
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving porous properties of activated carbon from carbon gel by the OTA method†
Panuwat Lawtae *ab,
Shintaroh Nagaishib,
Chaiyot Tangsathitkulchaia,
Shinichiroh Iwamurac and
Shin R. Mukai*c
*ab,
Shintaroh Nagaishib,
Chaiyot Tangsathitkulchaia,
Shinichiroh Iwamurac and
Shin R. Mukai*c
aSchool of Chemical Engineering, Suranaree University of Technology, Nakhon Ratchasima 30000, Thailand. E-mail: pnwlawtae@gmail.com
bGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo 060-8628, Japan
cDivision of Applied Chemistry, Faculty of Engineering, Hokkaido University, Sapporo 060-8628, Japan. E-mail: smukai@eng.hokudai.ac.jp
First published on 9th May 2023
Abstract
High-surface-area microporous–mesoporous carbons were produced from carbon gel by applying the three consecutive steps of air oxidation, thermal treatment, and activation (the OTA method) to the gel. The formation of mesopores occurs both inside and outside the nanoparticles which form the carbon gel, while micropores are predominantly created within the nanoparticles. The OTA method offered a greater increase in pore volume and BET surface area of the resulting activated carbon in comparison with conventional CO2 activation either under the same activation conditions or at the same degree of carbon burn-off. Under the best preparation conditions, the maximum values of micropore volume, mesopore volume, and BET surface area achievable using the OTA method were found to be 1.19 cm3 g−1, 1.81 cm3 g−1, and 2920 m2 g−1, respectively at a 72% carbon burn-off. The larger increase in porous properties of activated carbon gel prepared by the OTA method over those based on conventional activation stems from the effects of the oxidation and heat treatment steps of the OTA method that could produce a large number of reaction sites which lead to efficient pore formation during the following CO2 activation process.
1. Introduction
Continuous technological advancements have evoked efforts to apply various techniques for improving material properties as well as exploring new materials with superior properties. Because of the particular characteristics of the carbon element and the diversity of carbon–carbon bonds, carbon materials offer special benefits and are possibly one of the most versatile materials in terms of properties.1 From the view of controlling material properties, synthetic carbon materials with high purity and controllable chemistry have attracted much attention because of the flexibility in modifying their final properties. Carbon gels are porous carbon materials, which have a hierarchical and easily accessible porous structure. Carbon gels possess predominant properties as a controllable pore structure with a high porosity, low electrical resistivity as well as a high machinability.2 These outstanding properties provide a remarkable potential for the design and tailoring of this material for specific applications such as electric double-layer capacitors,3–5 lithium-ion batteries,6–8 electrodes for fuel cells,9–11 catalyst supports,12–14 gas adsorption,15–17 and water treatment.18–20Pekala and co-workers21–23 were the first to prepare organic gels which can be used as the precursor for carbon gels by applying the sol–gel method in 1989. The most common way to synthesize such organic gels is through the polymerization reaction between resorcinol (R) and formaldehyde (F) with the use of a basic catalyst as the reaction promoter and water as the solvent. Since then, numerous publications on various synthesis and processing conditions for specific applications have appeared in the literature.24–27 However, the RF polymerization reaction is typically a prolonged process that must be carried out over several days. Thus, to reduce the processing time, fast synthesis has been the focal point of several investigations.28–30 Normally, organic gels obtained after the drying step are usually rich in oxygen surface chemistry and have a BET surface area (SBET) of less than 500 m2 g−1.31,32 Once the organic gels have been obtained, the carbonization step must be performed to increase the carbon content so that thermally stable gels called carbon gels can be obtained. Carbonization temperatures between 800–1000 °C are generally applied with a slow heating rate to prevent the excessive shrinkage of the structure.33 Indeed, the obtained carbon gels have a similar mesoporosity to their parent organic gels, but with more developed microporosity in the polymeric nodules which form it, hence the BET surface area is slightly increased to 500–700 m2 g−1. Obviously, the utilization of carbon gels in many applications often requires a relatively large surface area with controlled proportions of micropore and mesopore volumes.19 The physical activation process is one of the most widely used methods to increase surface area due to its simplicity and low cost. Activation processes can increase surface area to values higher than 2000 m2 g−1,3 which results mainly from the development of microporosity in the carbon gels. Narrow mesopores can also be introduced during the activation process, but introduction usually occurs at a relatively high carbon burn-off through the coalescence of some small pores developed during gasification.34
Recently, a new physical activation method for activated carbon synthesis has been proposed that is capable of producing a relatively large amount of micropores and mesopores in carbons through a process of modified physical activation with CO2, being referred to as the OTA method.35 This method consists of three consecutive steps: (1) air oxidation of an initial microporous carbon to a relatively low burn-off level for the introduction of additional surface oxygen functional groups, (2) thermal destruction of the existing functional groups in an inert atmosphere at a high temperature to enhance the surface reactivity through bond disruption, and (3) CO2 activation of the treated carbon to widen its original micropores as well as to form new micropores. Overall, the advantages of this innovative method are that it is easy to operate, eco-friendly, low cost, and is not associated with any liquid oxidizing agent, since the process is conducted using only air, inert gas, and CO2. The maximum values of the porous properties of activated carbon obtained from longan seed biomass through three treatment cycles were found to be 0.600 cm3 g−1 of micropore volume, 0.474 cm3 g−1 of mesopore volume, and 1770 cm2 g−1 of BET surface area. This activated carbon was proven to be applicable to the removal of methylene blue from an aqueous solution, both in view of adsorption efficiency and capacity.36 In this work, the OTA method was applied to improve the porous properties of activated carbon produced from carbon gel.
Therefore, the present work aims to prepare high-surface-area mesoporous carbons from organic gels by applying the OTA method. This work focused on investigating the effect of the surface reactivity of carbon prior to CO2 activation on improving the porous properties of the activated carbon produced via the OTA method. Surface reactivity was controlled by adjusting conditions of the air oxidation/thermal treatment steps, as well as varying the porous properties of the initial carbon. The underlying mechanism of pore development by the OTA method for the carbon gel and experimental evidence which supports the mechanism were presented.
