
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsopropenyl phosphate as an atom efficient reagent for phosphorylation of alcohols with catalytic base†
Jens Wéry ,
Igor Beckers and
Dirk E. De Vos*
,
Igor Beckers and
Dirk E. De Vos*
cMACS, KU Leuven, Celestijnenlaan 200F, Leuven 3001, Belgium. E-mail: dirk.devos@kuleuven.be
First published on 10th May 2023
Abstract
The atom efficient transesterification of phosphate esters with catalytic base was investigated using an isopropenyl leaving group, generating acetone as the only by-product. The reaction proceeds in good yields at room temperature, with excellent chemoselectivity towards primary alcohols. Mechanistic insights were obtained by obtaining kinetic data using in operando NMR-spectroscopy.
Phosphate esters (PEs) are of vital importance in biological systems as a main constituent of genetic material, phospholipid membranes and as key signalling molecules.1,2 Due to their biological importance, PEs are also desired synthetic targets, either in the pharmaceutical industry for drug delivery (e.g. pronucleotides or phospholipids) or in the chemical industry for flame retardants, plasticizers or agricultural products.3–5 In traditional organic chemistry, the synthesis relies on reactive P(III) intermediates (e.g. PCl3) which readily undergo nucleophilic substitution towards phosphite esters; the latter can be converted into phosphate esters via a subsequent oxidation step. Consequently, the air sensitivity of phosphites and thus the need for protecting groups or mild oxidation conditions limit the efficiency of these reactions. Therefore, in contrast to the P(III) chemistry, the redox-neutral conversion of less reactive P(V) compounds has recently gained interest as a direct and more controllable synthetic approach.6–9 Within P(V) chemistry, synthesis mainly relies on phosphorus pentoxide, pyrophosphates and chlorophosphates. Even though multiple reagents exist, industry opts for the use of chlorophosphates (e.g. POCl3) which exhibit high reactivity.10 However, due to their high reactivity, stepwise substitution is challenging and difficult to control.6 This concern illustrates the need for better and greener alternatives for selective transesterification reactions. Therefore, reports in literature demonstrate the use of organic leaving groups (LGs) instead of chlorine (Fig. 1).11–14
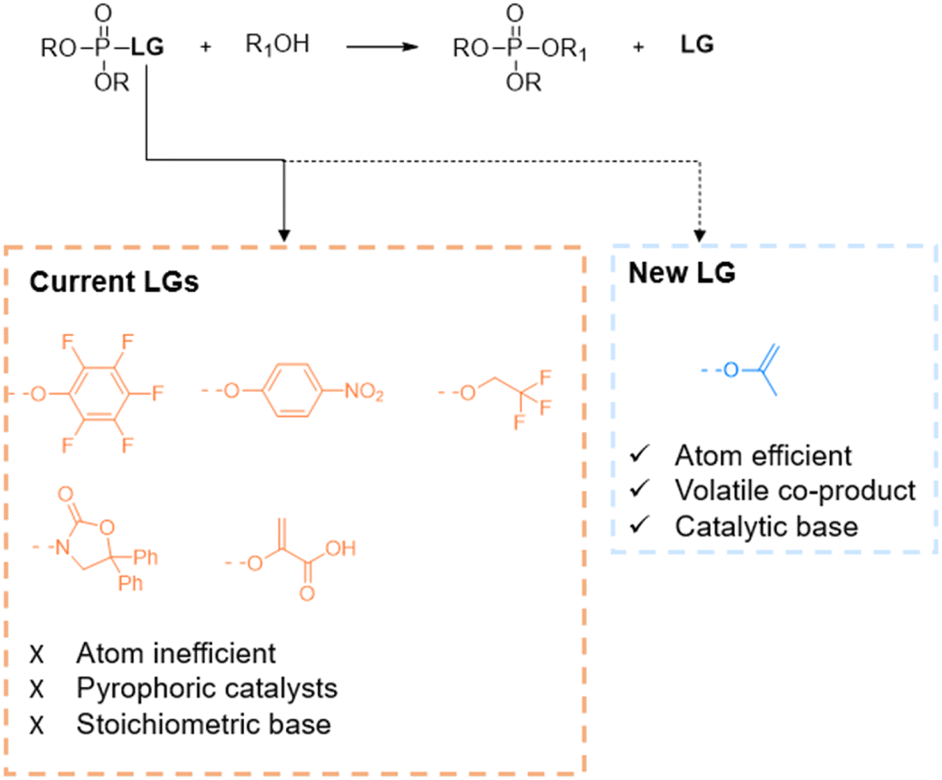 | ||
| Fig. 1 Leaving group strategies in the phosphorylation of alcohols. | ||
Current alternative leaving groups are para-nitrophenyl, pentafluorophenyl, trifluoroethyl, enol pyruvate or N-phosphorylated oxazolidinones (Fig. 1).11–15 Although aforementioned LGs lead to good yields and selectivity, the phosphorylation conditions require mostly equimolar amounts of bases and/or pyrophoric catalysts. Moreover, while being large and often polyfluorinated compounds, these LGs produce stoichiometric amounts of waste which demands additional purification steps and thus decreases the sustainability of the reaction. In terms of green chemistry, these transesterification reactions exhibit poor atom efficiency.16
