
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the radical scavenging potential of vanillin-based pyrido-dipyrimidines: experimental and in silico approach
Nenad Janković *a,
Julijana Tadićb,
Emilija Milovića,
Zoran Markovićac,
Svetlana Jeremićc,
Jelena Petronijevićd,
Nenad Joksimovićd,
Teona Teodora Boroviće and
Syed Nasir Abbas Bukharif
*a,
Julijana Tadićb,
Emilija Milovića,
Zoran Markovićac,
Svetlana Jeremićc,
Jelena Petronijevićd,
Nenad Joksimovićd,
Teona Teodora Boroviće and
Syed Nasir Abbas Bukharif
aUniversity of Kragujevac, Institute for Information Technologies, Department of Sciences, Jovana Cvijića bb, 34000 Kragujevac, Serbia. E-mail: nenad.jankovic@uni.kg.ac.rs
bVinča Institute of Nuclear Sciences – National Institute of the Republic of Serbia, University of Belgrade, Mike Petrovića Alasa 12-14, 11351 Vinča, Belgrade, Serbia
cThe State University of Novi Pazar, 36300 Novi Pazar, Serbia
dUniversity of Kragujevac, Faculty of Science, Department of Chemistry, Radoja Domanovića 12, Kragujevac, Serbia
eFaculty of Sciences, University of Novi Sad, Trg Dositeja Obradovića 3, 21000 Novi Sad, Serbia
fDepartment of Pharmaceutical Chemistry, College of Pharmacy, Jouf University, Sakaka, Al Jouf 72388, Saudi Arabia
First published on 18th May 2023
Abstract
Antioxidants have a significant contribution in the cell protection against free radicals which may induce oxidative stress, and permanently damage the cells causing different disorders such as tumors, degenerative diseases, and accelerated aging. Nowadays, a multi-functionalized heterocyclic framework plays an important role in drug development, and it is of great importance in organic synthesis and medicinal chemistry. Encouraged by the bioactivity of the pyrido-dipyrimidine scaffold and vanillin core, herein, we made an effort to thoroughly investigate the antioxidant potential of the vanillin-based pyrido-dipyrimidines A–E to reveal novel promising free radical inhibitors. The structural analysis and the antioxidant action of the investigated molecules were performed in silico by DFT calculations. Studied compounds were screened for their antioxidant capacity using in vitro ABTS and DPPH assays. All the investigated compounds showed remarkable antioxidant activity, especially derivative A exhibiting inhibition of free radicals at the IC50 value (ABTS and DPPH assay 0.1 mg ml−1 and 0.081 mg ml−1, respectively). Compound A has higher TEAC values implying its stronger antioxidant activity compared to a trolox standard. The applied calculation method and in vitro tests confirmed that compound A has a strong potential against free radicals and may be a novel candidate for application in antioxidant therapy.
Introduction
Antioxidants have gained attention in numerous research fields comprising medicinal chemistry, biomedicine, and clinical trials, mainly due to their therapeutic properties and beneficial effects on human health and wellbeing.1 Oxidative stress – an imbalance of free radicals (RONS – reactive oxygen and nitrogen species) and antioxidants in cells – causes damage to biomolecules (DNA, protein, lipids) resulting in pathophysiological changes of cells.2 These changes can be associated with certain conditions such as allergic diseases,3 cancer,4 cardiovascular,5 immune,6 and age-related diseases including neurodegenerative disorders such as Alzheimer's disease (AD)7 and Parkinson's disease (PD).8 However, molecules with antioxidant activity inhibit free radical reactions and delay or prevent cellular damage.9 Recent studies have suggested that antioxidant-based treatments can reduce the effect of RONSs and counteract oxidative stress providing promising therapy for patients suffering from cancer, AD, PD, and other degenerative conditions, and subsequently promoting healthy longevity.10 Therefore, development of redox medicine and application of antioxidants are an emergency need.11–14Among the N-containing heterocycles, pyrimidine derivatives are in particularly important building blocks in pharmaceuticals' design exhibiting a remarkable therapeutic potential.15 Moreover, when pyrimidine moiety is fused with other heterocycles it results in the compounds with even more powerful activity. Such the compounds are the pyrido[2,3-d]pyrimidines which have broad spectrum of medicinal applications.16 Namely, pyridopyrimidine molecule is a notable pharmacophore in drug design due to its various pharmacological actions including anticancer,17 antioxidant,18 analgesic,19 anti-inflammatory,20 and antimicrobial activity.21 Furthermore, some pyridopyrimidine derivatives have been under the clinical trials for treatment of different cancers, and 22 of them are being used in the clinic for cancer treatment (FDA approval).22,23
On the other hand, vanillin is a natural compound, produced from the renewable sources on industrial level, and presenting a significant tool in the synthesis of fine chemicals.24 This phenolic molecule possesses strong biological potential and highlighted antioxidant properties.25 Furthermore, vanillin derivatives are reported to be promising multi-target drugs for the treatment of degenerative diseases having adequate pharmacokinetic and pharmacodynamic characteristics.25
In the light of the studies, and in the extension of our recently published work24 on the design of bioactive vanillin-based pyrido-dipyrimidines, in presented study, we thoroughly investigated the antioxidant capability of the synthetized molecular hybrids using in silico (DFT) and in vitro approaches (ABTS and DPPH assays), in order to discover much needed new candidates for treatment of different redox-related conditions.
