
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem addition of nucleophilic and electrophilic reagents to vinyl phosphinates: the stereoselective formation of organophosphorus compounds with congested tertiary carbons†
Zhu Lin,
De-Hua Zhai,
Yong-Ming Sun,
Hong-Xing Zheng,
Qiang Li ,
Yan-Lan Wang,
Jing-Hong Wen* and
Chang-Qiu Zhao*
,
Yan-Lan Wang,
Jing-Hong Wen* and
Chang-Qiu Zhao*
College of Chemistry and Chemical Engineering, Liaocheng University, No. 1, Hunan Road, Liaocheng, Shandong 252059, China. E-mail: literabc@hotmail.com; wenjing-hong@lcu.edu.cn
First published on 9th May 2023
Abstract
Carbon anions formed via the addition of Grignard reagents to SP-vinyl phosphinates were modified with electrophilic reagents to afford organophosphorus compounds with diverse carbon skeletons. The electrophiles included acids, aldehydes, epoxy groups, chalcogens and alkyl halides. When alkyl halides were used, bis-alkylated products were afforded. Substitution reactions or polymerization occurred when the reaction was applied to vinyl phosphine oxides.
Organophosphorus compounds play important roles in the fields of medicine,1 biology,2 materials3 and synthetic chemistry.4 Because the performance of these compounds is highly dependent on their carbon skeleton, the construction of C–P bonds is of great significance in modern organic synthesis.5
Traditionally, C–P bonds were constructed using various phosphorus sources. For example, P–Cl species react with organometallic reagents to form C–P bonds.6 Electrophilic organohalides and PIII species undergo rearrangements also affording C–P bonds (Michaelis–Arbuzov reactions).7 The alkylation of P–H bonds with various electrophilic species forms C–P bonds, and these reactions are promoted by bases (Michaelis–Becker reactions)8 or transition metals.9 The carbon skeletons of organophosphorus compounds can be diversified via these various modifications.10
The formation of C–C bonds is an important theme in organic synthesis. One such effective approach, nucleophilic addition to unsaturated C–C bonds (Michael addition), has been extensively studied and utilized.11 Vinyl phosphorus can also be applied to this addition.10c–e However, apart from a few bifunctionalization or carbometallation reactions, the simultaneous or tandem formation of two C–C bonds by means of an addition has been quite limited (Chart 1).12
 | ||
| Chart 1 Comparison of reported reactions to our current work. | ||
Herein, we present a novel method to construct C–C bonds where nucleophilic and electrophilic alkyl groups are introduced via a tandem process onto vinyl phosphorus. The electrophilic reagents include, but are not limited to, alkyl halides. A carbon anion was proposed as an intermediate, and P,C-stereogenic phosphinates with congested tertiary carbons were obtained, in many cases in optically pure states.
The research began with SP-menthyl phenylphosphinate (SP-3), a compound with a vinyl link on a chiral phosphorus atom. SP-3 was obtained from epimeric menthyl phosphinate 1 and acetophenone. Two of four possible diastereomers of 2 were obtained via recrystallization, which were converted to SP-3a and SP-3b via chlorination and elimination (Scheme 1).
 | ||
| Scheme 1 The preparation of optically pure SP-3. | ||
When the reaction of SP-3a with ethyl magnesium bromide 4b was quenched with acetic acid 5, two major signals at 40.0 and 38.6 ppm were observed in the 31P NMR spectrum, in a ratio of 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :62 (run 1 of Scheme 2). The two signals were assigned as the two diastereomers 6ba/6ba′, respectively, as derived from the chiral α-carbon. 1H NMR spectrum of isolated 6ba confirmed the structure. The menthoxyl group of SP-3a was not replaced by the Grignard reagent.13
:62 (run 1 of Scheme 2). The two signals were assigned as the two diastereomers 6ba/6ba′, respectively, as derived from the chiral α-carbon. 1H NMR spectrum of isolated 6ba confirmed the structure. The menthoxyl group of SP-3a was not replaced by the Grignard reagent.13
 | ||
| Scheme 2 Examination of the addition of ethyl magnesium bromide to SP-3a. | ||
Addition of 4b at −80 °C did not improve the ratio of 6ba/6ba′, and led to the observation of multiple signals in the range 43 to 39 ppm (run 2). The signals were ascribed to 7a a species that has two indefinite chiral carbon atoms, and hence theoretically gives four diastereomers and eight P-signals (Scheme 2). The integration on the 1H NMR spectrum supported the structure of 7a. When the reaction was carried out at −60 °C for 8 hours, 7a was detected as the major product (run 3).