In this work, we report on the catalytic transesterification of enol phosphates, bearing small leaving groups that allow stringent control of the reaction selectivity. In particular, dimethyl isopropenyl phosphate (iPP), which is commercially available, is applied for phosphorylation of alcohols. Enol phosphates can also easily be prepared by a Perkow reaction of trimethyl phosphite and α-haloacetone or halogen free alcohol-derived leaving groups as recently reported by Shi et al.17 Enol phosphates (EPs) are either used in organic chemistry for (hydro)alkenylation reactions through C–H activation18 or as reagent for epoxidations with dimethyl dioxirane as a mild oxidant.19,20 EPs also occur naturally as important biological intermediates such as phosphoenolpyruvate which plays an important role in ATP synthesis.21 Although vinyl esters are well known reactants for acylation reactions with tunable selectivity for primary or secondary alcohols,22–24 enol phosphates remain underexplored as phosphorylating reagent.15 The advantage of vinyl or isopropenyl esters relies on the formation of the alcohols after transesterification, which tautomerize into acetaldehyde or acetone respectively, and this makes the reaction irreversible. Next to improved atom efficiency, the leaving group can be easily separated by evaporation after reaction. We moreover show that the leaving group enables the use of catalytic amounts of base in the transesterification reaction, thereby avoiding the need for stoichiometric base additives. In operando NMR spectroscopy enabled us to study in depth the reaction kinetics and the catalytic mechanism.
To investigate the desired transesterification, iPP (Scheme 1) was dissolved in the polar aprotic solvent THF in the presence of different bases and 1-butanol at room temperature, as shown in Table 1. When different nucleophilic N-containing bases were added in equimolar amounts, no conversion of the starting material was observed (entries 1–3). When the base was changed to a non-nucleophilic N-containing base like DBU (entry 4) low conversion to the desired product dimethylbutyl phosphate (dMBP) was obtained. Increasing the nucleophilicity of 1-butanol by deprotonation with a stronger base like potassium or sodium tert-butoxide in substoichiometric (5 mol%) amount led to major improvement of conversion, with 68% of product formed with potassium tert-butoxide (entry 5). In contrast, sodium tert-butoxide did not result in good conversion (entry 6). Slightly increasing the concentration of t-BuOK (7.5 mol%) resulted in almost full conversion of the P-donor with 79% product yield (entry 7). This base screening illustrates the need for a strong non-nucleophilic base like t-BuOK which creates a highly nucleophilic alkoxide that can consecutively attack the isopropenyl phosphate and transesterify the leaving group. Since THF is a suitable solvent for dissolving t-BuOK, analogous ether solvents were tested; these led to similar conversions as obtained in THF (entries 8–9). In contrast, a less polar ether like cyclopentyl methyl ether (CPME) lowered the conversion and product yields probably due to lower base solubility (entry 10).