Results and discussion
The antioxidant activity of selected compounds (Fig. 1) was further evaluated by determination of the IC50 values. IC50 represent the concentration of sample able to scavenge 50% of ABTS or DPPH radicals in the solution, and high IC50 values generally suggest low antioxidant activity. The solutions of pyrido-dipyrimidine in methanol/PBS solution, as well as solutions of ABTS or DPPH were prepared and obtained IC50 were compared (Table 1). The tests were performed in triplicate.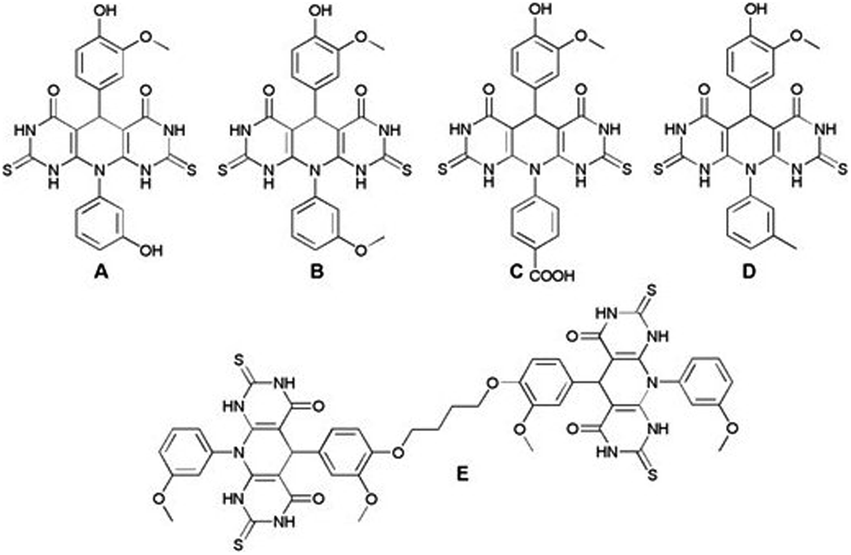 | ||
| Fig. 1 Structures of investigated pyrido-dipyrimidines A–E. | ||
| ABTS | DPPH | |||||
|---|---|---|---|---|---|---|
| I (%) | IC50 | TEAC | I (%) | IC50 | TEAC | |
| A | 99.9 ± 0.01 | 0.100 ± 0.003 | 2.54 ± 0.09 | 95.92 ± 0.14 | 0.081 ± 0.005 | 2.884 ± 0.161 |
| B | 99.99 ± 0.01 | 0.200 ± 0.006 | 1.27 ± 0.04 | 93.15 ± 0.13 | 0.199 ± 0.013 | 1.174 ± 0.066 |
| C | 99.12 ± 0.13 | 0.370 ± 0.013 | 0.69 ± 0.02 | 92.26 ± 0.13 | 0.144 ± 0.009 | 1.622 ± 0.090 |
| D | 99.99 ± 0.01 | 0.240 ± 0.008 | 1.06 ± 0.04 | 96.20 ± 0.15 | 0.124 ± 0.008 | 1.884 ± 0.106 |
| E | 99.99 ± 0.01 | 0.390 ± 0.013 | 0.65 ± 0.02 | 94.22 ± 0.18 | 0.195 ± 0.013 | 1.198 ± 0.067 |
| Trolox | 93.16 ± 0.12 | 0.250 ± 0.009 | 1 ± 0 | 96.17 ± 0.17 | 0.234 ± 0.015 | 1 ± 0 |
Compounds A–E have been evaluated for their IC50 and TEAC (Trolox Equivalent Antioxidant Capacity) values. Solutions of A–D, in concentration range from 0.0125–1 mg ml−1, were prepared and IC50 values were established, and consequently, the TEAC values were calculated (Table 1). The antioxidant activity based on IC50 and TEAC values of tested compounds is presented in Table 1. All investigated compounds exhibited remarkable antioxidant potential. Namely, analysed compounds exhibited excellent antioxidant activity (the inhibition was up to 99%), three molecules expressed good ability to scavenge the ABTS˙+ radical cation while all tested compounds showed very good antioxidant properties in comparison with DPPH radical (Table 1). Considering the results presented in Table 1 it can be determined that investigated compounds not just exhibited remarkable capacity to inhibit ABTS˙+ or DPPH radical, but also molecules A–D showed higher TEAC than Trolox standard indicating a strong potential against free radicals. In comparison to Trolox, compounds A–D exhibited high inhibition of DPPH radicals up to 96% confirming strong antioxidant potential. TEAC values for DPPH were in range 2.88 to 1.17. Molecules A and B showed the best TEAC values for ABTS. The presence additional hydroxy group at m-position could be crucial for such a high activity of molecule A in both assays.
Taking into account presented results, it can be seen that investigated compounds displayed powerful antioxidant activity indicating their promising potential for application as novel antioxidants.
In addition, compounds A–E were exposed to cytotoxic activity against normal fibroblast MRC-5. Delivered IC50 values for all investigated compounds (IC50 >200 μM) were showed significant non-toxic effect to MRC-5 cell lines.
In order to explore antioxidative mechanism at molecular level for two most active compounds (A and B) further investigation was performed by using computational approach. Conformational analyse, performed to find the most stable conformation of the estimated compounds, give the appropriate torsion angles values.
Values of the τ1, τ2, τ3, τ4 and τ5 dihedral angles that correspond to the energy minima for molecule A are 8.73°, 3.59°, 81.34°, 100.08° and 0.16° respectively, and for molecule B they are 0.02°, 0.01°, 60.85°, 88.68° and 0.07° respectively (Fig. 2). The calculated angle values indicate significant deviation from the planarity of both investigated molecules.