2. Experimental section
2.1. Materials
Resorcinol (R, 99.0+%), aqueous formaldehyde solution (F, 37.0 wt%, containing 5–10% of methanol as a stabilizer), and sodium carbonate (C, 99.8%) were purchased from Wako Pure Chemical Industries, Ltd. N2 gas of high purity grade (UHP, 99.99%), CO2 gas of high purity grade (99.99%), and air (99.99%) were supplied in gas cylinders by Air Water Inc. (Japan).2.2. Carbon gel preparation
An organic xerogel was prepared through the sol–gel polymerization of resorcinol (R) and formaldehyde (F) in water (W), using sodium carbonate (C) as the catalyst. The preparation procedure described by Pekala and co-workers was adopted.21,22 Briefly, the molar ratios of R/C, R/F, and the concentration of R were 1000 mol mol−1, 0.5 mol mol−1, and 0.5 g mL−1, respectively. All precursors were mixed by magnetic stirring and first kept under a constant temperature of 30 °C for 26 h, and then kept at 85 °C for 48 h in an electric oven to complete the gelation and curing stages. The resulting cross-linked gel was then dried at 150 °C for 24 h to obtain the xerogel product. After drying, the xerogel was carbonized in a quartz tube reactor (inner diameter: 50 mm, length: 1000 mm) that was inserted in a vertical electric tube furnace (Asahi Rika Co., Ltd., ARF-30K, Japan). The xerogel was heated in a stream of N2 (100 cm3 min−1) from room temperature to 250 °C at a heating rate of 3.75 °C min−1, followed by heat treatment at 250 °C for 2 h, then was subsequently heated up to 800 °C at a heating rate of 4.17 °C min−1, and held at 800 °C for 2 h. Finally, the produced carbon gel was kept in a constant-temperature vacuum dryer (Tokyo Rikakikai Co., Ltd., VOS-210C) for subsequent experiments.2.3. Activated carbon production
High surface area mesoporous carbons were prepared by applying the OTA method35 to the carbon gel. Briefly, the derived carbon gels were oxidized in a quartz tube reactor by heating them in an air stream (50 cm3 min−1) from room temperature to the required oxidation temperature of 350 °C and held at this temperature for 6 h. The purpose of this step was to create additional oxygen functional groups within the carbons. Then, the oxidized carbon was heated at 1000 °C for 1 h under a flow of N2 (50 cm3 min−1) to remove oxygen functional groups. By this high-temperature thermal treatment, the surface reactivity of the treated carbons was expected to increase due to the production of a large number of unpaired electrons caused by the thermal destruction of the bonding of the functional groups. After that, the sample was activated at 1000 °C for 1 h under a flow of CO2 (50 cm3 min−1) which resulted in the production of large amounts of mesopores and micropores in the gel matrix. The activated carbon prepared from the carbon gel by the OTA method was denoted as CG-OTAx, where CG stands for carbon gel, and the symbol x represents the activation time varying from 1–3 h. The activated carbon prepared by the conventional activation method using CO2 was denoted as ACx, where AC stands for activated carbon derived under the activation temperature of 1000 °C, and the symbol x also represents the activation time (1–4 h). The carbon burn-off for the activation step was calculated by eqn (1), where Wi and Wf denote the weight of carbon gels before and after the activation process, respectively.
 | (1) |
2.4. Characterization
The porous properties of the derived carbon gels and activated carbons were determined from their N2 adsorption isotherms measured at −196 °C using an adsorption analyzer (BEL Japan, BELSORP-mini). The BET surface area (SBET) was calculated from the N2 adsorption isotherm data by applying the Brunauer–Emmett–Teller (BET) equation.37 The micropore volume (Vmi) was determined from the Dubinin–Radushkevich equation.38 Using Vmi and total pore volume calculated at the relative pressure (P/P0) of 0.96 (V0.96), the mesopore volume (Vme = V0.96 − Vmi) was then obtained. The total pore volume (VT) was estimated from the amount of N2 gas adsorbed at P/P0 of 0.99 by converting it to the corresponding volume of N2 in the liquid state at −196 °C. The mesopore size distributions were determined by applying the Dollimore–Heal (DH) method.39The surface functional groups of the carbon samples were analyzed by the TPD method. The sample was weighed and loaded into a test-tube-shaped quartz reactor and then heated up under vacuum at a heating rate of 10 °C min−1. The gases emitted from the samples were then continuously analyzed by a quadrupole mass spectrometer (Qulee MGM-102, Ulvac). The desorption rates of the emitted gases were calculated from the area under the curves of each gas in the TPD spectra. The amount of each type of oxygen-containing functional group was determined through peak separation of the TPD spectra of CO2 and CO, as described in the ESI.†
In addition, a thermogravimetric analyzer (NETZSCH STA 2500, Japan) was applied to determine the reactivity of the mesoporous carbons toward the CO2 gasification reaction. The carbon sample was loaded into a platinum crucible (70 μL capacity) of the thermogravimetric analyzer and heated from room temperature to 1000 °C at a heating rate of 20 °C min−1 under a constant flow of N2 (10 cm3 min−1). When the final gasification temperature was reached, the gas flow was switched from N2 to CO2 (10 cm3 min−1), and the sample was held at this temperature for 1 h. The sample weight loss via the gasification reaction was continuously recorded as a function of time and the data collected were used for the computation of carbon reactivity. The thermal decomposition behavior of the xerogel precursor was also studied using the thermogravimetric analyzer (NETZSCH STA 2500, Japan) by non-isothermal heating from room temperature to the final temperature of 1000 °C, using a heating rate of 5 °C min−1 under a constant flow of N2 (10 cm3 min−1).
3. Results and discussions
3.1. Thermal decomposition behavior of precursor organic xerogels
Fig. 1 shows the TG curve recorded during the non-isothermal heating of the xerogel precursor under an N2 flow (10 cm3 min−1) in the TGA from room temperature to the final temperature of 1000 °C, using the heating rate of 5 °C min−1. Weight loss of about 0.5 wt% was observed when the sample was heated from room temperature to about 100 °C, and this was attributed mainly to the release of residual moisture remaining in the precursor. Then, significant weight loss started to occur, which continued with the increase in pyrolysis temperature to about 750 °C. Two peaks, one at around 380 °C and the other at around 540 °C, occurred in the DTG curve derived from the TG curve, showing the main pyrolysis decomposition of the xerogels progressing over the range between these two temperatures. The first peak probably corresponds to the elimination of H2O formed from the condensation of OH groups, while the second peak could correspond to the release of CO2 and CH4 or other organic molecules as well as the desorption of adsorbed organic compounds.31 The second region of the TG curve for the temperature above 750 °C showed a slow decrease in the remaining weight and finally approached a constant solid carbon yield of about 55 wt%. These results are consistent with the findings of other investigators.40–42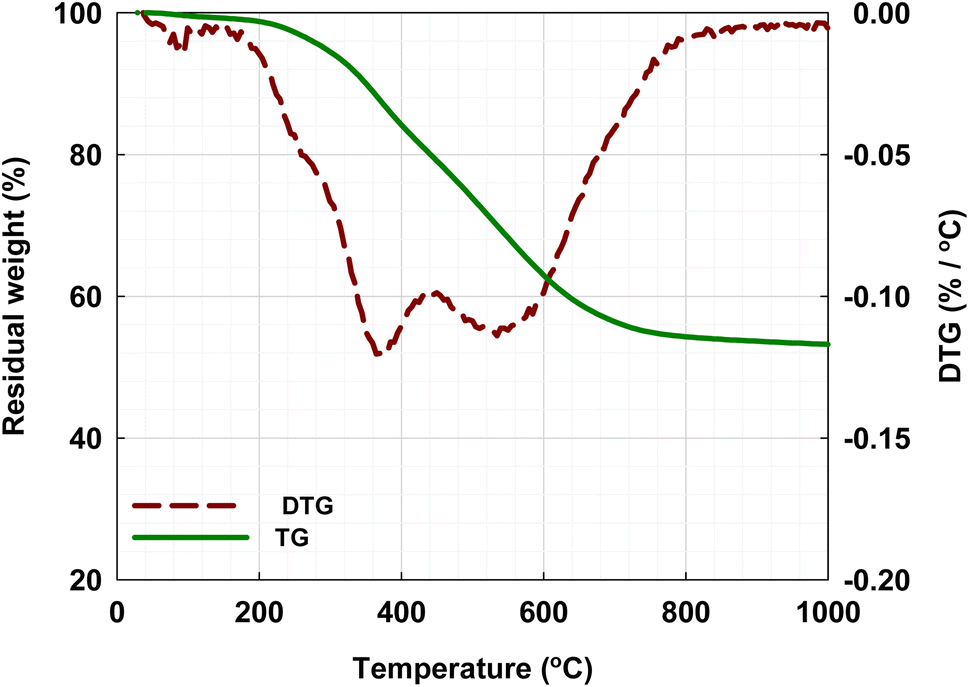 | ||