The formation of 7a demonstrated that α-carbon anion 8 was generated as an intermediate (vide infra, Scheme 5), and was converted to 6ba when quenched with 5a. It was supposed that the addition of 4a to SP-3a became slow at low temperature, and unconsumed SP-3a reacted with 8 to afford 7a (run 3).
The formation of 6 and 7 inspired us to modify 8 with various electrophilic reagents (El), such as aldehydes, epoxy groups, and chalcogen compounds (Table 1). When paraformaldehyde (5b) was added to the mixture of SP-3a and 4b, hydroxyl-substituted 6bb was obtained in a 95% yield and 56:44 dr (entry 2 of Table 1). Isolated 6bb/6bb′ (in 64:36 dr) resulted in a distinct 31P NMR spectrum whilst integration of the 1H NMR spectrum supported the structures of the two diastereomers. The existence of a stereogenic P-atom led to a complicated 13C NMR spectrum.
|
||||
|---|---|---|---|---|
| Entry | R | El, 5 | Yield of 6%a (dr) | Isolated 6 yield %a (dr) |
| a Typical procedure: to a solution of SP-3a (80 mg, 0.192 mmol) in THF (1 mL), was added the solution of Grignard reagent (384 μL, 1 M in THF, 0.384 mmol). After stirring at room temperature for 30 min, El (1.5 eq. to SP-3a) was added. The reaction was quenched with aqueous ammonium chloride after stirring at r.t. for 3 h. The yield and dr were estimated from the 31P{1H} NMR spectrum, and dr was assigned as SP-6/RP-6′.b The stripe of the compounds in the preparative TLC gathered in a narrow range. | ||||
| 1 | Et | HOAc | 95 (38:62) |
6ba, 33 (97:3)b |
| 6ba′, 37 (26:74)b |
||||
| 2 | (CH2O)n | 95 (56:44) |
6bb, 55 (64:36) |
|
| 3 | 2-Me-oxirane | 87 (23:31:26:20) |
6bc, 17 (>99:1)b |
|
| 6bc′, 46 (35:65) |
||||
| 4 | Me2S2 | 89 (54:46) |
6bd, 62 (48:52) |
|
| 5 | Ph2S2 | 75 (55:45) |
6be, 56 (54:46) |
|
| 6 | iPr | HOAc | 93 (49:51) |
6ca, 21 (56:44)b |
| 7 | Me2S2 | 73 (61:39) |
6cd, 50 (48:52) |
|
| 8 | Ph2S2 | 75 (65:35) |
6ce, 53 (71:29) |
|
| 9 | Ph | HOAc | 88 (52:48) |
6da, 38 (98:2)b |
| 6da′, 35 (20:80)b |
||||
| 10 | (CH2O)n | 89 (42:58) |
6db, 70 (36:64) |
|
| 11 | 2-Me-oxirane | 95 (6:41:42:11) |
6dc, 67 (5:81:5:9) |
|
| 12 | Me2S2 | 95 (81:19) |
6dd, 68 (79:21) |
|
| 13 | Ph2S2 | 91 (73:27) |
6de, 68 (75:25) |
|

When racemic 2-methyloxirane (5c) was used, four diastereomers of 6bc were generated, with an isolated P-signal at 46.0 ppm (entry 3). The diastereomeric ratio of 6 could be improved after isolation.14 Sulfur could be introduced on the α-carbon by means of dimethyl disulfide (5d) and diphenyl disulfide (5e), affording 6bd and 6be, respectively, in excellent yields (entries 4 and 5).
