DOI:
10.1039/D3RA02300A
(Paper)
RSC Adv., 2023,
13, 20105-20112
Structural diversity of cocrystals formed from acridine and two isomers of hydroxybenzaldehyde: 3-hydroxybenzaldehyde and 4-hydroxybenzaldehyde†
Received
6th April 2023
, Accepted 27th June 2023
First published on 4th July 2023
Abstract
Cocrystals formed from acridine and two isomers of hydroxybenzaldehyde: 3-hydroxybenzaldehyde (1) and 4-hydroxybenzaldehyde (2) were synthesized and structurally characterized. Single-crystal X-ray diffraction measurements show that compound 1 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, whereas compound 2 crystallizes in the monoclinic P21/n space group. In the crystals of title compounds, the molecules interact via O–H⋯N and C–H⋯O hydrogen bonds, and C–H⋯π and π–π interactions. DCS/TG measurements indicate that compound 1 melts at a lower temperature than the separate cocrystal coformers, whereas compound 2 melts at a higher temperature than acridine but at a lower temperature than 4-hydroxybenzaldehyde. The FTIR measurements reveal that the band attributed to the stretching vibrations of the hydroxyl group of hydroxybenzaldehyde disappeared, but several bands appeared in the range of 3000–2000 cm−1.
space group, whereas compound 2 crystallizes in the monoclinic P21/n space group. In the crystals of title compounds, the molecules interact via O–H⋯N and C–H⋯O hydrogen bonds, and C–H⋯π and π–π interactions. DCS/TG measurements indicate that compound 1 melts at a lower temperature than the separate cocrystal coformers, whereas compound 2 melts at a higher temperature than acridine but at a lower temperature than 4-hydroxybenzaldehyde. The FTIR measurements reveal that the band attributed to the stretching vibrations of the hydroxyl group of hydroxybenzaldehyde disappeared, but several bands appeared in the range of 3000–2000 cm−1.
1. Introduction
The preparation, identification, and structural characterization of multicomponent crystals, such as cocrystals, salts or solvates involving Active Pharmaceutical Ingredients (APIs) is one of the most active areas of crystal engineering.1–8 Multicomponent crystals containing API combinations have great pharmacological potential because their different forms can exhibit different physicochemical properties, such as solubility, dissolution rate, thermal stability, or bioavailability compared with their individual pure components.2–9 Interesting coformers of pharmaceutical significance, acridine and two natural isomers of hydroxybenzaldehyde: 3-hydroxybenzaldehyde and 4-hydroxybenzaldehyde, were selected for the study.
Acridines belong to a class of compounds with high biological activity and wide therapeutic potential as an antimalarial, antiseptic, antitumor, antiviral, antifungal and other agents.10–14 This is due to their ability to intercalate into the DNA double strand, causing various disorders and commonly leading to the deactivation of the genetic material.15–18 Acridines are also chemiluminescent indicators and chemiluminogenic fragments of chemiluminescent labels with analytical importance.19,20
In turn, 3-hydroxybenzaldehyde is a novel therapeutic agent with vasculoprotective potential because it has therapeutic effect in atherosclerosis,21 whereas 4-hydroxybenzaldehyde sensitizes bacteria to antibiotics,22 accelerates acute wound healing,23 shows beneficial effects on insulin resistance in an animal model of type II diabetes24 and significantly increases specific protein production in infected host cells, causing an enhanced defence response to invasion by Toxoplasma gondii parasite.25 Furthermore, both 3-hydroxybenzaldehyde and 4-hydroxybenzaldehyde are potential therapeutic agents for the treatment of human angiostrongyliasis.26
A search of the Cambridge Structure Database (CSD version 5.44, update March 2022)27 for structures formed from acridines and benzaldehyde derivatives shows that there is only one crystal structure of cocrystal of acridine with vanillin.28
In this article, as a continuation of our recent work concerning multicomponent crystals formed from acridines,29–31 we synthesized and structurally characterized cocrystals of acridine with two isomers of hydroxybenzaldehyde: 3-hydroxybenzaldehyde (1) and 4-hydroxybenzaldehyde (2) (Scheme 1).
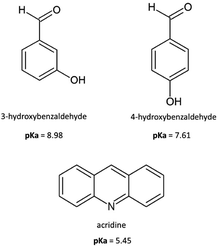 |
| | Scheme 1 Molecular structures of acridine and hydroxybenzaldehydes reported in the study. | |
2. Experimental
2.1. Materials and methods
All the chemicals were purchased from Sigma-Aldrich and used without further purification.
2.2. Synthesis of compounds 1–2
2.2.1. Cocrystal of acridine with 3-hydroxybenzaldehyde (1). Acridine (0.050 g, 0.279 mmol) and 3-hydroxybenzaldehyde (0.034 g, 0.279 mmol) were dissolved in 20 mL of a methanol/dichloromethane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and heated for 20 min to dissolve the sample. The solution was allowed to evaporate at 4 °C for a few days to give yellow crystals of 1 (yield > 95%; mp = 96.5 °C); analysis calculated/found for C20H15NO2: C 79.72/79.50, H 5.02/5.02, N 4.65/4.59.
:1 v/v) and heated for 20 min to dissolve the sample. The solution was allowed to evaporate at 4 °C for a few days to give yellow crystals of 1 (yield > 95%; mp = 96.5 °C); analysis calculated/found for C20H15NO2: C 79.72/79.50, H 5.02/5.02, N 4.65/4.59.
2.2.2. Cocrystal of acridine with 4-hydroxybenzaldehyde (2). Acridine (0.050 g, 0.279 mmol) and 4-hydroxybenzaldehyde (0.034 g, 0.279 mmol) were dissolved in 20 mL of an ethanol/water mixture (4:1 v/v) and heated for 20 min to dissolve the sample. Once the heating was complete, there was added 10 cm3 of isobutanol to the mixture. The solution was allowed to evaporate at room temperature for a few days to give pale yellow crystals of 2 (yield > 95%; mp = 118.5 °C); analysis calculated/found for C20H15NO2: C 79.72/79.64, H 5.02/5.02, N 4.65/4.56.
2.3. Single-crystal X-ray diffraction (SCXRD)
Diffraction data were collected on an Oxford Diffraction Gemini R ULTRA Ruby CCD diffractometer (λMo = 0.71073 Å, T = 293(2) K). The lattice parameters were obtained using CrysAlis CCD, whereas data were reduced using CrysAlis RED32 programs. The structures were solved and refined using the SHELX programs.33 H-Atoms from hydroxyl groups were located on a difference Fourier map and refined freely. All H-atoms bound to C–atoms were placed geometrically and refined using a riding model with C–H = 0.93 Å and Uiso(H) = 1.2Ueq(C). All interactions were found using the PLATON program.34 The ORTEPII,35 PLUTO-78 (ref. 36) and Mercury37 programs were used to prepare the molecular graphics.
2.4. Thermogravimetry (TG) and differential scanning calorimetry (DSC)
Thermal analysis was performed using simultaneous TG-DSC analyzer Netzsch STA 449 F3 Jupiter. The samples (3–5 mg) were heated with the heating rate of 10 °C min−1 from 30 to 450 °C in nitrogen atmosphere.
2.5. Fourier-transform infrared spectroscopy (FTIR)
FTIR spectra were measured on a PerkinElmer Spectrum 3TM instrument (PerkinElmer, Waltham, USA) equipped with attenuated total reflectance (ATR) accessory. The spectra were recorded at room temperature in reflective mode from 4000 to 500 cm−1 at a resolution of 4 cm−1 averaging 16 scans for each measurement. The FTIR spectra were processed and referred to their baseline using PerkinElmer Spectrum IR version 10.7.2 software.