| Entry | Solvent | Base | Productb (%) | Conversionc (%) |
|---|---|---|---|---|
| a Reaction conditions: 0.2 mmol dimethyl isopropenyl phosphate, 400 μl solvent, 100 μl 1-butanol, base, 3 h, room temperature, argon atmosphere.b 31P NMR yield based on quantity of iPP supplied.c Conversion of iPP.d After 24 h. | ||||
| 1 | THF | Et3N (1 eq.) | 0 | <1 |
| 2 | THF | DIPEA (1 eq.) | 0 | <1 |
| 3 | THF | DABCO (1 eq.) | 0 | <1 |
| 4 | THF | DBU (1 eq.) | <5 | 20 |
| 5 | THF | t-BuOK (5 mol%) | 68 | 85 |
| 6 | THF | t-BuONa (5 mol%) | 48 | 51d |
| 7 | THF | t-BuOK (7.5 mol%) | 79 | 98 |
| 8 | MeTHF | t-BuOK (7.5 mol%) | 81 | 99 |
| 9 | 1,4-Dioxane | t-BuOK (7.5 mol%) | 80 | 100 |
| 10 | CPME | t-BuOK (7.5 mol%) | 52 | 82 |
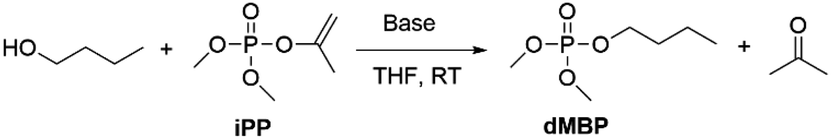 | ||
| Scheme 1 Phosphorylation of 1-butanol with dimethyl isopropenyl phosphate (iPP). | ||
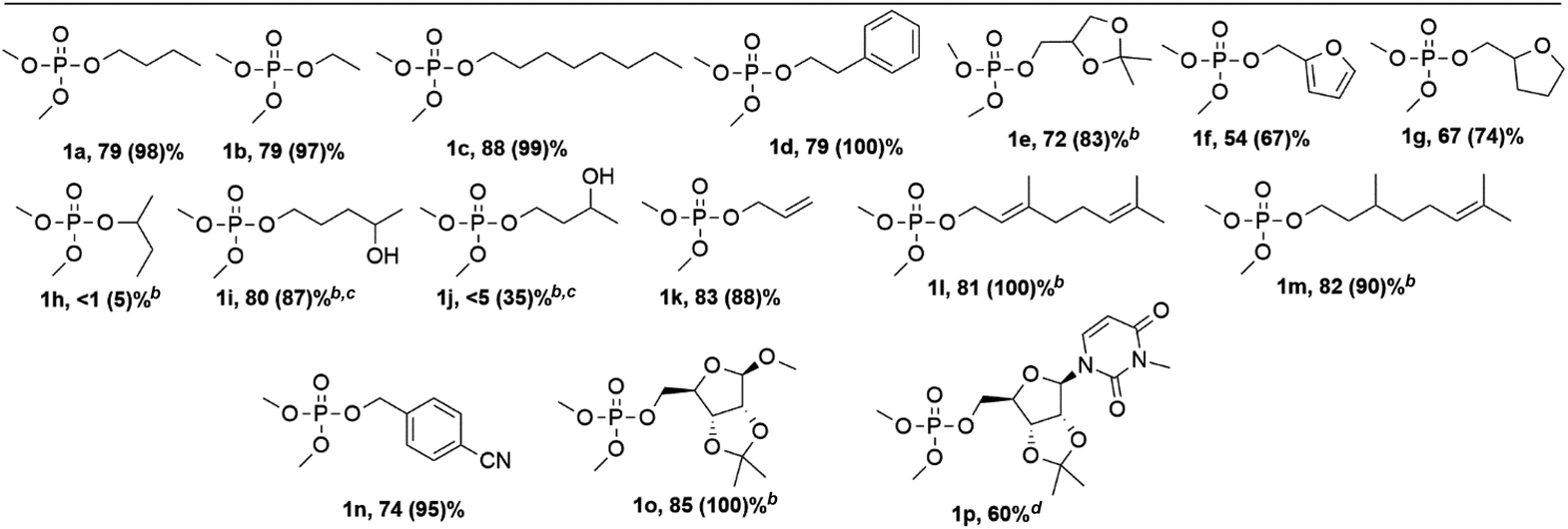
After optimization of the reaction conditions, we moved on to the phosphorylation of various alcohol reactants (Table 2). Next to phosphorylation of 1-butanol (1a), also shorter and longer primary aliphatic alcohols (1b and 1c respectively) could be phosphorylated by iPP. A primary alcohol bearing an aromatic ring like phenethyl alcohol (1d) also showed high conversion and yield. Phosphorylation of saturated as well as unsaturated cyclic ethers was achieved successfully as indicated by compounds 1e–g. After successful phosphorylation of primary hydroxyl groups, also secondary alcohols were tested. However, 2-butanol (1h) did not show any conversion with our conditions. Intrigued by this result, selectivity was investigated with molecules combining both primary and secondary hydroxyl groups. This was demonstrated with 1,4-pentanediol (1i) where 92% selectivity was achieved on the primary hydroxyl function. However, when coupling 1,3-butanediol to iPP, only 5% of the desired product 1j is detected while moderate conversion is obtained. Based on the multiple peaks in the 31P NMR spectrum as well as on the detection of methanol with 1H NMR, it is concluded that several products were formed during the reaction where presumably methoxy groups are expelled next to the leaving group of iPP. A plausible explanation for this observation might