Secondary alkyl Grignard reagent 4c was also added to SP-3a. When quenched with 5a, 6ca was afforded in a 93% yield and 49:51 dr (entry 6). Alkylthio-substituted 6cd and 6ce were formed from 5d or 5e, respectively (entries 7 and 8). When phenyl magnesium bromide 4d was used, the modifications with 5a–e afforded 6da–de (entries 9–13). The reaction with 2-methyl-oxirane afforded four diastereomers that were not separated.
The structures of Sα-C-6db and Sα-C-6ca were confirmed using X-ray diffraction (Fig. 1). Given that Sα-C-6ca resulted in the upfield signal in the 31P NMR spectrum (39.0 ppm), the signal located downfield (40.5 ppm) was assigned to Rα-C-6ca. The structures of the two diastereomers of 6 were inferred based on that of 6ca.
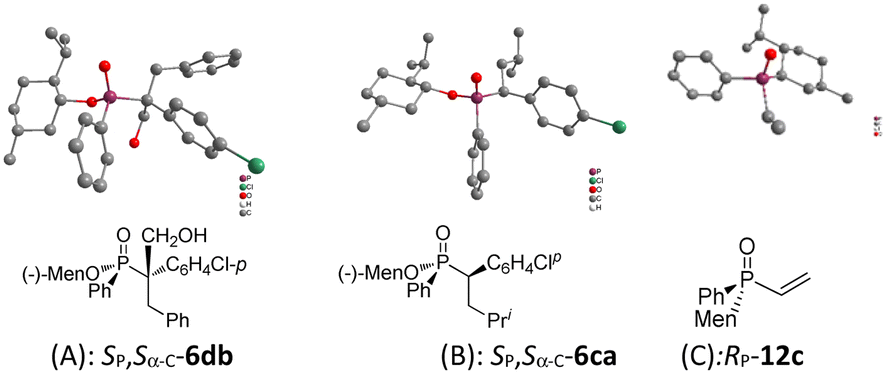 | ||
| Fig. 1 The structures of X-ray diffraction. (A) SP,Sα-C-6db; (B) SP,Sα-C-6ca; (C) RP-12c. | ||
It was interesting that alkyl halides 9 could be used as the El to modify 8, affording the 1,2-bisalkylated compounds 10. After SP-3a was mixed with 4b, methyl iodide 9a was added, and 10aba was afforded in a 78% yield and 60:40 dr, as confirmed from the signals at 43.4 and 40.0 ppm in the 31P NMR spectrum (entry 2 of Table 2). The former diastereomer was isolated and confirmed using 1H NMR spectroscopy when methyl magnesium bromide 4a was used and the mixture was treated with ethyl bromide 9b, 10aab, which has two α-ethyl groups and a symmetric α-carbon, was obtained as a single isomer (entry 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R on Ar | R1 | R2 | Yield of 10% | dra | Isolated yield of 10% (dr) |
| a The yield and dr were estimated from the 31P{1H} NMR spectrum, and the dr was determined form the ratio of the two diastereomers with at upfield and downfield signals in the 31P-NMR spectrum.b Alkyl halide 9 and SP-3 were mixed, then the Grignard reagent was added.c The stripe of the compounds in the preparative TLC gathered in a narrow range.d PhMgBr was added at 0 °C to a solution of the starting material, then BnCl was added at room temperature. Entry 10 gave a similar result to entry 9 and could not be isolated.e The product could not be separated from 6 (direct acidification with acetic acid). | ||||||
| 1 | Cl | Me | Me | 63 | NA | 10aab, 50 |
| 2 | Cl | Et | H | 78 | 60:40 |
10aba, 58 (99:1) |
| 3 | Cl | Et | Me | 77 | 56:44 |
10abb, 58 (>99:1)b |
| 4 | Cl | Et | Et | 66 | NA | 10abc, 50 |
| 5 | Cl | Et | nPr | 47 | 47:53 |
10abd, 35 (49:51) |
| 6 | Cl | iPr | H | 87 | 62:38 |
10aca, 13 (99:1)c |
| 7 | Cl | Ph | H | 76 | 62:38 |