| Fig. 1 The residual weight (TG) and the weight loss rate (DTG) curves for non-isothermal pyrolysis in N2 of the organic xerogels using a thermogravimetric analyzer (TGA). | ||
3.2. Porous properties of prepared activated carbons
A mesoporous carbon gel was synthesized at a high R/C ratio of 1000 mol mol−1. Typically, the use of a low concentration of the catalyst would lead to the formation of large resorcinol–formaldehyde (RF) particles leading eventually to the formation of a network akin to a string-of-pearls,43 which has interparticle spaces corresponding to mesopores, while micropores are formed within the precursor RF nanoparticles. Fig. 2 shows the N2 adsorption isotherms at −196 °C for the activated carbons prepared by the conventional activation method and the OTA method. All samples exhibited Type I adsorption isotherms with a sharp rise at extra-low relative pressures, indicating the presence of abundant micropores.44 The higher relative pressure region was characterized by a combination of Type I and Type IV isotherms, indicating a typical microporous/mesoporous structure.45,46 Additionally, the hysteresis loop shape resembled Type H2(b) of the IUPAC classification of hysteresis loops,47 which indicates the presence of interconnected networks of pores with similar shapes in the adsorbent. | ||
| Fig. 2 N2 adsorption isotherms (−196 °C) of mesoporous activated carbons prepared by (A) the conventional activation method (AC series) and (B) the OTA method (OTA series) at the same activation temperature of 1000 °C. | ||
The N2 isotherms in Fig. 2B indicate that the OTA samples adsorbed larger amounts of N2 when compared with the AC samples (Fig. 2A) prepared under the same activation conditions of time and temperature or activated to a comparable carbon burn-off. These results imply that the OTA method offers an increase in the number of active sites for the CO2 gasification reaction resulting in improved porous properties under comparable activation conditions. The formation and destruction of oxygen functional groups prior to CO2 gasification lead to a significant increase in carbon reactivity due to the increasing number of unpaired electrons generated by bond disruption.35 The carbon reactivity for CO2 gasification will be discussed in Section 3.5.
Carbon xerogels have a hierarchical porous structure, which is a unique porous structure that is completely distinct from most porous carbons. Due to their polymeric structure of interconnected nodules, the carbon xerogel essentially includes both micropores and mesopores. Basically, the interparticle spaces of the carbon gel form its mesoporous structure which is relatively stable even when a subsequent activation process is applied.48 On the other hand, the activation process is directly responsible for the generation of micropores within the nanoparticles which form the carbon. Table 1 compares the porous properties of activated carbons prepared from the carbon gel by the conventional method and by the OTA method. If we consider first the results of activated carbons produced by the conventional activation method (AC1–AC4), the BET surface area, the micropore volume, the mesopore volume, and the total pore volume increased with the increase in activation time from 1 to 4 h. Concerning the effect of gasification time on the development of activated gel surface area, it was found from Table 1 that the rate of increase of surface area for both the AC series and the OTA series tended to decrease with time. This means that the surface area will finally reach a constant value at a limiting gasification time. We fitted the data in Table 1 with a second-order polynomial function and found that the limiting times were approximately 4.96 and 3 hours, respectively, for the AC and OTA sample series. Therefore, the carbon gel burn-off will be less than 100% even if a very long gasification time is used. This could occur because only part of the total surface area was available for gasification since the carbon gel was gasified in the packed mode in a tube reactor, thus resulting in the presence of a dead spot (inaccessible surface area) at the point of particle contact. If, in another case, all the particle surface area is available for gasification, the increase in the gasification time may not be able to gasify the carbon to the condition of complete conversion. This could occur if a certain number of reaction sites on the carbon surface has a lower energy level than the activation energy of the reaction. Furthermore, the mesopore volume was higher than the micropore volume for all activation times which emphasizes the unique particle-network structure of the carbon gel. By increasing the activation time, the carbon burn-off increased from 24.5 to 89.0% which results from the removal of more carbon atoms through the CO2 gasification reaction leading to the improvement of porous properties. AC4 prepared by the conventional activation had the maximum BET surface area of 2160 m2 g−1 with a mesopore volume of 1.292 cm3 g−1 corresponding to about 58.7% of the total pore volume.
| Sample name | SBET (m2 g−1) | Vmi (cm3 g−1) (%) | Vme (cm3 g−1) (%) | VT (cm3 g−1) | Burn-off (%) | Yield (%) |
|---|---|---|---|---|---|---|
| CG | 632 | 0.254 (27.5) | 0.657 (71.3) | 0.922 | — | — |
| AC1 | 1120 | 0.447 (37.5) | 0.735 (61.7) | 1.19 | 24.5 | 75.5 |
| AC2 | 1700 | 0.677 (39.1) | 1.04 (59.9) | 1.73 | 51.9 | 48.1 |
| AC3 | 1990 | 0.818 (40.8) | 1.17 (58.2) | 2.01 | 70.1 | 29.9 |
| AC4 | 2160 | 0.879 (40.0) | 1.29 (58.7) | 2.20 | 89.0 | 11.0 |
| CG-OTA1 | 1750 | 0.719 (40.9) | 1.03 (58.5) | 1.76 | 48.3 | 51.7 |
| CG-OTA2 | 2310 | 0.925 (42.7) | 1.22 (56.5) | 2.17 | 67.1 | 32.9 |
| CG-OTA3 | 2540 | 0.987 (41.2) | 1.38 (57.4) | 2.40 | 86.9 | 13.1 |
Next, the porous properties of activated carbons produced by the OTA method (CG-OTA1–3) are presented. It was noticed that the activated carbons from the OTA method showed remarkably higher porous properties, that is, micropore volume, mesopore volume, and BET surface area for the same activation time as compared to those obtained through conventional activation, for example, by comparing sample AC1 with CG-OTA1, AC2 with CG-OTA2 or even for the same % carbon burn-off (see Fig. 3). The OTA method using 3 h of activation time produced activated carbon (sample CG-OTA3) with the highest porous properties of 2540 m2 g−1 BET surface area, 0.987 cm3 g−1 of micropore volume (41.2%), and 1.38 cm3 g−1 of mesopore volume (57.4%). These results clearly illustrate that under the same activation conditions (time and temperature), the OTA method was able to remove more carbon atoms via the CO2 gasification reaction than the conventional method. Therefore, this finding strongly indicates that the introduction of the oxidation and thermal treatment steps prior to the activation step could produce a larger number of reactive sites that favor pore development in the subsequent gasification reaction. The OTA method is an effective means for increasing both the micropore volume and mesopore volume of activated carbons derived from carbon gels.