be the activation of the secondary alcohol after the primary hydroxyl group is already phosphorylated. This leads to products containing a six membered ring after intramolecular attack, or to different pyrophosphate type products after intermolecular attack (Scheme S1†). Allyl alcohol (1k) and terpenes such as geraniol (1l) and citronellol (1m) show high conversion and good yields as well. Also, cyano functionalized alcohols could be phosphorylated as shown by compound 1n. Lastly, an isopropylidene protected ribofuranose functionalized with varying substituents could be successfully phosphorylated as illustrated by compounds 1o and 1p. This last compound has been successfully isolated (see ESI†); this demonstrates that the method is potentially useful for the synthesis of phosphorylated nucleosides, which are one of the focal points of current drug development.25
In order to gain insight in the mechanism of the reaction, we focused on the phosphorylation kinetics of 1-butanol with iPP and t-BuOK as base. Therefore, we monitored the reaction via in operando 31P-NMR spectroscopy. The resulting time profile (Fig. 2A) shows that the conversion of iPP and formation of dMBP could be followed with excellent time resolution by obtaining data every 2 minutes. When plotting ln[iPP]/ln[iPP]0 as a function of time (Fig. S3†), a linear curve is obtained indicating that the reaction order in iPP is one, as could be expected from Fig. 2A. Additional kinetic experiments were performed by varying the concentrations of base and alcohol to evaluate their influence on the overall rate constant k. In order to investigate the influence of base concentration, conversion of iPP over time was measured with varying amounts of t-BuOK while all other reactant concentrations were kept constant (Fig. S4†). Rate constants k were determined with the method of initial rates by plotting ln[iPP]/ln[iPP]0 as a function of time (Fig. S5†). In Fig. 2B, the obtained k-values are plotted as a function of base concentration. From this graph it is clear that at higher catalyst concentrations, the reaction rate increases linearly with base concentration. As the t-BuOK concentration increases, the concentration of the nucleophilic butoxide is parallelly raised.