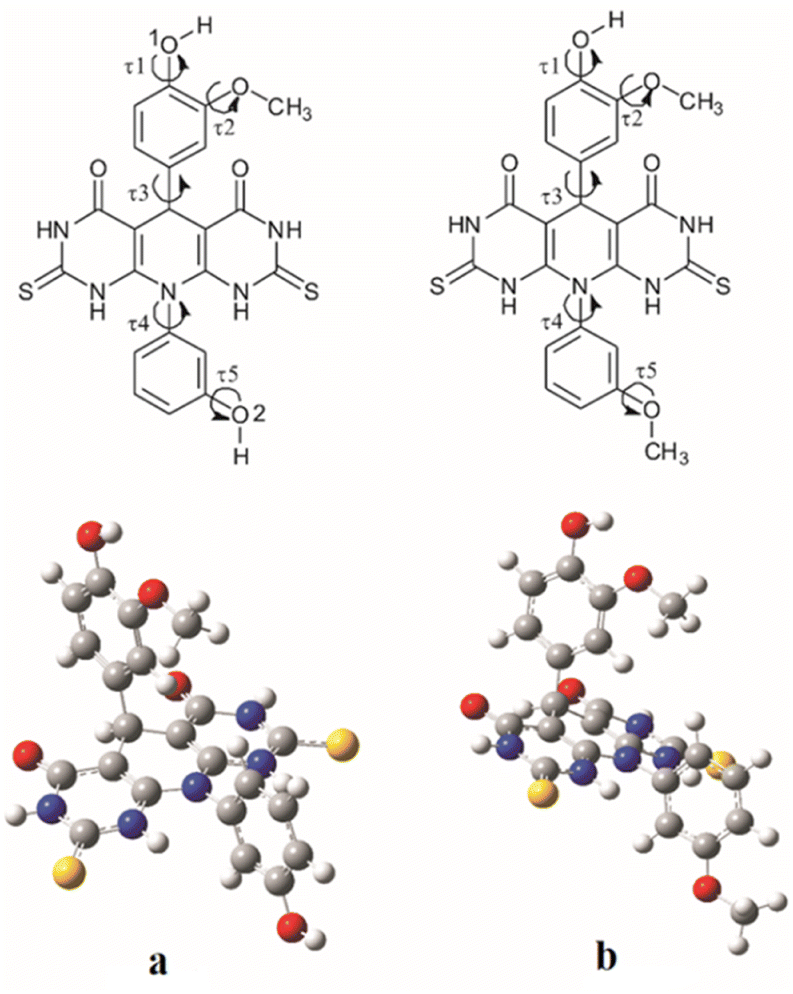 | ||
| Fig. 2 Investigated torsion angles (τ1, τ2, τ3, τ4 and τ5) for molecules A (a) and B (b) (upper) and their optimized geometries (down). | ||
Antioxidant activity of some molecule can be manifested by the easiness of releasing the hydrogen atom, and the formation of a more stable radical than the starting. The values of thermodynamic parameters can be used to determine which of the investigated reaction mechanisms is the most likely reaction path for free radical scavenging. Table 2 shows the values of these parameters for the molecules A and B computed in water, methanol and benzene as the solvents.
| HAT | SPLET | SET-PT | ||||
|---|---|---|---|---|---|---|
| BDE | PA | ETE | IP | PDE | ||
| Water | A–O1 | 350 | 178 | 349 | 506 | 21 |
| A–O2 | 466 | 140 | 503 | 506 | 137 | |
| B | 352 | 181 | 350 | 509 | 21 | |
| Methanol | A–O1 | 350 | 173 | 361 | 525 | 9 |
| A–O2 | 466 | 133 | 517 | 525 | 125 | |
| B | 353 | 175 | 362 | 528 | 9 | |
| Benzene | A–O1 | 359 | 452 | 330 | 634 | 148 |
| A–O2 | 465 | 369 | 520 | 634 | 254 | |
| B | 359 | 455 | 327 | 634 | 149 | |
Radicals and anions of the A and B molecules
The homolytic breaking of the O–H bonds in A and B results in formation of the corresponding radicals (Fig. 4). The stability of the formed radicals in methanol plays the main role in determining the antioxidant activity of the investigated molecules. The obtained values of BDE are given in Table 2, and the lower the BDE, the more stable the radical. Molecule A can form two radicals since it can undergo to the homolytic cleavage of the two O–H groups. According to the BDE values from Table 2, the radical formed in position 1 (A–O1 radical) is more stable than the radical in position 2 (A–O2 radical). Comparing spin density