
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature-dependent tailoring of the pore structure based on MOF-derived carbon electrodes for electrochemical capacitors†
Yuru Wanga and
Qing Zhang *ab
*ab
aInstitutes of Physical Science and Information Technology, Anhui University, Hefei, 230601, China. E-mail: zhangq@ahu.edu.cn
bAnhui Graphene Engineering Research Center, Anhui University, Hefei, 230601, China
First published on 15th June 2023
Abstract
The pore structures of carbon play a critical role in the charge storage process of electrochemical capacitors; however, the involvement of other varying characteristics, such as electrical conductivity and surface functionalities, complicate the research of the pore size effects on various electrochemical phenomena. In this study, by carbonizing MOF-5 at a selected temperature range of 500–700 °C, a series of MOF-derived carbon materials were obtained with pore size distribution concentrated in different size ranges while admitting similar results in the graphitization degree and surface functionalities. The related morphological changes of ZnO were systematically investigated by changing the carbonization temperature and dwelling time, demonstrating a “from thin to thick, from inside to outside” growth routine of ZnO crystals. With the pore size approximated as the sole variable, the as-assembled electrochemical capacitors present a linear relationship between the 1–10 nm pores and the impedance resistance, which for the first time demonstrate how 1–10 nm pores is beneficial to ion diffusion. The results of this study not only provide a useful approach to manipulating the pore structure in carbon electrodes but also pave the way to establish the numerical relationship between the pore structure and various phenomena in electrochemistry or other related areas.
1. Introduction
Electrochemical capacitors (ECs), as an efficient tool for energy storage and management with high power density and reliable cyclability, are one of the promising solutions for achieving the stable and sustainable use of renewable energy sources.1,2 Carbons (activated carbon, aerogels, xerogels, carbon nanotubes, and graphene) are the primary materials used to prepare EC electrodes owing to their chemical stability, natural abundance, and feasibility by engineering their structures/textures and surface chemistry to adapt to various application scenarios.2–5 The electrochemical performance of carbon materials depends on various material characteristics, such as the pore size structure, structural crystallinity, and surface functionalities.2,3,6–9 Adjusting the pore structure is a common and effective way to optimize the electrochemical performance of carbon-based ECs, especially electrochemical double-layer capacitors, where the properties of the as-formed double layer structure depend greatly on the pore size and curvature with respect to the chosen electrolytic ions.10–12Template carbonization is one of the effective methods for controlling the pore size of carbon materials and is classified into hard- and soft-templating methods.6,13–15 Metal–organic frameworks, composed of metal units and organic ligands with intrinsic porosity, are ideal soft-templating precursors for the construction of porous carbon materials,15–17 eliminating the use of dangerous and toxic hydrogen fluoride usually required in the hard-templating methods.13,18 Moreover, carbons produced by hard-templating methods are usually advanced with a narrow pore size distribution,6,13,18 while in practice especially for industrialized products, carbon materials with a wide pore size distribution and hierarchical porous structure are the common case.19
So far, there has been extensive research on synthesizing porous carbons using MOFs as precursors ever since their first report in 2008 by Liu et al.,17 utilizing Zn-based MOF-5 (consists of the metal unit Zn4O linked to the organic ligand p-benzenedioic acid),20 to prepare nanoporous carbon at 530 °C with ZnO removed by acid washing, and at 1000 °C (boiling point of Zn metal 908 °C), showing excellent hydrogen absorption capacity and electrochemical properties. Further research by Zhang et al. revealed that the thermal decomposition of MOF-5 occurred at 400 °C and above involved breaking the carboxyl bridge between the benzene ring and the Zn4O cluster and producing amorphous carbon along with CO2, benzene, and ZnO crystals.21 Effects of carbonization temperature on the properties of MOF-derived carbons have been repeatedly demonstrated where the resulting pore structure concentrated in the mesoporous range at temperature as low as 500 °C or higher than 700 °C;22,23 a morphological change of ZnO was also observed upon increasing the carbonization temperature from 600 °C to 1000 °C,23,24 showing that the conversion of Zn4O clusters to ZnO is quite sensitive to the carbonization temperature and demonstrating the possibility to control the pore size distribution through controlling the morphology of ZnO.
Herein, in order to locate the proper treatment conditions for tailoring the pore size distribution, the effects of carbonization temperature as well as the dwelling time have been systematically investigated using MOF-5 as the precursor. Varied pore size distributions were achieved with other critical features affecting the electrochemical performance changing little between 570 °C to 700 °C. Based on the obtained MOF-derived carbon with pore size distribution approximated as the sole variable, a linear relationship has been revealed between the 1–10 nm pores and the impedance resistance, which would offer fundamental insight into the pore size-ion diffusion relation for developing advanced electrochemical devices.