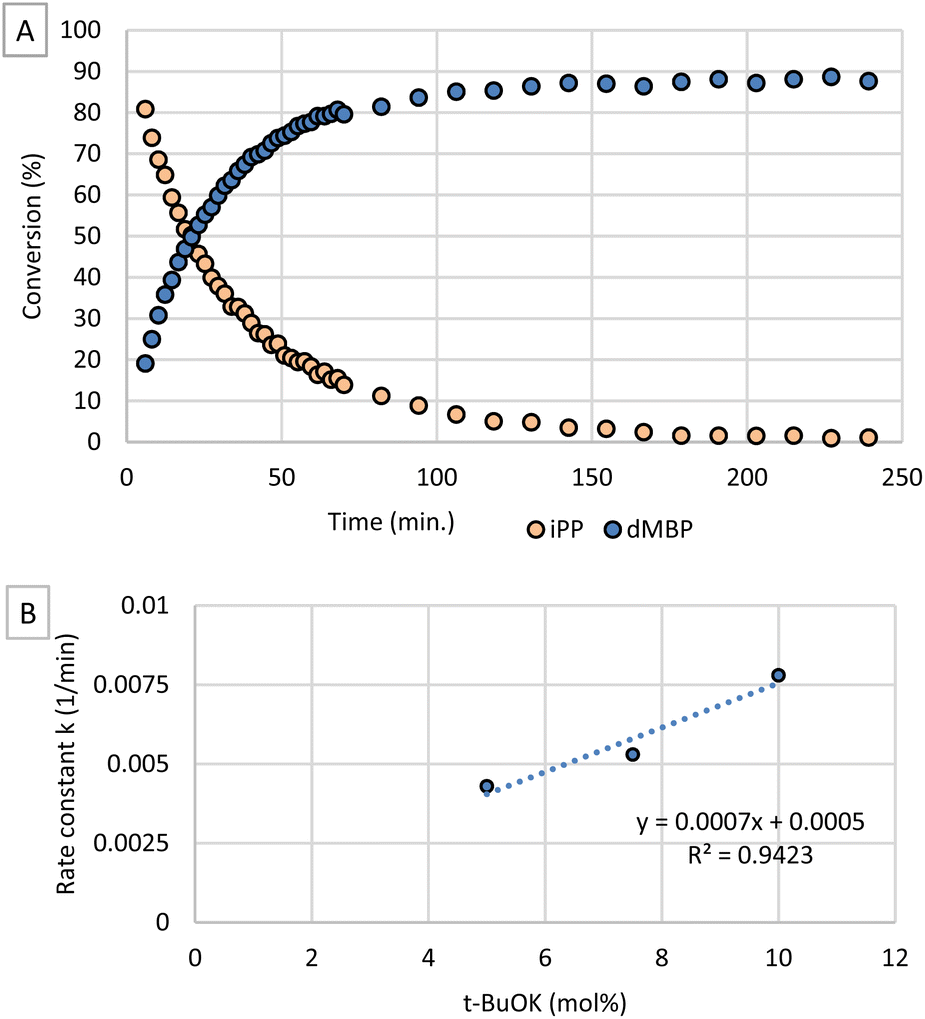 | ||
| Fig. 2 (A) Concentrations of iPP and dMBP as a function of time. Reaction conditions: 0.2 mmol dimethyl isopropenyl phosphate, 850 μl THF, 150 μl 1-BuOH, 7.5 mol% t-BuOK, 25 °C. (B) Rate constant (k) as a function of catalyst concentration. | ||
Secondly, different concentrations of 1-butanol were tested while concentrations of t-BuOK and iPP were held constant. When plotting the conversion of iPP as shown in Fig. 3A, it is clear that the transesterification reaction proceeds at a remarkably slower rate when applying a higher alcohol concentration. Therefore, the order of the reaction in 1-butanol was investigated by applying the method of initial rates to determine the rate constants k for each concentration (Fig. S6†). The logarithmic plot of k as a function of 1-butanol concentration (Fig. 3B) shows an apparent negative order in 1-butanol. This negative order is rather unexpected considering this type of reaction but can be explained by ‘caging’ of the butoxide anions.26 As the concentration of base is significantly lower than the alcohol concentration, the butoxide concentration cannot exceed the t-BuOK concentration. For this reason, butoxide anions will be surrounded with 1-butanol molecules which shield the negatively charged oxygen atom by forming hydrogen bonds with their alcohol functionalities. Consequently, nucleophilic attack on the iPP electrophile is inhibited causing a lower reaction rate. Higher concentrations of 1-butanol will increase the caging effect and inhibit the reaction to a higher extent explaining the observed negative order.