3. Results and discussion
3.1. Crystal structure and analysis of intermolecular interactions
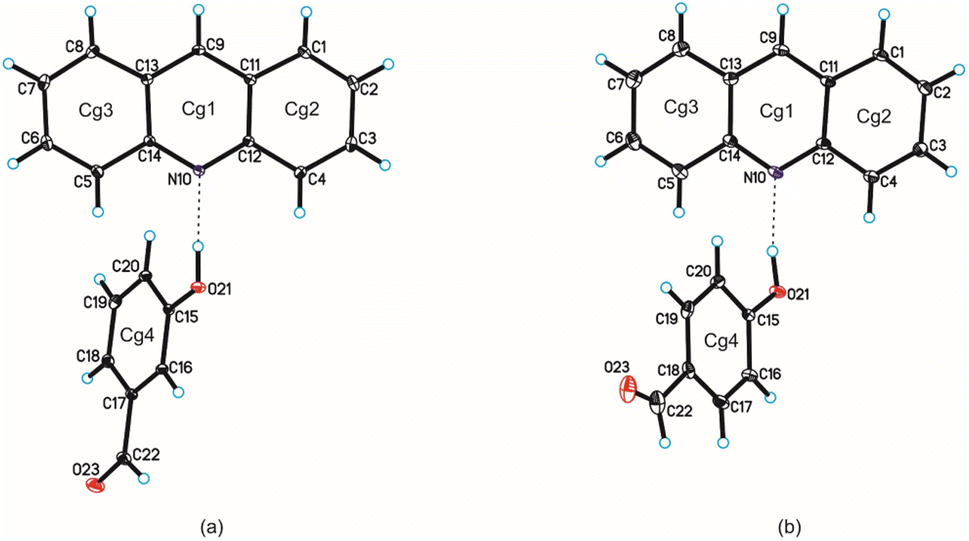
Single-crystal X-ray diffraction measurements show that compound 1 crystallizes in the triclinic P space group with one acridine and one 3-hydroxybenzaldehyde molecules in the asymmetric unit (Fig. 1 and Table 1), whereas compound 2 crystallizes in the monoclinic P21/c space group with one acridine and one 4-hydroxybenzaldehyde molecules in the asymmetric unit (Fig. 1, Table 1).
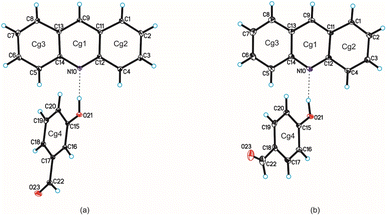 |
| | Fig. 1 Molecular structures of compounds 1 and 2 are shown in (a) and (b), respectively, with the atom-labelling scheme. Cg1–Cg4 denote the ring centroids. Displacement ellipsoids are drawn at the 25% probability level and H atoms are shown as small spheres of arbitrary radius (hydrogen bonds are represented by dashed line). | |
Table 1 Crystal data and structure refinement parameters for title compounds
| Compound |
1 |
2 |
| Chemical formula |
C13H9N·C7H6O2 |
C13H9N·C7H6O2 |
| FW/g mol−1 |
301.33 |
301.33 |
| Crystal system |
Triclinic |
Monoclinic |
| Space group |
P |
P21/c |
| a/Å |
7.2502(8) |
11.5433(8) |
| b/Å |
9.0739(11) |
10.6297(10) |
| c/Å |
12.1019(10) |
12.9034(12) |
| α/° |
100.594(9) |
90 |
| β/° |
94.574(8) |
96.365(9) |
| γ/° |
95.976(10) |
90 |
| V/Å3 |
774.34(15) |
1573.5(2) |
| Z |
2 |
4 |
| T/K |
293(2) |
293(2) |
| λMo/Å |
0.71073 |
0.71073 |
| ρcalc/g cm−3 |
1.292 |
1.272 |
| F(000) |
316 |
628 |
| μ/mm−1 |
0.084 |
0.082 |
| θ range/° |
3.43–25.00 |
3.46–25.00 |
| Completeness of θ/% |
99.7 |
99.7 |
| Reflections collected |
4840 |
10810 |
| Reflections unique |
2728 |
2769 |
| [Rint = 0.0276] |
[Rint = 0.0854] |
| Data/restraints/parameters |
2728/0/211 |
2769/0/211 |
| Goodness of fit on F2 |
1.021 |
1.073 |
| Final R1 value (I > 2σ(I)) |
0.0511 |
0.0791 |
| Final wR2 value (I > 2σ(I)) |
0.1221 |
0.1635 |
| Final R1 value (all data) |
0.0782 |
0.1410 |
| Final wR2 value (all data) |
0.1411 |
0.1971 |
| CCDC number |
2253867 |
2253868 |
Confirmation that proton transfer does not occur between hydroxyl group of benzaldehyde and endocyclic N-atom of acridine results from an analysis of geometric parameters characterizing of both molecules.28,29 The C–O bond lengths in the hydroxyl group of the hydroxybenzaldehyde molecule are 1.36 Å and 1.35 Å, whereas the C12–N10–C14 valence angles in the acridine molecule are 119.1° and 118.8°, for compounds 1 and 2 respectively. In the cocrystal of acridine with vanillin these values are similar (1.35 Å and 118.3°, respectively).28 As we previously shown,29 above mentioned C12–N10–C14 angle ranges from 118 to 120° for acridine cocrystals, whereas is in the range 123 ÷ 124° for acridinium salts.