2. Experimental methods
2.1 Preparation of Zn-MOF-5
MOF-5 was synthesized following the method first reported by Yaghi et al.20 7.76 mmol of zinc acetate dihydrate (Zn(CH3COO)2·2H2O, Alfa Aesar, 98%) was dissolved in a conical flask containing 50 ml of N,N-dimethylformamide (DMF, Alfa Aesar, 99.8%); 3.05 mmol of terephthalic acid (1,4-H2BDC, Alfa Aesar, 98%) was dissolved in 40 ml of DMF with 0.85 ml of triethylamine (TEA, Alfa Aesar, 99%) added afterwards. Then, the two solutions were mixed together under stirring at 600 rpm for 2.5 h. White precipitates of Zn-MOF-5 were washed three times with anhydrous ethanol (Sinopharm, AR grade), collected by centrifugation, and dried under vacuum at 80 °C for 3 hours.2.2 Carbonization of Zn-MOF-5
The as-synthesized Zn-MOF-5 was loaded into a ceramic boat and placed in a quartz tube under Ar flow. The temperature was first raised from room temperature to 400 °C at a heating rate of 10 °C min−1 and then dwelled for 1 h. Afterwards, the temperature was increased at a heating rate of 5 °C min−1 to the set target temperature T and dwelling for a set period of time t (Table S1†). The carbonized MOFs were labeled as ZnO/C-Tx-ty (where x is the target temperature, and y is the dwelling time). ZnO/C-Tx-ty were then soaked in 6 M HCl (aq) at room temperature for 24 h to remove ZnO, followed by washing with copious deionized water until reaching neutral (pH ∼ 7) and then dried at 100 °C for 12 h. The as-produced MOF-5-derived carbons were denoted as C-Tx-ty.2.3 Material characterization
Thermogravimetric analysis (TGA) was carried out using a TA instrument (HR-1) from room temperature to 1000 °C at a heating rate of 10 °C min−1 under nitrogen protection. X-ray diffraction (XRD) data were collected on a Rigaku SmartLab diffractometer (D/MAX-γA, λ = 1.54 Å). The scanning electron microscopic (SEM) images were obtained on a Zeiss Crossbeam 550, and the transmission electron microscopic (TEM) images, scanning transmission electron microscopic (STEM) images, and the corresponding elemental mapping results on a JEM-F200. Raman spectra were collected using a Renishaw InVia Reflex confocal microscope with an excitation light source of 532 nm. The laser power used was 0.15 mW to minimize burning the samples. X-ray photoelectron spectroscopy (XPS) spectra were collected on an ESCALAB 250Xi and calibrated by setting the C 1s photoemission peak for sp2-hybridized carbons to 284.8 eV. N2 adsorption/desorption isotherms were acquired using a Quadrasorb Instrument (Quantachrome, Autosorb IQ-MP-MP) at 77 K, where the surface area and pore size distributions were calculated by the quenched solid density functional theory (QSDFT) method.2.4 Electrochemical tests
85 wt% C-Tx-ty, 6 wt% carbon black, and 9 wt% polyvinylidene fluoride (PVDF, Arkema-HSV900, 99%) binder were mixed and ground with a mortar and pestle in N-methylpyrrolidone (NMP, Alfa Aesar, 99%). The slurry was then coated onto circular disks of aluminum foil (14 mm in diameter). The mass loading of each electrode was around 1 mg. ECs were assembled in a coin cell (CR2032-SUS316L, Japan). A Whatman glass microfiber filter was used as the separator immersed in 1 M tetraethylammonium tetrafluoroborate (Alfa Aesar, 99%)–polycarbonate (Alfa Aesar, 99%) (TEABF4/PC) as the organic electrolyte.The cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were performed on a Solartron 1470E electrochemical testing system. CV was performed at scan rates ranging from 2 to 500 mV s−1 within a voltage window of 0 to 2 V. EIS was recorded at 0 V over a frequency range from 100 kHz to 10 mHz with a bias amplitude of 5 mV. Galvanostatic charge–discharge (GCD) was measured using an Arbin instrument (LBT-21084) from 0 to 2 V at a series of current densities from 10 to 1000 mA g−1. The specific capacitances C (F g−1) based on CV tests were calculated using the following equation:
 | (1) |
The specific capacitance based on the GCD tests was calculated based on the following equation:
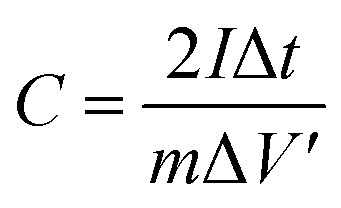 | (2) |
3. Results and discussion
Fig. 1a schematically presents the synthesis procedure of MOF-5-derived carbon. In order to obtain porous carbon with different pore size distributions, the synthesized microcubes of Zn-MOF-5 (identified with XRD result, Fig. 1b) were treated with different carbonization temperatures T and dwelling time t to control the morphology of the ZnO formed. The temperature was chosen based on TGA results. As shown in Fig. 1c, the weight loss of Zn-MOF-5 during the heating from room temperature to 1000 °C could be divided into four main stages: (1) the first stage (weight loss: ∼1%) as located below ∼110 °C could be ascribed to the removal of water adsorbed on the surface of the material;21,24,25 (2) the second stage (weight loss: ∼9%) between ∼(120–300) °C was mainly due to the dissipation of the residual solvent molecules (DMF here) incorporated in the frameworks;24,25 (3) significant weight loss (∼40%) occurred around ∼(400–550) °C of the third stage, which should be caused by the thermal decomposition of the Zn-MOF-5 skeleton. During this stage, carboxylic bridges between benzene rings and Zn4O clusters broke, releasing CO2 and benzene, and Zn-MOF-5 crystals were pyrolyzed into a composite of ZnO and carbon;21,22,24 (4) as the temperature continued to reach around 750 °C, carbonaceous materials started to deoxidize ZnO to Zn with itself being oxidized mainly into CO2 and CO;22,24 when reaching the boiling point of the zinc metal monomer (908 °C), the gaseous zinc would begin to evaporate under the nitrogen current flow. This reaction was the most intense at 950 °C,22–24, and the weight loss of this stage was ∼28%. Therefore, the temperature zone to tune the ZnO morphology was selected between (500–700) °C, where carbon was produced from the decomposition of organic components but little consumption of carbon to reduce ZnO. | ||
| Fig. 1 (a) The schematic illustration of the synthesis of MOF-derived carbon C-Tx-ty. (b) XRD pattern with four characteristic peaks at 6.9°, 9.8°, 13.8°, and 15.4° corresponding to (200), (220), (400), and (420) of Zn-MOF-5, respectively.21,23 (c) TGA curve of Zn-MOF-5 under nitrogen atmosphere (the purple dotted line indicates the differential result of TGA curve). | ||
As shown in Fig. 2a–g, the carbonized MOFs (named ZnO/C-Tx-ty, where x denotes the carbonization temperature and y the dwelling time) exhibited a morphological transition of ZnO crystals (as demonstrated with the XRD results, Fig. 2h) upon increasing the temperature and the dwelling time. Besides, the carbon cubes presented different morphologies as well. All ZnO/C-Tx-ty samples retained the cubic skeleton without collapse. Differences in the ZnO crystals are reflected not only in the shape and size but also in the location. ZnO/C-T500-t0.5 treated at the lowest 500 °C showed a sponge-like porous structure (Fig. 2a, the SEM) with fine ZnO nanoparticles (∼10–20 nm in diameter) and nanoneedles (∼10–30 nm in diameter and ∼300–500 nm in length) embedded inside the cube and some protruding out (Fig. 2a, TEM) since the lower temperature is prone to the formation of fine ZnO nanoparticles/nanoneedles.26 On prolonging the treatment time to 2 h, ZnO/C-T500-t2 exhibited a much smoother surface with some ZnO aggregates grown onto the external surface (Fig. 2b, SEM); nanoneedles disappeared and were replaced by nanospheres (∼10–20 nm in diameter, Fig. 2b, TEM).27 ZnO/C-T570-t0.5 with the higher temperature at 570 °C but a shorter dwelling time of 0.5 h presented a sponge-like porous structure similar to that of ZnO/C-T500-t0.5, and were filled with ZnO nanoparticles (∼10–20 nm in diameter) and nanoneedles as well (∼10–30 nm in diameter and ∼300–500 nm in length, Fig. 2c); further increasing the dwelling time at this temperature to 2 h and 4 h lead to ZnO/C-T570-t2 and ZnO/C-T570-t4, respectively, both exhibiting similar morphology to that of ZnO/C-T500-t2 enriched with ZnO nanospheres (∼30–60 nm, Fig. 2d and e). As the temperature was further raised to 700 °C, nearly all ZnO was extracted out of the cubic frameworks and grew into large ZnO rods (∼100–200 nm in diameter), possibly as a result of that temperature up to 700 °C would melt the nanospheres and favor the growth of nanorods due to the carbon reduction of ZnO, where the resulting gaseous zinc reacted with the fine unreduced ZnO nanoneedles/nanospheres within the cubes to form ZnO rods that extended outwards.23,24,28 ZnO/C-T700-t0.5 and ZnO/C-T700-t2 both presented clean structures inside and smooth surfaces except for some large holes probably marked by the ZnO rods (Fig. 2f and g). Conclusively, within the (500–700) °C temperature range, the morphology of ZnO could be controlled from nanoneedles to nanospheres, and eventually diffusing out of the cubic frameworks and forming rod-like ZnO crystals by increasing the carbonization temperature and adjusting the dwelling time.