 | ||
| Fig. 3 Effect of carbon burn-off on the BET surface area of carbon products by the OTA and conventional preparation methods. | ||
Now, consider only the development of mesopore size in the size distributions of activated carbons determined by the DH method. The results are shown in Fig. 4. For the sake of discussion, the developed mesopores are divided into four size ranges, that is, 2–5, 5–10, 10–20, and 20–50 nm. In terms of pore volume, the presence of macropores (pores with sizes larger than 50 nm) is considered insignificant. First, for activated carbons of both the AC series (see Fig. 4A) and the OTA series (Fig. 4B), each sample (for each activation time) showed approximately a normal pore size distribution with a peak occurring at a pore size in the range of 10–20 nm. Thus, it may be inferred that the gasification reaction of carbon atoms with CO2, leading to pore development in activated carbon, should occur in a random manner. In other words, the active sites (reaction sites) for gasification should be distributed randomly on the carbon surface with a distribution of reaction activation energy. The extent of the gasification reaction, therefore, is dependent on both the number and reactivity of the active sites. Second, the highest production of mesopores in the pore size range of 10–20 nm indicates that the active sites probably concentrate mostly in this pore size range. Finally, for pore sizes in the range of 2–10 nm, there is a clear tendency for the mesopore volume to increase with the increase in activation time for both the AC series and the OTA series. It is probable that the distribution of activation energy of the active sites in this pore size range is relatively wide, thus the increase in activation time would help increase the probability of carbon–CO2 reaction to occur, leading to mesopore development. There is no general definite trend for the variation of pore volume as a function of activation time for pore sizes in the range of 10–50 nm for both carbon series, except that the pore volume of mesopores remained relatively unchanged for the pore size range of 20–50 nm in both carbon series. This has to do with the nature of the distribution of both the number and activation energy of the active sites which are governed by the preparation conditions of the precursor carbon for activated carbon production. Overall, for the same activation time, the OTA method could produce activated carbons with a higher mesopore volume for all pore size ranges as compared to the conventional activation method.
 | ||
| Fig. 4 Pore size distributions of activated carbon gels determined by the DH method for (A) the AC series and (B) the OTA series. | ||
3.3. The effect of porous properties of the initial carbon precursor
Obviously, the porous properties of activated carbons produced by the OTA method from a given precursor will depend on the preparation and treatment conditions including oxidation and heat treatment conditions, activation conditions, porous properties of the initial carbon precursor, etc. This section reports the effect of porous properties of the initial carbon precursor on mesopore development by the OTA method.Activated carbons were prepared using the OTA method from three different carbon precursors with increasing porosity, namely CG, AC1, and AC2, giving the corresponding activated carbon products designated as CG-OTA, AC1-OTA, and AC2-OTA, respectively. Fig. 5 shows the N2 adsorption isotherms of the three activated carbon products measured at −196 °C. The derived isotherms display Type IV isotherms of the IUPAC classification, typical of mesoporous adsorbents with a distribution of pore sizes. The sizes of the hysteresis loops are approximately the same but the relative pressure at which the desorption branch closes the loops tended to decrease in the order AC2-OTA < AC1-OTA < CG-OTA. The amount of N2 adsorbed increased in the following order, AC2-OTA > AC1-OTA > CG-OTA, which is indicative of the increase in the surface area and pore volume of the respective carbon samples. The porous properties of activated carbon products, calculated from the adsorption isotherms, are listed in Table 2. All porous properties of activated carbons produced from each precursor by the OTA method were significantly higher than those of the original precursors. For example, increases in BET surface area of about 177, 120, and 72% were observed for activated carbons produced from CG, AC1, and AC2, respectively. Fig. 6 shows the correlation between the porous properties of the initial precursors and those of the corresponding activated carbons, from which a linear relationship can be observed. This signifies that the number of reaction sites for CO2 gasification increased proportionally with the increase in the surface area of the precursors. Again, the OTA method has proved to be an excellent process for increasing the porous properties of activated carbon produced from carbon gel.
 | ||
| Fig. 5 N2 adsorption isotherms (−196 °C) of mesoporous activated carbons prepared under the same activation conditions by the OTA method from carbon precursors having different initial porous properties. | ||
| Sample name | SBET (m2 g−1) | Vmi (cm3 g−1) | Vme (cm3 g−1) | VT (cm3 g−1) | Burn-off (%) | Yield (%) |
|---|---|---|---|---|---|---|
| CG | 632 | 0.254 (27.5) | 0.657 (71.3) | 0.922 | — | — |
| CG-OTA | 1750 | 0.719 (40.9) | 1.03 (58.5) | 1.76 | 48.3 | 51.7 |
| AC1 | 1120 | 0.447 (37.5) | 0.735 (61.7) | 1.19 | 24.5 | 75.5 |
| AC1-OTA | 2460 | 0.994 (41.7) | 1.35 (57.4) | 2.38 | 62.1 | 37.9 |
| AC2 | 1700 | 0.677 (39.1) | 1.04 (59.9) | 1.73 | 51.9 | 48.1 |
| AC2-OTA | 2920 | 1.19 (39.2) | 1.81 (59.6) | 3.03 | 76.3 | 23.7 |
 | ||
| Fig. 6 Effect of the surface area of initial precursors on the surface area of activated carbon produced by the OTA method. | ||
Next, the development of micropores and mesopores in activated carbons produced by the OTA method from carbon precursors with different porous properties (CG, AC1, and AC2) is further explored in more detail. We studied the pore development in terms of the pore size distribution for the following carbon samples, CG, AC1, AC2, CG-OTA, AC1-OTA, and AC2-OTA. The N2 adsorption isotherms of the carbon samples were measured from a low relative pressure (P/P0) of 1 × 10−6 to 1 by using a high-performance adsorption analyzer (BELMax, Belsorp, Japan), and the derived information was analyzed by the NLDFT method to obtain micropore and mesopore size distribution data. The obtained results are presented graphically and numerically in Fig. 7 and Table 3, respectively, for the following pore size ranges: <0.7 nm (ultra-micropores), 0.7–2 nm (super-micropores), and 2–10 nm (lower-size mesopores) and 10–50 nm (upper-size mesopores).