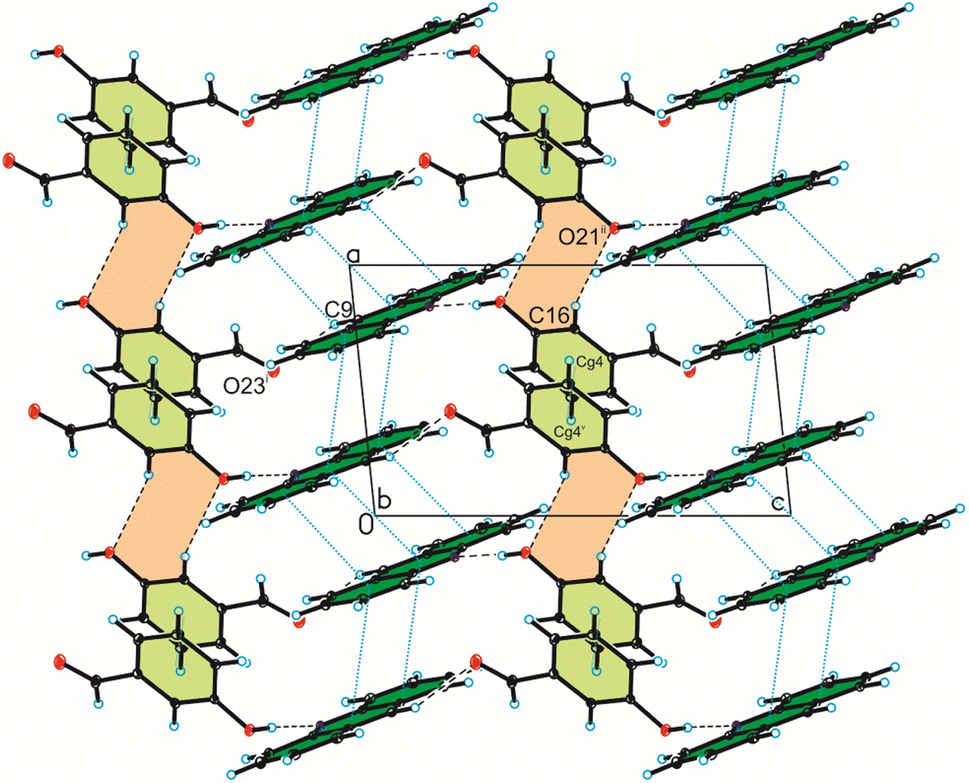
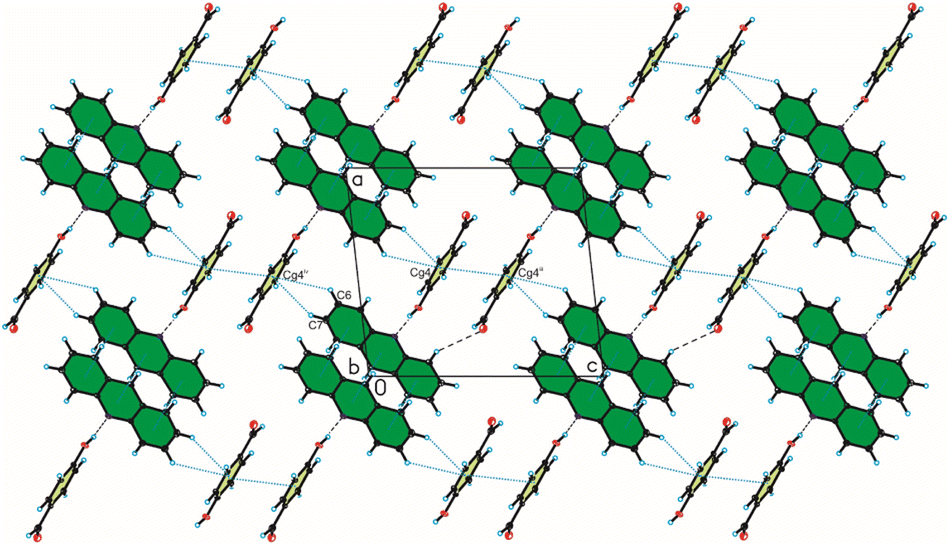
Analysis of interactions in the crystal packing of compound 1 shows that the acridine molecule interact with the 3-hydroxybenzaldehyde molecule through O(3HBA)–H⋯N(ACR) hydrogen bond [d(H21⋯N10) = 1.81(3) Å, and ∠(O21–H21⋯N10) = 175(3)°] to form heterodimer (Fig. 1a and 2, Table 2), and then homodimers are linked via C(ACR)–H⋯O(3HBA) = C hydrogen bond [d(H9⋯O23) = 2.58 Å, and ∠(C9–H9⋯O23) = 158°] to produce chains along [0 0 1] direction (Fig. 2, Table 2). The adjacent antiparallel chains interact by weak C(ACR)–H⋯O(3HBA) hydrogen bond [d(H16⋯O21) = 2.61 Å, and ∠(C16–H16⋯O21) = 146°] and π(ACR)–π(ACR) interactions between aromatic rings of acridine molecules [d(Cg⋯Cg) = 3.730(2) ÷ 3.813(3) Å] and π(3HBA)–π(3HBA) interactions between aromatic rings of 3-hydroxybenzaldehyde molecules [d(Cg⋯Cg) = 3.885(3) Å] building blocks along [0 1 1] direction (Fig. 2, Tables 2 and 4). In these blocks the acridine molecules are arranged in π-stacked columns (Fig. 3, the blocks are shown as green rectangles).
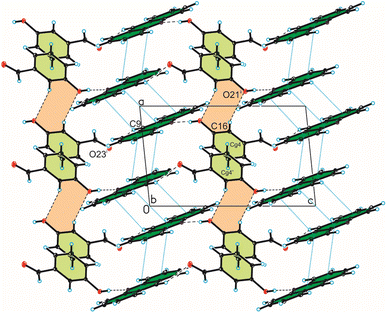 |
| | Fig. 2 Crystal packing of compound 1 viewed along the b-axis (hydrogen bonds are represented by black dashed lines, whereas π⋯π interactions by blue dotted lines). | |
Table 2 Hydrogen bonds geometry for compounds 1 and 2
| |
D–H⋯A |
d(D–H) [Å] |
d(H⋯A) [Å] |
d(D⋯A) [Å] |
∠D–H⋯A [°] |
| 1 |
O21–H21⋯N10 |
0.90(3) |
1.81(3) |
2.703(2) |
175(3) |
| C9–H9⋯O23i |
0.93 |
2.58 |
3.453(3) |
158 |
| C16–H16⋯O21ii |
0.93 |
2.61 |
3.431(5) |
146 |
| Symmetry code: (i) x, 1 + y, 1 + z; (ii) −x, −y, 1 − z |
| 2 |
O21–H21⋯N10 |
0.93(3) |
1.80(4) |
2.721(4) |
171(3) |
| C1–H1⋯O23i |
0.93 |
2.58 |
3.470(6) |
160 |
| Symmetry code: (i) −1 + x, 3/2 − y, −1/2 + z |
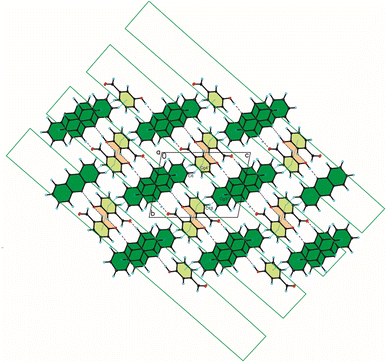 |
| | Fig. 3 Crystal packing of compound 1 viewed along the a-axis (hydrogen bonds are represented by black dashed lines, whereas C–H⋯π and π⋯π interactions by blue dotted lines). | |
The weak C(ACR)–H⋯π(3HBA) and C(3HBA)–H⋯π(ACR) interactions between aromatic rings of both acridine and 3-hydroxybenzaldehyde molecules [d(H⋯Cg) = 3.10 ÷ 3.12 Å] linked these blocks (Table 3, Fig. 3).