 | ||
| Fig. 2 SEM, TEM, and STEM images with elemental mapping results of (a) ZnO/C-T500-t0.5, (b) ZnO/C-T500-t2, (c) ZnO/C-T570-t0.5, (d) ZnO/C-T570-t2, (e) ZnO/C-T570-t4, (f) ZnO/C-T700-t0.5, and (g) ZnO/C-T700-t2. (h) XRD patterns of ZnO/C-Tx-ty show distinct peaks at 31.6°, 34.5°, 36.2°, 47.5°, 56.6°, 62.8°, 66.3°, 67.9° and 69°, representing ZnO.23,27 Note: the scale bar in the STEM images represents 500 nm. | ||
After acid washing, Zn-MOF-5 derived carbons (Fig. 3a–g, denoted as C-Tx-ty) were easily obtained. C-Tx-ty inherited the cubic shape and the morphology of the precedent ZnO/C-Tx-ty with ZnO being completely removed (as evidenced with only broad XRD peaks at ∼23° and ∼44° for amorphous carbon, Fig. 3h).17 C-T500-t0.5 and C-T570-t0.5 exhibited sponge-like porous structures while the rest showed a relatively smoother surface. TEM images of the C-Tx-ty presented distinct pore structures, where C-T500-t0.5 and C-T570-t0.5 were filled with large pores (up to 20–30 nm in diameter) while pores in C-T700-t0.5 and C-T700-t2 were too small to be appreciable (Fig. S2†).
 | ||
| Fig. 3 SEM images of (a) C-T500-t0.5, (b) C-T500-t2, (c) C-T570-t0.5, (d) C-T570-t2, (e) C-T570-t4, (f) C-T700-t0.5, and (g) C-T700-t2. (h) XRD patterns of C-Tx-ty. | ||
Further analysis with N2 adsorption–desorption measurement demonstrated that the varied pore size distribution was achieved by controlling the morphology of ZnO. The QSDFT model was applied as an effective method for the pore size distribution calculation in micro-mesoporous carbons with heterogeneous structures.29–31 The N2 adsorption–desorption isotherms and the corresponding pore size distribution are given in Fig. 4a–f. C-T500-t0.5 and C-T570-t0.5 not only presented similar textures of carbon microcubes (Fig. 3a and c) but also admitted similar isotherms (as a combination of type I (micropore), type IV (mesopore), and type II (macropore) isotherms, Fig. 4a and b), suggesting a hierarchical porous structure with a wide pore size distribution (Fig. 4d and e).32 The sharp uptakes at a low relative pressure (p/p0 < 0.1) indicated the existence of micropores and the hysteresis loop in the region (p/p0 ∼ 0.5 to 0.9) the presence of mesopores; the sharp uptakes at a high relative pressure (p/p0 > 0.9) indicated the presence of a large amount of macropores.33,34 The rest five C-Tx-ty treated either at higher carbonization temperature or longer dwelling time also possessed hierarchical porous structures as indicated by a combination of type I and type IV isotherms (Fig. 4a–c), where the upward tail at a high relative pressure (p/p0 > 0.9) disappeared except for a small tail of C-T700-t2. Hence, within the temperature range (500–700) °C, the higher the carbonization temperature or a longer period of dwelling time, the fewer macropores existed; while at the upper-temperature limit of 700 °C, with the longer dwelling time of 2 h, macropores started to show up again.23
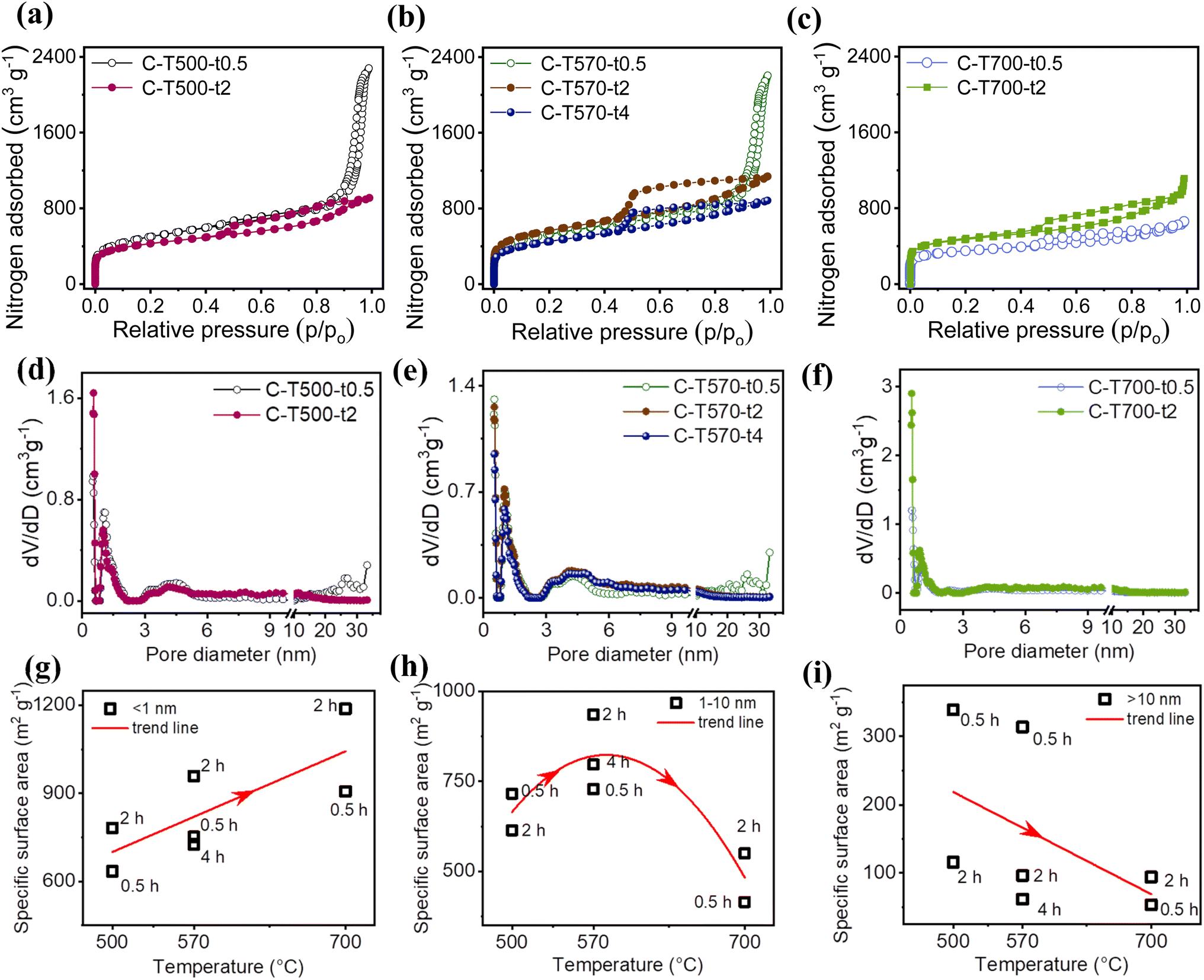 | ||
| Fig. 4 N2 adsorption–desorption isotherms and pore size distribution of (a and d) C-T500-t0.5 and C-T500-t2, (b and e) C-T570-t0.5, C-T570-t2 and C-T570-t4, and (c and f) C-T700-t0.5, C-T700-t2. Relationship between carbonization temperature and the partial SSA contributed by (g) (<1 nm) pores, (h) (1–10) nm pores, and (i) (>10 nm) pores in C-Tx-ty materials with trend line. | ||