distributions of A–O1 and A–O2 radicals (Fig. 3), it can be concluded that the unpaired electron in radical A–O1 is delocalized not only over the atoms of the corresponding benzene ring, but also through some atoms of the central tricyclic part of the molecule. On the other hand, the delocalization of the unpaired electron of radical A–O2 involves only the atoms of the corresponding benzene ring. The spin density distribution of B is similar to the spin density distribution of A–O1, which is in accordance with the similar values of their BDEs.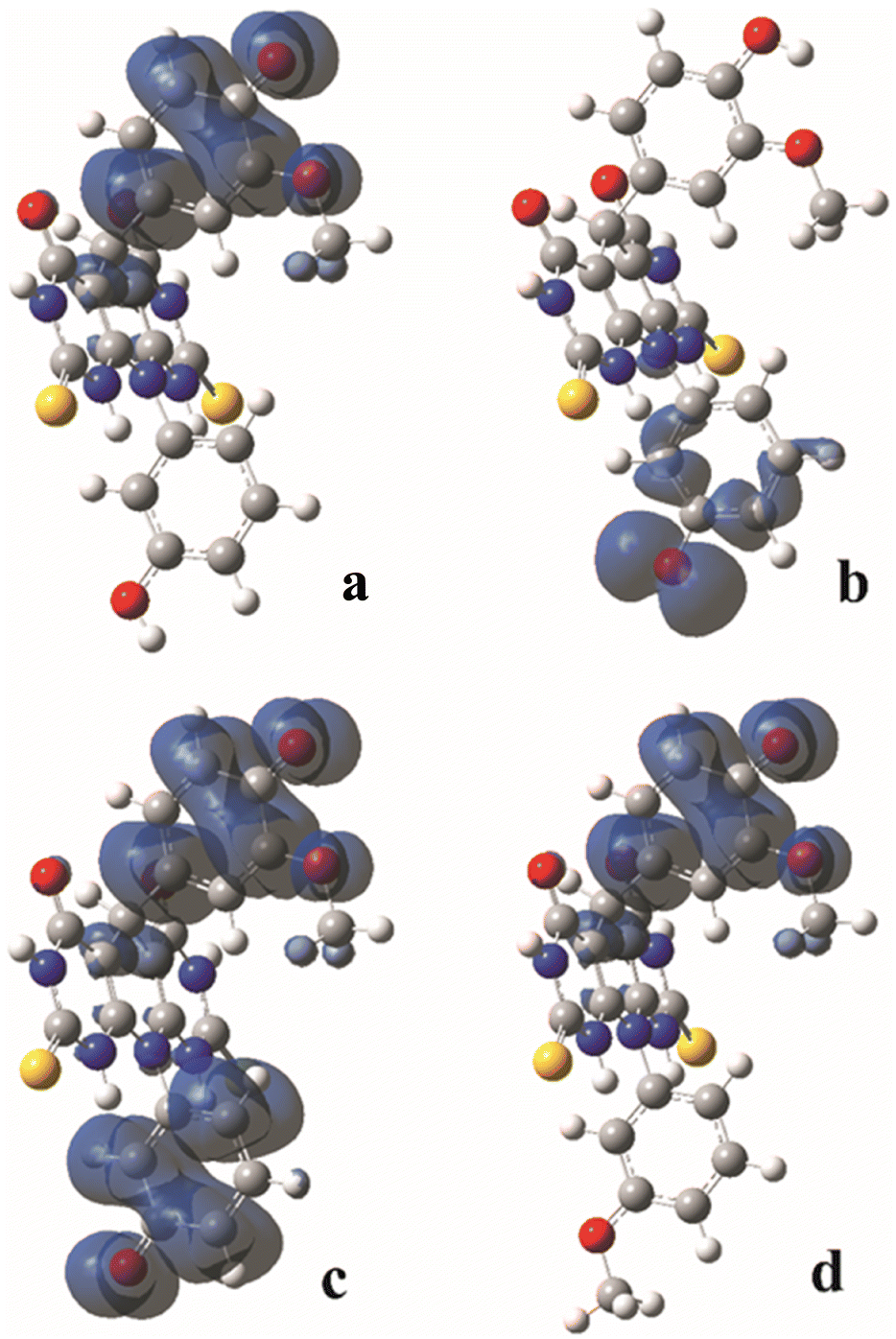 | ||
| Fig. 3 Spin density distribution (only positive isovalues) in the corresponding radical species of the A and B molecules calculated in methanol: A–O1 radical (a); A–O2 radical (b); A diradical (c); B radical (d). | ||
It must be mentioned that molecule A can homolytic release hydrogen atoms at both active positions simultaneously. This leads to the formation of a diradical species. Having on mind that A–O1 is more stable, the higher tendency to undergo to the further stabilisation due to diradical formation possess A–O2 radical.26,27 The enthalpies of the diradical formation following homolytic breaking of the remaining O–H bond starting from the A–O2 are: 270 kJ mol−1, 271 kJ mol−1 and 285 kJ mol−1 in water, methanol and benzene respectively. When the diradical is formed starting from the A–O1 radical, the values of the corresponding enthalpies are: 386 kJ mol−1, 386 kJ mol−1, and 391 kJ mol−1 in water, methanol and benzene respectively. The delocalization of unpaired electrons in this diradical forms include both benzenoid parts of the molecule (Fig. 4). The spin density distribution excludes the central tricyclic part of the molecule, which is a consequence of the non-planarity of the system.