Table 3 C–H⋯π interactions geometry for compounds 1 and 2
| |
C–H⋯Cg |
d(H⋯Cg) [Å] |
d(C⋯Cg) [Å] |
∠C–H⋯Cg [°] |
| 1 |
C6–H6⋯Cg4vi |
3.12 |
3.771(3) |
129 |
| C18–H18⋯Cg3vii |
3.10 |
3.974(5) |
158 |
| Symmetry code: (vi) −x, 1 − y, 1 + z; (vii) 1 − x, 1 − y, 1 − z |
| 2 |
C6–H6⋯Cg4iv |
3.25 |
3.886(5) |
127 |
| C7–H7⋯Cg4iv |
3.23 |
3.871(5) |
128 |
| C8–H8⋯Cg2v |
2.86 |
3.784(5) |
176 |
| C17–H17⋯Cg3vii |
2.79 |
3.696(3) |
166 |
| Symmetry code: (iv) 1 − x, −1/2 + y, ½ − z; (v) x, 3/2 − y, −1/2 + z; (vii) 1 − x, 1/2 + y, 1/2 − z |
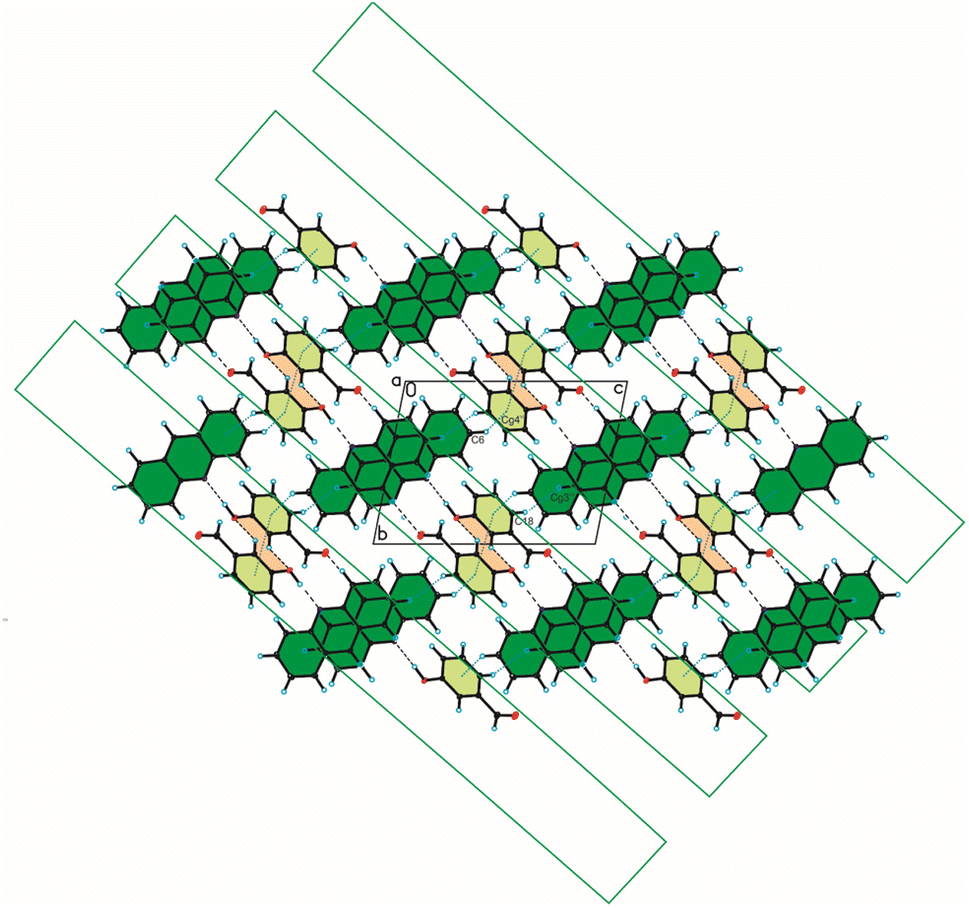
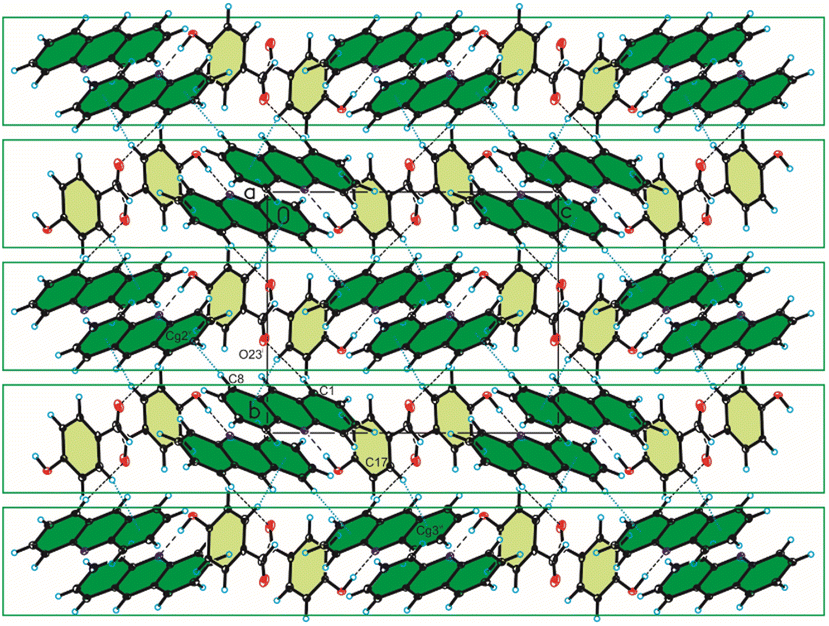
In the crystal packing of compound 2, the acridine molecule interact with the 4-hydroxybenzaldehyde molecule by O(4HBA)–H⋯N(ACR) hydrogen bond [d(H21⋯N10) = 1.80(4) Å, and ∠(O21–H21⋯N10) = 171(3)°] to form heterodimer (Fig. 1a, Table 2), but neighbouring homodimers are connected via π(ACR)–π(ACR) interactions between acridine molecules [d(Cg⋯Cg) = 3.677(2) ÷ 4.156(2) Å] to form heterotetramers (Fig. 4, Table 4). However, the acridine molecules do not form the π-stacked columns. The adjacent heterotetramers interact by π(4HBA)–π(4HBA) interaction between the aromatic rings of 4-hydroxybenzaldehyde molecules [d(Cg⋯Cg) = 3.905(2) Å] and weak C(ACR)–H⋯π(4HBA) interactions [d(H⋯Cg) = 3.23 ÷ 3.25 Å] building blocks along c-axis (Fig. 4 (Fig. 3, the blocks are shown as green rectangles, Tables 3 and 4). Finally, the neighbouring blocks are linked by weak C(ACR)–H⋯O(4HBA) = C hydrogen bond [d(H1⋯O23) = 2.58 Å, and ∠(C1–H1⋯O23) = 160°] and short C(ACR)–H⋯π(4HBA) and C(ACR)–H⋯π(ACR) interactions involving aromatic rings of 4-hydroxybenzaldehyde and acridine molecules, respectively [d(X⋯Cg) = 2.79÷2.86 Å] (Fig. 5, Tables 2 and 4).