The analyzed textural characteristics of the C-Tx-ty carbons are summarized in Tables 1 and 2, where the carbon materials presented a significant increase in specific surface area (SSA) as compared to that of the Zn-MOF-5 (only 897 m2 g−1, Fig. S3 and Table S3†). To further analyze the porous structure with respect to its effects on electrochemical performance, the pores were divided into three categories based on the electrolyte used in the following study (1 M TEABF4/PC, where the ion size of TEA+ is 0.68 nm, and 1.30 nm with the solvated shell, and the ion size of BF4− is 0.48 nm and 1.16 nm with the solvated shell.11,35), which were small pores of (<1 nm), medium pores of (1–10) nm, and large pores of (>10 nm). Desolvation or partial desolvation is required for electrolytic ions to diffuse into the small pores;11,35 for the medium pores the pore curvature is significant while for the large pores the pore wall can be approximated as a planar surface with respect to the ions.12
| Sample | Vtotal (cm3 g−1) | V<1 nm (cm3 g−1) | V1–10 nm (cm3 g−1) | V>10 nm (cm3 g−1) | V<1 nm (%) | V1–10 nm (%) | V>10 nm (%) |
|---|---|---|---|---|---|---|---|
| a Vtotal is the total pore volume. | |||||||
| C-T500-t0.5 | 2.84 | 0.19 | 0.61 | 2.04 | 6.6 | 21.6 | 71.8 |
| C-T500-t2 | 1.29 | 0.24 | 0.63 | 0.42 | 18.6 | 48.8 | 32.6 |
| C-T570-t0.5 | 2.69 | 0.23 | 0.64 | 1.82 | 8.5 | 24.0 | 67.5 |
| C-T570-t2 | 1.62 | 0.29 | 0.97 | 0.36 | 17.9 | 59.9 | 22.2 |
| C-T570-t4 | 1.27 | 0.21 | 0.83 | 0.23 | 16.9 | 65.0 | 18.1 |
| C-T700-t0.5 | 0.90 | 0.27 | 0.42 | 0.21 | 29.8 | 46.6 | 23.6 |
| C-T700-t2 | 1.36 | 0.36 | 0.64 | 0.36 | 26.5 | 46.9 | 26.6 |
| Sample | SSA (m2 g−1) | SSA<1 nm (m2 g−1) | SSA1–10 nm (m2 g−1) | SSA>10 nm (m2 g−1) | SSA<1 nm (%) | SSA1–10 nm (%) | SSA>10 nm (%) |
|---|---|---|---|---|---|---|---|
| C-T500-t0.5 | 1689 | 634 | 716 | 339 | 37.5 | 42.4 | 20.1 |
| C-T500-t2 | 1512 | 782 | 614 | 116 | 51.7 | 40.6 | 7.7 |
| C-T570-t0.5 | 1794 | 752 | 728 | 314 | 41.9 | 40.6 | 17.5 |
| C-T570-t2 | 1991 | 959 | 936 | 96 | 48.2 | 47.0 | 4.8 |
| C-T570-t4 | 1585 | 726 | 797 | 62 | 45.8 | 50.3 | 3.9 |
| C-T700-t0.5 | 1376 | 907 | 415 | 54 | 65.9 | 30.2 | 3.9 |
| C-T700-t2 | 1832 | 1188 | 550 | 94 | 64.8 | 30.0 | 5.2 |
A further comparison of the pore size distribution based on the above three categories (Tables 1 and 2) showed that C-T500-t0.5 and C-T570-t0.5 both held a considerable amount of large pores of (>10 nm) while C-T700-t0.5 and C-T700-t2 possessed a fairly large amount of small pores of (<1 nm). Large pores of (>10 nm) took up a pore volume ratio (the partial pore volume V>10 nm contributed by large pores divided by the total pore volume Vtotal) of 71.8% and 67.5% in C-T500-t0.5 and C-T570-t0.5 (Table 1), hence C-T500-t0.5 and C-T570-t0.5 could be considered predominantly composed of large pores. Small pores of (<1 nm) took up an SSA ratio (the partial SSA SSA<1 nm contributed by small pores divided by the total SSA) of 65.9% and 64.8% in C-T700-t0.5 and C-T700-t2, respectively (Table 2), hence C-T700-t0.5 and C-T700-t2 could be considered predominantly composed of small pores. As for C-T570-t4, the medium pores of (1–10) nm took up relatively higher ratios by 50.3% in SSA ratio and 65.0% in pore volume ratio. C-T500-t2 and C-T570-t2 did not present a major type of pores but a relatively evenly distributed combination of different-sized pores. More interestingly, as vividly shown by the trend lines (Fig. 4g–i), SSA<1 nm increased with the carbonization temperature, while SSA>10 nm decreased with the carbonization temperature, and SSA1–10 nm reached a maximum at 570 °C. Hence, though there was no clear relation existed between the total SSA and the carbonization temperature, partial SSA did relate to the temperature; based on this, it is possible to realize fine-tuning to desirable pore size distributions by designing the treatment conditions within the temperature range of (500–700) °C.