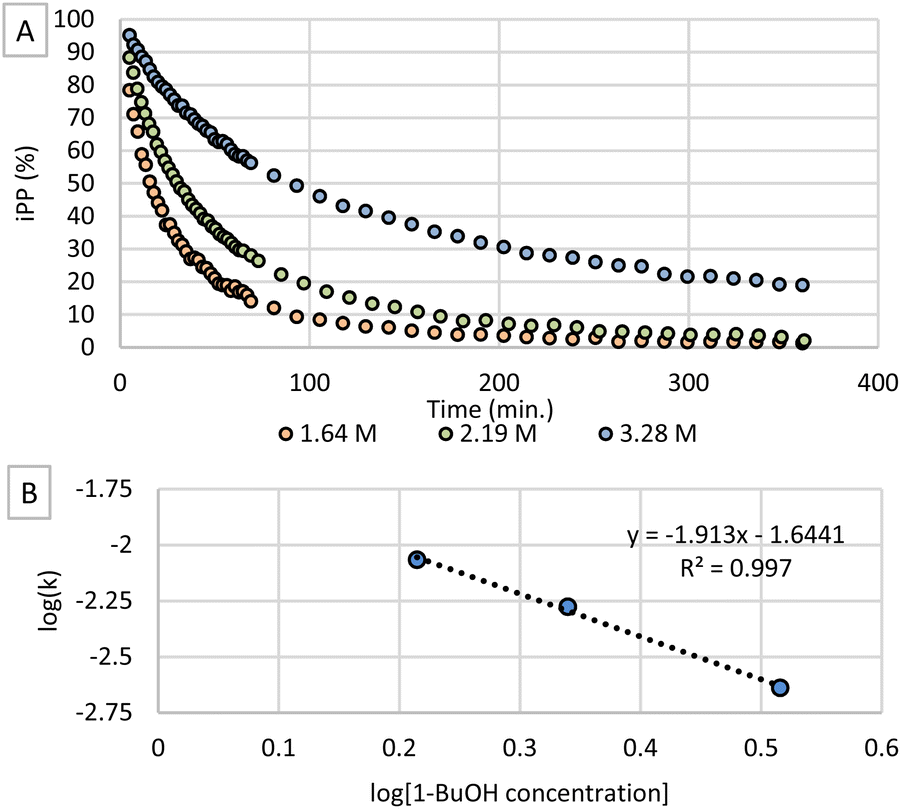 | ||
| Fig. 3 (A) Conversion of iPP at different concentrations of 1-butanol with 7.5 mol% t-BuOK. (B) Logarithmic plot of rate constant k as a function of alcohol concentration. | ||
After investigating the kinetics thoroughly, we suggest a two-step mechanism as shown in Scheme 2 in which the first step is reversible and the second step is rate determining. The first step involves the nucleophilic attack of the generated butoxide anion which occurs on the enol phosphate and creates the phosphorane intermediate which can also proceed in the opposite direction, with the intermediate converting back to iPP. After dissociation of the LG from this intermediate, the desired product dMBP is formed. After protonation of the enolate leaving group, the enol tautomerizes into acetone thereby closing the catalytic cycle. Due to the tautomerization, the last step is irreversible in contrast to the first step.
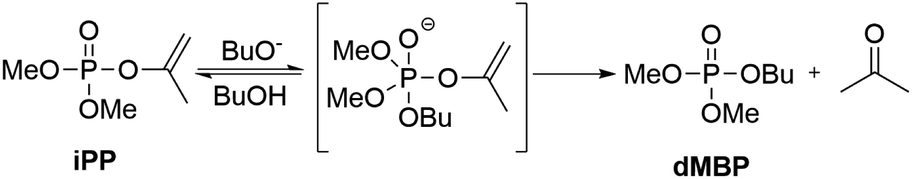 | ||
| Scheme 2 Alcohol phosphorylation reaction mechanism with iPP. | ||
In conclusion, we have developed the selective phosphorylation of primary alcohols at room temperature, relying on the isopropenyl leaving group as an atom efficient alternative to more complex groups. The reaction was achieved using base catalysis, avoiding the need for stoichiometric base additives. With this reaction, a remarkable selectivity for primary vs. secondary alcohols can be achieved. The catalytic mechanism was thoroughly investigated via in operando NMR spectroscopic kinetic experiments. The insights acquired on the catalytic conversion of phosphates described in this work facilitate the atom efficient synthesis of phosphate esters and provide inspiration for the development of new catalytic methods in the field of phosphorus chemistry.
Author contributions
All authors contributed to the presented work. J. W. performed and designed all the experiments. J. W. wrote the article with critical input and feedback from I. B. and D. D. V. I. B. and D. D. V. contributed to the interpretation and processing of NMR kinetic data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. D. V. is grateful to FWO for project funding.Notes and references
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1–132 CrossRef CAS PubMed.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CrossRef CAS PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- A. Blum, M. Behl, L. S. Birnbaum, M. L. Diamond, A. Phillips, V. Singla, N. S. Sipes, H. M. Stapleton and M. Venier, Environ. Sci. Technol. Lett., 2019, 6, 638–649 CrossRef CAS PubMed.