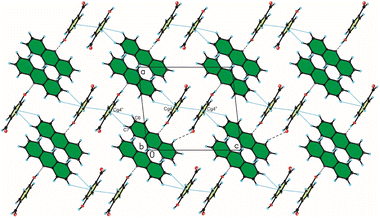 |
| | Fig. 4 Crystal packing of compound 2 viewed along the b-axis (hydrogen bonds are represented by black dashed lines, whereas C–H⋯π and π⋯π interactions by blue dotted lines). | |
Table 4 π–π interactions geometry for compounds 1 and 2
| |
CgIa |
CgJa |
CgI⋯CgJb [Å] |
Dihedral anglec [°] |
Interplanar distanced [Å] |
Offsete [Å] |
| Cg represents the centre of gravity of the rings. Cg⋯Cg is the distance between ring centroids. The dihedral angle is that between the mean planes of CgI and CgJ. The interplanar distance is the perpendicular distance from CgI to ring J. The offset is the perpendicular distance from ring I to ring J. |
| 1 |
1 |
1iii |
3.790(1) |
0.0(1) |
3.447(1) |
1.574 |
| 1 |
2iii |
3.813(1) |
1.5(1) |
3.452(1) |
1.619 |
| 1 |
2iv |
3.730(1) |
1.5(1) |
3.416(1) |
1.499 |
| 2 |
1iii |
3.813(1) |
1.5(1) |
3.422(1) |
1.685 |
| 2 |
1iv |
3.731(1) |
1.5(1) |
3.438(1) |
1.448 |
| 2 |
2iv |
3.794(1) |
0.0(1) |
3.440(1) |
1.600 |
| 2 |
3iii |
3.812(1) |
3.2(1) |
3.423(1) |
1.677 |
| 3 |
2iii |
3.812(1) |
3.2(1) |
3.481(1) |
1.553 |
| 4 |
4v |
3.885(1) |
0.0(1) |
3.425(1) |
1.834 |
| Symmetry code: (iii) −x, 1 − y, 2 − z, (iv) 1 − x, 1 − y, 2 − z, (v) 1 − x, −y, 1 − z |
| 2 |
1 |
1ii |
3.677(2) |
0.0(2) |
3.492(2) |
1.153 |
| 1 |
2ii |
4.156(2) |
0.2(2) |
3.489(2) |
2.256 |
| 2 |
3ii |
3.698(2) |
0.5(2) |
3.486(2) |
1.234 |
| 4 |
4iii |
3.905(2) |
0.0(2) |
3.495(2) |
1.742 |
| Symmetry code: (ii) −x, 2 − y, −z, (iii) 1 − x, 2 − y, 1 − z |
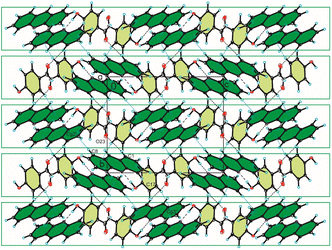 |
| | Fig. 5 Crystal packing of compound 2 viewed along the a-axis (hydrogen bonds are represented by black dashed lines, whereas C–H⋯π interactions by blue dotted lines). | |
3.2. TG/DSC studies
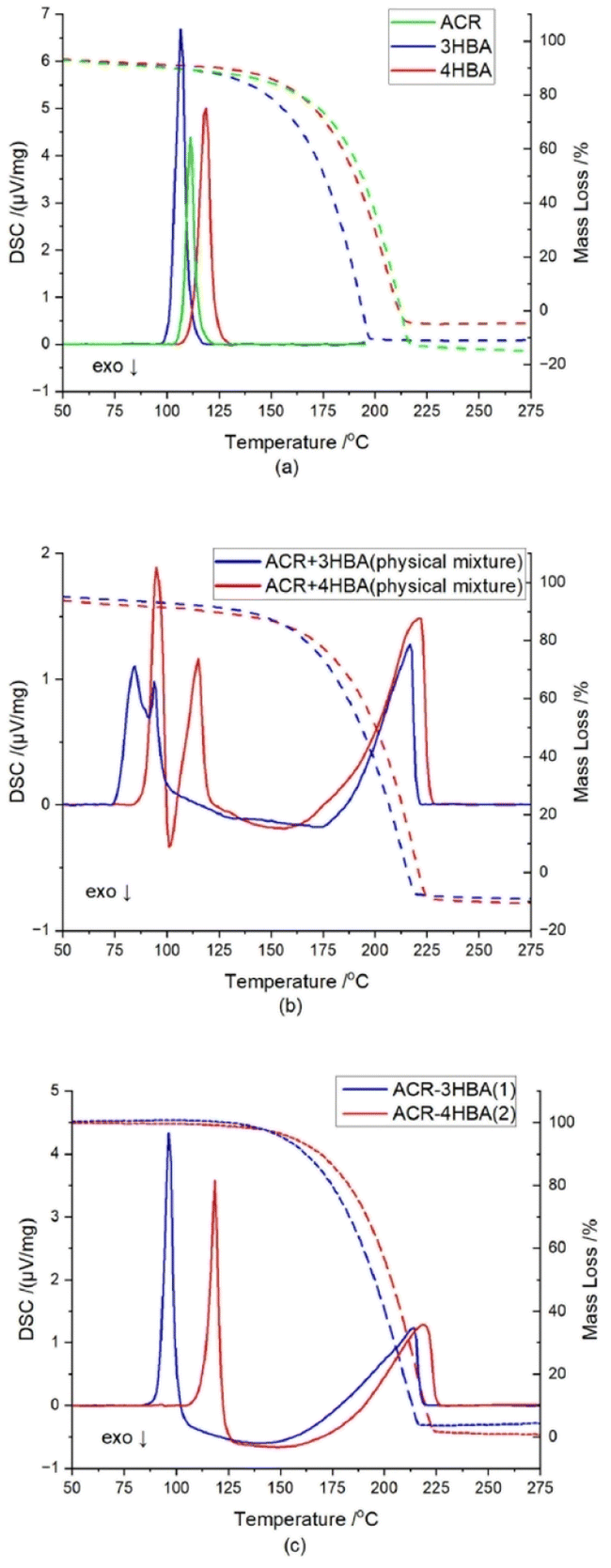
The thermal behaviour of a pure coformers, physical mixture of coformers at 1:1 molar ratio, and cocrystals of 1 and 2 are shown in Fig. 6. An analysis of TG and DSC diagrams for acridine, 3-hydroxybenzaldehyde and 4-hydroxybenzaldehyde shows that endothermic peaks are observed at ∼111.2 °C, ∼106.5 °C, and ∼119.0 °C, respectively, which represent their melting points. For a physical mixture of acridine and 3-hydroxybenzaldehyde, the first endothermic peak is observed at ∼84.5 °C, immediately followed by an exothermic peak at ∼91.5 °C, whereas the second endothermic peak is detected at ∼94.0 °C. In the case physical mixture, the endotherms are attributed to eutectic and cocrystal melting respectively, while exothermic peak suggesting cocrystal formation.40–42 In the cocrystal 1, these melting temperatures are close to each other, thus, probably there was a merge of melting temperatures of eutectic and cocrystal.40–44 Similar profile is observed for the curve obtained for the physical mixture of acridine and 4-hydroxybenzaldehyde. The first endothermic peak is observed at ∼95.0 °C, which is followed by an exothermic peak at ∼101.5 °C, whereas the second endothermic peak is observed at ∼115.0 °C. On the DSC curves for the cocrystals of 1 and 2, sharp endothermic peaks are observed at ∼96.5 °C, and ∼118.5 °C, respectively, which represent their melting points.