 | ||
| Fig. 7 Pore size distributions of carbon samples obtained by using the NLDFT method. | ||
| Sample name | Micropores (cm3 g−1) | Mesopores (cm3 g−1) | ||
|---|---|---|---|---|
| 0–0.7 nm | 0.7–2 nm | 2–10 nm | 10–50 nm | |
| CG | 0.032 (3.4%) | 0.162 (17.5%) | 0.150 (16.2%) | 0.564 (61.2%) |
| AC1 | 0.034 (2.9%) | 0.338 (28.4%) | 0.216 (18.1%) | 0.596 (50.1%) |
| AC2 | 0.131 (7.6%) | 0.445 (25.7%) | 0.375 (21.6%) | 0.760 (43.9%) |
| CG-OTA | 0.096 (5.5%) | 0.493 (28.0%) | 0.388 (22.0%) | 0.755 (42.9%) |
| AC1-OTA | 0.078 (3.3%) | 0.742 (31.1%) | 0.666 (27.9%) | 0.878 (36.8%) |
| AC2-OTA | 0.136 (4.5%) | 0.726 (23.9%) | 1.229 (40.5%) | 0.898 (29.6%) |
Starting from the CG precursor, Table 3 indicates that most pores are concentrated in the mesopore size range of 2–50 nm (77.4 volume%) which in part reflects the mesoporous structure of the carbon gel. For the activated carbon AC1, derived by activating the CG sample with CO2 for 1 h at 1000 °C, the proportion of its upper-size mesopores notably decreased from 61.2 to 50.1% while those of smaller pores (0.7–10 nm) tended to increase. For the pore volume, the percentage increase of pore volume was 6.2, 108, 44, and 5.7% for the ultra-micropores, the super-micropores, the lower-size mesopores, and the upper-size mesopores, respectively. The large percentage increase of the super-micropores indicated that the micropores were created inside the particles at a much faster rate as compared to the development of mesopores. This could be attributed to the large internal surface area of the nanoparticles which favors the creation of micropores by gasification. It is further noted that the percentage increase of the upper-size mesopore volume was much lower than that of the lower-size mesopore volume (5.7% vs. 44%). This appears to confirm that the upper-size mesopores are possibly created outside the carbon gel particles (inter-particle pore formation), while the lower-size mesopores are developed inside the particles (intra-particle pore formation) by the enlargement of smaller micropores via the gasification reaction. It should be noted that the average size of interparticle spaces of carbon gels in the range of 17–80 nm has been reported30,49 that approximately covers the range of the upper-size mesopores in this work. The AC2 sample, which was derived by activating the AC1 sample for 1 h or by activating the CG sample for 2 h, showed a decrease in the proportional percentage of the upper-size mesopores from 50.1% to 43.9% but an increase from 18.1% to 21.6% was observed for the lower-size mesopores.
The CG-OTA sample, which was prepared from the CG precursor by the OTA method, showed a significant increase in the pore volume for all pore sizes, on average by about twofold. Furthermore, the order of increase in the proportional percentage of pore volumes of CG-OTA was the same as that of the CG sample, that is, upper-size mesopores > super-micropores > lower-size mesopores > ultra-micropores. However, a drop in the percentage of upper-size mesopore volume from 61.2 to 42.9% was observed which further substantiates that pore development mostly occurs inside the nanoparticles which form the carbon gel. Similar results were obtained when comparing AC1 with AC1-OTA and AC2 with AC2-OTA. In view of carbon preparation methods, both micropore and mesopore volumes of CG-OTA prepared by the OTA method were higher than those of the sample prepared by the conventional method (AC1) on the average by 50%, while similar results were obtained when comparing AC1-OTA with AC2. When the surface area of the precursor series increased (CG < AC1 < AC2), the volume percent of the upper-size mesopores in the activated carbons produced by the OTA method decreased from 42.9 to 29.6% but an increase in the volume percent of the lower-size mesopores from 22 to 40.5% was attained, possibly at the expense of the micropore volume by the pore enlargement mechanism. It is interesting to examine further, from Table 3, the formation of mesopores from the enlargement of original micropores by the OTA method. As an example, comparing the porous properties of the CG and CG-OTA samples. The super-micropores volume of the CG-OTA carbon is 0.493 cm3 g−1, while that of the original CG sample is 0.162 cm3 g−1. Therefore, the increase in the volume of newly formed micropores = (0.493 − 0.162)(100)/(0.162) = 204%. The lower-size mesopores volume (2–10 nm) of the CG and the CG-OTA are 0.150 and 0.388 cm3 g−1, respectively. From these numerical values, the percentage of micropores of the original CG that is enlarged by CO2 gasification to form the lower-size mesopores = (0.388 − 0.150)(100)/(0.162) = 147% which is greater than 100%. This indicates that the widening of micropores to form mesopores could occur with the newly-formed micropores as well.
Overall, the obtained results confirm that the preparation of activated carbon from carbon gel by the OTA method is superior to the conventional activation method for improving the porous properties of the resulting carbon. This is due principally to the higher gasification rate of the OTA-treated carbon, resulting from a larger number of reaction sites in the gel particles.
3.4. Role of surface functional groups in the OTA method
The key success of the OTA method is the inclusion of oxidation and heat treatment steps prior to the final activation of a carbon. The oxidation step increases the amount of surface functional groups, while the following heat treatment removes such groups. Thus, a large number of active reaction sites for the subsequent activation step are produced that substantially help the improvement of the porous properties of the derived carbon. Thermal destruction can control the reactivity of carbon gel by controlling the degree of destruction of functional groups, for example, by controlling the level of heat treatment temperature. The higher the thermal treatment temperature, the larger amounts of functional groups being removed, and hence the increase in the carbon gel reactivity for gasification.In this work, the amounts of oxygen-containing functional groups of the carbon samples were analyzed by a TPD (Temperature Programmed Desorption) method conducted under vacuum. Typical TPD spectra, which show the desorption rate of CO, CO2, and H2 as a function of temperature, of carbon samples collected after each step of the sequence of the OTA method, namely the initial carbon obtained through CO2 activation for 1 h (AC), the air oxidized sample (AC-O), the sample after heat treatment (AC-OT), and the final reactivated carbon (AC-OTA) are shown in Fig. 8. The total amounts of CO, CO2, and H2 desorbed from the samples in the unit of μmol g−1 are presented in Table 4. For the initial activated carbon (AC), the profile of CO2 showed a maximum at around 340 °C, while the profile of CO displayed a maximum at about 1000 °C. In the literature, the CO2 peak at low temperatures is usually attributed to carboxylic groups, while CO2 desorption above 400 °C is assigned to acid anhydrides, and lactones.50 CO desorption is ascribed to the decomposition of phenol, ethers, and carbonyls and the desorption temperature of carbonyls is higher than those of the others.50,51 For the air-oxidized carbon (AC-O), two sharp peaks appear in its CO2 spectra which are at around 300 °C for the smaller one and about 670 °C for the larger one. The maximum CO desorption rate for the air-oxidized sample can be observed at around 700 °C, which is thought to be due to the decomposition of phenolic groups.52 However, it should be noted that larger amounts of CO2 and CO desorbed from AC-O than from AC, by about 10 and 25 times, respectively. It is obvious that the air oxidation of activated carbon has led to the creation of large amounts of oxygen-containing functional groups on the graphene sheets which form the carbon.
 | ||
| Fig. 8 (A) CO (B) CO2 and (C) H2 desorption spectra of the OTA samples. | ||
| Sample code | CO2 (μmol g−1) | CO (μmol g−1) | H2 (μmol g−1) |
|---|---|---|---|
| AC | 63.8 | 191 | 2026 |
| AC-O | 723 | 5520 | 1989 |
| AC-OT | 56.1 | 122 | 1805 |
| AC-OTA | 55.9 | 169 | 1887 |
After heating the oxidized carbon at 1000 °C in an N2 flow, the heat-treated sample (AC-OT) showed similar results to those of the initial activated carbon (AC) for both the CO2 and CO spectra but with lower amounts of emitted gases. This indicates that most of the oxygen-containing functional groups were removed from the heat-treated carbon, thus the number of unpaired electrons due to bond disruption was enhanced. Slightly larger amounts of gases were emitted from the carbon reactivated by CO2 (AC-OTA) than from the heat-treated sample (AC-OT), which implies that functional groups are readily formed by CO2 during the activation step. On the other hand, AC-OTA also showed similar spectra to the initial activated carbon (AC) but the amounts of emitted gases were slightly lower than those of the initial activated carbon. It could be possible that the CO2 gasification reaction removes more of the reactive edge sites in the carbon structure, leading to fewer functional groups at the edge sites as well.