10ada, 60 (>99:1) |
| 8 | Cl | Ph | Me | 57 | 72:28 |
10adb, 40 (84:16) |
| 9 | Cl | Ph | Ph | 79 | NA | 10ade, 63b |
| 10 | Cl | Ph | Ph | 66 | NA | 10aded |
| 11 | H | Me | Me | 43 | NA | 10bab, 33 |
| 12 | H | Et | H | 48 | 58:42 |
10bbae |
| 13 | H | Et | Me | 94 | 67:33 |
10bbb, 52 (59:41) |
| 23 (88:12)c |
||||||
| 14 | H | Et | Et | 77 | NA | 10bbc, 61 |
| 15 | H | Ph | Ph | 68 | NA | 10bde, 50 |
| 16 | OMe | Et | H | 77 | >99:1 |
10cba, 58 (>99:1) |
| 17 | OMe | Et | Me | 23 | 65:35 |
10cbb, 5 (96:4)c |
| 13 (67:33)c |
||||||
| 18 | OMe | Et | nPr | 42 | 43:57 |
10cbd, 34 (40:60)c |
| 19 | OMe | Ph | Ph | 90 | NA | 10cde, 80 |

The reaction of SP-3a with 4b and several primary alkyl halides (9b to 9d) afforded 10abb to 10abd (entries 3 to 5). After isolation, one of the two diastereomers of 10abb was obtained whilst 10abc was formed as a single isomer.
The Nu-El pair of 4c/9a and SP-3a afforded 10aca in an 87% yield, and a single diastereomer was also obtained after isolation (entry 6). Phenyl Grignard 4d, 9a–9b similarly afforded 10ada–10adb, respectively (entries 7 and 8). When benzyl chloride 9e was used, 10ade was obtained in a similar yield, irrespective of the order of addition (entries 9 and 10).
Similarly to SP-3a, SP-3b reacted with the Nu-El pair of 4a/9b, affording 10bab as a single isomer (entry 11). The pair of 4b/9a gave crude 10bba that contained a by-product of 6 (entry 12). The isolation of 10bbb gave a different variation of two diastereomers (entry 13). 10bbc and 10bde, both with symmetric α-carbons, were smoothly obtained as single isomers (entries 14–15).
p-Methoxyl substituted SP-3c was obtained via chlorination of SP-2c and elimination with LiH/DMSO (refer to Scheme 1). It was interesting that SP-3c and the Nu-El pair of 4b/9a afforded 10cba in an excellent dr (entry 16). When 9b and 9d were used, both 10cbb and 10cbd were obtained in unsatisfactory yields (entries 17 and 18). α-Symmetric 10cde was obtained in a good yield (entry 19).
SP-3d has a 2-propenyl group and was prepared via addition of RP-1 to acetone, followed by chlorination and elimination (refer to Scheme 1). When SP-3d was mixed with a Grignard reagent, multiple signals around 45.0 ppm were observed in the 31P NMR spectrum, which were assigned as dimer 7 or polymer 11 (Scheme 3). The structure of 7 or 11a was not confirmed. It was supposed that the smaller steric hindrance of the vinyl moiety led to further addition of the carbon anion to SP-3d, to afford the product with multiple signals at 45.0 ppm.