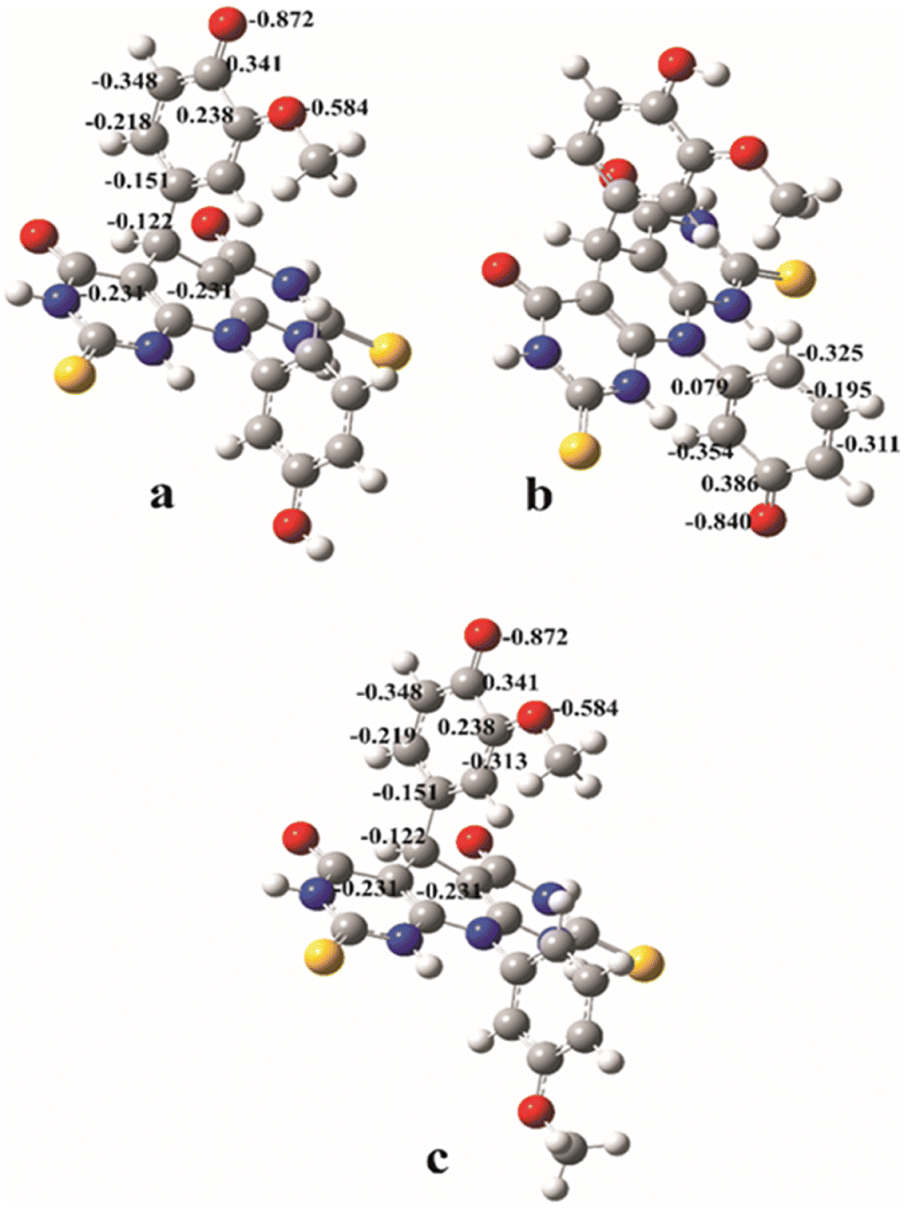 | ||
| Fig. 4 The natural charge distributions in the most stable anions formed from the A and B molecules calculated in methanol: anion A–O1 (a); anion A–O2 (b); anion B (c). | ||
Therefore, the higher antioxidant capacity of molecule A can be explained by the easiness of formation of triplet diradicals from monoradical species.26,27 In addition, the fact is that on this molecule antioxidant processes can take place in two positions simultaneously. This is in agreement with the previously established fact that the antioxidant capacity of the compound increases with the increase in the number of hydroxyl groups.28,29 The molecule B can achieve its antioxidant activity only through one active position, forming monoradical moiety.
The most plausible mechanism for the stable antioxidant radical formation can be discussed based on the BDE, PA and IP values. These values correspond to the enthalpy of the first reaction step of the appropriate mechanisms. The lowest enthalpy value corresponds to the most plausible mechanistic pathway. In order to be able to discuss the different behaviour of antioxidants depending on the polarity of the environment, the corresponding thermodynamic parameters were calculated in water (ε = 78.36), methanol (ε = 32.61) and benzene (ε = 2.27). Water was chosen as a typical polar medium, and also as the dominant solvent in which the DPPH analysis was performed. Methanol is a polar solvent, the molecules of which are small in size. Therefore, its solvation effect, which is a consequence of the formation of intermolecular hydrogen bonds with the dissolved molecule, is similar to that of water.30,31 In addition, the ABTS test was performed in this solvent, so for the sake of comparability of theoretical and experimental results, part of the calculation was performed in methanol. Benzene was chosen as a typical non-polar solvent, in order to simulate the behaviour of antioxidants in a lipid environment. It was observed that the values of thermodynamic parameters calculated in benzene corresponded most closely to the experimentally obtained results that indicate the antioxidative activities in a lipid environment.32
Since PA values are significantly lower than BDEs and IPs, one can conclude that SPLET is the most favourable reaction mechanism for both estimated molecules in polar solvents. The product of the first step of the SPLET is an anionic species. The resulting anions can be surrounded by water or methanol molecules, and thus stabilized by the solvation effect. Because of this, in polar solvents, SPLET appears as the dominant reaction mechanism. The lowest PA value corresponds to the anion of A molecule, indicating its higher antioxidant capacity. This is in accordance with the experimentally obtained results. A has two hydroxyl groups and thus two potential positions for antioxidant reactions. The PA value that corresponds to the cleavage of the O–H bond in position 2 is lower (A–O2 anion), and it indicates that it is more reactive position for antioxidative action over SPLET than the O–H bond in position 1 (A–O1 anion). The explanation of this phenomenon lies in the natural charge distribution of the appropriate anionic species (Fig. 4). The value of the natural charge at the O1 atom of the anion A–O1 is lower (−0.872) and more localised than negative charge at the atom O2 of the anion A–O2 (−0.840), calculated in methanol. It indicates better natural charge distribution at anion A–O2 over the neighbour atoms, and at the same time, its higher stability. It can be noticed that the values of the natural charges of anions A–O1 and B are almost equal. This indicates almost equal stability of these two anions, and it agrees with the similarities of the corresponding PA values from Table 2.