To investigate the influence of oxygen functional groups on the main role of the OTA method, the amount of each type of oxygen functional group was determined by peak separation of the TPD spectra of CO2 and CO (refer to Fig. S1†). The desorption temperature and full width at half-maximum of the oxygen-containing functional groups were determined based on previous work.49 Table 5 presents the amount of oxygen-containing functional groups in the carbon samples. The order of oxygen-containing functional group content in the carbon sample after the oxidation step (AC-O) was as follows: phenol/ether > lactone > carbonyl > carboxylic > anhydride. These results indicate that air oxidation of carbon primarily led to the formation of phenol/ether groups. The acidic groups of the AC-OT sample were almost completely removed as compared to those of the oxidized carbons, with their amounts being slightly lower than those in the starting activated carbon (AC). On the other hand, after heat treatment, the carbonyl groups content was reduced to the value of about 50% as compared to that of the AC sample (0.128 vs. 0.064 mmol g−1), while the contents of other functional groups remained almost unchanged. Interestingly, the AC-OTA sample showed a slight increase in the functional groups formed by CO2 during the activation step, except for the carbonyl groups which increased up to 95%. This suggests that the thermal destruction of the carbonyl groups during gasification could play a stronger role in the generation of reaction sites for gasification, and hence the pore development in activated carbon.
| Sample code | Carboxylic (mmol g−1) | Anhydride (mmol g−1) | Lactone (mmol g−1) | Phenol/ether (mmol g−1) | Carbonyl (mmol g−1) |
|---|---|---|---|---|---|
| AC | 0.036 | 0.019 | 0.010 | 0.044 | 0.128 |
| AC-O | 0.072 | 0.000 | 0.651 | 4.987 | 0.532 |
| AC-OT | 0.034 | 0.012 | 0.009 | 0.035 | 0.064 |
| AC-OTA | 0.038 | 0.009 | 0.009 | 0.046 | 0.125 |
3.5. Determination of carbon reactivity towards CO2 gasification
TGA analysis was employed to illustrate the importance of the OTA method in increasing the reactivity of active sites for CO2 gasification. The carbon reactivity for CO2 gasification (Rc) was defined here as the rate of fractional conversion according to eqn (2):
 | (2) |
 | ||
| Fig. 9 (A) The carbon conversion profiles for CO2 gasification at 1000 °C of the activated carbons prepared by conventional activation (AC) and by the OTA method (AC-OT) and (B) the effect of carbon conversion on the carbon reactivity towards CO2 gasification. | ||
The carbon conversion data with respect to the gasification time are presented in Fig. 9A. The results indicated that the carbon conversion of AC-OT increased at a much higher rate than that of AC, which can be observed from the steeper slope of the curve. This could be associated with the larger number of reactive sites available during the activation step. Fig. 9B shows the effect of carbon conversion on carbon reactivity. The reactivity curve of AC showed a relatively constant rate, while the AC-OT curve stayed above the AC curve and showed a characteristic of continuous rising with the increase in the fractional conversion, meaning higher reactivity at all degrees of carbon conversion. The rising trend of the reactivity curve is attributed to the increase in surface area with increasing carbon conversion that provided more reactive sites for the gasification, leading to a higher reaction rate. As mentioned above, the reactivity of carbon from the OTA method was higher than that derived through the conventional activation method, as observed from the comparison between the curves of AC and AC-OT at the same carbon conversion (X). These results support the fact that the OTA method is capable of increasing the number of active sites for the gasification reaction by CO2, resulting in the rapid development of pores and the corresponding increase in surface area.
In summary, the OTA method is a modified method of the conventional physical activation method for producing mesoporous-activated carbon by incorporating two consecutive steps of air oxidation and thermal destruction of the developed oxygen-containing functional groups before CO2 gasification. Mesopores are created not by the coalescence of micropores at a high degree of carbon burn-off, but as a result of micropore widening caused by the increased surface reactivity of micropores for CO2 gasification. Therefore, when the OTA method is used, small mesopores can occur within the carbon nanoparticles at any level of carbon burn-off. This would be advantageous for applications such as capacitors due to a large specific surface area and mesopores that develop not only in the interparticle spaces but also within the nanoparticles of the activated carbon produced from carbon gel.
4. Conclusions
Carbon gels were used as precursors for the synthesis of mesoporous activated carbon by a modified physical activation method, called the OTA method, consisting of three consecutive steps of air oxidation of the starting carbon, high-temperature heating of the oxidized carbon, and CO2 activation of the heat-treated product. It was found that the activated carbons derived from the OTA procedure provided much improved porous properties when compared with those prepared through the traditional activation method under the same activation conditions and/or at the same carbon burn-off value. The maximum values of the porous properties of activated carbon gel obtained by the OTA method in this work were 2920 m2 g−1, 3.03 cm3 g−1, 1.19 cm3 g−1, and 1.81 cm3 g−1 for the BET surface area, total pore volume, micropore volume, and mesopore volume, respectively. The superiority of the OTA method is its ability to generate a larger number of reaction sites for CO2 gasification which results from the bond disruption of the created surface functional groups. The produced activated carbon had a larger volume of mesopores as compared to that of micropores, which substantiates the highly porous nature of the carbon gel. As a result, the overall structure of the carbon gel is relatively stable along the course of the activation process. Furthermore, increasing the activation time tended to decrease the percentage of mesopores and a corresponding increase in the volume percent of micropores. It is also evident that the upper-size mesopores (10–50 nm) are probably formed outside the carbon particles in the carbon gel, while the lower-size mesopores (2–10 nm) and micropores (<2 nm) are created inside the particles. It is believed that the activated carbon gel produced by the OTA method with a large surface area and pore volume can be applied in the adsorption field for the effective removal of organic compounds and metal ions from aqueous solutions as well as the storage of such gases as carbon dioxide, hydrogen, and methane.Author contributions
Panuwat Lawtae: conceptualization, methodology, validation, formal analysis, investigation, data curation, visualization, writing – original draft, writing – review & editing. Shintaroh Nagaishi: investigation, formal analysis, validation. Chaiyot Tangsathitkulchai: writing – review & editing, supervision. Shinichiroh Iwamura: validation, resources. Shin R. Mukai: writing – review & editing, supervision.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We gratefully acknowledge the financial support of this work in the form of a full study scholarship to PL provided by the Royal Golden Jubilee (RGJ) PhD Program, batch number 20 (PHD/0057/2560) under the National Research Council of Thailand (NRCT) and Thailand Research Fund (TRF) in collaboration with Suranaree University of Technology. We also thank the Research Group of Chemical Engineering, Division of Applied Chemistry, Hokkaido University for the facilities and laboratories as well as for providing resources that have mainly contributed to the research results reported in this work.References
- F. Kang, M. Inagaki and H. Itoi, Porous Carbons: Syntheses and Applications, Elsevier Science, 2021 Search PubMed.