 | ||
| Scheme 3 The polymerization of 2-propenyl phosphinate SP-3d with 4b. | ||
Vinyl phosphine oxide 12 was prepared from the reaction of phosphine chloride with a vinyl Grignard reagent (Scheme 4). Optically pure RP-12c was obtained via recrystallization, and its structure was confirmed using X-ray diffraction (Fig. 1). When 12b was stirred with 4b at room temperature, a signal at 21 ppm, accompanied with multiple peaks around 36–38 ppm, were observed in the 31P NMR spectrum. The signal at 21 ppm was ascribed to diphenyl phosphine oxide 13. After treatment with methyl iodide, 13 was converted to diphenyl methylphosphine oxide 14a. The multiple peaks were assigned as polymerized 11b (Scheme 3, part B of Scheme 4). The reaction of 12a similarly formed 13 and 14.
 | ||
| Scheme 4 The preparation of 12 and their reactions with Grignard reagents/alkyl halides. | ||
When 12b was added to a mixture of 4b and 9b, the formation of 11b was suppressed and 14b was afforded as major product. Meanwhile a small signal at 36 ppm was assigned as 15a, whose molecular ion peak could be observed in the mass spectrum (vide infra).
The reaction of 12c with 4b/9b also gave multiple signals. After isolation, dimer 16 was obtained as a mixture of diastereomers (part C of Scheme 4). The reaction of 12d with 4b did not occur, even at elevated temperature.
The reactivity shown in Scheme 4 is proposed to proceed via the coordination of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O to magnesium to form 17, which is converted to carbon anion 8 or a P-anion (as 13) (Scheme 5). When a p-tolyl Grignard reagent reacted with 12b, the vinyl-H signals of 21 were not detected, which rules out the generation of 13 via route A. Simultaneously, a yellow alkynyl copper solid was obtained when the reaction mixture was treated with CuI/ammonia, the structure of which was confirmed from the alkynyl signal (2254 cm−1) in the infrared spectrum. The results indicated that the elimination of a β-proton afforded 22, as well as 13 (route B). Alternatively, addition of a Nu to the β-carbon formed 20, which was converted to 8 then to 15 (route C). The molecular ion peak of 15b (R = p-tolyl) at 334.2 could be observed in the mass spectrum.
O to magnesium to form 17, which is converted to carbon anion 8 or a P-anion (as 13) (Scheme 5). When a p-tolyl Grignard reagent reacted with 12b, the vinyl-H signals of 21 were not detected, which rules out the generation of 13 via route A. Simultaneously, a yellow alkynyl copper solid was obtained when the reaction mixture was treated with CuI/ammonia, the structure of which was confirmed from the alkynyl signal (2254 cm−1) in the infrared spectrum. The results indicated that the elimination of a β-proton afforded 22, as well as 13 (route B). Alternatively, addition of a Nu to the β-carbon formed 20, which was converted to 8 then to 15 (route C). The molecular ion peak of 15b (R = p-tolyl) at 334.2 could be observed in the mass spectrum.
 | ||
| Scheme 5 Proposed reaction of vinyl phosphine oxide 12 with a Grignard reagent. | ||
Conclusions
In summary, the addition of Grignard reagents to the vinyl bonds of a series of SP-menthyl phenylphosphinates (3) formed carbon anion intermediates (8), which were modified with various electrophilic reagents (El) to afford organophosphorus compounds with diverse carbon skeletons. In many cases, single stereoisomers were obtained. The El reagents (5) included acids, aldehydes, epoxy groups, and chalcogen compounds. When alkyl halide 9 was used as the El, bis-alkylated product 10 was obtained via a tandem addition process. Aliphatic and aromatic alkyl groups could be introduced as nucleophiles, whilst primary alkyl and benzyl groups were introduced as the El. For vinyl diphenylphosphine oxides, a replacement of the vinyl group occurred via base-promoted elimination. Vinyl menthyl phenylphosphine oxide tended to form polymerized products. This research provides a novel approach to construct organophosphorus compounds with diverse carbon skeletons, which it is hoped will have important applications in the fields of organophosphorus chemistry and asymmetric catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the Natural Science Foundation of China (grant no. 21802062, 21901097) and the Natural Science Foundation of Shandong Province (grant no. ZRZR2021MB072 and ZR2020QB020).Notes and references
- (a) M. Sawa, T. Kiyoi, K. Kurokawa, H. Kumihara, M. Yamamoto, T. Miyasaka, Y. Ito, R. Hirayama, T. Inoue, Y. Kirii, E. Nishiwaki, H. Ohmoto, Y. Maeda, E. Ishibushi, Y. Inoue, K. Yoshino and H. Kondo, J. Med. Chem., 2002, 454, 919–929 CrossRef PubMed; (b) X. Chen, D. J. Kopecky, J. Mihalic, S. Jeffries, X. Min, J. Heath, J. Deignan, S. Lai, Z. Fu and C. Guimaraes, J. Med. Chem., 2012, 55, 3837–3851 CrossRef CAS PubMed.