Besides the pore structures, Raman, and XPS were applied to further analyze the effects of carbonization temperature and dwelling time on the chemical properties of C-Tx-ty. As shown in Fig. 5a, the Raman spectra of all the C-Tx-ty materials were fitted by four Gaussian peaks: TPA peak (at ∼1200 cm−1, attributed to heteroatoms), D peak (at ∼1350 cm−1, corresponding to A1g vibrations of sp3 defects), A peak (at ∼1490 cm−1, from defects in graphite layer stacks), and G peak (at ∼1595 cm−1, the vibrations of sp2 hybridized carbon structures).36,37 The smaller the intensity ratio of ID/IG (the ratio of the deconvoluted peak areas), the higher the degree of graphitization.16,37 C-T500-t0.5 showed the largest ID/IG of 2.33, indicating the low degree of graphitization probably due to the low carbonization temperature.38 Increasing the dwelling time to 2 h, the ID/IG of C-T500-t2 decreased to 1.42. The rest of ID/IG for higher carbonization temperatures of 570 °C and 700 °C were all around ∼1.2, suggesting no significant change in the crystallinity of the MOF-derived carbons when the temperature fell between 570 °C and 700 °C.
 | ||
| Fig. 5 (a) Raman spectra of C-Tx-ty. High-resolution spectra of (b) C 1s, and (c) O 1s of C-T570-t2. (d) Comparison of atomic ratios of sp2 carbon, sp3 carbon, and functional groups as calculated from the fitting results of C 1s spectra of C-Tx-ty. | ||
Chemical identification of C-Tx-ty carbons was addressed by XPS. All survey scans showed a dominant peak around 284.8 eV for C 1s and one around 532.9 eV for O 1s,16,39 while no peaks were observed for the Zn element (Fig. S4†), again demonstrating that ZnO was completely removed after acid washing. Fitting results of high-resolution scans gave the same combination of deconvoluted peaks (Fig. S5 and S6†). Taking C-T570-t2 as an example (Fig. 5b and c), the high-resolution C 1s region could be deconvoluted into four peaks: sp2 carbon (∼284.8 eV), sp3 carbon (285.4 ± 0.3 eV), C–O(C–OH) (286.6 ± 0.3 eV) and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O(H2Oads) (289.6 ± 0.3 eV);16,36 while the high-resolution O 1s region could be deconvoluted into two peaks: C–O(C–OH) (532.6 ± 0.3 eV) and OC–O(H2Oads) (533.6 ± 0.3 eV).16,39,40 Comparing the calculated atomic ratios of different carbons (Fig. 5d and Table S4†), the ratio of sp2 carbon, sp3 carbon, and the functional groups varied little after 570 °C, demonstrating similar chemical compositions. Taking sp2 carbon as an example, C-T500-t0.5 and C-T500-t2 treated at 500 °C admitted lower ratios of only 52.62 at% and 53.33 at%, respectively. After increasing the carbonization temperature to 570 °C, the ratio increased to ∼64 at% where prolonging the dwelling time from 0.5 h to 4 h affected the ratio little. The sp2-C ratio decreased a little to ∼57 at% after further increasing the carbonization temperature to 700 °C, probably due to the reduction of ZnO that might have taken place at this temperature.
C–O(H2Oads) (289.6 ± 0.3 eV);16,36 while the high-resolution O 1s region could be deconvoluted into two peaks: C–O(C–OH) (532.6 ± 0.3 eV) and OC–O(H2Oads) (533.6 ± 0.3 eV).16,39,40 Comparing the calculated atomic ratios of different carbons (Fig. 5d and Table S4†), the ratio of sp2 carbon, sp3 carbon, and the functional groups varied little after 570 °C, demonstrating similar chemical compositions. Taking sp2 carbon as an example, C-T500-t0.5 and C-T500-t2 treated at 500 °C admitted lower ratios of only 52.62 at% and 53.33 at%, respectively. After increasing the carbonization temperature to 570 °C, the ratio increased to ∼64 at% where prolonging the dwelling time from 0.5 h to 4 h affected the ratio little. The sp2-C ratio decreased a little to ∼57 at% after further increasing the carbonization temperature to 700 °C, probably due to the reduction of ZnO that might have taken place at this temperature.
Conclusively, control of the pore size distribution can be achieved by tuning the carbonization temperature and the dwelling time; moreover, as the chemical properties varied little within the temperature range (570–700) °C, pore size distribution could be approximated as a single variable to investigate its effects on various applications.