- A. Celzard, V. Fierro and G. Amaral-Labat, Chapter 7 - Adsorption by Carbon Gels, in Novel Carbon Adsorbents, ed. J. M. D. Tascón, Elsevier, Oxford, 2012, pp. 207–244 Search PubMed.
- T. Tsuchiya, T. Mori, S. Iwamura, I. Ogino and S. R. Mukai, Binderfree synthesis of high-surface-area carbon electrodes via CO2 activation of resorcinol–formaldehyde carbon xerogel disks: analysis of activation process, Carbon, 2014, 76, 240–249 CrossRef CAS.
- T. Mori, S. Iwamura, I. Ogino and S. R. Mukai, Cost-effective synthesis of activated carbons with high surface areas for electrodes of non-aqueous electric double layer capacitors, Sep. Purif. Technol., 2019, 214, 174–180 CrossRef CAS.
- J. Yoo, I. Yang, D. Kwon, M. Jung, M.-S. Kim and J. C. Jung, Low-Cost Carbon Xerogels Derived from Phenol–Formaldehyde Resin for Organic Electric Double-Layer Capacitors, Energy Technol., 2021, 9(4), 2000918 CrossRef CAS.
- E. Wang, C. Shao, S. Qiu, H. Chu, Y. Zou, C. Xiang, F. Xu and L. Sun, Organic carbon gel assisted-synthesis of Li1.2Mn0.6Ni0.2O2 for a high-performance cathode material for Li-ion batteries, RSC Adv., 2017, 7(3), 1561–1566 RSC.
- V. Velez, G. Ramos-Sánchez, B. Lopez, L. Lartundo-Rojas, I. González and L. Sierra, Synthesis of novel hard mesoporous carbons and their applications as anodes for Li and Na ion batteries, Carbon, 2019, 147, 214–226 CrossRef CAS.
- M. Kakunuri and C. S. Sharma, Resorcinol-formaldehyde derived carbon xerogels: a promising anode material for lithium-ion battery, J. Mater. Res., 2018, 33(9), 1074–1087 CrossRef CAS.
- J. L. Figueiredo and M. F. R. Pereira, Synthesis and functionalization of carbon xerogels to be used as supports for fuel cell catalysts, J. Energy Chem., 2013, 22(2), 195–201 CrossRef CAS.
- C. Alegre, D. Sebastián, M. E. Gálvez, R. Moliner and M. J. Lázaro, Sulfurized carbon xerogels as Pt support with enhanced activity for fuel cell applications, Appl. Catal., B, 2016, 192, 260–267 CrossRef CAS.
- W. Kiciński, S. Dyjak and W. Tokarz, Carbon gel-derived Fe–N–C electrocatalysts for hydrogen-air polymer electrolyte fuel cells, J. Power Sources, 2021, 513, 230537 CrossRef.
- O. Gómez-Cápiro, A. Hinkle, A. M. Delgado, C. Fernández, R. Jiménez and L. E. Arteaga-Pérez, Carbon Aerogel-Supported Nickel and Iron for Gasification Gas Cleaning. Part I: Ammonia Adsorption, Catalysts, 2018, 8(9), 347 CrossRef.
- S. Mishra, A. Yadav and N. Verma, Carbon gel-supported Fe-graphene disks: synthesis, adsorption of aqueous Cr(VI) and Pb(II) and the removal mechanism, Chem. Eng. J., 2017, 326, 987–999 CrossRef CAS.
- H. Guo, D.-H. Si, H.-J. Zhu, Q.-X. Li, Y.-B. Huang and R. Cao, Ni single-atom sites supported on carbon aerogel for highly efficient electroreduction of carbon dioxide with industrial current densities, eScience, 2022, 2(3), 295–303 CrossRef.
- O. Czakkel, B. Nagy, G. Dobos, P. Fouquet, E. Bahn and K. László, Static and dynamic studies of hydrogen adsorption on nanoporous carbon gels, Int. J. Hydrogen Energy, 2019, 44(33), 18169–18178 CrossRef CAS.
- R. U. Soni, V. A. Edlabadkar, D. Greenan, P. M. Rewatkar, N. Leventis and C. Sotiriou-Leventis, Preparation of Carbon Aerogels from Polymer-Cross-Linked Xerogel Powders without Supercritical Fluid Drying and Their Application in Highly Selective CO2 Adsorption, Chem. Mater., 2022, 34(11), 4828–4847 CrossRef CAS.
- H. Majedi Far, P. M. Rewatkar, S. Donthula, T. Taghvaee, A. M. Saeed, C. Sotiriou-Leventis and N. Leventis, Exceptionally High CO2 Adsorption at 273 K by Microporous Carbons from Phenolic Aerogels: The Role of Heteroatoms in Comparison with Carbons from Polybenzoxazine and Other Organic Aerogels, Macromol. Chem. Phys., 2019, 220(1), 1800333 CrossRef.
- G. Wei, Y.-E. Miao, C. Zhang, Z. Yang, Z. Liu, W. W. Tjiu and T. Liu, Ni-Doped Graphene/Carbon Cryogels and Their Applications As Versatile Sorbents for Water Purification, ACS Appl. Mater. Interfaces, 2013, 5(15), 7584–7591 CrossRef CAS PubMed.
- A. Pillai and B. Kandasubramanian, Carbon Xerogels for Effluent Treatment, J. Chem. Eng. Data, 2020, 65(5), 2255–2270 CrossRef CAS.
- R. Ganesamoorthy, V. K. Vadivel, R. Kumar, O. S. Kushwaha and H. Mamane, Aerogels for water treatment: a review, J. Cleaner Prod., 2021, 329, 129713 CrossRef CAS.
- R. W. Pekala, Aerogels and Xerogels from Organic Precursors, Lawrence Livermore National Lab. (LLNL), Livermore, CA (United States), 1989 Search PubMed.
- R. W. Pekala, Organic aerogels from the polycondensation of resorcinol with formaldehyde, J. Mater. Sci., 1989, 24(9), 3221–3227 CrossRef CAS.
- R. Pekala and F.-M. Kong, A synthetic route to organic aerogels-mechanism, structure, and properties, J. Phys. Colloq., 1989, 50(C4), 33–40 Search PubMed.
- S. A. Al-Muhtaseb and J. A. Ritter, Preparation and Properties of Resorcinol–Formaldehyde Organic and Carbon Gels, Adv. Mater., 2003, 15(2), 101–114 CrossRef CAS.