- (a) L. Bialy and H. Waldmann, Angew. Chem., Int. Ed., 2005, 44, 3814–3839 CrossRef CAS PubMed; (b) A. George and A. Veis, Chem. Rev., 2008, 108, 4670–4693 CrossRef CAS PubMed; (c) R. K. Haynes, W. A. Loughlin and T. W. Hambley, J. Org. Chem., 1991, 56, 5785–5790 CrossRef CAS; (d) R. K. Haynes, S. C. Vonwiller and T. W. Hambley, J. Org. Chem., 1989, 54, 5162–5170 CrossRef CAS.
- (a) H. R. Allcock, M. A. Hofmann, C. M. Ambler and R. V. Morford, Macromolecules, 2002, 35, 3484–3489 CrossRef CAS; (b) H. Onouchi, T. Miyagawa, A. Furuko, K. Maeda and E. Yashima, J. Am. Chem. Soc., 2005, 127, 2960–2965 CrossRef CAS; (c) C. Queffelec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef CAS PubMed.
- (a) J. Boutagy and R. Thomas, Chem. Rev., 1974, 74, 87–99 CrossRef CAS; (b) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS; (c) M. Kitamura, M. Tokunaga and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2931–2932 CrossRef CAS.
- (a) C. Baillie and J. Xiao, Curr. Org. Chem., 2003, 7, 477–514 CrossRef CAS; (b) F. M. Tappe, V. T. Trepohl and M. Oestreich, Synthesis, 2010, 2010, 3037–3062 CrossRef; (c) C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed; (d) S. Hore and R. P. Singh, Org. Biomol. Chem., 2022, 20, 498–537 RSC; (e) J. Yang, J. Xiao, Y. Zhou, T. Chen, S. Yin and L. Han, Chin. J. Chem., 2017, 37, 1055–1068 CrossRef CAS; (f) Y. Huang and Q. Chen, Chin. J. Chem. Eng., 2021, 41, 4138–4153 CAS.
- B. Zhang, C. G. Daniliuc and A. Studer, Org. Lett., 2014, 16, 250–253 CrossRef CAS PubMed.
- (a) P. Y. Renard, P. Vayron, E. Leclerc, A. Valleix and C. Mioskowski, Angew. Chem., Int. Ed., 2003, 115, 2491–2494 CrossRef; (b) A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415–430 CrossRef CAS; (c) X. Ma, Q. Xu, H. Li, C. Su, L. Yu, X. Zhang, H. Cao and L.-B. Han, Green Chem., 2018, 20, 3408–3413 RSC.
- (a) K. M. Kem, N. V. Nguyen and D. J. Cross, J. Org. Chem., 1981, 46, 5188–5192 CrossRef CAS; (b) S. Hore and R. P. Singh, Org. Biomol. Chem., 2022, 20, 498–537 RSC.