The electrochemical properties of C-Tx-ty materials were characterized for EDLC applications using the organic electrolyte 1 M TEABF4/PC. As shown in Fig. 6a, C-T500-t0.5 showed poor electrochemical performance with significantly distorted CV curves even at a moderate scan rate of 10 mV s−1, and the specific capacitance was only 2.1 F g−1 at 500 mV s−1, exhibiting low rate capability (Table S5†). After increasing the dwelling time to 2 h, the CV curves were approaching rectangular with only slight distortion, and the specific capacitance was raised to 15.1 F g−1 at 500 mV s−1 with a rate capability of 31.9% for C-T500-t2 (Fig. 6b, and Table S5†). Optimal performance was obtained for C-T570-t2 (Fig. 6d), admitting a specific capacitance of 48.1 F g−1 at 500 mV s−1 with a rate capability of 60.2%, while further increasing the carbonization temperature and the dwelling time resulted in a slight reduction of the as-measured specific capacitance (Table S5†). The improved electrochemical performance could be ascribed to the enhanced crystallinity and electrical conductivity at temperature ≥ 570 °C (Fig. 6h). The shape of all the CV curves presented a quasi-rectangular geometry (Fig. 6c–g), corroborating the enhanced crystalline structure (ID/IG ∼ 1.2) and chemical composition (sp2 carbon ∼ 60 at%) within the range of (570–700) °C. Moreover, there was quite a difference in charge capacitance. Taking the scan rate of 2 mV s−1 as an example, the specific capacitance admitted the highest 79.9 F g−1 for C-T570-t2 and the lowest 56.7 F g−1 for C-T700-t0.5. As displayed in Fig. 6i (Fig. S9†), the specific capacitance increased with increasing SSA, hence SSA remained as one of the critical characteristics affecting the electrochemical energy storage in our carbon-based EDLCs mainly depending on the double-layer formation to store energy (as indicated with the nearly straight charge/discharge line, Fig. S10†);1,2 however, as circled in Fig. 6i, three samples, C-T570-t0.5, C-T570-t2, and C-T700-t2 with similar SSAs of 1794 m2 g−1, 1991 m2 g−1 and 1832 m2 g−1, respectively, (with a difference ratio of 9.88%) showed an 18.99% difference in specific capacitance. Tracing back the structural characteristics of the three samples, it has been noticed that they had distinct pore size distributions (Tables 1 and 2), where C-T570-t0.5 majored with large pores of a pore volume ratio up to 67.5%, C-T700-t2 majored with small pores of an SSA ratio up to 64.8%, and C-T570-t2 possessed a relatively evenly-distributed pore size distribution with the highest ratio of medium pores among the three. Therefore, it is reasonable to conjecture that varied pore size distributions contributed to the difference in specific capacitance and medium pores would be more beneficial for storing energy compared to the other two.6,8,10,41
 | ||
| Fig. 6 (a–g) CV curves of C-Tx-ty at the scan rates of 10 and 500 mV s−1. (h) The as-calculated specific capacitance values. (i) The specific capacitance of C-Tx-ty at 2 mV s−1 versus the specific surface area with a fitted trend line in red (with the correlation coefficient R2 ∼ 0.7). | ||
To validate the above conjecture, EIS measurements were carried out from the viewpoint of ion diffusion kinetics. Nyquist plots of all MOF-derived carbons are given in Fig. 7a, all composed of a semi-circle (or incomplete semi-circle for C-T500-t0.5 and C-T500-t2) at the high-frequency range, followed by a straight diffusion line at the low-frequency range related to the mass transfer process. All the C-Tx-ty exhibited a nearly vertical straight line except that C-T500-t0.5 presented a jumbled curve with a slope of ∼45°, suggesting distinct and significantly poor ion diffusion resistance, possibly due to the low degree of graphitization (Fig. 5a).37,40 All spectra as measured except for that of C-T500-t0.5 (Fig. S11†) were fitted with the equivalent circuit consisting of two parallel circuits (Fig. 7b). The existence of the two circuits in series instead of one could be explained by a possible interference of surface functional groups (C–O and O–CO, which would intervene between the electrolytic ions and the electrode/electrolyte interface and create variation in the electrostatic bonding strength), and will be explored in the future study.42 The fitting parameters are summarized (Table S7†). Series resistance Rs is mainly contributed by the electrolyte, carbon electrodes, their interfacial resistance, and the contact resistance between the active material and the current collector, while the charge transfer resistance Rct is mainly affected by the surface functionalities.40 C-T500-t2 admitted relatively higher series resistance Rs, higher charge transfer resistance Rct-1 and Rct-2 as well as substantially high diffusion impedance Wo–R and the diffusion interpretation Wo–T, demonstrating that the carbonization temperature of 500 °C, although could successfully pyrolyze Zn-MOF-5, restricted it from the poor crystallinity and was not suitable for application as the electrode material. For the other five 570 °C and 700 °C treated samples, their series resistance Rs and charge transfer resistance Rct varied but showed no significant difference, which was consistent with their similar ratios of ID/IG, and of sp2-C, sp3-C, and functional groups (except for the Rct-1 of C-T570-t0.5, which might be related to the pore structure dominated by >10 nm pores). The effects of the size of pores that serve as ion transporting channels on the Rct were also reflected by the elevated Rct-2 of C-T700-t0.5 and C-T700-t2 majored with <1 nm pores. Interestingly, Wo–R (Wo–T) admitted the smallest values for C-T570-t2 and C-T570-t4 possessing the highest ratios of medium pores, and the largest values for C-T700-t0.5 and C-T700-t2 possessing the lowest ratios of medium pores. As illustrated in Fig. 7c and d (Fig. S12†), Wo–R (Wo–T) decreased nearly linearly upon increasing the partial surface area SSA1–10 nm contributed by 1–10 nm pores, in both terms of absolute value and the relative ratio. This result directly proved that with respect to the electrolyte 1 M TEABF4/PC, 1–10 nm pores provided the most suitable transporting channel, as the pores can be accessed freely and quickly by electrolyte ions and facilitated the formation of more stable electrical double layer structures;41,43,44 mostly importantly, it demonstrated the linear relationship between the structural characteristic and performance parameter by controlling the pore size distribution to be approximated as the sole variable. The roles of small pores or large pores were not presented in this study and could be studied in further research.