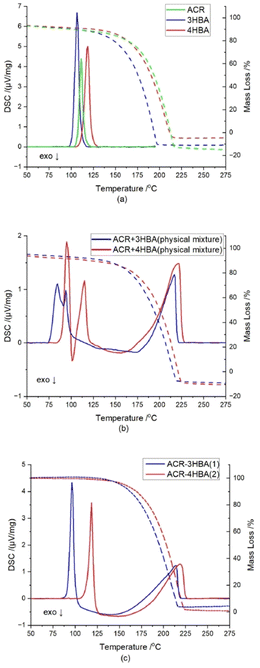 |
| | Fig. 6 TG/DSC curves of (a) acridine (green lines), 3-hydroxybenzaldehyde (blue lines) and 4-hydroxybenzaldehyde (red lines); (b) physical mixtures of acridine and 3-hydroxybenzaldehyde (blue lines) and acridine and 4-hydroxybenzaldehyde (red lines) at 1:1 molar ratio; (c) cocrystal of acridine with 3-hydroxybenzaldehyde (1) (blue lines) and cocrystal of acridine with 4-hydroxybenzaldehyde (2) (red lines). | |
3.3. FTIR spectroscopic studies
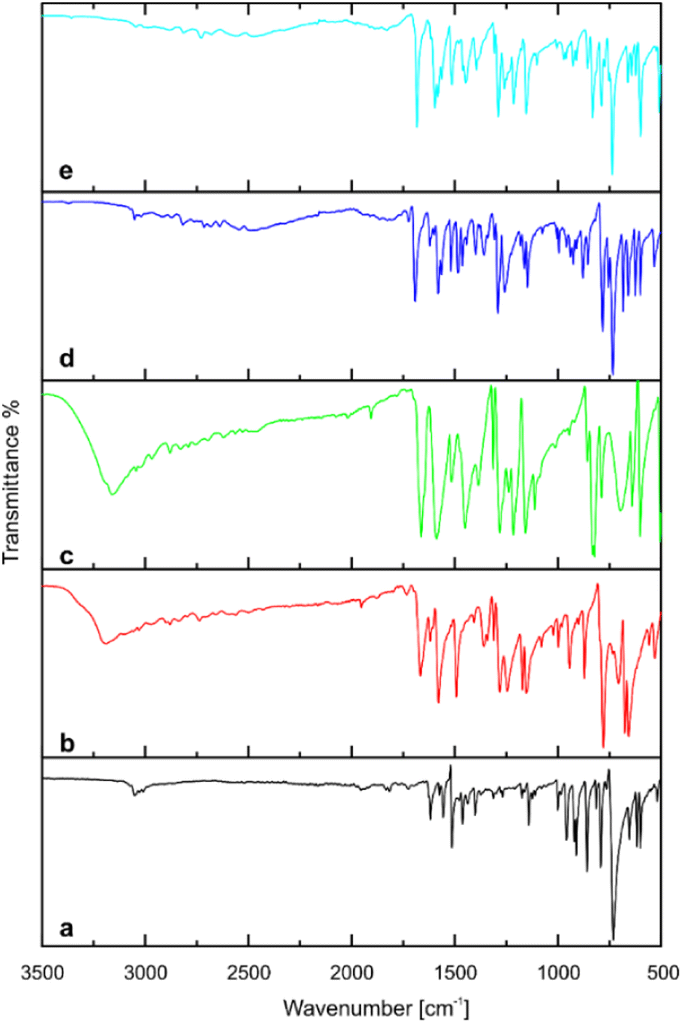
FTIR spectra of pure coformers and title compounds are displayed at Fig. 7. For acridine (spectrum a), the bands at 3052.6, 1620–1400, 731.5 cm−1 correspond to aromatic C–H stretching vibrations, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN skeleton stretching vibrations, and C–H out-of-plane bending vibrations, respectively. The FTIR spectrum of 3-hydroxybenzaldehyde (spectrum b) shows characteristic bands at 3195.0 cm−1 (broad OH stretch), 1666.3 cm−1 (CO stretch) and 780.1 cm−1 (C–H out-of-plane bend). The corresponding bands in the spectrum of 4-hydroxybenzaldehyde (spectrum c) appears at 3160.0 cm−1, 1663.0 cm−1 and 822.5 cm−1. The position of the last one is related to the aromatic ring substitution. In both aldehydes, the stretching band of OH partially overlaps with C–H stretching vibrations of aldehyde group (2900–2650 cm−1). With cocrystals 1 and 2, the band assigned to the carbonyl group of hydroxybenzaldehyde component shifts to 1691.8 and 1682.8 cm−1, respectively (spectra d and e), suggesting its less involvement in hydrogen bonding compared to pure hydroxybenzaldehydes. Interestingly, the band attributed to the stretching vibrations of the hydroxyl group (A-type) of hydroxybenzaldehydes vanished, but several bands appeared in the range of 3000–2000 cm−1. These bands can be attributed to Fermi resonance between stretch and in-plane bending of hydrogen bonded O–H groups (B-type) and are usually determined for strong hydrogen-bonded complexes.38,39
C and CN skeleton stretching vibrations, and C–H out-of-plane bending vibrations, respectively. The FTIR spectrum of 3-hydroxybenzaldehyde (spectrum b) shows characteristic bands at 3195.0 cm−1 (broad OH stretch), 1666.3 cm−1 (CO stretch) and 780.1 cm−1 (C–H out-of-plane bend). The corresponding bands in the spectrum of 4-hydroxybenzaldehyde (spectrum c) appears at 3160.0 cm−1, 1663.0 cm−1 and 822.5 cm−1. The position of the last one is related to the aromatic ring substitution. In both aldehydes, the stretching band of OH partially overlaps with C–H stretching vibrations of aldehyde group (2900–2650 cm−1). With cocrystals 1 and 2, the band assigned to the carbonyl group of hydroxybenzaldehyde component shifts to 1691.8 and 1682.8 cm−1, respectively (spectra d and e), suggesting its less involvement in hydrogen bonding compared to pure hydroxybenzaldehydes. Interestingly, the band attributed to the stretching vibrations of the hydroxyl group (A-type) of hydroxybenzaldehydes vanished, but several bands appeared in the range of 3000–2000 cm−1. These bands can be attributed to Fermi resonance between stretch and in-plane bending of hydrogen bonded O–H groups (B-type) and are usually determined for strong hydrogen-bonded complexes.38,39
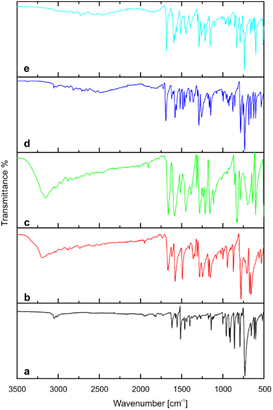 |
| | Fig. 7 FTIR spectra of (a) acridine, (b) 3-hydroxybenzaldehyde, (c) 4-hydroxybenzaldehyde, (d) cocrystal of acridine with 3-hydroxybenzaldehyde (1), (e) cocrystal of acridine with 4-hydroxybenzaldehyde (2). | |
3.4. An analysis of crystal packing and melting points
The arrangement of coformer molecules in the crystals of the title compounds result in different degree of filling the crystal space. Compound 1 have a higher Kitaigorodskii type of packing index (with the percentage of filled space equal to 68.1 and 66.8% for cocrystal 1 and 2, respectively) and crystal density (1.292 and 1.272 g cm−3, for cocrystal 1 and 2, respectively) than compound 2.34 This confirms that multicomponent crystals in which acridine molecules are arranged in π-stacked columns have a higher degree of crystal packing and density, than crystals in which acridine molecules are in the other arrangement.29–31 The influence of the crystal packing and type and number of interactions in the crystals on their melting points have been also described in the literature.45–48 Analysis of melting points shows that the compound 1 melts at a temperature lower than its separate components (mp = 96.5 °C), whereas the compound 2 melts at a temperature higher than acridine, but at lower temperature than 4-hydroxybenzaldehyde (mp = 118.5 °C, Table 5).