Since IP values are very high, the SET-PT mechanism is the lowest plausible mechanistic pathway in considering conditions. This can be explained by the fact that in the first step of SET-PT radical-cation is formed. When the reaction leads to the formation of the relative stable product, the appropriate thermodynamical parameters are low. On the other hand, the formation of unstable species is characterized by high values of thermodynamic parameters. Bearing that in mind, one can conclude that the radical-cation, created in the first step of the SET-PT is unstable specie. It is also known that the radical-cation is a highly reactive species, with high oxidation potentials,29,30 so this explains the difficulty of its formation.33,34 The instability and high reactivity of here formed radical-cations can be explained by the high localisation of the positive charge at the carbon atom linked to the hydroxyl-group of interest for radical reaction (Fig. 5).
 | ||
| Fig. 5 Total spin density distribution in the radical cations of the A (left) and B (right) molecules calculated in methanol. | ||
Conclusions
Theoretical investigations included two molecules with the highest experimentally determined antioxidative capacity. Molecule A has two, and molecule B has one active position for antioxidative reaction. Both molecules show a tendency to follow a SPLET mechanism as the dominant mechanism of antioxidant action in polar environment, while in non-polar conditions predominant mechanism is HAT. In the first step of SPLET, the corresponding anion is formed. This anion can be stabilized in methanol and water as the solvents by the solvation effect. The greater stability of the A–O2 anion than that of anion of B is in agreement with the experimentally determined the highest antioxidant capacity of A molecule and is the consequence of the better natural charge distribution at anion A–O2 over the neighbours atoms. By comparing BDE values and spin density distributions, conclusions can be brought about the stability of antioxidant radicals, and the easiness of their formation by homolytic breaking of the O–H bond. It was found that the BDEs and spin density distributions for the monoradical forms of both molecules are similar. On the other hand, the BDE value corresponding to the formation of diradical of molecule A is significantly lower than all BDEs for the formation of monoradicals. Also, the distribution of unpaired electrons of the resulting diradical covers the largest surface area of the molecule. Taking it on mind one can conclude that molecule A achieves its antioxidant activity through both active positions simultaneously, forming a diradical. Moreover, here obtained theoretical results are in accordance with the experimental ones.Experimental section
Antioxidant activity
| Inhibition (%) = (Ac − As)/Ac × 100 |
| TEAC = IC50 (Trolox)/IC50 (tested compound) |
| Inhibition (%) = [1 − (As − Ab)/Ac)] × 100 |
TEAC values were calculated as described above.
DFT study
The DFT method was utilized to gain a better knowledge of the antioxidant capability of the studied compounds. According to the experimental findings, the compounds A and B were shown to be more potent antioxidants than the other evaluated substances. As a result, these two compounds were used for in silico investigations. The conformational analysis of the A and B molecules was done. For this purpose, different conformations were obtained by the rotations around the single bonds of interest. Each torsion angle was scanned in steps of 10° with no constraints on any other geometrical parameters. The geometry corresponding to the energy minimum for each analysed torsion angle was then used to optimize the geometry with the minimal energy values for investigated molecules, as well as to calculate the appropriate thermodynamically parameters. The geometry optimizations and frequency calculations of investigated molecules were performed using DFT/M06-2X functional37 in conjunction with the 6-311++G(d,p) basis set,38 implemented in the Gaussian 09 program package.39 This functional gives satisfactory results in the thermochemical and kinetic calculations and has been used widely by numerous authors.40–42 The restricted calculations are used for moieties with paired electrons, while the unrestricted calculations are performed for moieties with unpaired electrons. Therefore unrestricted calculations applied for radicals and diradicals in a doublet and a triplet state can give us reliable and comparable values. To obtain the results comparable with the experimentally ones, all calculations were carried out in water, methanol and benzene, using the CPCM solvation model.43 Spin density distribution and natural charge distribution in radical and ionic species were determined in methanol (ε = 32.61) as a solvent with an average value of the dielectric constant among the three solvents used here. To determine distribution of natural charge in ionic moieties, as well as to determine the spin density distribution in radical forms, NBO analysis is performed.44,45 Potential energy minima for all the optimized species are verified by the absence of the imaginary frequencies. All calculations were done at 298.15 K.The scavenging of free radicals appears to play an essential role in phenolic compounds' antioxidant action. The capacity of phenolic compounds to transfer their phenolic H-atom to a free radical is associated with their antiradical abilities. Several mechanistic pathways can be used to describe this process. Among the most investigated are HAT (Hydrogen Atom Transfer), SPLET (Sequential Proton Loss Electron Transfer) and SET-PT (Single-Electron Transfer followed by Proton Transfer).46–49 The process describing the homolytic breakage of the O–H bond is known as HAT (eqn (1)). The capabilities of this mechanism are estimated based on BDE (Bond Dissociation Enthalpy) values (eqn (1a)).
| A–OH → A–O˙ + H˙ | (1) |
| BDE = H(A–O˙) + H(H˙) − H(A–OH) | (1a) |
In the previous equations A–OH, A–O˙ and H˙ denote antioxidant molecule, antioxidant radical and hydrogen atom, while H(A–OH), H(A–O˙) and H(H˙) present their enthalpies respectively.
The SPLET mechanism is a two-step process. Dissociation happens in the first stage of the reaction, resulting in the formation of a proton (H+) and an antioxidant anion (A–O−). The second phase involves electron transfer (e−), which is followed by the formation of the appropriate radical (eqn (2)). The PA value (Proton Affinity, eqn (2a)) may be used to describe the first step of the reaction, and the ETE value can be used to describe the second step (Electron Transfer Enthalpy, eqn (2b)).