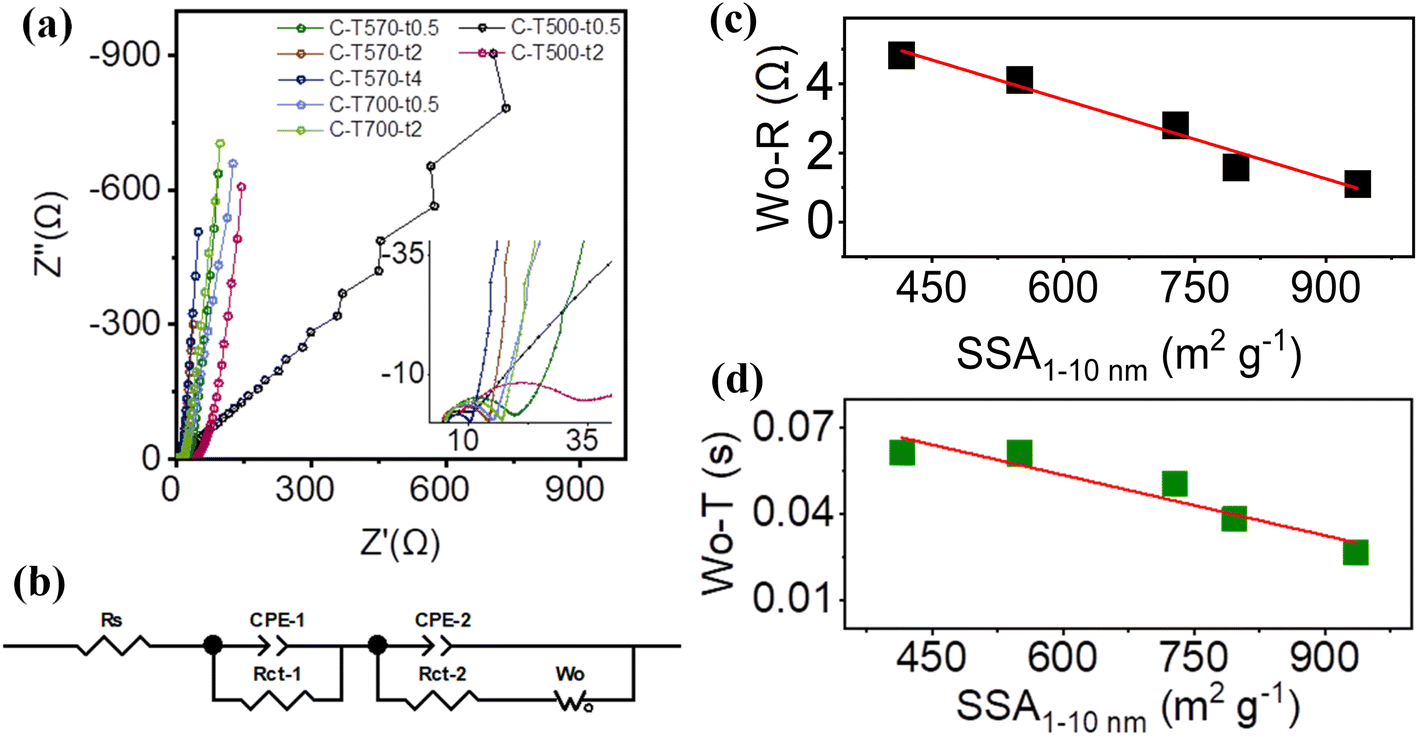 | ||
| Fig. 7 (a) Nyquist plots of C-Tx-ty, (b) the equivalent circuit for all C-Tx-ty except C-T500-t0.5. (c) Wo–R and (d) Wo–T versus the partial SSA1–10 nm with a fitted trend line in red (with the correlation coefficient R2 ∼ 0.97 and 0.90, respectively). | ||
4. Conclusions
We reported the exploration of a synthesis route to prepare porous carbon materials with a pore size distribution approximated as a single variable, demonstrating that a temperature zone of (570–700) °C is suitable to carbonize Zn-MOF-5 with similar chemical properties. Studying the electrochemical performance of the MOF-derived carbons reveals a linear relationship between the 1–10 nm pores and the diffusion impedance Wo–R, which is the first numerical relation built empirically between the electrode structural feature and ion diffusion characteristic. The results of this study enrich the understanding of pore size effects on the electrochemical process within the porous medium, and the method to tailor the pore size as a single variable could be extended to various electrochemical or electrocatalysis-related areas to quantitatively evaluate the effects of pore size distribution.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (Grant No. 22002001), the Natural Science Foundation of Anhui Province (Grant No. 1908085QE175), and the Innovation Program of Anhui Province (Grant No. 2019LCX009).References
- F. Béguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef PubMed
.