Table 5 Melting points of pure coformers and compounds 1 and 2
| Compound |
Melting point [°C] |
| Acridine |
111.2 |
| 3-Hydroxybenzaldehyde |
106.5 |
| 4-Hydroxybenzaldehyde |
119.0 |
| Cocrystal of acridine with 3-hydroxybenzaldehyde (1) |
96.5 |
| Cocrystal of acridine with 4-hydroxybenzaldehyde (2) |
118.5 |
Even though the same types of interactions occur in the crystal packing of compounds 1 and 2, the number of interactions between adjacent blocks, especially C–H⋯π interactions, is greater for compound 2 than for compound 1, which may explain the higher melting point of cocrystal acridine with 4-hydroxybenzaldehyde. This also confirms the other rule observed in our previous studies, that ortho-substituted benzoic acids have a lower melting point than acridine, while meta-substituted benzoic acids have a higher melting point than acridine.29
4. Conclusions
Based on experiments carried out using SCXRD, FTIR, TG/DSC methods it was demonstrated that acridine forms cocrystals with two isomers of hydroxybenzaldehyde: 3-hydroxybenzaldehyde and 4-hydroxybenzaldehyde in a molar ratio of 1:1. The crystals of compounds 1 and 2 were obtained using a slow evaporation of solvents at room temperature. Analysis of the melting points shows that the compound 1 melts at a lower temperature than its separate coformers, whereas the compound 2 melts at a higher temperature than acridine, but at lower temperature than 4-hydroxybenzaldehyde. The cocrystal of acridine with 3-hydroxybenzaldehyde (1) crystallizes in the triclinic P space group whereas the cocrystal of acridine with 4-hydroxybenzaldehyde (2) crystallizes in the monoclinic P21/c space group. In both cocrystals O–H⋯N heterodimers occur, however differences in the crystal packing of both cocrystals are observed. In the cocrystal of 1, the molecules of acridine and 3-hydroxybenzaldehyde are arranged in the pattern of antiparallel chains which are linked through C(ACR)–H⋯O(3HBA) hydrogen bond and π(ACR)–π(ACR) and π(3HBA)–π(3HBA) interactions, to form blocks along [0 1 1] direction. In these blocks the acridine molecules are arranged in π-stacked columns. In turn, in the cocrystal of 2, the acridine and 4-hydroxybenzaldehyde molecules are arranged in heterotetramers, but the acridine molecules do not form the π-stacked columns. The adjacent heterotetramers interact by π(4HBA)–π(4HBA) and C(ACR)–H⋯π(4HBA) interactions building blocks along [0 0 1] direction. In the crystal packing of compound 1, the adjacent blocks are linked via C(ACR)–H⋯π(3HBA) and C(3HBA)–H⋯π(ACR) interactions, whereas in the crystal packing of compound 2 the neighbouring blocks are linked by C(ACR)–H⋯O(4HBA) = C hydrogen bond, and C(ACR)–H⋯π(4HBA) and C(ACR)–H⋯π(ACR) interactions. Differences in the arrangement of coformers molecules in both cocrystals result in higher degree of filling the crystal space and crystal density for cocrystal of 1. The FTIR measurements shows that the band attributed to the stretching vibrations of the hydroxyl group of hydroxybenzaldehydes vanished, but several bands appeared in the range of 3000–2000 cm−1. These bands can be attributed to Fermi resonance between stretch and in-plane bending of hydrogen bonded O–H groups and are usually determined for strong hydrogen-bonded complexes.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This research was funded by: Research of Young Scientists grant (BMN) no. 539-T080-B056-23 (University of Gdańsk) and DS/530-8228-D738-23 (University of Gdańsk).
Notes and references
- G. R. Desiraju, Crystal engineering: a holistic view, Angew. Chem., Int. Ed. Engl., 2007, 46(44), 8342–8356 CrossRef CAS PubMed.
- D. Braga, L. Casali and F. Grepioni, The Relevance of Crystal Forms in the Pharmaceutical Field: Sword of Damocles or Innovation Tools?, Int. J. Mol. Sci., 2022, 23(16), 9013 CrossRef CAS PubMed.
- Q. Zhou, Z. Tan, D. Yang, J. Tu, Y. Wang, Y. Zhang, Y. Liu and G. Gan, Improving the Solubility of Aripiprazole by Multicomponent Crystallization, Crystals, 2021, 11(4), 343 CrossRef CAS.
- P. J. Grobelny, A. Mukherjee and G. R. Desiraju, Drug-drug co-crystals: Temperature-dependent proton mobility in the molecular complex of isoniazid with 4-aminosalicylic acid, CrystEngComm, 2011, 13, 4358–4364 RSC.
- M. L. Cheney, D. R. Weyna, N. Shan, M. Hanna, L. Wojtas and M. J. Zaworotko, Coformer Selection in Pharmaceutical Cocrystal Development: a Case Study of a Meloxicam Aspirin Cocrystal That Exhibits Enhanced Solubility and Pharmacokinetics, J. Pharm. Sci., 2011, 100(6), 2172–2181 CrossRef CAS PubMed.
- N. Shan and M. J. Zaworotko, The role of cocrystals in pharmaceutical science, Drug Discovery, 2008, 13(9–10), 440–446 CAS.
- D. J. Good and N. Rodriguez-Hornedo, Solubility advantage of pharmaceutical cocrystals, Cryst. Growth Des., 2009, 9(5), 2252–2264 CrossRef CAS.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Pharmaceutical cocrystals: Along the path to improved medicines, Chem. Commun., 2016, 52(4), 640–655 RSC.
- D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Synthesis and structural characterization of cocrystals and pharmaceutical cocrystals: mechanochemistry vs. slow evaporation from solution, Cryst. Growth Des., 2009, 9(2), 1106–1123 CrossRef CAS.
- M. M. Patel, M. D. Mali and S. K. Patel, Bernthsen synthesis, antimicrobial activities and cytotoxicity of acridine derivatives, Bioorg. Med. Chem. Lett., 2010, 20(21), 6324–6326 CrossRef CAS PubMed.
- M. Tonelli, G. Vettoretti, B. Tasso, F. Novelli, V. Boido, F. Sparatore, B. Busonera, A. Ouhtit, P. Farci, S. Blois, G. Giliberti and P. La Colla, Acridine derivatives as anti-BVDV agents, Antiviral Res., 2011, 91(2), 133–141 CrossRef CAS PubMed.
- S. Hassan, D. Laryea, H. Mahteme, J. Felth, M. Fryknäs, W. Fayad, S. Linder, L. Rickardson, J. Gullbo, W. Graf, L. Påhlman, B. Glimelius, P. Larsson and P. Nygren, Novel activity of acriflavine against colorectal cancer tumor cells, Cancer Sci., 2011, 102(12), 2206–2213 CrossRef CAS PubMed.
- P. Belmont, J. Bosson, T. Godet and M. Tiano, Acridine and Acridone Derivatives, Anticancer Properties and Synthetic Methods: Where Are We Now?, Anti-Cancer Agents Med. Chem., 2007, 7(2), 139–169 CrossRef CAS PubMed.
- W. A. Denny, Acridine derivatives as chemotherapeutic agents, Curr. Med. Chem., 2002, 9(18), 1655–1665 CrossRef CAS PubMed.
- L. R. Ferguson and W. A. Denny, The genetic toxicology of acridines, Mutat. Res., Rev. Genet. Toxicol., 1991, 258, 123–160 CrossRef CAS PubMed.
- G. P. Moloney, D. P. Kelly and P. Mack, Synthesis of Acridine-based DNA Bis-intercalating Agents, Molecules, 2001, 6(3), 230–243 CrossRef CAS.
- S. Nafisi, A. A. Saboury, N. Keramat, J. F. Neault and H. A. Tajmir-Riahi, Stability and structural features of DNA intercalation with ethidium bromide, acridine orange and methylene blue, J. Mol. Struct., 2007, 827(1–3), 35–43 CrossRef CAS.
- G. P. Moloney, D. P. Kelly and P. Mack, Synthesis of acridine-based DNA bis-intercalating agents, Molecules, 2001, 6(3), 230–243 CrossRef CAS.