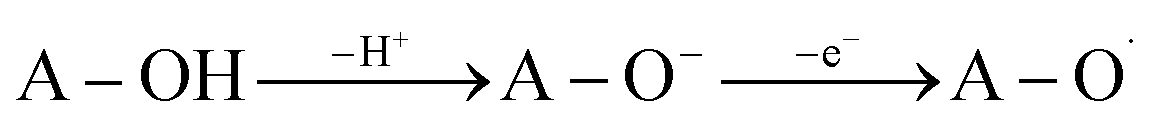 | (2) |
| PA = H(A–O−) + H(H+) − H(A−OH) | (2a) |
| ETE = H(A–O˙) + H(e−) − H(A–O−) | (2b) |
SET-PT is another mechanistic pathway that describes heterolytic OH-cleavage (eqn (3)). In the first step of this mechanism electron leaves molecule forming antioxidant radical-cation specie (A–O˙+), and in the second step, this radical-cation releases the proton, forming corresponding radical. The first mechanistic step can be estimated based on IP values (Ionization Potential, eqn (3a)), and the second step can be estimated based on PDE values (Proton Dissociation Enthalpy, eqn (3b)). The appropriate enthalpy values for the proton and electron in methanol as a solvent were taken from the literature.50
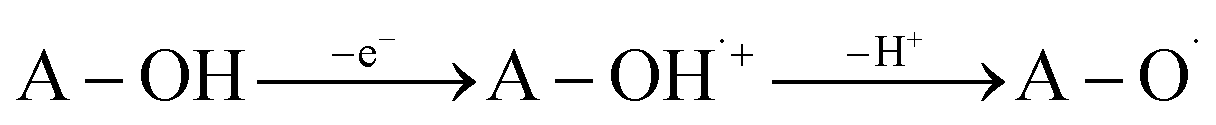 | (3) |
| IP = H(A–OH˙+) + H(e−) − H(A–OH) | (3a) |
| PDE = H(A–O˙) + H(H+) − H(A–OH˙+) | (3b) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the Ministry of Science, Technological Development and Innovation of the Republic of Serbia for the support through agreements no. 451-03-47/2023-01/200252, 451-03-47/2023-01/200378 and 451-03-68/2022-14/200017.Notes and references
- B. Halliwell and J. M. M. Gutteridge, Free Radicals in Biology and Medicine, 5th edn; Oxford University Press, 2015 Search PubMed.
- I. Liguori, G. Russo, F. Curcio, G. Bulli, L. Aran, D. Della-Morte, G. Gargiulo, G. Testa, F. Cacciatore, D. Bonaduce and P. Abete, Clin. Interventions Aging, 2018, 13, 757–772 CrossRef CAS PubMed.
- C. Sackesen, H. Ercan, E. Dizdar, O. Soyer, P. Gumus, B. N. Tosun, Z. Büyüktuncer, E. Karabulut, T. Besler and O. A. Kalayci, J. Allergy Clin. Immunol., 2008, 122, 78–85 CrossRef CAS PubMed.
- S. George and H. Abrahamse, Antioxidants, 2020, 9, 1156 CrossRef CAS PubMed.
- A. Taleb, K. Ali Ahmad, A. Ullah Ihsan, J. Qu, N. Lin, K. Hezam, N. Koju, L. Hui and D. Qilong, Biomed. Pharmacother., 2018, 102, 689–698 CrossRef CAS PubMed.
- M. Sharifi-Rad, N. V. Anil Kumar, P. Zucca, E. M. Varoni, L. Dini, E. Panzarini, J. Rajkovic, P. V. Tsouh Fokou, E. Azzini, I. Peluso, A. Prakash Mishra, M. Nigam, Y. El Rayess, M. E. Beyrouthy, L. Polito, M. Iriti, N. Martins, M. Martorell, O. Docea Anca, W. N. Setzer, D. Calina, W. C. Cho and J. Sharifi-Rad, Front. Physiol., 2020, 11 DOI:10.3389/fphys.2020.00694.
- M. Bortolami, F. Pandolfi, V. Tudino, A. Messore, V. N. Madia, D. De Vita, R. Di Santo, R. Costi, I. Romeo, S. Alcaro, M. Colone, A. Stringaro, A. Espargaró, R. Sabatè and L. Scipione, Pharmaceuticals, 2022, 15, 673 CrossRef CAS PubMed.
- M. Scipioni, G. Kay, I. Megson and P. K. T. Lin, Eur. J. Med. Chem., 2018, 143, 745–754 CrossRef CAS PubMed.
- S. B. Nimse and D. Pal, RSC Adv., 2015, 5, 27986–28006 RSC.
- M. L.-H. Huang, S. Chiang, D. S. Kalinowski, D.-H. Bae, S. Sahni and D. R. Richardson, Oxid. Med. Cell. Longevity, 2019, 2019, 26 Search PubMed.
- V. Thao-Vi Dao, A. I. Casas, G. J. Maghzal, T. Seredenina, N. Kaludercic, N. Robledinos-Anton, F. Di Lisa, R. Stocker, P. Ghezzi, V. Jaquet, A. Cuadrado and H. H. H. W. Schmidt, Pharmacology and Clinical Drug Candidates in Redox Medicine, Antioxid. Redox Signaling, 2015, 1113–1129 Search PubMed.
- H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS PubMed.
- H. Sies, C. Berndt and D. P. Jones, Annu. Rev. Biochem., 2017, 20(86), 715–748 CrossRef PubMed.
- D. P. Jones and H. Sies, The Redox Code, Antioxid. Redox Signaling, 2015, 734–746 CrossRef CAS PubMed.
- M. R. Bhosle, L. D. Khillare, I. R. Mali, A. P. Sarkate, D. K. Lokwani and S. V. Tiwari, New J. Chem., 2018, 42, 18621–18632 RSC.
- F. Buron, J. Y. Mérour, M. Akssira, G. Guillaumet and S. Routier, Eur. J. Med. Chem., 2015, 95, 76–95 CrossRef CAS PubMed.