- A. M. ElKhatat and S. A. Al-Muhtaseb, Advances in Tailoring Resorcinol-Formaldehyde Organic and Carbon Gels, Adv. Mater., 2011, 23(26), 2887–2903 CrossRef CAS PubMed.
- N. Rey-Raap, J. Angel Menéndez and A. Arenillas, RF xerogels with tailored porosity over the entire nanoscale, Microporous Mesoporous Mater., 2014, 195, 266–275 CrossRef CAS.
- N. Rey-Raap, A. Arenillas and J. A. Menéndez, A visual validation of the combined effect of pH and dilution on the porosity of carbon xerogels, Microporous Mesoporous Mater., 2016, 223, 89–93 CrossRef CAS.
- E. G. Calvo, E. J. Juárez-Pérez, J. A. Menéndez and A. Arenillas, Fast microwave-assisted synthesis of tailored mesoporous carbon xerogels, J. Colloid Interface Sci., 2011, 357(2), 541–547 CrossRef CAS PubMed.
- E. Isaacs Páez, M. Haro, E. J. Juárez-Pérez, R. J. Carmona, J. B. Parra, R. Leyva Ramos and C. O. Ania, Fast synthesis of micro/mesoporous xerogels: textural and energetic assessment, Microporous Mesoporous Mater., 2015, 209, 2–9 CrossRef.
- N. Job, F. Panariello, J. Marien, M. Crine, J.-P. Pirard and A. Léonard, Synthesis optimization of organic xerogels produced from convective air-drying of resorcinol–formaldehyde gels, J. Non-Cryst. Solids, 2006, 352(1), 24–34 CrossRef CAS.
- N. Job, R. Pirard, J. Marien and J.-P. Pirard, Porous carbon xerogels with texture tailored by pH control during sol–gel process, Carbon, 2004, 42(3), 619–628 CrossRef CAS.
- S. J. Taylor, M. D. Haw, J. Sefcik and A. J. Fletcher, Gelation Mechanism of Resorcinol-Formaldehyde Gels Investigated by Dynamic Light Scattering, Langmuir, 2014, 30(34), 10231–10240 CrossRef CAS PubMed.
- A. Arenillas, J. A. Menéndez, G. Reichenauer, A. Celzard, V. Fierro, F. J. M. Hodar, E. Bailón-Garcia and N. Job, Organic and Carbon Gels: From Laboratory Synthesis to Applications, Springer, 2019 Search PubMed.
- H. Marsh and F. Rodríguez-Reinoso, Activated Carbon, Elsevier Science Ltd, Oxford, 2006 Search PubMed.
- P. Lawtae and C. Tangsathitkulchai, A New Approach for Controlling Mesoporosity in Activated Carbon by the Consecutive Process of Air Oxidation, Thermal Destruction of Surface Functional Groups, and Carbon Activation (the OTA Method), Molecules, 2021, 26(9), 2758 CrossRef CAS PubMed.
- P. Lawtae and C. Tangsathitkulchai, The Use of High Surface Area Mesoporous-Activated Carbon from Longan Seed Biomass for Increasing Capacity and Kinetics of Methylene Blue Adsorption from Aqueous Solution, Molecules, 2021, 26(21), 6521 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60(2), 309–319 CrossRef CAS.
- M. M. Dubinin, Physical Adsorption of Gases and Vapors in Micropores, in Progress in Surface and Membrane Science, ed. D. A. Cadenhead, J. F. Danielli and M. D. Rosenberg, Elsevier, 1975, pp. 1–70 Search PubMed.
- D. Dollimore and G. R. Heal, An improved method for the calculation of pore size distribution from adsorption data, J. Appl. Chem., 1964, 14(3), 109–114 CrossRef CAS.
- C. Lin and J. A. Ritter, Carbonization and activation of sol–gel derived carbon xerogels, Carbon, 2000, 38(6), 849–861 CrossRef CAS.
- A. H. Moreno, A. Arenillas, E. G. Calvo, J. M. Bermúdez and J. A. Menéndez, Carbonisation of resorcinol–formaldehyde organic xerogels: effect of temperature, particle size and heating rate on the porosity of carbon xerogels, J. Anal. Appl. Pyrolysis, 2013, 100, 111–116 CrossRef CAS.
- A. Mohaddespour, S. Atashrouz and S. J. Ahmadi, Nanostructured Carbon Xerogels by Super-Fast Carbonization, Ind. Eng. Chem. Res., 2017, 56(21), 6213–6220 CrossRef CAS.
- R. W. Pekala and D. W. Schaefer, Structure of organic aerogels. 1. Morphology and scaling, Macromolecules, 1993, 26(20), 5487–5493 CrossRef CAS.
- Z. Song, L. Miao, L. Ruhlmann, Y. Lv, D. Zhu, L. Li, L. Gan and M. Liu, Lewis Pair Interaction Self-Assembly of Carbon Superstructures Harvesting High-Energy and Ultralong-Life Zinc-Ion Storage, Adv. Funct. Mater., 2022, 32(48), 2208049 CrossRef CAS.
- J. Yan, L. Miao, H. Duan, D. Zhu, Y. Lv, L. Li, L. Gan and M. Liu, High-energy aqueous supercapacitors enabled by N/O codoped carbon nanosheets and “water-in-salt” electrolyte, Chin. Chem. Lett., 2022, 33(5), 2681–2686 CrossRef CAS.
- X. Zheng, L. Miao, Z. Song, W. Du, D. Zhu, Y. Lv, L. Li, L. Gan and M. Liu, In situ nanoarchitecturing of conjugated polyamide network-derived carbon cathodes toward high energy-power Zn-ion capacitors, J. Mater. Chem. A, 2022, 10(2), 611–621 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87(9–10), 1051–1069 CrossRef CAS.
- M. Canal-Rodríguez, J. A. Menéndez and A. Arenillas, Carbon Xerogels: The Bespoke Nanoporous Carbons, in Porosity-Process, Technologies and Applications, IntechOpen, London, UK, 2017, pp. 69–89 Search PubMed.
- S. Nagaishi, S. Iwamura, T. Ishii and S. R. Mukai, Clarification of the Effects of Oxygen Containing Functional Groups on the Pore Filling Behavior of Discharge Deposits in Lithium–Air Battery Cathodes Using Surface-Modified Carbon Gels, J. Phys. Chem. C, 2023, 127(5), 2246–2257 CrossRef CAS.
- T. Ishii and T. Kyotani, Chapter 14 - Temperature Programmed Desorption, in Materials Science and Engineering of Carbon, ed. M. Inagaki and F. Kang, Butterworth-Heinemann, 2016, pp. 287–305 Search PubMed.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Modification of the surface chemistry of activated carbons, Carbon, 1999, 37(9), 1379–1389 CrossRef CAS.
- U. Zielke, K. J. Hüttinger and W. P. Hoffman, Surface-oxidized carbon fibers: I. Surface structure and chemistry, Carbon, 1996, 34(8), 983–998 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01647a |
| This journal is © The Royal Society of Chemistry 2023 |