- Y. Takuma, S. Kato and T. Kojima, Stud. Surf. Sci. Catal., 2006, 159, 829–832 CrossRef CAS.
- K. W. Knouse, D. T. Flood, J. C. Vantourout, M. A. Schmidt, I. M. McDonald, M. D. Eastgate and P. S. Baran, ACS Cent. Sci., 2021, 7, 1473–1485 CrossRef CAS PubMed.
- K. W. Knouse, J. N. deGruyter, M. A. Schmidt, B. Zheng, J. C. Vantourout, C. Kingston, S. E. Mercer, I. M. Mcdonald, R. E. Olson, Y. Zhu, C. Hang, J. Zhu, C. Yuan, Q. Wang, P. Park, M. D. Eastgate and P. S. Baran, Science, 2018, 361, 1234–1238 CrossRef CAS PubMed.
- D. Xu, N. Rivas-Bascón, N. M. Padial, K. W. Knouse, B. Zheng, J. C. Vantourout, M. A. Schmidt, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 5785–5792 CrossRef CAS PubMed.
- M. Ociepa, K. W. Knouse, D. He, J. C. Vantourout, D. T. Flood, N. M. Padial, J. S. Chen, B. B. Sanchez, E. J. Sturgell, B. Zheng, S. Qiu, M. A. Schmidt, M. D. Eastgate and P. S. Baran, Org. Lett., 2021, 23, 9337–9342 CrossRef CAS PubMed.
- M. B. Geeson and C. C. Cummins, ACS Cent. Sci., 2020, 6, 848–860 CrossRef CAS PubMed.
- B. Simmons, Z. Liu, A. Klapars, A. Bellomo and S. M. Silverman, Org. Lett., 2017, 19, 2218–2221 CrossRef CAS PubMed.
- M. Uchiyama, A. Yoshio, R. Noyori and Y. Hayakawa, J. Org. Chem., 1993, 58, 373–379 CrossRef CAS.
- S. Jones and C. Smanmoo, Tetrahedron Lett., 2004, 45, 1585–1588 CrossRef CAS.
- K. Tsubaki, H. Shimooka, M. Kitamura and T. Okauchi, Org. Lett., 2019, 21, 9779–9783 CrossRef CAS PubMed.
- K. Domon, M. Puripat, K. Fujiyoshi, M. Hatanaka, S. A. Kawashima, K. Yamatsugu and M. Kanai, ACS Cent. Sci., 2020, 6, 283–292 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- H. Guo, Y. Zhang, Z. Li, P. Zhao, N. Li and E. Shi, RSC Adv., 2022, 12, 14844–14848 RSC.
- X.-H. Hu, X.-F. Yang and T.-P. Loh, Angew. Chem., 2015, 127, 15755–15759 CrossRef.
- W. Adam, L. Hadjiarapoglou, V. Jager, J. Klicik, B. Seidel, X. Wang, W. Adam, L. Hadjiarapoglou, V. Jager, J. Klicieib, B. Seidel and X. Wang, Chem. Ber., 1991, 124, 2361–2368 CrossRef CAS.
- W. Adam, L. Hadjiipoglou and J. Klicic, Tetrahedron Lett., 1990, 31, 6517–6520 CrossRef CAS.
- J. A. Barnett, Yeast, 2003, 20, 509–543 CrossRef CAS PubMed.
- G. A. Grasa, R. M. Kissling and S. P. Nolan, Org. Lett., 2002, 4, 3583–3586 CrossRef CAS.
- T. Kano, K. Sasaki and K. Maruoka, Org. Lett., 2005, 7, 1347–1349 CrossRef CAS PubMed.
- Y. Wang, J. Lalonde, M. Momongan, D. Bergbreiter and C. Wong, J. Am. Chem. Soc., 1987, 110, 7200–7205 CrossRef.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS PubMed.
- J. H. Exner and E. C. Steiner, J. Am. Chem. Soc., 1968, 69, 915 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, NMR analysis and kinetic data. See DOI: https://doi.org/10.1039/d3ra02293e |
| This journal is © The Royal Society of Chemistry 2023 |