- (a) T. Hirao, T. Masunaga, N. Yamada, Y. Ohshiro and T. Agawa, Bull. Chem. Soc. Jpn., 1982, 55, 909–913 CrossRef CAS; (b) A. L. Schwan, Chem. Soc. Rev., 2004, 33, 218–224 RSC; (c) H. Rao, Y. Jin, H. Fu, Y. Jiang and Y. A. Zhao, Chem.–Eur. J., 2006, 12, 3636–3646 CrossRef CAS PubMed; (d) F. M. Tappe, V. T. Trepohl and M. Oestreich, Synthesis, 2010, 2010, 3037–3062 CrossRef; (e) I. Beletskaya and M. Kazankova, Russ. J. Org.Chem., 2002, 38, 1391–1430 CrossRef CAS; (f) M. Andaloussi, J. Lindh, J. Sävmarker, P. JR Sjöberg and M. Larhed, Chem.–Eur. J., 2009, 15, 13069–13074 CrossRef CAS PubMed; (g) R. Zhuang, J. Xu, Z. Cai, G. Tang, M. Fang and Y. Zhao, Org. Lett., 2011, 13, 2110–2113 CrossRef CAS PubMed; (h) H.-Y. Zhang, M. Sun, Y.-N. Ma, Q.-P. Tian and S.-D. Yang, Org. Biomol. Chem., 2012, 10, 9627–9633 RSC; (i) Z.-Y. Wang, Q. Guo, S. Xu and K.-K. Wang, Synthesis, 2021, 53, 3683–3698 CrossRef CAS.
- (a) S. Montel, T. Jia and P. J. Walsh, Org. Lett., 2014, 16, 130–133 CrossRef CAS PubMed; (b) S. Montel, L. Raffier, Y. He and P. J. Walsh, Org. Lett., 2014, 16, 1446–1449 CrossRef CAS PubMed; (c) H.-J. Cristau, K. EL Hamad and E. Torreilles, Phosphorus, Sulfur, Silicon Relat. Elem., 1992, 66, 47–58 CrossRef CAS; (d) K. Afarinkia, H. M. Binch and E. De Pascale, Synlett, 2000, 1769–1770 CAS; (e) K. Afarinkia, H. M. Binch and I. Forristal, Synlett, 2000, 1771–1772 CAS.
- B. C. Ranu and S. Banerjee, Org. Lett., 2005, 7, 3049–3052 CrossRef CAS PubMed.
- (a) S. Das, C. G. Daniliuc and A. Studer, Org. Lett., 2016, 18, 5576–5579 CrossRef CAS PubMed; (b) V. Coeffard, A. Desmarchelier, B. Morel, X. Moreau and C. Greck, Org. Lett., 2011, 13, 5778–5781 CrossRef CAS PubMed; (c) S.-Y. Yang, W.-Y. Han, C. He, B.-D. Cui, N.-W. Wan and Y.-Z. Chen, Org. Lett., 2019, 21, 8857–8860 CrossRef CAS PubMed; (d) S. Maejima, E. Yamaguchi and A. Itoh, J. Org. Chem., 2020, 85, 10709–10718 CrossRef CAS PubMed; (e) Q.-F. Bao, M. Li, Y. Xia, Y.-Z. Wang, Z.-Z. Zhou and Y.-M. Liang, Org. Lett., 2021, 23, 1107–1112 CrossRef CAS; (f) X. Ye, B. Xu, J. Sun, L. Dai, Y. Shao, Y. Zhang and J. Chen, J. Org. Chem., 2020, 85, 13004–13014 CrossRef CAS.
- L.-J. Liu, W.-M. Wang, L. Yao, F.-J. Meng, Y.-M. Sun, H. Xu, Z.-Y. Xu, Q. Li, C.-Q. Zhao and L.-B. Han, J. Org. Chem., 2017, 82, 11990–12002 CrossRef CAS PubMed.
- (a) S. Pindi, J. Wu and G. Li, J. Org. Chem., 2013, 78, 4006–4012 CrossRef CAS PubMed; (b) G. An, C. Seifert and G. Li, Org. Biomol. Chem., 2015, 13, 1600–1617 RSC; (c) Z. Y. Zhou, H. Zhang, L. Yao, J. H. Wen, S. Z. Nie and C. Q. Zhao, Chirality, 2016, 28, 132–135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2216572, 2216573 and 2217203. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra02409a |
| This journal is © The Royal Society of Chemistry 2023 |