- A. Sikorski, K. Krzymiński, A. Konitz and J. Błażejowski, 2-Ethylphenyl acridine-9-carboxylate and 2,5-dimethylphenyl acridine-9-carboxylate, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o52 Search PubMed.
- A. Sikorski, K. Krzymiński, A. Niziołek and J. Błażejowski, 9-(2,6-Difluorophenoxycarbonyl)-10-methylacridinium trifluoromethane sulfonate and its precursor 2,6-difluorophenyl acridine-9-carboxylate: C-H⋯O, C-F⋯π, S-O⋯π and π-π stacking interactions, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o690–o694 CrossRef PubMed.
- B. S. Kong, S. J. Im, Y. J. Lee, Y. H. Cho, J. W. Byun, C. R. Ku and E. J. Lee, Vasculoprotective Effects of 3-hydroxybenzaldehyde against VSMCs proliferation and ECs inflammation, PLoS One, 2016, 11(3), e0149394 CrossRef PubMed.
- B. Shin, C. Park, J. A. Imlay and W. Park, 4-Hydroxybenzaldehyde sensitizes Acinetobacter baumannii to amphenicols, Appl. Microbiol. Biotechnol., 2018, 102(5), 2323–2335 CrossRef CAS PubMed.
- C. W. Kang, Y. E. Han, J. Kim, J. H. Oh, Y. H. Cho and E. J. Lee, 4-Hydroxybenzaldehyde accelerates acute wound healing through activation of focal adhesion signalling in keratinocytes, Sci. Rep., 2017, 7(1), 14192 CrossRef PubMed.
- S. Park, D. S. Kim and S. Kang, Gastrodia elata blume water extracts improve insulin resistance by decreasing body fat in diet-induced obese rats: Vanillin and 4-hydroxybenzaldehyde are the bioactive candidates, Eur. J. Nutr., 2011, 50(2), 107–118 CrossRef CAS PubMed.
- J. Lee, J. W. Choi, H. Y. Han, W. S. Kim, H. Y. Song, E. B. Byun, Y. H. Lee and J. M. Yuk, 4-hydroxybenzaldehyde restricts the intracellular growth of Toxoplasma gondii by inducing SIRT1-mediated autophagy in macrophages, Korean J. Parasitol., 2020, 58(1), 7–14 CrossRef CAS PubMed.
- K. Y. Chen, Y. J. Chen, C. J. Cheng, K. Y. Jhan, C. Chiu and L. C. Wang, 3-Hydroxybenzaldehyde and 4-Hydroxybenzaldehyde enhance survival of mouse astrocytes treated with Angiostrongylus cantonensis young adults excretory/secretory products, Biomed. J., 2021, 44(6 Suppl 2), S258–S266 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- D. Braga, F. Grepioni, L. Lucia Maini, P. P. Mazzeo and K. Rubini, Solvent-free preparation of co-crystals of phenazine and acridine with vanillin, Thermochim. Acta, 2010, 507(08), 1–8 CrossRef.
- K. Kowalska, D. Trzybiński and A. Sikorski, Influence of the halogen substituent on the formation of halogen and hydrogen bonding in co-crystals formed from acridine and benzoic acids, CrystEngComm, 2015, 17, 7199–7212 RSC.
- A. Sikorski and D. Trzybiński, Synthesis and structural characterization of a cocrystal salt containing acriflavine and 3,5-dinitrobenzoic acid, Tetrahedron Lett., 2014, 55, 2253–2255 CrossRef CAS.
- D. Trzybiński and A. Sikorski, Solvent-bridged frameworks of hydrogen bonds in crystals of 9-aminoacridinium halides, CrystEngComm, 2013, 15, 6808–6818 RSC.
- CrysAlis CCD and CrysAlis RED, Version 1.171.36.24, Oxford Diffraction Ltd., Yarnton, UK, 2012 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. K. Johnson, ORTEP II, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1976 Search PubMed.
- S. Motherwell and S. Clegg, PLUTO-78, Program for Drawing and Molecular Structure, University of Cambridge, Cambridge, UK, 1978 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. Martínez-Felipe, A. G. Cook, M. J. Wallage and C. T. Imrie, Hydrogen bonding and liquid crystallinity of low molar mass and polymeric mesogens containing benzoic acids: a variable temperature Fourier transform infrared spectroscopic study, Phase Transitions, 2014, 87(12), 1191–1210 CrossRef.
- S. E. Odinokov and A. V. Iogansen, Torsional γ(OH) vibrations, Fermi resonance [2γ(OH) ⇐ ν(OH)] and isotopic effects in i.r. spectra of H-complexes of carboxylic acids with strong bases, Spectrochim. Acta, Part A, 1972, 28(12), 2343–2350 CrossRef CAS.
- E. Lu, L. N. Rodriguez-Hornedob and R. Suryanarayanan, A rapid thermal method for cocrystal screening, CrystEngComm, 2008, 10, 665–668 RSC.
- H. Yamashita, Y. Hirakura, M. Yuda, T. Teramura and K. Terada, Detection of Cocrystal Formation Based on Binary Phase Diagrams Using Thermal Analysis, Pharm. Res., 2013, 30, 70–80 CrossRef CAS PubMed.
- Z. Zhou, H. M. Chan, H. H. Y. Sung, H. H. Y. Tong and Y. Zheng, Identification of New Cocrystal Systems with Stoichiometric Diversity of Salicylic Acid Using Thermal Methods, Pharm. Res., 2016, 33, 1030–1039 CrossRef CAS PubMed.
- P. P. Mazzeo, M. Prencipe, T. Feiler, F. Emmerling and A. Bacchi, On the Mechanism of Cocrystal Mechanochemical Reaction via Low Melting Eutectic: A Time-Resolved In Situ Monitoring Investigation, Cryst. Growth Des., 2022, 22(7), 4260–4267 CrossRef CAS PubMed.
- A. N. Manin, A. P. Voronin, K. V. Drozd, N. G. Manin, A. Bauer-Brandl and G. L. Perlovich, Cocrystal screening of hydroxybenzamides with benzoic acid derivatives: A comparative study of thermal and solution-based methods, Eur. J. Pharm. Sci., 2014, 65, 56–64 CrossRef CAS PubMed.
- D. Ejarque, T. Calvet, M. Font-Bardia and J. Pons, Cocrystals Based on 4,4′-bipyridine: Influence of Crystal Packing on Melting Point, Crystals, 2021, 11, 191 CrossRef CAS.
- D. Cinčić, T. Friščić and W. Jones, Isostructural Materials Achieved by Using Structurally Equivalent Donors and Acceptors in Halogen-Bonded Cocrystals, Chem.–Eur. J., 2008, 14, 747–753 CrossRef PubMed.
- L. Orola, M. V. Veidis, I. Mutikainen and I. Sarcevica, Neutral and Ionic Supramolecular Complexes of Phenanthridine and Some Common Dicarboxylic Acids: Hydrogen Bond and Melting Point Considerations, Cryst. Growth Des., 2011, 11, 4009–4016 CrossRef CAS.
- R. Montis, M. B. Hursthouse, H. C. S. Chan, J. Kendrick and F. J. Leusen, Experimental and theoretical investigations of the polymorphism of 5-chloroacetoxybenzoic acid (5-chloroaspirin), CrystEngComm, 2012, 14, 1672–1680 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Artur Sikorski
and
Artur Sikorski