- Q. Wang, J. Yan and Z. J. Fan, Energy Environ. Sci., 2016, 9, 729–762 RSC
- X. Li, Y. Tang, J. H. Song, W. Yang, M. S. Wang, C. Z. Zhu, W. G. Zhao, J. M. Zheng and Y. H. Lin, Carbon, 2018, 129, 236–244 CrossRef CAS
- S. C. Wang, Y. Gao, Q. Y. Huang, X. Y. Guo, A. N. Yang, Y. K. Zhang, Q. N. Zhuang, D. D. Chen, L. N. Chen, X. Ju, H. Hu, S. D. Zhang, Y. Zhu, F. Yan and Z. J. Zheng, Adv. Funct. Mater., 2022, 32, 2203730 CrossRef CAS
- W. W. Liu, M. Li, G. P. Jiang, G. R. Li, J. B. Zhu, M. L. Xiao, Y. F. Zhu, R. Gao, A. P. Yu, M. Feng and Z. W. Chen, Adv. Energy Mater., 2020, 10, 2001275 CrossRef CAS
- H. J. Liu, J. Wang, C. X. Wang and Y. Y. Xia, Adv. Energy Mater., 2011, 1, 1101–1108 CrossRef CAS
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., 2008, 120, 379–382 CrossRef
- S. Z. Chen, Y. X. Deng, X. H. Cao, W. X. Zhou, Y. X. Feng, L. M. Tang and K. Q. Chen, J. Mater. Chem. A, 2019, 7, 21976–21984 RSC
- L. Jiang, J. Wang, X. Y. Mao, X. Y. Xu, B. Zhang, J. Yang, Y. F. Wang, J. Zhu and S. F. Hou, Carbon, 2017, 111, 207–214 CrossRef CAS
- G. Salitra, A. Soffer, L. Eliad, Y. Cohen and D. Aurbach, J. Electrochem. Soc., 2000, 147, 2486 CrossRef CAS
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Angew. Chem., Int. Ed., 2008, 47, 3392–3395 CrossRef CAS PubMed
- J. S. Huang, B. G. Sumpter and V. Meunier, Chem.–Eur. J., 2008, 14, 6614–6626 CrossRef CAS PubMed
- L. L. Xie, Z. H. Jin, Z. D. Dai, Y. L. Chang, X. Jiang and H. L. Wang, Carbon, 2020, 170, 100–118 CrossRef CAS
- Y. J. Du, J. Gao, L. Y. Zhou, L. Ma, Y. He, X. F. Zheng, Z. H. Huang and Y. J. Jiang, Adv. Sci., 2019, 6, 1801684 CrossRef PubMed
- Z. Q. Xie, W. W. Xu, X. D. Cui and Y. Wang, ChemSusChem, 2017, 10, 1645–1663 CrossRef CAS PubMed
- I. A. Khan, A. Badshah, I. Khan, D. Zhao and M. A. Nadeem, Microporous Mesoporous Mater., 2017, 253, 169–176 CrossRef
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed
- H. L. Wang, Q. M. Gao, J. Hu and Z. Chen, Carbon, 2009, 47, 2259–2268 CrossRef CAS
- R. Muzyka, E. Misztal, J. Hrabak, S. W. Banks and M. Sajdak, Energy, 2023, 263, 126128 CrossRef CAS
- H. L. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS
- L. Zhang and Y. H. Hu, J. Phys. Chem. C, 2010, 114, 2566–2572 CrossRef CAS
- L. M. Chang, J. R. Li, X. Y. Duan and W. Liu, Electrochim. Acta, 2015, 176, 956–964 CrossRef CAS
- S. Shin, H. Yoon, Y. Yoon, S. Park and M. W. Shin, Microporous Mesoporous Mater., 2021, 311, 110726 CrossRef CAS
- K. Cendrowski, P. Skumial, P. Spera and E. Mijowska, Mater. Des., 2016, 110, 740–748 CrossRef CAS
- B. Chen, X. J. Wang, Q. F. Zhang, X. Y. Xi, J. J. Cai, H. Qi, S. Shi, J. Wang, D. Yuan and M. Fang, J. Mater. Chem., 2010, 20, 3758–3767 RSC
- M. Vaghayenegar, A. Kermanpur and M. H. Abbasi, Sci. Iran., 2011, 18, 1647–1651 CrossRef CAS
- S. Hajiashrafi and N. M. Kazemi, Heliyon, 2019, 5, e02152 CrossRef CAS PubMed
- A. B. Hartanto, X. Ning, Y. Nakata and T. Okada, Appl. Phys. A, 2004, 78, 299–301 CrossRef CAS
- A. V. Neimark, Y. Z. Lin, P. I. Ravikovitch and M. Thommes, Carbon, 2009, 47, 1617–1628 CrossRef CAS
- P. I. Ravikovitch and A. V. Neimark, Stud. Surf. Sci. Catal., 2007, 160, 9–16 CrossRef CAS
- H. L. Wang, Z. W. Xu, A. Kohandehghan, Z. Li, K. Cui, X. H. Tan, T. J. Stephenson, C. K. King’ondu, C. M. B. Holt, B. C. Olsen, J. K. Tak, D. Harfield, A. O. Anyia and D. Mitlin, ACS Nano, 2013, 7, 5131–5141 CrossRef CAS PubMed
- K. A. Cychosz and M. Thommes, Engineering, 2018, 4, 559–566 CrossRef CAS
- M. Demir and M. Doguscu, ChemistrySelect, 2022, 7, e202104295 CAS
- W. Chaikittisilp, M. Hu, H. J. Wang, H. S. Huang, T. Fujita, K. C. W. Wu, L. C. Chen, Y. Yamauchi and K. Ariga, Chem. Commun., 2012, 48, 7259–7261 RSC
- H. Banda, B. Daffos, S. Périé, Y. Chenavier, L. Dubois, D. Aradilla, S. Pouget, P. Simon, O. Crosnier, P. L. Taberna and F. Duclairoir, Chem. Mater., 2018, 30, 3040–3047 CrossRef CAS
- Y. Dong, S. Zhang, X. Du, S. Hong, S. G. Zhao, Y. X. Chen, X. H. Chen and H. H. Song, Adv. Funct. Mater., 2019, 29, 1901127 CrossRef
- D. He, Y. Gao, Z. P. Wang, Y. C. Yao, L. Wu, J. Zhang, Z. H. Huang and M. X. Wang, J. Colloid Interface Sci., 2021, 581, 238–250 CrossRef CAS PubMed
- B. Liu, H. Shioyama, H. L. Jiang, X. B. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS
- B. Liu, Y. J. Liu, H. B. Chen, M. Yang and H. M. Li, J. Power Sources, 2017, 341, 309–317 CrossRef CAS
- Z. X. Li, B. L. Yang, K. Y. Zou, L. G. Kong, M. L. Yue and H. H. Duan, Carbon, 2019, 144, 540–548 CrossRef CAS
- E. Raymundo-Pinero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2016, 44, 2498–2507 CrossRef
- Q. Zhang, C. Cai, J. W. Qin and B. Q. Wei, Nano Energy, 2014, 4, 14–22 CrossRef CAS
- R. Y. Yan, T. Heil, V. Presser, R. Walczak, M. Antonietti and M. Oschatz, Adv. Sustainable Syst., 2018, 2, 1700128 CrossRef
- L. Qie, W. M. Chen, H. H. Xu, X. Q. Xiong, Y. Jiang, F. Zou, X. L. Hu, Y. Xin, Z. L. Zhang and Y. H. Huang, Energy Environ. Sci., 2013, 6, 2497–2504 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02451b |
| This journal is © The Royal Society of Chemistry 2023 |