2016, 71(10), 2713–2722, DOI:10.1093/jac/dkw239.
- S. Marshall, A. M. Hujer and L. J. Rojas, et al., Can Ceftazidime-Avibactam and Aztreonam Overcome β-Lactam Resistance Conferred by Metallo-β-Lactamases in Enterobacteriaceae?, Antimicrob. Agents Chemother., 2017, 61(4) DOI:10.1128/aac.02243-16.
- E. Wenzler, M. F. Deraedt and A. T. Harrington, et al., Synergistic activity of ceftazidime-avibactam and aztreonam against serine and metallo-β-lactamase-producing gram-negative pathogens, Diagn. Microbiol. Infect. Dis., 2017, 88(4), 352–354, DOI:10.1016/j.diagmicrobio.2017.05.009.
- M. Biagi, T. Wu and M. Lee, et al., Searching for the Optimal Treatment for Metallo- and Serine-β-Lactamase Producing Enterobacteriaceae: Aztreonam in Combination with Ceftazidime-avibactam or Meropenem-vaborbactam, Antimicrob. Agents Chemother., 2019, 63(12) DOI:10.1128/aac.01426-19.
- B. Davido, L. Fellous and C. Lawrence, et al., Ceftazidime-Avibactam and Aztreonam, an Interesting Strategy To Overcome β-Lactam Resistance Conferred by Metallo-β-Lactamases in Enterobacteriaceae and Pseudomonas aeruginosa, Antimicrob. Agents Chemother., 2017, 61(9) DOI:10.1128/aac.01008-17.
- L. M. Avery and D. P. Nicolau, Assessing the in vitro activity of ceftazidime/avibactam and aztreonam among carbapenemase-producing Enterobacteriaceae: defining the zone of hope, Int. J. Antimicrob. Agents, 2018, 52(5), 688–691, DOI:10.1016/j.ijantimicag.2018.07.011.
- A. Yoshizumi, Y. Ishii and S. Kimura, et al., Efficacies of calcium–EDTA in combination with imipenem in a murine model of sepsis caused by Escherichia coli with NDM-1 β-lactamase, J. Infect. Chemother., 2013, 19(5), 992–995, DOI:10.1007/s10156-012-0528-y.
- P. P. Bose, U. Chatterjee and I. Hubatsch, et al., In vitro ADMET and physicochemical investigations of poly-N-methylated peptides designed to inhibit Abeta aggregation, Bioorg. Med. Chem., 2010, 18(16), 5896–5902, DOI:10.1016/j.bmc.2010.06.087.
- S. B. Falconer, S. A. Reid-Yu and A. M. King, et al., Zinc Chelation by a Small-Molecule Adjuvant Potentiates Meropenem Activity in Vivo against NDM-1-Producing Klebsiella pneumoniae, ACS Infect. Dis., 2015, 1(11), 533–543, DOI:10.1021/acsinfecdis.5b00033.
- T. Wang, K. Xu and L. Zhao, et al., Recent research and development of NDM-1 inhibitors, Eur. J. Med. Chem., 2021, 223, 113667, DOI:10.1016/j.ejmech.2021.113667.
- R. Azumah, J. Dutta and A. M. Somboro, et al., In vitro evaluation of metal chelators as potential metallo- β -lactamase inhibitors, J. Appl. Bacteriol., 2016, 120(4), 860–867, DOI:10.1111/jam.13085.
- K. F. Omolabi, N. Reddy and S. Mdanda, et al., The in vitro and in vivo potential of metal-chelating agents as metallo-beta-lactamase inhibitors against carbapenem-resistant Enterobacterales, FEMS Microbiol. Lett., 2023, 370 DOI:10.1093/femsle/fnac122.
- B. K. Peters, N. Reddy and M. Shungube, et al., In Vitro and In Vivo Development of a β-Lactam-Metallo-β-Lactamase Inhibitor: targeting Carbapenem-Resistant Enterobacterales, ACS Infect. Dis., 2023, 9(3), 486–496, DOI:10.1021/acsinfecdis.2c00485.
- N. Reddy, L. Girdhari and M. Shungube, et al., Neutralizing Carbapenem Resistance by Co-Administering Meropenem with Novel β-Lactam-Metallo-β-Lactamase Inhibitors, Antibiotics, 2023, 12(4), 633, DOI:10.3390/antibiotics12040633.
- J. Dutta, P. K. Chinthakindi and P. I. Arvidsson, et al., A Facile Synthesis of NODASA-Functionalized Peptide, Synlett, 2016, 27(11), 1685–1688, DOI:10.1055/s-0035-1561970.
- P. Nordmann, L. Poirel and L. Dortet, Rapid detection of carbapenemase-producing Enterobacteriaceae, Emerging Infect. Dis., 2012, 18(9), 1503–1507, DOI:10.3201/eid1809.120355.
- C. M. Baldwin, K. A. Lyseng-Williamson and S. J. Keam, Meropenem: a review of its use in the treatment of serious bacterial infections, Drugs, 2008, 68(6), 803–838, DOI:10.2165/00003495-200868060-00006.
- B. Peters, H. G. Kruger, T. Govender and T. Naicker, Metallo-β-Lactamase Inhibitors, World Intellectual Property Organisation, 2023, PCT/IB2022/056748 Search PubMed.
- South Africa Antimicrobial Resistance National Strategy Framework; A One Health Approach 2018–2024, available from: https://www.knowledgehub.org.za/system/files/elibdownloads/2020-03/AMR_National_Action_Plan_2018_-_2024.pdf Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02490c |
| This journal is © The Royal Society of Chemistry 2023 |