- N. Kahriman, K. Peker, V. Serdaroğlu, A. Aydin, A. Usta, S. Fandaklı and N. Yaylıd, Bioorg. Chem., 2020, 99, 103805 CrossRef CAS PubMed.
- A. R. Saundane, K. Vijaykumar, A. V. Vaijinath and P. Walmik, Med. Chem. Res., 2013, 22, 806–817 CrossRef CAS.
- C.-H. Lee, M. Jiang, M. Cowart, G. Gfesser, R. Perner, K. H. Kim, Y. G. Gu, M. Williams, M. F. Jarvis, E. A. Kowaluk, A. O. Stewart and S. S. Bhagwat, J. Med. Chem., 2001, 44, 2133–2138 CrossRef CAS PubMed.
- M. A. Abdelgawad, M. M. Al-Sanea, A. Musa, M. Elmowafy, A. K. El-Damasy, A. A. Azouz, M. M. Ghoneim and R. B. Bakr, J. Inflammation Res., 2022, 15, 451–463 CrossRef CAS PubMed.
- Z. Hussain, M. A. Ibrahim, N. M. El-Gohary and A.-S. Badran, J. Mol. Struct., 2022, 1269, 133870 CrossRef CAS.
- P. Yadav and K. Shah, Chem. Biol. Drug Des., 2020, 97, 633–648 CrossRef PubMed.
- S. Wang, X.-H. Yuan, S.-Q. Wang, W. Zhao, X.-B. Chen and B. Yu, Eur. J. Med. Chem., 2021, 214, 113218 CrossRef CAS PubMed.
- E. Milović, N. Janković, M. Vraneš, S. Stefanović, J. Petronijević, N. Joksimović, J. Muškinja and Z. Ratković, Environ. Chem. Lett., 2021, 19, 729–736 CrossRef.
- L. Blaikie, G. Kay and P. K. Thoo Lin, Bioorg. Med. Chem. Lett., 2020, 30, 127505 CrossRef CAS PubMed.
- W. T. Borden, R. Hoffmann, T. Stuyver and B. Chen, J. Am. Chem. Soc., 2017, 139, 9010–9018 CrossRef CAS PubMed.
- Ž. Milanović, J. Tošović, S. Marković and Z. Marković, RSC Adv., 2020, 10, 43262–43272 RSC.
- G. Mazzone, N. Malaj, A. Galano, N. Russo and M. Toscano, RSC Adv., 2015, 5, 565–575 RSC.
- Z. Marković, M. Filipović, N. Manojlović, A. Amić, S. Jeremić and D. Milenković, Chem. Pap., 2018, 72, 2785–2793 CrossRef.
- S. Hwang, Q. Shao, H. Williams, C. Hilty and Y. Q. Gao, J. Phys. Chem. B, 2011, 115, 6653–6660 CrossRef CAS PubMed.
- C. Caratelli, J. Hajek, S. M. J. Rogge, S. Vandenbrande, E. J. Meijer, M. Waroquier and V. Van Speybroeck, ChemPhysChem, 2018, 19, 420–429 CrossRef CAS PubMed.
- S. Jeremić, A. Amić, M. Stanojević-Pirković and Z. Marković, Org. Biomol. Chem., 2018, 16, 1890–1902 RSC.
- N. H. Attanayake, T. M. Suduwella, Y. Yan, A. P. Kaur, Z. Liang, M. S. Sanford and S. A. Odom, J. Phys. Chem. C, 2021, 125, 14170–14179 CrossRef CAS.
- N. H. Attanayake, A. P. Kaur, T. M. Suduwella, C. F. Elliott, S. R. Parkin and S. A. Odom, New J. Chem., 2020, 44, 18138–18148 RSC.
- T. Lunić, J. Lađarević, M. Mandić, V. Veruševski, B. Božić Nedeljković, D. Mijin and B. Božić, J. Mol. Struct., 2022, 1256, 132546 CrossRef.
- T. Kundu and A. Pramanik, Bioorg. Chem., 2020, 98, 103734 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannen-berg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev D.1 Gaussian Inc, Wallingford CT, 2013 Search PubMed.
- M. Spiegel, A. Gamian and Z. A. Sroka, Molecules, 2021, 26, 5058 CrossRef CAS PubMed.
- D. S. Dimić, D. A. Milenković, E. H. Avdović, Đ. J. Nakarada, J. M. D. Marković and Z. S. Marković, Chem. Eng. J., 2021, 424, 130331 CrossRef.
- D. A. Milenković, D. S. Dimić, E. H. Avdović, A. D. Amić, J. M. D. Marković and Z. S. Marković, Chem. Eng. J., 2020, 395, 124971 CrossRef.
- Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005, 1, 70–77 CrossRef PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.9. Theoretical Chemistry Institute, University of Wisconsin, Madison, 2009 Search PubMed.
- J. E. Carpenter and F. Weinhold, J. Mol. Struct.: THEOCHEM, 1988, 169, 41–62 CrossRef.
- Z. Marković, J. Serb. Soc. Comput. Mech., 2016, 10, 135–150 CrossRef.
- E. Klein, V. Lukeš and M. Ilčin, Chem. Phys., 2007, 336, 51–57 CrossRef CAS.
- A. Galano, J. Mex. Chem. Soc., 2015, 59, 231–262 Search PubMed.
- G. Mazzone, A. Galano, J. R. Alvarez-Idaboy and N. Russo, J. Chem. Inf. Model., 2016, 56, 662–670 CrossRef CAS PubMed.
- Z. Marković, J. Tošović, D. Milenković and S. Marković, Comput. Theor. Chem., 2016, 1077, 11–17 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |