
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelectivity of reaction pathways for green diesel production towards biojet fuel applications
Zeni Rahmawatia,
Liangga Santosoa,
Alan McCue b,
Nor Laili Azua Jamaric,
Sri Yayu Ninglasarid,
Triyanda Gunawana and
Hamzah Fansuria
b,
Nor Laili Azua Jamaric,
Sri Yayu Ninglasarid,
Triyanda Gunawana and
Hamzah Fansuria
aDepartment of Chemistry, Faculty of Science and Data Analytics, Institut Teknologi Sepuluh Nopember, Keputih, Sukolilo, Surabaya 60111, Indonesia. E-mail: zeni.rahmawati@its.ac.id
bDepartment of Chemistry, University of Aberdeen, Aberdeen AB24 3UE, UK
cDepartment of Chemistry & Biology, Centre of Defence Foundation Studies, National Defence University of Malaysia, Kem Sungai Besi, Kuala Lumpur 57000, Malaysia
dDepartment Business Management, Faculty of Creative Design and Digital Business, Institut Teknologi Sepuluh Nopember, Keputih, Sukolilo, Surabaya 60111, Indonesia
First published on 4th May 2023
Abstract
Green diesel is the second generation biofuel with the same structure as fossil fuels (alkanes), allowing this biofuel to provide excellent fuel properties over biodiesel such as higher energy content and lower hazardous gas emission. Generally, green diesel can be produced through the deoxygenation/hydrogenation of natural oil and/or its derivatives at 200–400 °C and 1–10 MPa over supported metal catalysts. This process comprises of three reaction pathways: hydrodeoxygenation, decarboxylation, and decarbonylation. The extent to which these three different pathways are involved is strongly influenced by the catalyst, pressure, and temperature. Subsequently, the determination of catalyst and reaction condition plays a significant role owing to the feasibility of the process and the economic point of view. This article emphasizes the reaction pathway of green diesel production as well as the parameters influencing the predominant reaction route.
1. Introduction
The consumption of fossil fuel has been increasing day by day, as shown by the use of 76 quadrillion British thermal units (Btu) of non-renewable fuel in 2021 and 33 quadrillion Btu per May 2022.1 By 2050, the global use of fossil fuel is forecasted to reach 800 quadrillion Btu where industrial sector accounts for 50.9% followed by transportation with 27.9%. This elevating consumption of fossil fuel causes an adverse effect on the environment and the depletion of petroleum storage.2As consequence, the high energy demand urges the development of renewable energy resource to address the issues related to fossil fuels. Many studies have been attempted to shift the non-renewable energy to the sustainable sources such as electrification, clean hydrogen and its derivatives, as well as renewable energy in the form of wind, solar, and biofuels. Among those alternatives, biofuel is projected as the most promising one for the reasons of feasible application and ability in particular areas such as aviation.3–5
Biofuel is classified into categories based upon the raw material: first, second, third, and fourth generation. The first generation is the conventional biofuel produced from food crops, including biodiesel through the esterification or transesterification of natural oil and animal fats. This generation also comprises bioethanol via fermentation process.6 Second generation biofuel is synthesized from non-edible feedstock and organic waste. This second generation involves the production of biofuel with the same structure as fossil fuel such as green diesel.7–11 The third generation biofuel refers to the biodiesel generated from microalgae.12 Lastly, the fourth generation biofuel refers to the metabolism of genetically modified algae.13
In terms of productivity, fourth generation biofuel exhibited a promising superiority with 20–300 folds production above traditional biomass crops in a shorter harvesting cycle. It also performs high photosynthesis efficiency and requires lower bioproductive land as well as freshwater supplies. Despite those advantages, this type of biofuel shows several drawbacks associated with the high production cost, insufficient biomass production, human health and environmental risk.14 The first and third generation biofuel provides several advantages in terms of the practicality of the process and the fuel properties. Transesterification reaction has very simple procedure under mild condition. Biodiesel is nontoxic, biodegradable, and having a high cetane number between 49–60.15 The exhaust gas from biodiesel combustion contains no SOx and relatively small amounts of CO.16 Aside of those excellent properties, biodiesel exhibits several downsides compared to fossil fuel. High oxygen content of biodiesel causes an incomplete combustion, leading to an accumulation of carbon in the engine, filter and nozzles. Therefore, biodiesel is unable to be applied directly to the engine without mixing with fossil fuel. In addition, degradation of properties proceeds during the storage due to oxidation and polymerization.17–19 These challenges can be addressed by green diesel or renewable hydrocarbon.
Green diesel is the second generation of biofuel having the same structure as fossil fuels (alkanes) and has the ability to reduce greenhouse gas emission.20 Green diesel increases the Green House Gas (GHG) saving up to 60% higher compared to the Renewable Energy Use Directive (RED) reduction target. This value has not been achieved by the first generation of biofuel.21
Green diesel has been extensively studied and developed globally. Multinational company called Neste has become the main capitol of green diesel production in Europe, with production capacity of 3.37 billion litres per year.5 USA produces about 960 million gallons of renewable diesel annually, which are used for Sustainable Aviation Fuel (SAF).22 In Indonesia, the development of green diesel for biojet fuel has just started recently even though the initiative has emerged since 2013 through Indonesia Aviation Biofuels and Renewable Energy Task Force.23 Biojet fuel performed a debut in Indonesia by the application of J.24 (Jet fuel containing 2.4% bioavtur from palm oil) to CN 235-22 plane that flew 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 feet above West Java in September 2021.24 However, this development was behind the time according to some media and global organisation focused on renewable energy. This opinion was derived upon the fact that many kinds of biojet feedstock such as natural oils and biodiesel abundantly present in Indonesia.
000 feet above West Java in September 2021.24 However, this development was behind the time according to some media and global organisation focused on renewable energy. This opinion was derived upon the fact that many kinds of biojet feedstock such as natural oils and biodiesel abundantly present in Indonesia.
Green diesel can be produced from vegetable oil through deoxygenation reaction. Mostly, the reaction is conducted at 200–400 °C and 1–10 MPa over supported metal catalysts.25 Fatty acids and fatty acid methyl esters (FAME or biodiesel) are also employed as the feedstock noting that natural oil deoxygenation comprises of complicated steps. Fatty acid methyl ester is preferable since the transesterification of natural oil to produce FAME is more feasible than oil splitting to generate fatty acid. Furthermore, deoxygenation of biodiesel enables the oxygen removal leading to excellent fuel properties.26
Green diesel production involves several reactions including hydrodeoxygenation (HDO), decarboxylation (DCO2) and decarbonylation (DCO). Hydrodeoxygenation removes oxygen via hydrogen insertion. This route produces water and hydrocarbons with the same carbon number as the feedstock.27 In decarboxylation pathway, oxygen is released by discharging CO2 resulting hydrocarbons with one less carbon atom. Meanwhile, decarbonylation eliminates oxygen from fatty acid through CO formation in the presence of hydrogen.28 The extent to which these three different pathways involved is strongly influenced by the catalyst, pressure, and temperature.29
The selectivity of HDO and DCO/DCO2 pathways is beneficial to observe since the route of desired hydrocarbon is associated to the reaction condition leading to the production cost and energy considerations.30,31 For example, HDO requires more hydrogen while DCO/DCO2 proceeds at higher temperature. Moreover, the products also meet the fuel properties: higher carbon in green diesel demonstrates higher cetane number and a lower ignition delay.
In this paper, the selectivity of HDO and DCO/DCO2 routes in green diesel production will be discussed based upon the catalyst design and reaction conditions. This review also provides a profound outlook through a bibliometric study as well as the future challenges in this area, which still unavailable in other reviews as far.
2. Green diesel outlook: a bibliometric study
The bibliometric analysis provides green diesel outlook and its development through the years. The data was collected from Scopus database with the following sequence of query: TOPIC = Green diesel, LANGUAGE = English, timespan: 1972–2023, and keyword: 5 words. Without refining the data, 3150 sources were obtained from three categories: articles, reviews, and proceedings. The bibliometric analysis result was defined on the number of articles per year, sources, country, and citation network. The VOSviewer was selected to analyse this result.The number of article related to green diesel per years was depicted in Fig. 1. The term green diesel was firstly considered through two articles published in 1972. Afterwards, this research area gained a very slight interest in a consistent number within 5 articles per year till 2000. Beyond this point, the number of published articles associated with green diesel continuously heightened up to 372 in 2022, affirming the enormous importance of green diesel as the promising renewable energy source.
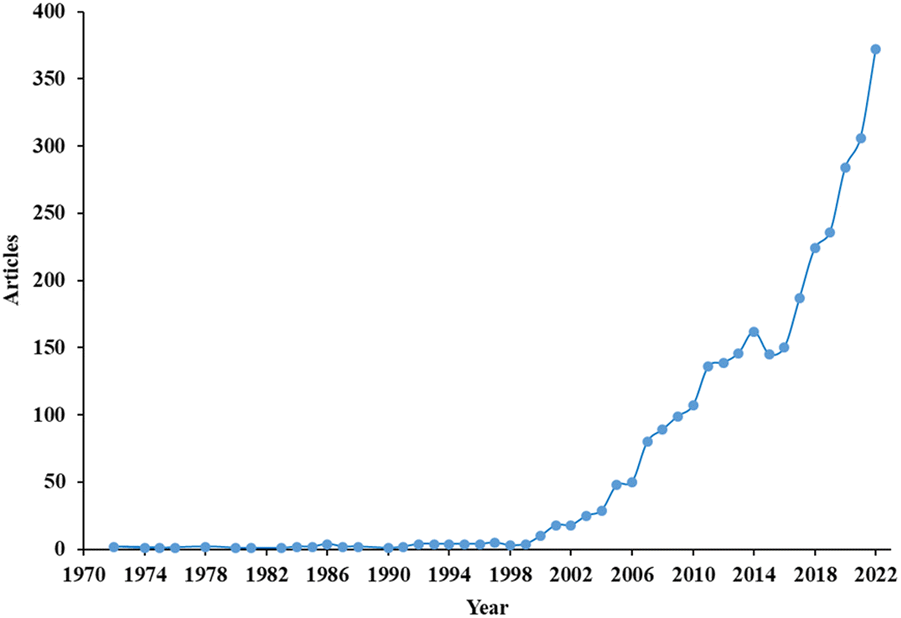 | ||
| Fig. 1 The number of articles on green diesel studies published in the span on 1972–2022. | ||
The enormous interest on green diesel research was spread all over the world as depicted in Fig. 2. Among 3150 articles, the highest studies (544) were conducted in India, followed by the US with 431 articles. China was positioned at third highest contributor with 372 articles and Malaysia run afterward with 197 articles. Below that, various countries also generated articles with variety of article numbers.
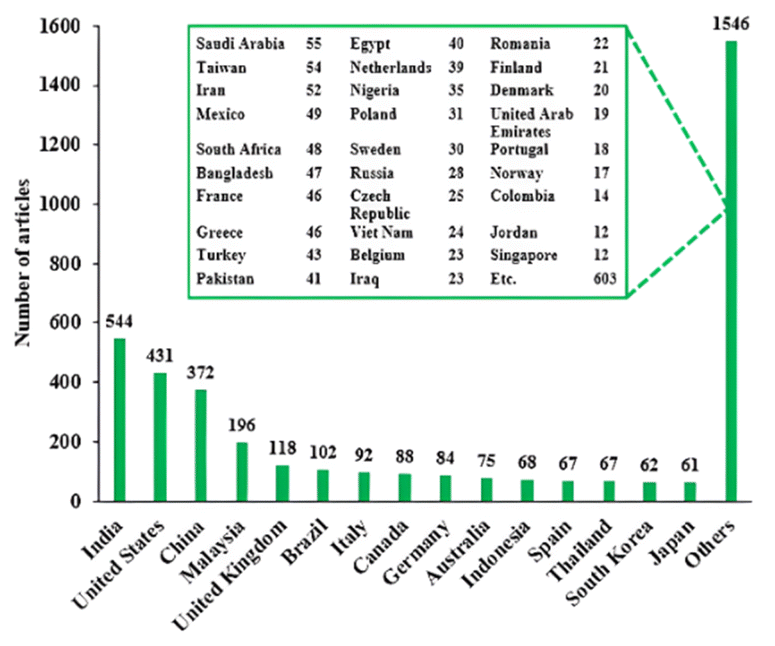 | ||
| Fig. 2 The origin of green diesel studies published in various journals. | ||
In the span of 50 years, these 3150 articles related to green diesel have been published in various journals. In general, the authors opted to report green diesel studies in several journals related to the fuel, sustainable energy, green chemistry, environment, biomass and bioconversion. The articles akin to green diesel without noting the exact term was not included for the purpose of simplification.
A profound bibliographical study based upon the abstract key words was conducted as illustrated in Fig. 3. In terms of green diesel, the most mentioned keywords were related to the reaction pathways, feedstock, catalyst, and its function (blue line and dots). Deoxygenation was the highest mentioned keyword since it is the main reaction to produce green diesel with several reaction pathways such as decarboxylation and decarbonylation. Similar term such as co-processing and hydrotreatment were also used in several articles with palm oil as well as stearic acid were usually employed as the feedstock. The supported nickel was the most employed catalyst in green diesel production and activated carbon was the common support for the catalyst. Green diesel function as the renewable diesel was confirmed by the interrelated keywords of green diesel–biofuel–biodiesel (blue-yellow-purple lines and dots). Together with biodiesel, green diesel is very promising biofuel as renewable energy source, where green diesel enables the upgrade the biodiesel to the second generation biofuel apart from other feedstocks such as natural oil and its derivatives. The specific application of green diesel as biojet fuel was also highlighted since long chain alkanes was the predominates result. Subsequently, it is beneficial to investigate the route of green diesel production and the parameter influencing the conversion as well as the alkane selectivity.
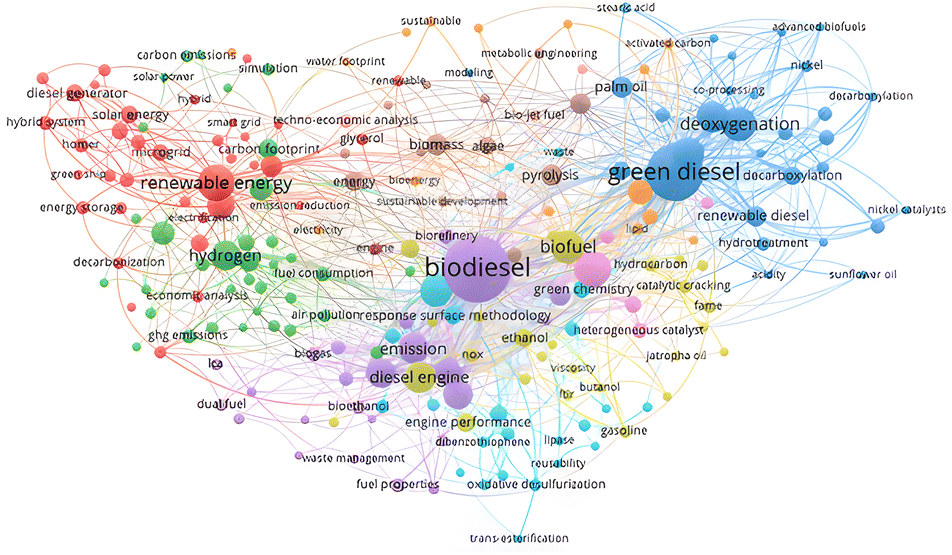 | ||
| Fig. 3 Citation network on bibliometric result of green diesel studies. | ||
3. Green diesel
Green diesel, a second generation of biofuel with an alkane structure of fossil fuels, meets all the criteria for biofuel which is renewable, widely available, well distributed around the world, and has the potential to lower the greenhouse gaseous emissions.26,32–35 The biofuel can be prepared from low quality feedstock with high fatty acid content that causes saponification reaction in biodiesel synthesis.The most advantage of green diesel lays on the superior fuel properties over biodiesel. It fulfills the standard of ASTM D-975 regarding the density, viscosity, and cetane number.31 Moreover, the study of emission and fuel consumption of green diesel and biodiesel tested by heavy duty diesel inferred that green diesel has better performance in the reduction of NOx and lower fuel consumption.6 Green diesel generated NOx emission of 6.64 g kW−1 h−1.36 Table 1 shows the properties comparison between biodiesel and green diesel.
| Fuel properties | Unit | Biodiesel | Green diesel |
|---|---|---|---|
| Density | kg m−3 | 880 | 790 |
| Viscosity at 40 °C | mm2 s−1 | 2.9–11 | 2–4 |
| Flash point | °C | 100–180 | 59–138 |
| Low heating value | MJ kg−1 | 37.2–38 | 43.7–44.5 |
| Cetane number | 45–65 | 70–90 | |
| Oxygen | wt% | 11.2 | 0 |
Generally, green diesel can be produced from natural oil through deoxygenation reaction at 200–400 °C and 1–10 MPa over metal catalysts.37–41 The initial step of deoxygenation is the hydrogenation of fatty ester or fatty acid containing double bonds, due to the reactivity of double bond being thermodynamically higher than the acid group.42 This step is followed by the cracking of hydrogenated triglycerides to produce free fatty acids (Fig. 4).
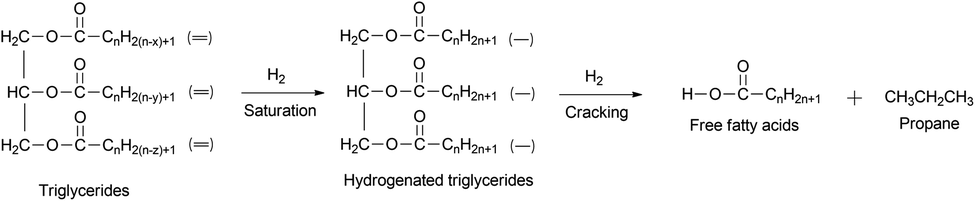 | ||
| Fig. 4 Reaction pathway for free fatty acids production from triglycerides. | ||
Afterwards, free fatty acid is converted to alkanes through the hydrodeoxygenation, decarboxylation and decarbonylation pathways (Fig. 5).43 The main reaction pathway is associated to the catalysts and reaction condition.
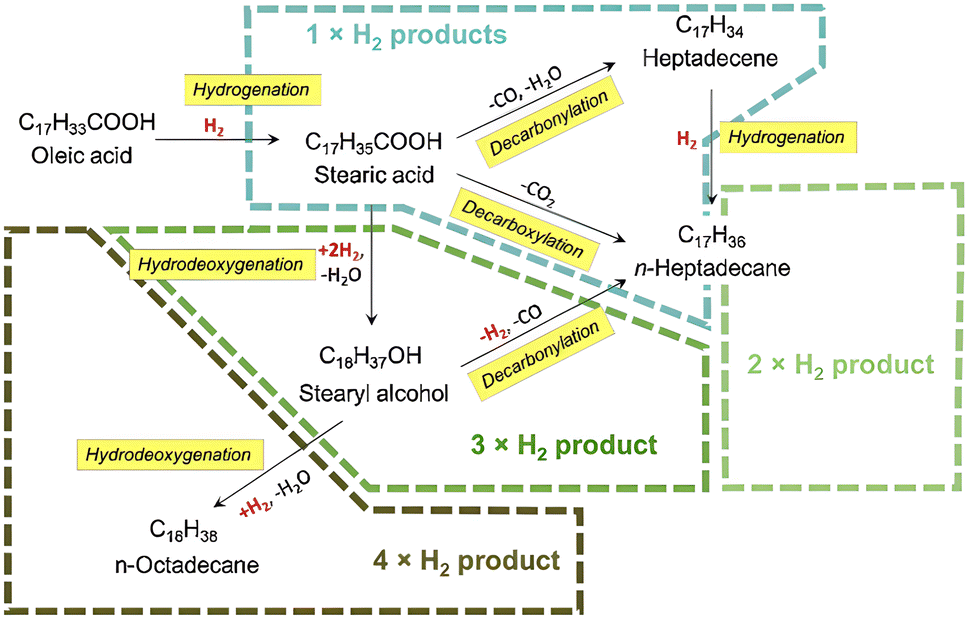 | ||
| Fig. 5 Reaction pathway for green diesel production from fatty acid. | ||
4. Hydrodeoxygenation
Hydrodeoxygenation (HDO) removes oxygen from fatty acids by insertion of hydrogen resulting hydrocarbons with the same carbon number as feedstock. This route is very efficient yet unpreferable sometimes, considering the requirement of high pressure of hydrogen.44 In addition, this pathway consists of several intermediates such as aldehydes and fatty alcohols (Fig. 6). Subsequently, this route is also adopted to produce fatty alcohol by a manipulation on certain reaction conditions and catalyst. Nevertheless, HDO is mostly adopted in green diesel production at industrial scale such as Neste oil from the refinery in Porvoo, Finland.45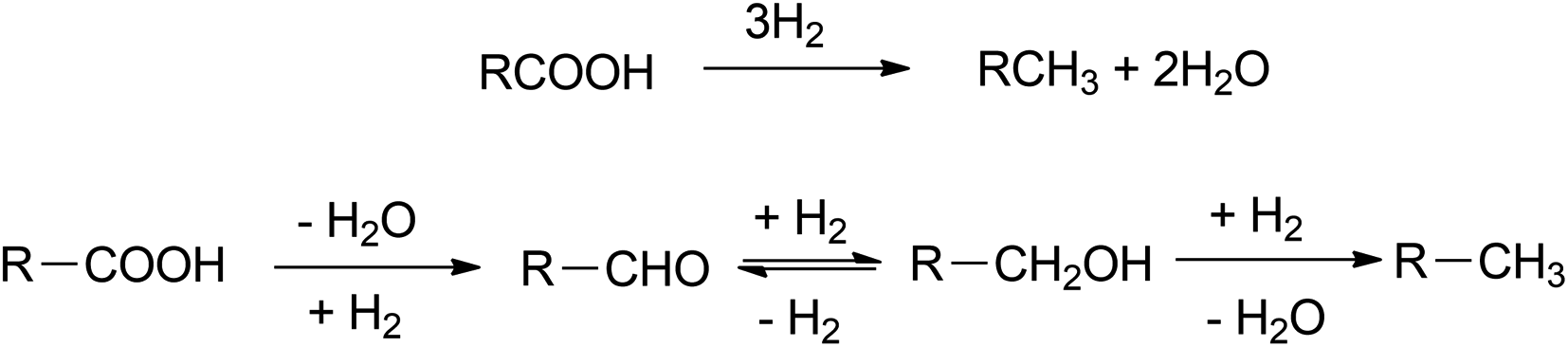 | ||
| Fig. 6 Several steps of hydrodeoxygenation route. | ||
5. Decarboxylation/decarbonylation
Deoxygenation term will be frequently use in this section to indicate the process of oxygen removal which reflecting hydrodeoxygenation, decarboxylation and decarbonylation pathways. In decarboxylation, oxygen is removed from fatty acids by releasing CO2 and producing hydrocarbons with one less carbon atom. Meanwhile, decarbonylation eliminates oxygen from fatty acid by forming CO in the presence of hydrogen (Fig. 7). Both routes occur at higher temperatures to allow C–C cleavage.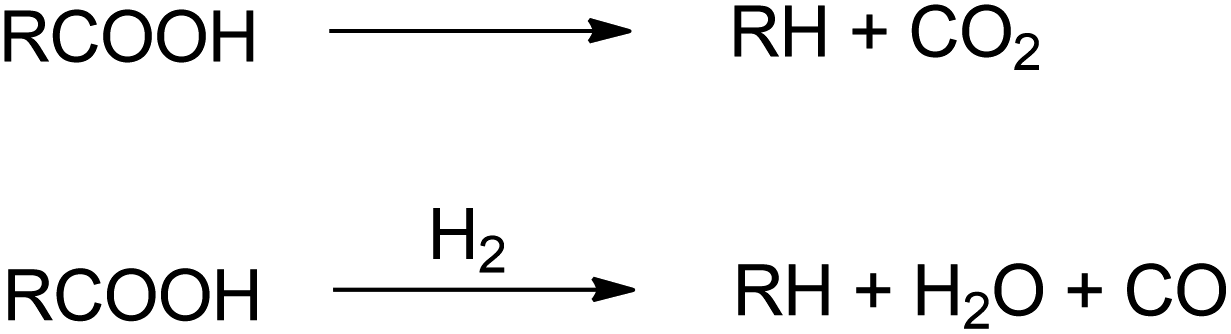 | ||
| Fig. 7 Decarboxylation and decarbonylation routes. | ||
Decarbonylation requires less hydrogen than hydrodeoxygenation, whereas decarboxylation proceeds in the absence of hydrogen.46,47 The excess of hydrogen initiates methanation or water gas shift reaction, where the CO or CO2 reacting with hydrogen as described in Fig. 8.48 Under hydrogen atmosphere, methanation and water gas shift might occur causing the decarboxylation and decarbonylation hardly to be distinguished.
 | ||
| Fig. 8 Methanation and the water gas shift reaction in DCO/DCO2. | ||
The deoxygenation process has been employed on the production of green diesel from several feedstocks such as natural oils, fatty acids, and FAME (biodiesel). Waste cooking oil was successfully converted to green diesel after two hours. The deoxygenation was conducted at 300 °C and produced heptadecane–octadecane as the main product and several intermediates including fatty alcohols, fatty esters, and fatty acids.49 Deoxygenation of oleic acid over Pt/P@MIL-101 exhibited 95% yield with 75.5% heptadecane selectivity, conveying the preferable decarboxylation/decarbonylation route.50 Šimáček and Kubička51 reported that rapeseed oil was completely converted at 280 °C after one hour reaction. Hexadecane, heptadecane, and octadecane were the main products with fatty alcohols, fatty esters, and fatty acids as the intermediates. Soybean was successfully deoxygenated over NiMo/Al2O3, Ni/Al2O3 and Pd/Al2O3.52 The highest conversion was reported to be 92.9% generated over a bimetallic catalyst NiMo/Al2O3. Decarboxylation was favoured over Ni/Al2O3 and Pd/Al2O3 indicated by the high selectivity of heptadecane up to 80%. Srifa et al.53 investigated the catalytic behaviour of Ni/γ-Al2O3 and Co/γ-Al2O3 on the hydrodeoxygenation of palm oil. Catalysts exhibited excellent performances with more than 90% yield. The selectivity of the product was influenced by the catalysts, where decarboxylation/decarbonylation was favourable over Ni/γ-Al2O3 and all routes were dominant over Co/γ-Al2O3.
The complicated steps of natural oil deoxygenation has lead many attempts to adopt simpler compound as feedstock such as fatty acid and FAME. A simple fatty acid was mostly applied as model compound to investigate the mechanism of deoxygenation, reaction steps, and some related parameters. In addition, saturated fatty acid was the most attractive model compound that avoid the competition between the hydrogenation of double bond and fatty acid. Since the enthalpy required to break C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond (614 kJ mol−1) is lower than a CO bond (799 kJ mol−1), the hydrogenation of CC double bond proceeds before deoxygenation,54 as confirmed by the study of oleic acid and palmitic deoxygenation. Oleic acid deoxygenation attained 92% conversion while palmitic acid was completely transformed at the same reaction condition.55 Similar result was observed on the deoxygenation of oleic acid over catalyst Ni2P/Al-SBA-15.56 Oliveira Camargo et al.56 investigated the deoxygenation of oleic acid over catalyst Ni2P/Al-SBA-15. Decarbonylation was proposed as the main route with heptadecane as the main product. Pimenta et al. claimed that hydrodeoxygenation was two folds more selective in the deoxygenation of stearic acid over NiMo catalysts. Other catalysts like NiMo/Al2O3, the selectively favoured decarboxylation.57
C bond (614 kJ mol−1) is lower than a CO bond (799 kJ mol−1), the hydrogenation of CC double bond proceeds before deoxygenation,54 as confirmed by the study of oleic acid and palmitic deoxygenation. Oleic acid deoxygenation attained 92% conversion while palmitic acid was completely transformed at the same reaction condition.55 Similar result was observed on the deoxygenation of oleic acid over catalyst Ni2P/Al-SBA-15.56 Oliveira Camargo et al.56 investigated the deoxygenation of oleic acid over catalyst Ni2P/Al-SBA-15. Decarbonylation was proposed as the main route with heptadecane as the main product. Pimenta et al. claimed that hydrodeoxygenation was two folds more selective in the deoxygenation of stearic acid over NiMo catalysts. Other catalysts like NiMo/Al2O3, the selectively favoured decarboxylation.57
A reaction mechanism study on fatty acid deoxygenation was reported in many studies. Wagenhofer et al. proposed a mechanism for decarbonylation of fatty acids over Ni–MoS2 catalyst as illustrated in Fig. 9.58 Partial replacement of Mo with Ni reduced the bonding energy of sulphur atoms, hence increasing the number of sulphur vacancies. These sulfur vacancies and metallic Ni acted as active sites in decarbonylation.
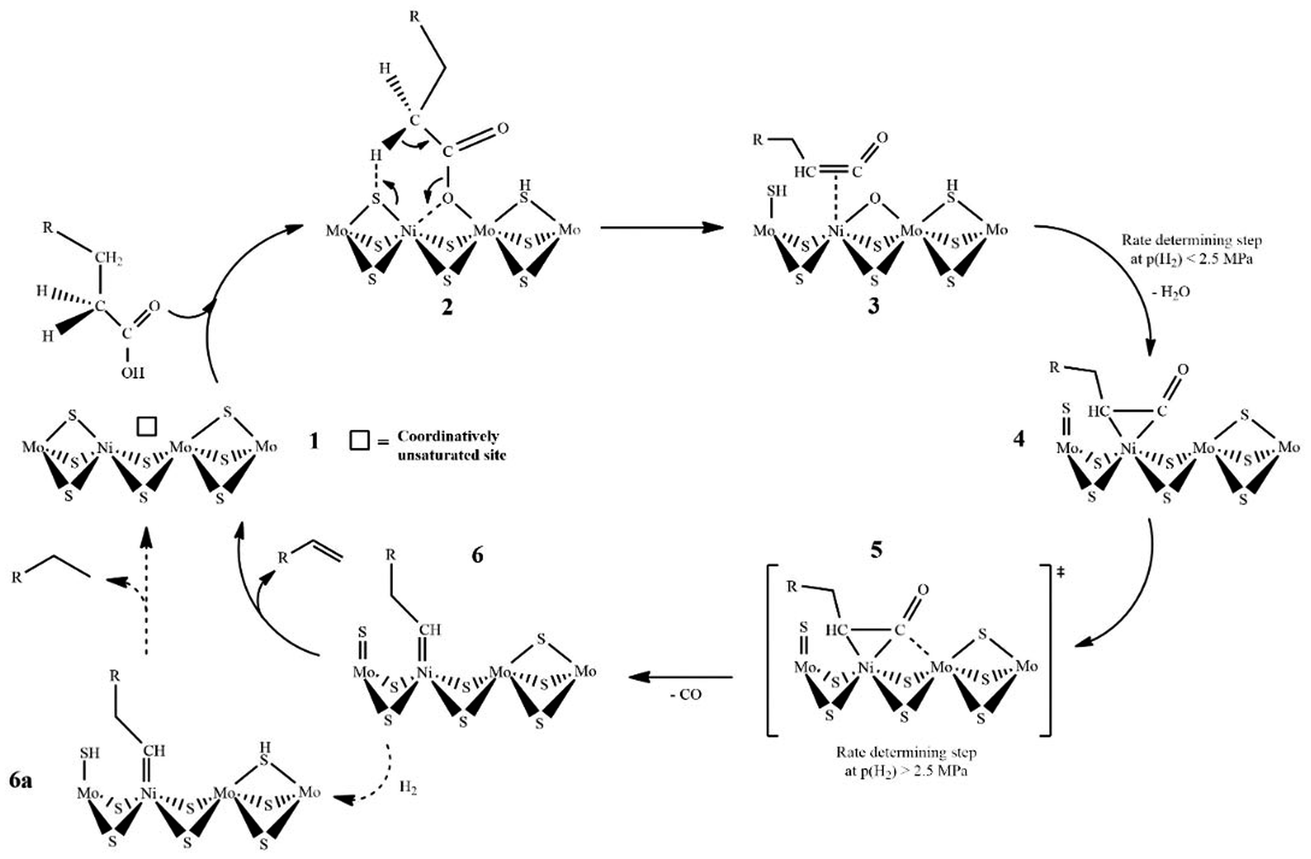 | ||
| Fig. 9 Proposed mechanism of fatty acid decarbonylation on Ni–MoS2 catalyst. | ||
The reduction of sulphur binding energy inclined electron density of neighbouring sulphur, consequently increasing the basicity of sulphur to deprotonate fatty acids. First, fatty acids are adsorbed onto sulphur and Ni to form Ni–O bonds and activated acyl compounds. Afterwards, the C–O bond scission was induced by a strong interaction with the oxophilic Mo cation and the sulphur atom catches the alpha hydrogen to produce the ketene intermediate. This ketene subsequently formed a η2(C–C) geometry complex. Furthermore, the C–C bond scission on the ketene generated CO gas. Finally, the removal of the substrate on the surface of catalyst generated alkanes or alkene (Cn−1).58
The mechanism of carboxylic acid decarboxylation was studied on a Pd/C in the form of α-phase palladium hydride (α-PdHx). The C–H bond of carboxylic acid was cleaved on the surface of Pd creating intermediate R(Cn−1)–CH*–COO*. Furthermore, the intermediate underwent C–COO bond cleavage resulting in R(Cn−1)–CH* and reacted with two adsorbed H to produce R(Cn−1)–CH3.59 A similar explanation can be seen in other studies.60,61
The last promising feedstock is biodiesel or fatty acid methyl ester (FAME). This feedstock is beneficial since biodiesel can be prepared from natural oil through a simple transesterification. Deoxygenation reaction allows the catalytic upgrade of methyl ester with associated fuel properties issues to alkanes. Several studies reported the successful transformation of FAME through deoxygenation. Yang et al.62 tested the activity of the catalyst Ni–ZrO2 for the deoxygenation of methyl laurate at 280 °C and 2 MPa. Undecane was the main product over dodecane confirming the dominance of decarboxylation/decarbonylation pathway over hydrodeoxygenation. Lauric acid appeared to be the primary intermediate followed by lauryl alcohol. Bie et al.63 investigated deoxygenation of methyl palmitate over catalyst Rh/ZrO2 at 270 °C and 8 MPa. Decarboxylation/decarbonylation was the major reaction route affirmed by higher selectivity of pentadecane than hexadecane. Several intermediates involved in the process including palmitic acid, hexadecanol, palmityl palmitate and hexadecanal. The deoxygenation of methyl stearate was accomplished using Ru/H-ZSM-5. This catalyst demonstrated high activity with 98.2% conversion after 8 hours reaction at 220 °C. Heptadecane and octadecane were the main product, resulted from several steps including hydrogenolysis and hydrolysis.64 Methyl palmitate was oxygenated to pentadecane over Ni/Al-SBA-15 catalyst through decarbonylation route, with 66% selectivity at 99.3% conversion.65
Regardless of the feedstock, the green diesel production as well as the main reaction pathway are determined by several parameters including temperature, pressure, and catalyst. For instance, the deoxygenation FAME over Co/ZrO2 where the reaction route and the product were influenced by the catalyst, reaction temperature and pressure (Fig. 10).66
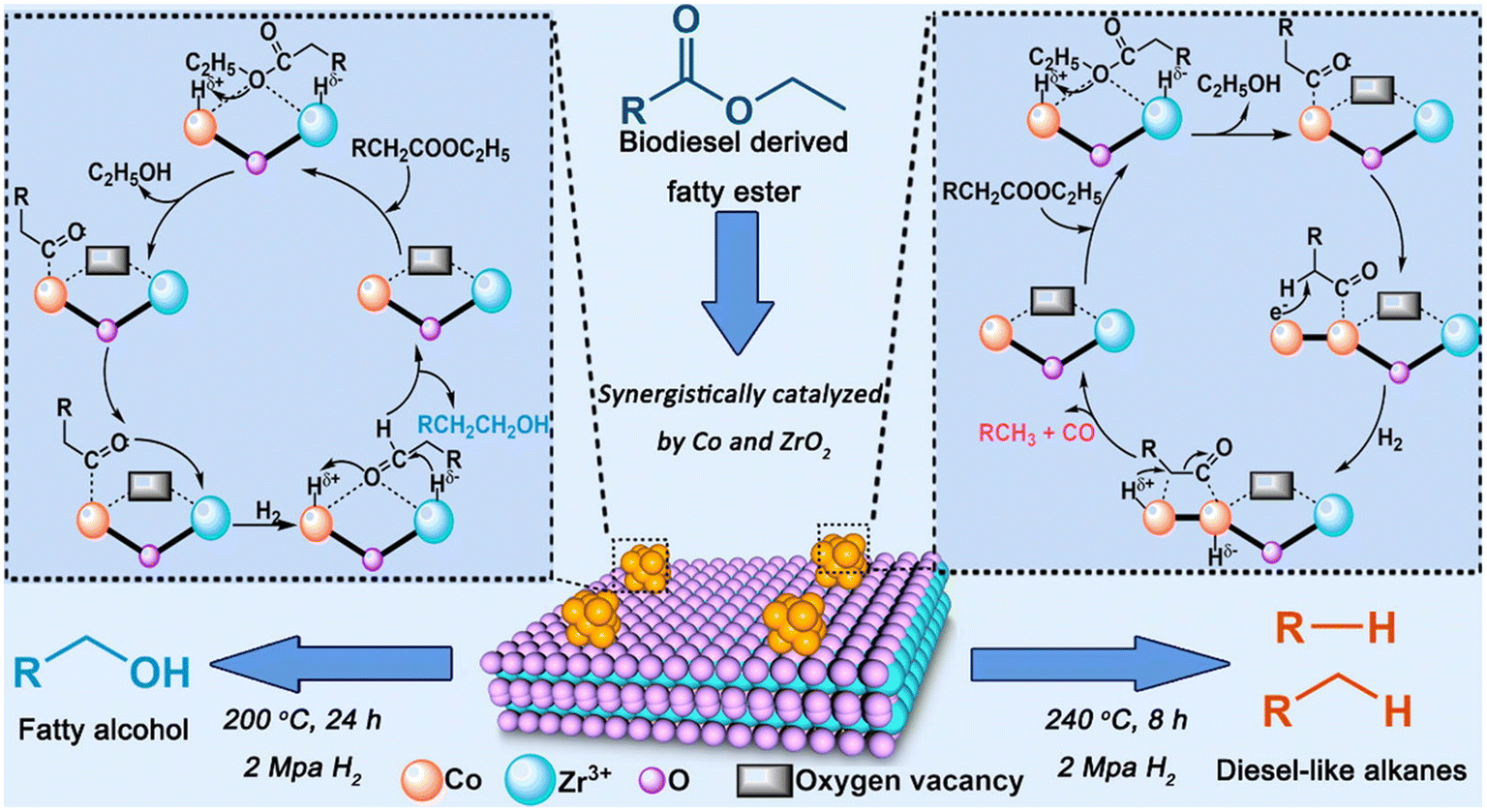 | ||
| Fig. 10 The influence of catalyst, temperature, and pressure on the reaction mechanism of FAME deoxygenation over Co/ZrO2. This figure has been adapted from ref. 65 with permission from Elsevier, copyright 2020. | ||
6. Influence of temperature
Reaction temperature significantly effects the green diesel production. In general, deoxygenation reaction proceeds at the temperature range of 200–400 °C. Higher temperature is not advisable due to the possibility of alkane cracking that result many compounds in various range leading to the lower selectivity of desired alkanes. In addition, catalysts mostly proned to deactivation due to coke formation, carbon deposition and sintering at high temperature. The collapsed structure due to an elevated temperature also lowered the catalytic activity.67,68 The increase of temperature improves conversion or yield of green diesel. The optimum yield of tall oil fatty acid deoxygenation was obtained at 350 °C.69,70 Altering the temperature from 260 °C to 300 °C permitted the completely converted of methyl stearate from initial conversion of 65%.71 The incline of conversion from 52% to 89% was also occurred on the deoxygenation of oleic acid when the temperature rose from 300 to 350 °C.72 The optimum yield of palm oil deoxygenation was obtained at 375 °C.67 Šimáček et al.70 declared that conversion of rapeseed oil increased with the raise of temperature where all reactants as well as intermediates were completely transformed at 310 °C. Similar result were also observed elsewhere.11,73–80Apart from conversion, reaction temperature also determines the reaction pathway assigned by the selectivity of the products. Higher temperature favours decarboxylation/decarbonylation since this condition allow C–C cleavage and carboxylic group release as CO2 or CO. The optimum temperature to break the carboxylic group was 375 °C.81
The influence of reaction temperature on the reaction route was mentioned in many studies. At an elevating temperature of 260 °C to 300 °C on the deoxygenation of methyl stearate over CoNi/HAP, the selectivity of heptadecane inclined, followed by the decline of octadecanol selectivity indicating the occurrence of DCO/DCO2.71 Deoxygenation of rapeseed oil at a span temperature of 260–340 °C affirmed that higher temperature favoured decarboxylation indicated by low C18/C17 ratio.82 Moreover, deoxygenation of palmitic acid produced CO2 as indicator of decarboxylation presented at above 290 °C.83 A similar result was obtained from numerous studies.70,84–87
Some studies suggested that the high temperature was not applicable due to sintering phenomenon. To address this issue, HZSM-5 was opted to hinder the movement of metal particles at high temperature.88 Another study proposed ionic liquid as coating material to stabilize single atom catalyst such as Pd1/HAP. Ionic liquid covered the metal atom increased the kinetic barrier for the formation of metal–metal bond on the catalyst surface. In addition, the presence of ionic liquid caused the formation of metal aggregates become thermodynamically less favorable.89
7. Influence of pressure
The presence of hydrogen is critical for deoxygenation, especially, oxygen removal and catalytic activity maintenance.90 Overall, green diesel production occurs at hydrogen pressure within the range of 1–10 MPa.17Comparable to reaction temperature, hydrogen pressure also has a significant role in both reaction results and pathways. In terms of oil feedstock, hydrogen is urgently required to transform triglycerides into fatty acids. Higher pressures of hydrogen generates higher conversions as reported in the deoxygenation of soybean oil, palmitic acid, and other feedstocks.69,91,92
Oleic acid was 55.5% converted over Ni–Fe/ZrO2 at 1 MPa and 97.98% at 3 MPa.93 Raising the hydrogen pressure from 0.7 to 3 MPa improved the stearic conversion from 53% to 99% at deoxygenation of stearic acid over Ni–γ-Al2O3 catalyst.94 Notably, high pressure provided sufficient hydrogen to completely transform the feedstock.95 The enhanced conversion due to hydrogen pressure can be observed in other studies.67,96–98
Aside from conversion, hydrogen pressure also regulates the reaction pathway. Under rich hydrogen environment, hydrodeoxygenation takes place predominantly.25,99,100 Low hydrogen availability encouraged decarbonylation route, however the presence of hydrogen unnecessarily required in the decarboxylation.
The influence of hydrogen pressure on the reaction pathway was confirmed in the deoxygenation of methyl palmitate over Co3Mo3N catalyst. Within the range of hydrogen pressure 1–4 MPa, the hydrodeoxygenation progressed as inferred by the improvement of hexadecane selectivity from 54% to 97%.101 The ratio of C18/C17 in the deoxygenation of rapeseed oil inclined with the rise of hydrogen pressure.100 Furthermore, higher hydrogen pressure inhibited decarboxylation in the decarboxylation of free fatty acids over 5 wt% Pd/C at 300 °C under 5% H2.99 The reaction pathway shifted from decarboxylation to decarbonylation was proven by the transformation of CO2 to CO with the increase of hydrogen pressure.
Contrary, the pentadecane was the main product in deoxygenation of methyl palmitate over B2O3/ZrO2 even when the hydrogen pressure was varied between 2-8 MPa.29 Similar result was observed in the deoxygenation of stearic acid over Ni/γ-Al2O3. Heptadecane selectivity shifted from 49% to 96% when the pressure increased.102 Furthermore, HDO pathway produces intermediates like aldehydes that enable DCO pathway to take over. These contradictive results underlined the other factor influencing the reaction route, especially the catalyst.
8. Influence of reaction time
Reaction time is the most common and unemphasized parameter in green diesel production. This parameter is automatically investigated in every study which the general result inferred that conversion inclined as the function of reaction time.103 Prolonged reaction time provided adequate interaction between reactant and catalyst. The value of reaction time is varied from 30 minutes to 48 hours and slightly beyond.104 Notably, the maximum reaction differed depends upon the other reaction condition such as feedstock, catalyst, reaction temperature and pressure.105The explicit reaction time influence was investigated on deoxygenation of waste cooking oil and its derivatives over NiCo/SBA-15.11 Within 2 hours, the product yield increased from 18% to 80% and hydrocarbon selectivity inclined from 31% to 70%. The study on methyl laurate over Co/ZSM-5 reported similar trend with the maximum conversion was attained after 4 hours.106
Nevertheless, excessive extended reaction time was inadvisable in regards to product selectivity. The products underwent side reactions such as cracking, isomerisation, cyclization, and dimerization in prolonged reaction time leading to low hydrocarbon selectivity.67,107
In the matter of reaction pathway, the influence of reaction time was reported negligible below the maximum region. The deoxygenation of oleic acid over Ni/MgO–Al2O3 claimed the raising conversion and hydrocarbon yield without any change on the selectivity profile. Octadecane was produced in a rapid rate compared to heptadecane affirming HDO as the min pathway. The shift of pathway was not observed within 3 hours of reaction.108
9. Influence of catalyst
Catalyst is the most important factor in the deoxygenation of vegetable oil and its derivatives. Supported metal catalysts are the most applied catalysts in most research. Several properties of catalyst are crucial for deoxygenation with the purpose of promoting hydrogenation and C–C cleavage, such as high acidity, small particle size with high metal area, excellent dispersion, superior thermal stability.46,109,110 In terms of hydrodeoxygenation, the ability to split H2 is inevitable for the catalyst. These beneficial characteristics are associated with the active metal site, metal concentration, catalyst amount and catalyst support. Hence, the employ of metal and support individually define the conversion as well as product selectivity. Fig. 11 explains the active metal and support material employed on deoxygenation of natural oil and its derivatives.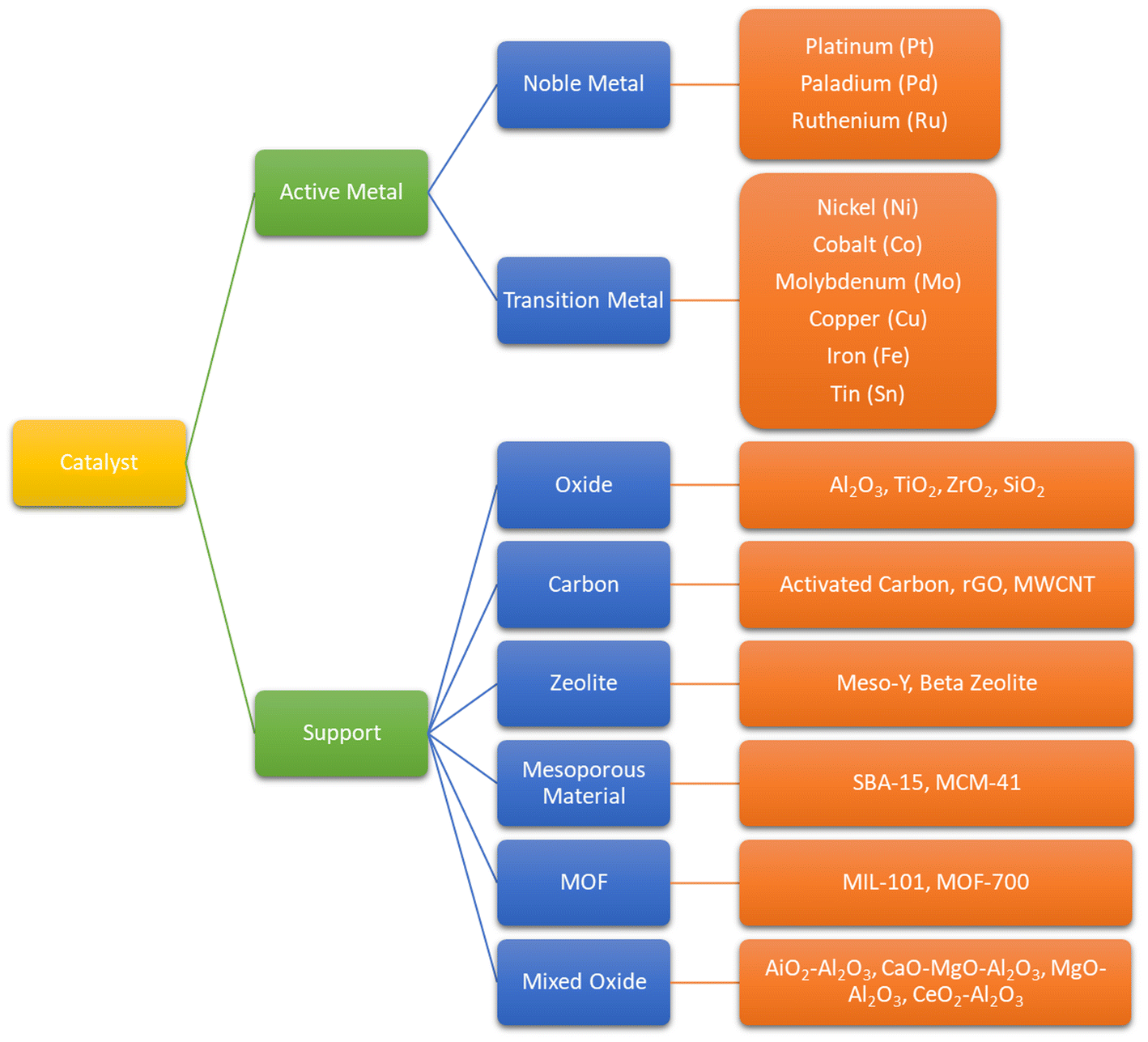 | ||
| Fig. 11 Active metal and support material commonly used in biojet production. | ||
9.1. Effect of metal
In supported metal catalyst, metal performs as the active site accommodating the hydrogen split, hydrogen insertion and C–C cleavage. Metal active sites determine the deoxygenation pathway (HDO, DCO, and DCO2). In general, metal species in catalysts is classified into noble metals and transition metals. Transition noble metals like platinum (Pt), palladium (Pd), and ruthenium (Ru) are the main metals extensively applied owing to their excellent performance in activity and product selectivity. Nevertheless, these metals demonstrated several issues including expensive cost, water poisoning, deactivation, and dependence on the support which driving these catalysts became less preferable.45,109,111 Alternatively, nickel (Ni), cobalt (Co) and molybdenum (Mo) were opted as the promising alternative.52,81,110–113 Pd@PPN catalyst performed a high activity on the deoxygenation of stearic acid with 90% conversion and 83% C17 selectivity. The study claimed that the excellent result was attributed to the electron rich palladium (Pd) that activated the interface bound H2. Another advantage of Pd was robustness to high temperature as claimed in the study of waste cooking oil deoxygenation at 400 °C. However, the maximum desired alkane selectivity only achieved 77% because the alkane underwent cracking process.114 Comparable result was observed in the activity of platinum (Pt) as deoxygenation catalyst. Generally, Pt catalyst converted about 90% feedstock with 60% main product selectivity.9,115–117 Excluding the hydrogen pressure, Pd and Pt drive the reaction pathway towards DCO/DCO2.Noble metal likes ruthenium (Ru) exhibits highest activity compared to Pt and Pd. Ru/C catalyst activated with ZnCl2 gave 100% conversion of stearic acid at low temperature (140 °C). The selectivity for C17 is 88% and C18 is 12%, indicating the DCO/DCO2 pathway is preferable to HDO.118
Another study used Ru/TiO2 for the conversion of ethyl stearate under moderate temperature of 220 °C. Ru/TiO2 catalyst performed 98.4% conversion with selectivity of C17 and C18 were 63.8% and 29.5%, respectively.119 These results indicated the high activity of Ru and its selectivity for the DCO/DCO2 pathway. Higher activity of Ru compared to Pt and Pd was supported by the high specific rate and turnover frequency (TOF) of the Ru catalyst followed by Pt and Pd. In addition, the surface acid–base characteristics greatly affected the Pt and Pd catalysts, where the presence of phosphate on the carbon support severely reduced the activity of DCO and DCO2. However, this phenomenon was hardly observed during the employ of Ru catalyst, confirming its superiority in the conversion of fatty acids.115
The high price of noble metal catalysts promoted many attempts to pursuit the cheaper and abundant materials such as transition metals. Study on the drubber seed oil deoxygenation claimed that Mo/γ-Al2O3 was capable to convert 95% of the oil. Combination of Mo6+ and Mo4+ active sites directed the reaction pathway toward hydrodeoxygenation with 47% contribution. It is worth noted that the addition of molybdenum (Mo) loading inclined the HDO selectivity.8 Other results confirmed the application of Mo within conversion in the range of 54–98% with various preference on HDO and/or DCO/DCO2.120–122
Nickel (Ni) and cobalt (Co) were reported as the most promising catalyst since these transition metals embodied excellent properties for deoxygenation reaction. A high conversion of 96.7% was attained from deoxygenation of oleic acid over Ni/MgO–Al2O3 at 300 °C and 2 MPa. The metal active site together with the acid site available on support engineered the reaction pathway towards HDO with C18 and C16 selectivity of 45.9%. The interface of Ni and support provided hydride and protons for reactants. Apart from that, the active site of Ni metal accommodated the hydrogen split and C–C cleavage.108 Interestingly, Ni also directed the reaction route to DCO/DCO2 in the deoxygenation of stearic acid at 250 °C and 3 MPa. It was claimed that metal area of Ni was responsible for the high heptadecane selectivity of 90%. Many similar results concluded that Ni catalyst favoured DCO/DCO2 pathway.8,116,128,130,132
In contrast, cobalt catalyst was infamous for proportionally navigating the deoxygenation towards all routes. The Co/ZrO2 successfully transformed ethyl palmitate to hexadecane and pentadecane at 240 °C and 2 MPa. DCO/DCO2 pathway was dominant referred by pentadecane selectivity of 54.3%.66 The cobalt in the form of Co3O4/SiO2–Al2O3 catalyzed methyl stearate at 250 °C and 3 MPa. The highest octadecane selectivity of 88% represented the HDO as the major pathway. High dispersion was claimed as the most influencing properties where Co3O4 undertook in situ reduction to metallic cobalt.134 The essential role of cobalt in conversion and reaction pathway can also be found elsewhere.85,106,129,135
In the quest of the promising transition metal catalyst, some metals have been attempted to provide high conversion with a compromising cost such as copper (Cu), ferum (Fe) and stannum (Sn). Monometallic Cu converted 96% oleic acid at 330 °C and 1 MPa N2 for 3 hours with tetralin as hydrogen source, where this result was claimed higher over monometallic nickel with 82% conversion at the same reaction conditions. This is because Cu has a better hydrogen liberation ability than Ni as seen from Cu ability to convert tetralin (47%) which is better than Ni (33%).43
The high catalytic activity of monometallic Cu was also reported in the production of green diesel from stearic acid.136 Supported by quantum-chemical simulation, DCO was claimed as the major pathway due to the lower activation energy where DCO2 required 77.0 kJ mol−1 while DCO within the gap of 61.5 kJ mol−1.137 Improvement on catalytic activity of Cu was attempted by the addition of CeO2 as promotor. Stearic acid was converted to 96.37% product with alkane selectivity of 88.79%. The presence of CeO2 initiate the oxygen vacancies on the surface of Cu permitting the two oxygens of carboxylic functional group to attach on the surface of Cu.105 Similar results were also observed elsewhere.138,139
The application of Fe was also aimed as a cost-effective catalyst in which the Fe3+ was expected to promote DCO2 pathway via the formation of Fe3+ complex with COO− as ligands.142 The Fe/HMS was tested on triolein deoxygenation at 380 °C for 2 hours. This reaction generated 81.4% conversion and 96.4% C8–C18 selectivity with DCO/DCO2 as the favourable route. This result was claimed as the result of excellent dispersion of Fe and the synergistic interaction of Fe–SiO bonds. Newly formed Fe–SiO bond created new acid site, thus increase the amount of weak-medium acid site leading to the high conversion and selectivity.140
In a contrast, lower alkane selectivity of 52% was reported on the deoxygenation of palmitic acid over Fe/AC at 450 °C for 30 minutes with hydrocarbon yield of 55%.141 A slightly higher alkane selectivity of 60.7% was also found in the palmitic acid over Fe2O3/Al-MCM-41.142 Despite the excellent alkane selectivity, the employ of Fe catalyst generated lower conversion compared to other catalyst such as nickel. The conversion remained within the range of 75–89%.143,144
Those phenomena commenced the idea of Ni application alongside with Fe as bimetallic catalyst. The Ni–Fe/ZrO2 catalyst elevated the conversion of oleic acid up to 98.7% over the following reaction conditions: 240 °C, 2 MPa and 3 hours.93 In terms of selectivity, the existence of Fe assisted the reaction pathway owing to the oxophilic Fe that particularly adsorbed and activated the carboxylic acid towards DCO/DCO2.145 Nevertheless, Fe behaved as promotor to Ni and assisted the reduction time by lowering the NiO reduction temperature leading to the smaller particle size and higher surface area.93 Similar results can be seen in other studies.48,146,147
Another less applicable metal in deoxygenation is Sn which commonly used in the form of bimetal catalyst. The sole Sn metal displayed low to moderate conversion and selectivity causing the role of Sn as the promotor metal. For instance, deoxygenation of rapeseed oil over Pt–Sn/Al2O3 achieved 87.37% conversion and promoted the HDO pathway.148,154,155 In monometallic Pt, two oxygen atoms of fatty acid formed a chelate with Pt on catalyst surface and underwent hydrogenolysis to produce aldehyde. Afterwards, the aldehyde undertook C–C scission and CO was released. The addition of Sn hindered the decarbonylation by adsorbing the oxygen atom of aldehyde carbonyl group permitting further hydrogenation to occur.149 This pathway was supported by the high activity of Pt in hydrogen split. Under a rich hydrogen environment, the aldehyde was transformed to alcohol and then alkanes. Moreover, Sn was also in charge of lowering the reduction temperature producing Pt with smaller particle size and improved surface area, as exhibited in the Pt–Sn/SAPO-11.150 Furthermore, the presence of Sn also increased the Lewis acid sites that enhanced the catalytic activity. Other studies can be found elsewhere.151–153
In general, the noble metal catalyst performed high activity at relatively lower temperature and effectively drove the pathway towards DCO/DCO2 in the absence of hydrogen. At rich hydrogen environment, noble metal catalysts favoured HDO route. On the other hand, transition metal required high temperature and depended on the associated reaction condition to engineer the reaction pathway. Table 2 summarizes the influence of catalyst and reaction condition of deoxygenation of various feedstocks.
| Catalysts | Feedstock | Condition | Conv. (%) | Selectivity | Ref. |
|---|---|---|---|---|---|
| Pd@PPN | Stearic acid | 150 °C, 2 MPa H2, 14 h, batch reactor | 90 | C17 = 83% | 123 |
| Pd/SBA-15 | Stearic acid | 300 °C, 1.7 MPa 5% H2, 5 h, semi batch reactor | 96 | C17 = 98% | 124 |
| Pd/C | Waste cooking oil | 400 °C, no H2, 2 h, batch reactor | 100 | n-Paraffin = 71% | 114 |
| Pd/C | Stearic acid | 300 °C, 1.7 MPa 5% H2, 3 h, semi batch reactor | 94 | C17 = 99% | 110 |
| Pd/C | Stearic acid | 300 °C, 1.5 MPa 10% H2, 0.5 h, batch reactor | 100 | C17 = 98% | 125 |
| Pd/C | Stearic acid | 300 °C, 1.5 MPa 5% H2, 3 h, batch reactor | 95 | C17 = 99% | 99 |
| Pd/C | Lauric acid | 300 °C, 2 MPa H2, 5 h, semi batch reactor | 70 | C11 = 90% | 126 |
| Pd/C | Tall oil fatty acid | 300 °C, 1.7 MPa 1% H2, 3 h, batch reactor | 90 | C17 = 95% | 69 |
| Pd/C | Castor oil | 340 °C, 2.5 MPa H2, 7 h, batch reactor | 95 | C17 = 87% | 127 |
| C18 = 9% | |||||
| Pd/AC | Tristearin | 326 °C, 3 h, batch reactor | 100 | C17 = 54% | 115 |
| Pd | Soybean oil | 400 °C, 9.2 MPa H2, 2 h, batch reactor | 91 | C17 = 96% | 128 |
| Pd/Al2O3 | Palm oil | 330 °C, 4 MPa H2, flow reactor | 100 | C17 = 79.5% | 129 |
| C18 = 4.7% | |||||
| Pt/AC | Tristearin | 326 °C, 3 h, batch reactor | 100 | C17 = 64% | 115 |
| Pt/Al2O3 | Palm oil | 330 °C, 4 MPa H2, flow reactor | 100 | C17 = 72% | 129 |
| C18 = 7.6% | |||||
| Pt/Al2O3 | Tristearin | 260 °C, 4 MPa H2, 3 h, semi batch reactor | 5 | C17 = 65% | 116 |
| Pt/θ-Al2O3 | Soybean oil | 360 °C, 5 MPa H2, 2 h, fixed bed reactor | 81 | C13,15,17 = 63% | 117 |
| Pt/C | Crude palm kernel oil | 420 °C, 3.5 MPa H2, 10 h, fixed bed reactor | 59 | C8–16 = 28% | 9 |
| Ru/C | Stearic acid | 140 °C, 5 MPa H2, 6 h, batch reactor | 100 | C17 = 88% | 118 |
| C18 = 12% | |||||
| Ru/C | Corn stover oil | 3000 °C, 12.5 MPa H2, 4 h, batch reactor | 66 | — | 84 |
| Ru/TiO2 | Ethyl stearate | 220 °C, 10 MPa H2, 6 h, batch reactor | 98 | C17 = 64% | 119 |
| C18 = 30% | |||||
| Mo/γ-Al2O3 | Rubber seed oil | 350.15 °C, 3.5 MPa H2, 3 h, fixed bed reactor | 95 | C17 = 20% | 8 |
| C18 = 32% | |||||
| Mo/Al2O3 | Rapeseed oil | 270 °C, 3.5 MPa H2, 4 h, fixed bed reactor | 100 | C18 = 90% | 100 |
| Mo/AC | Palm fatty acid distillate | 350 °C, no H2, 1 h, batch reactor | 54 | C15+17 = 45% | 120 |
| Pt–MoOx/ZrO2 | Oleic acid | 220 °C, 2 MPa H2, 5 h, batch reactor | 94 | C17 = 14% | 121 |
| C18 = 63% | |||||
| MoS2/γ-Al2O3 | Oleic acid | 400 °C, 4 MPa H2, 3 h, batch reactor | 97.91 | C17 = 65% | 122 |
| C18 = 35% | |||||
| Ni/CeO2–Al2O3 | Methyl laurate | 300 °C, 3 MPa 10% H2/N2, 4 h, semi batch reactor | 100 | C11 = 99% | 130 |
| C12 = 2% | |||||
| Ni/Al2O3 | Tristearin | 260 °C, 4 MPa H2, 3 h, semi batch reactor | 2 | C17 = 83% | 116 |
| Ni/Al2O3 | Palm oil | 330 °C, 4 MPa H2, flow reactor | 100 | C17 = 80% | 129 |
| C18 = 2% | |||||
| Ni/γ-Al2O3 | Rubber seed oil | 350.15 °C, 3.5 MPa H2, 3 h, fixed bed reactor | 95 | C17 = 22% | 8 |
| C18 = 5% | |||||
| C15 = 23% | |||||
| Ni/MgO–Al2O3 | Oleic acid | 300 °C, 2 MPa H2, 3 h, batch reactor | 96.7 | C16–18 = 46% | 108 |
| NiMo/Al2O3 | Rapeseed oil | 260 °C, 7 MPa H2, 2 h, flow reactor | — | C18 = 39.6% | 70 |
| C17 = 4.7% | |||||
| NiMoC/Al-SBA-15 | Soybean oil | 400 °C, 4.5 MPa H2, 2 d, fixed bed reactor | 96 | C15–18 = 97% | 131 |
| Ni/HPS | Stearic acid | 250 °C, 3 MPa N2, 1 h, fixed bed reactor | 100 | C17 = 90% | 132 |
| Co/ZrO2 | Ethyl palmitate | 240 °C, 2 MPa H2, 8 h, batch reactor | 100 | C15 = 54% | 133 |
| C16 = 12% | |||||
| Co/Al2O3 | Palm oil | 330 °C, 4 MPa H2, flow reactor | 100 | C17 = 34% | 129 |
| C18 = 50% | |||||
| Co3O4/SiO2–Al2O3 | Methyl stearate | 250 °C, 3 MPa H2, 6 h, batch reactor | 100 | C17 = 10% | 134 |
| C18 = 88% | |||||
| CoMo/Al2O3 | Rapeseed oil | 270 °C, 5 MPa H2, batch reactor | 100 | C18/C17 = 20 | 85 |
| Co@SiO2 | Palmitic acid | 300 °C, 2 MPa H2, 4 h, batch reactor | 100 | C16 = 71% | 135 |
| Co-MOF-700 | Methyl laurate | 280 °C, 2 MPa H2, 4 h, batch reactor | 100 | C11 = 68% | 106 |
| C12 = 18% | |||||
| Cu | Oleic acid | 330 °C, 1 MPa N2, 3 h, batch reactor | 96 | C17 = 28% | 43 |
| Cu/γ-Al2O3 | Stearic acid | 350 °C, 1.4 MPa H2, 6 h, batch reactor | 95,5 | C17 = 78% | 136 |
| Cu/γ-Al2O3 | Stearic acid | 350 °C, 1.4 MPa H2, 1 h, batch reactor | 45 | C17 = 67% | 137 |
| CuO–CeO2/γ-Al2O3 | Stearic acid | 300 °C, 4 MPa H2, 12 h, batch reactor | 96 | — | 105 |
| CuCo/CNT | Stearic acid | 260 °C, 3 MPa H2, 4 h, batch reactor | 100 | C17 = 88% | 138 |
| C18 = 6% | |||||
| Cu–Ni/ZrO2 | Oleic acid | 350 °C, 3 h, batch reactor | 100 | C17 = 67% | 139 |
| Fe/HMS | Triolein | 380 °C, 2 h, batch reactor | 82 | C8–18 = 97% | 140 |
| Fe/AC | Waste cooking oil | 450 °C, 0.5 h, fixed bed reactor | 94 | C15+17 = 52% | 141 |
| Fe2O3/Al-MCM-41 | Palmitic acid | 350 °C, 2 h, batch reactor | 77 | C15 = 61% | 142 |
| CFeAl | Oleic acid | 300 °C, 3 h, batch reactor | 89 | C13–20 = 77% | 143 |
| Fe/CMD900 | Waste cooking oil | 390 °C, 0.5 h, batch reactor | 55 | C13–20 = 38% | 144 |
| Ni–Fe/ZrO2 | Oleic acid | 240 °C, 2 MPa H2, 3 h, batch reactor | 99 | — | 93 |
| FeNi/C | Stearic acid | 330 °C, 3 h, batch reactor | 100 | C17 = 77% | 145 |
| NiO–Fe2O3/MWCNT | Jatropha curcas oil | 350 °C, 1 h, semi-batch reactor | 73 | C15+17 = 63% | 48 |
| Ni–Fe/Al2O3 | Waste cooking oil | 375 °C, 24 h, fixed bed reactor | 100 | C10–20 = 95% | 146 |
| Ni–Fe/SBA-15 | Waste cooking oil | 350 °C, 2 h, semi batch reactor | 73 | C15–17 = 41% | 147 |
| Pt–Sn/Al2O3 | Rapeseed oil | 400 °C, fixed bed reactor | 100 | C18 = 84% | 148 |
| PtSn/SAPO-11 | Methyl palmitate | 375 °C, 3 MPa H2, 3 h, fixed bed reactor | 87 | C15+16 = 79% | 149 |
| Pt–Sn/SAPO-11 | Waste lard oil | 380 °C, 5 MPa H2, fixed bed reactor | 97 | C15–18 = 65% | 150 |
| Pt–Sn/Al2O3 | Rapeseed oil | 400 °C, 5 MPa H2, fixed bed reactor | 100 | C18 = 84% | 151 |
| Ni–Sn/C | Methyl palmitate | 330 °C, 1 MPa N2, 6 h, batch reactor | 99 | C15 = 88% | 152 |
| C16 = 5% | |||||
| Ni–Sn/Al2O3 | Tristearin | 350 °C, 4 MPa N2, 6 h, semi batch reactor | 99 | C17 = 55% | 153 |
As far, metal concentration and dispersion were highlighted as the main effecting properties followed by the acidity of the support. The increase of Ni loading from 1 wt% to 15 wt% generated higher conversion of FAME from 24.8% to 89.3%.156 The effect of metal loading was also confirmed by Miao et al.83 Higher conversion of palmitic acid was obtained with the increase of metal loading from 0 wt% to 20 wt%. Conversion of soybean oil over Ni/Al2O3 sharply inclined from 60.8% to 95.9% by the addition of catalyst amount.52 Kwon et al. investigated the impact of catalyst amount in deoxygenation of canola oil over NiMo/γ-Al2O3 within the catalyst mass range of 0.026–0.102 g.157 The result described that heptadecane and octadecane simultaneously increased as a function of the catalyst amount.157
Another influential factor of catalyst was the catalyst support that steer the reaction leaning towards particular pathway as described in the following section.
9.2. Effect of support
Catalyst support embodies excellent criteria in terms of porosity, metal–support interaction, acidity and thermal acidity. The nature of porosity supplies fundamental characteristic of heterogeneous catalyst including surface area, pore size and pore volume.46,109 These characteristics address the availability of the internal surface to accommodate the active sites, and the accessibility of reactant toward active sites.158 Thermal stability was high necessity since the impregnation method involves calcination and deoxygenation process carried out at elevated temperature. The other catalyst support characteristics display the important function to direct the reaction pathway in green diesel production.Pore size was claimed as the main property of the Ni/zeolite Y on the deoxygenation of microalgae biodiesel. The large pore size of zeolite Y (3.9 nm) prevented mass transfer limitation since methyl palmitate molecule size was smaller (2.5 nm). Expectedly, high conversion of 91.5% was attained at 275 °C and 2 MPa.159 The critical function of pore size was also implied by the deoxygenation of triglycerides over beta zeolite supported catalysts. Low conversion was reported as the result of pore accessibility issues as beta zeolite pore diameter was 5.7 nm while triglyceride size was 7.5 nm.160
Apart from the porosity, acidity of zeolite also plays significant role as elaborated in the deoxygenation of palm fatty acid with 84.8% conversion and 78.2% selectivity of C15–17. The acidity of zeolite that influenced by the Si/Al, addressed the availability of Lewis and Brønsted acid sites.161 The addition of metal into zeolite replaced the proton through an ion exchange, lowering number of Brønsted acid sites and increasing Lewis acid sites.47 Lowered Brønsted acid sites inhibited the cracking and coking process.
Brønsted acid site enabled proton mobility and improved the contact between the external acid site with metal particle that leads to the DCO/DCO2 pathway.162 On the other hand, Lewis acid sites activated carboxylate and carbonyl group, promoting HDO route.134 Other studies claimed zeolite acid sites improved metal dispersion and metal–support interaction.163
The second common catalyst support is Al2O3 which associated with metal–support interaction properties and metal dispersion. Higher dispersion provides more active site exposed on the surface of catalyst. Al2O3 support inclined the dispersion of Ni–Mo–S metal on the NiMoS2/Al2O3 catalyst leading to high conversion and hydrocarbon yield on the deoxygenation of palm oil at 300 °C and 4 MPa.164
Profound study of Al2O3 crystal structure on Pt dispersion in the deoxygenation of triglyceride had been carried out by Oh et al. Gamma and beta alumina displayed the highest Pt dispersion due to the availability of pentacoordinate site. In terms of catalytic activity Pt/γ-Al2O3 displayed highest paraffin yield of 78% at higher conversion due to the larger pore size over θ-Al2O3.117 In case of Ni/Al2O3, the strong Ni interaction with Al2O3 created NiAl2O4, a new active site as DCO/DCO2 driving force.165
The next common catalyst support is SiO2 which is less acidic compared to Al2O3. Many studies reported the employ of SiO2 support combined with various metals. Deoxygenation of oleic acid over CoxNi1−xP/SiO2 at 320 °C and 2 MPa generated 98% conversion and 86% heptadecane selectivity.166 The favored DCO/DCO2 pathway was also found in the deoxygenation of palmitic acid over Co/SiO2 at 260 °C and 2 MPa with 51.9% C15 selectivity. This high result was due to the high surface area (315 m2 g−1) and large pore diameter (36 nm). Notably, the acidity level of this catalyst was very low (0.01 mmol NH3 per g) that susceptible for further hydrogenation to occur.167 Other studies also mentioned the application of SiO2 as catalyst support.135
Other studies attempt to combine Al2O3 and SiO2. An improvement in terms of conversion and selectivity was reported in the deoxygenation of tristearin over Ni–Pd/Al2O3–SiO2. About 99.5% tristearin was achieved over this following reaction conditions: 260 °C, 4 MPa for 3 hours. The application of this catalyst shifted the reaction pathway towards HDO from 0% to 31% selectivity of C18.168
Another support mentioned in the literature is ZrO2. For the sake of comparison, Papageridis et al., investigated the influence of catalyst supports (Al2O3, SiO2, ZrO2) on palm oil conversion. The Ni/ZrO2 exhibited the highest dispersion and more abundance of acid sites. This superior characteristic rendered the ZrO2 as the most effective support in the lowest optimal temperature.169 Furthermore, ZrO2 support provided oxygen vacancies acting as active site for ethyl palmitate adsorption, activating the carbonyl groups and stabilizing intermediates.66
The same phenomenon was also observed in TiO2 support as proposed in the mechanism of deoxygenation over Pd/TiO2. Nanoparticle Pd facilitated H2 dissociation where the hydrogen was able to migrate TiO2 and induced the reduction of Ti4+ to Ti3+. This hydrogen spill over caused some defect in TiO2 structure and oxygen vacancy was formed. The reduction induced the feasible movement of electron from reduced TiO2 to Pd. Subsequently, electron rich Pd effectively activate oxygen in carboxylic/carbonyl group. In addition, the defects on TiO2 provide anchorage sites for Pd particles thereby preventing particle size growth, leading to higher Pd dispersion.170 In spite of high selectivity, the conversion generated from deoxygenation metal supported TiO2 was still below the common support such as Al2O3, SiO2 and zeolite. The WO3/Pt/TiO2 transformed 86% of jatropha fatty acids at 410 °C and 4 MPa (10% H2), with the C15+17 selectivity of 65%.171 The use of TiO2 as catalyst support was also mentioned in other studies.28,172
In respect of surface area, activated carbon attracted many attempts to be applied in deoxygenation reaction. For instance, Ni/AC contain surface area of 584 m2 g−1 with acid sites of 19120 μmol g−1. This trait was in charge of C–O activation and C–C cleavage. Moreover, Ni/AC possessed low to moderate basic sites of 4960 μmol g−1, enhancing the C–O scission in DCO pathway. This characteristic enable to convert 90% of waste cooking oil to 89% of C15 and C17, indicating the DCO/DCO2 route. Although Ni catalyst was commonly prone to deactivation due to coke formation, the high surface area of activated carbon contain sufficient space for adsorption of hydrocarbons formed during deoxygenation.173 The role of basic sites on directing the reaction route was also investigated in the conversion of chicken fat oil over Ni–Mn/MWCNT. The CO2 production inclined as the function of the number of basic sites. The preferable decarboxylation route was acclaimed by the selectivity of C15+17 up to 83%.174 Contrary, very low conversion of 6% was resulted from deoxygenation of methyl laurate over Co/C due to the high strength adsorption capacity of C, making it hardly released the products.106 This was the reason activated carbon rarely used as support material in deoxygenation. Similar result can be seen in other studies.175,176
10. Future outlook
Paris Agreement that set a goal to limit the global emissions to zero and temperature increases to 1.5 °C by 2050, reflects a heightened level of concern about the negative effects of climate change.177 This ultimate goal can be achieved by focusing on the elevation of the existing solutions involving six technological avenues to reach 36.9 giga tonnes CO2 emission cut off (Fig. 12).178 Renewable energy was positioned in the same level priority as electricity efficiency with renewable sustainable biomass as the primary driver.178 In addition, rapid decarbonising has been achieved through the electricity efficiency as well as electrification generated from green resource involving solar, wind and hydro. However, long distance transport particularly aviation remained as challenging issue and takes decade away to commercialise.5 This matter has urged many attempts to fine the solution since aviation sector contributed on 2% of global CO2 emission and about 671 metric tonnes in 2022 (Fig. 13).177 Therefore, the application of biojet/sustainable aviation fuel is beneficial.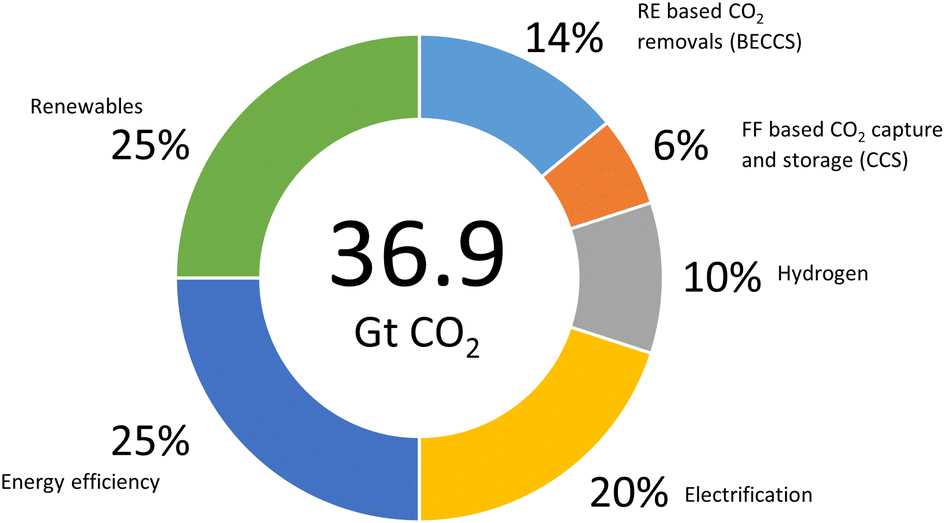 | ||
| Fig. 12 Reducing emissions by 2050 through six technological avenues (redraw from ref. 177) | ||
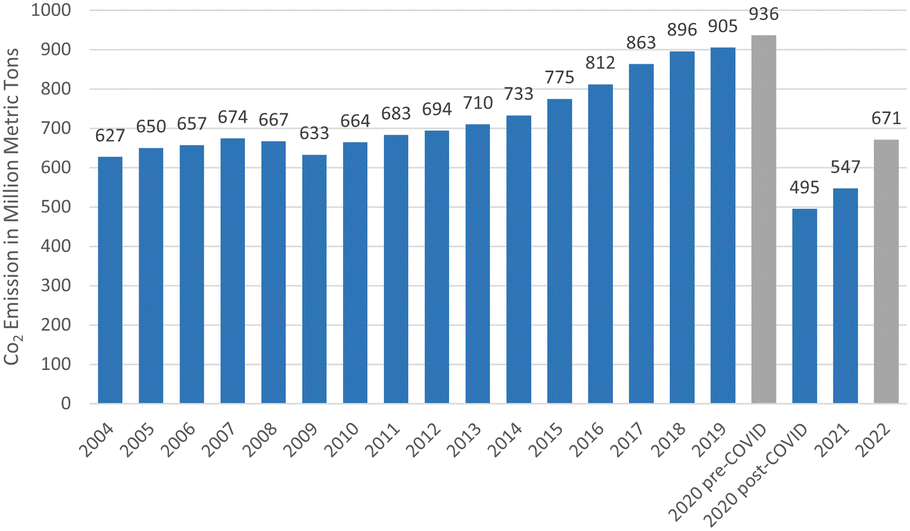 | ||
| Fig. 13 Carbon dioxide emissions from commercial aviation worldwide from 2004 to 2022 (https://www.statista.com/statistics/1186820/co2-emissions-commercial-aviation-worldwide). | ||
The current development of sustainable aviation fuel has been reported by The International Civil Aviation Organization by 2022. About 350000 commercial flights have used biojet fuel which distributed in 45 airports. About 21.6 billion liters of SAF is under offtake agreements and 9 conversion processes has been certified. Moreover, 22 policies associated to sustainable aviation fuel has been adopted or under development.179 Nevertheless, ongoing improvement seems hardly achieve the net zero target and the acceleration is urgently required.178 In consequence, the biojet demand will rapidly increase for the following future.
On a net zero trajectory, biofuel demand is estimated to reach 14 EJ in 2040 and acceleration up to 30% in 2027.180 In more detail, the demand of biodiesel together with biojet fuel is estimated to incline by 44% or 21 billion litres in the span of 2022–2027 (Fig. 14). This consumption growth is the result of increasing demand in major countries such as The United States, Europe, Brazil and Indonesia. In United State, the biofuel requirement is fulfilled by the domestic production using various feedstocks including soybean oil, rapeseed oil, corn oil, used cooking oil and animal fats. The most increasing demand of renewable diesel and biojet occurs in Europe, which trying to shift the feedstock from palm oil to wastes, residues and rapeseed oil, while Brazilian biojet relies on soybean oil. In Indonesia, there is a blending mandates entitled B30 consisting of fossil fuel and biofuel in 70:30 proportion, which boosts the biofuel consumption that generated majorly from palm oil.
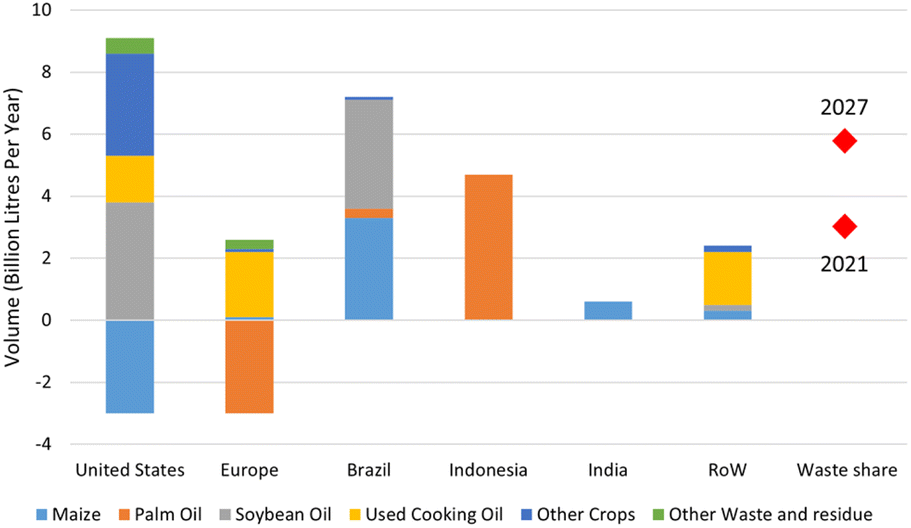 | ||
| Fig. 14 Total biofuel growth from 2021 to 2027 (estimation).179 | ||
In summary, the biojet fuel is forecasted to develop swiftly in the near future, followed by the research improvement in this area. This phenomenon is also supported by the result of bibliometric analysis showing the increasing number of articles aligned to green diesel. For instance, about 33 articles was published in the beginning of 2023.
11. Conclusions
The favoured pathway on green diesel production through deoxygenation depended upon several factors including temperature, hydrogen pressure and catalyst. High temperature preferred DCO/DCO2 owing to the high energy requirement for C–C cleavage and rich hydrogen environment directed the reaction towards HDO. In regards of catalyst, the reaction route was determined by two main categories: metal catalyst and support. Type of metal, metal area, and dispersion involved in the selectivity of the route. The characteristic of catalyst support was represented by the availability of Lewis acid sites, porosity and metal–support interaction.Author contributions
Conceptualization: Hamzah Fansuri. Writing – original draft: Zeni Rahmawati, Liangga Santoso, Nor Laili Azua Jamari. Writing-review and editing: Triyanda Gunawan and Alan McCue.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Ministry of Education, Culture, Research, and Technology Republik Indonesia for the research grant through the Penelitian Dasar Kompetitif Nasional scheme under main contract No. 084/E5/PG.02.00.PT/2022, and Researcher's Contract No. 1371/PKS/ITS/2022.References
- D. R. Dunn, J. Zaidi, M. Kopalek, G. Long, R. Berry G. McGrath, T. Shear, M. Klein, A. Gorski, E. Harrison, L. Jamison and K. Nakolan, Monthly Energy Review- August 2022, Energy Information Administration, United States, 2022 Search PubMed.
- F. Millo, T. Vlachos and A. Piano, Fuel, 2021, 285, 119092 CrossRef CAS.
- S. Manikandan, S. Vickram, R. Sirohi, R. Subbaiya, R. Y. Krishnan, N. Karmegam, C. Sumathijones, R. Rajagopal, S. W. Chang, B. Ravindran and M. K. Awasthi, Bioresour. Technol., 2023, 372, 128679 CrossRef CAS PubMed.
- S. Ding, M. J. Hülsey, J. Pérez-Ramírez and N. Yan, Joule, 2019, 3, 2897–2929 CrossRef CAS.
- IRENA, Reaching zero with renewables: Biojet fuels, International Renewable Energy Agency, Abu Dhabi, 2022 Search PubMed.
- H. I. Mahdi, A. Bazargan, G. McKay, N. I. W. Azelee and L. Meili, Chem. Eng. Res. Des., 2021, 174, 158–187 CrossRef CAS.
- M. K. Shahid, A. Batool, A. Kashif, M. H. Nawaz, M. Aslam, N. Iqbal and Y. Choi, J. Environ. Manage., 2021, 297, 113268 CrossRef CAS PubMed.
- M. Ameen, M. T. Azizan, A. Ramli, S. Yusup and B. Abdullah, Catal. Today, 2020, 355, 51–64 CrossRef CAS.
- M. Makcharoen, A. Kaewchada, N. Akkarawatkhoosith and A. Jaree, Energy Convers. Manage.: X, 2021, 12, 100125 CAS.
- B. Fattahi, M. Haghighi, B. Rahmanivahid and N. Vardast, Chem. Eng. Res. Des., 2022, 183, 411–423 CrossRef CAS.
- N. A. Abdul Razak, N.-A. Mijan, Y. H. Taufiq-Yap and D. Derawi, J. Cleaner Prod., 2022, 366, 132971 CrossRef CAS.
- P. S. D. Priya, Y. Verma, R. A. Muhal, C. Goswami and T. Singh, Mater. Today: Proc., 2022, 48, 1178–1184 CAS.
- M. M. A. Aziz, K. A. Kassim, Z. Shokravi, F. M. Jakarni, H. Y. Liu, N. Zaini, L. S. Tan, A. B. M. S. Islam and H. Shokravi, Renewable Sustainable Energy Rev., 2020, 119, 109621 CrossRef CAS.
- H. Shokravi, Z. Shokravi, M. Heidarrezaei, H. C. Ong, S. S. Rahimian Koloor, M. Petrů, W. J. Lau and A. F. Ismail, Chemosphere, 2021, 285, 131535 CrossRef CAS PubMed.
- C. Ngamcharussrivichai, P. Totarat and K. Bunyakiat, Appl. Catal., A, 2008, 341, 77–85 CrossRef CAS.
- R. Piloto-Rodríguez, Y. Sánchez-Borroto, M. Lapuerta, L. Goyos-Pérez and S. Verhelst, Energy Convers. Manage., 2013, 65, 255–261 CrossRef.
- C. Kordulis, K. Bourikas, M. Gousi, E. Kordouli and A. Lycourghiotis, Appl. Catal., B, 2016, 181, 156–196 CrossRef CAS.
- V. T. da Silva and L. A. Sousa, in The Role of Catalysis for the Sustainable Production of Bio-fuels and Bio-chemicals, Elsevier, 2013, pp. 67–92 Search PubMed.
- S. N. Naik, V. V. Goud, P. K. Rout and A. K. Dalai, Renewable Sustainable Energy Rev., 2010, 14, 578–597 CrossRef CAS.
- G. Di Vito Nolfi, K. Gallucci and L. Rossi, Int. J. Environ. Res. Public Health, 2021, 18, 13041 CrossRef CAS PubMed.
- H. K. Jeswani, A. Chilvers and A. Azapagic, Proc. R. Soc. A, 2020, 476, 20200351 CrossRef PubMed.
- U.S. Department of Energy, Sustainable Aviation Fuel: Review of Technical Pathways Executive, 2020 Search PubMed.
- S. Widiyanto, presented in part at the ICAO Seminar on Alternative Fuels, ICAO Headquarters, Montreal, 2017 Search PubMed.
- EBTKE Public Relation, Ground Run Test of Bioavtur J2.4: Well Accomplished, The Ministry of Energy and Mineral Resource Republic of Indonesia, 2021 Search PubMed.
- K. W. Cheah, S. Yusup, A. C. M. Loy, B. S. How, V. Skoulou and M. J. Taylor, Mol. Catal., 2022, 523, 111469 CrossRef CAS.
- N. Hongloi, P. Prapainainar and C. Prapainainar, Mol. Catal., 2022, 523, 111696 CrossRef CAS.
- F. T. Yani, H. Husin, Darmadi, S. Muhammad, F. Abnisa, Nurhazanah, F. Nasution and Erdiwansyah, J. Cleaner Prod., 2022, 354, 131704 CrossRef CAS.
- T. K. Vo, W.-S. Kim, S.-S. Kim, K. S. Yoo and J. Kim, Energy Convers. Manage., 2018, 158, 92–102 CrossRef CAS.
- Z. Cai, R. Liang, P. Yu, Y. Liu, Y. Ma, Y. Cao, K. Huang, L. Jiang and X. Bao, Fuel Process. Technol., 2022, 226, 107091 CrossRef CAS.
- P. Hellier, M. Talibi, A. Eveleigh and N. Ladommatos, Proc. Inst. Mech. Eng., Part D, 2018, 232, 90–105 CrossRef CAS.
- Y. Zhou, J. Remón, Z. Jiang, A. S. Matharu and C. Hu, Green Energy Environ., 2022, S2468025722000528 Search PubMed.
- Y. S. M. Altarazi, A. R. Abu Talib, J. Yu, E. Gires, M. F. Abdul Ghafir, J. Lucas and T. Yusaf, Energy, 2022, 238, 121910 CrossRef CAS.
- N. Asikin-Mijan, J. M. Ooi, G. AbdulKareem-Alsultan, H. V. Lee, M. S. Mastuli, N. Mansir, F. A. Alharthi, A. A. Alghamdi and Y. H. Taufiq-Yap, J. Cleaner Prod., 2020, 249, 119381 CrossRef CAS.
- R. G. Kukushkin, P. M. Yeletsky, C. T. Grassin, B.-H. Chen, O. A. Bulavchenko, A. A. Saraev and V. A. Yakovlev, Chem. Eng. J., 2020, 396, 125202 CrossRef CAS.
- Y. Wongnongwa, S. Jungsuttiwong, M. Pimsuta, P. Khemthong and M. Kunaseth, Chem. Eng. J., 2020, 399, 125586 CrossRef CAS.
- D. Singh, K. A. Subramanian and S. K. Singal, Appl. Energy, 2015, 155, 440–446 CrossRef CAS.
- K. B. Baharudin, N. Abdullah, Y. H. Taufiq-Yap and D. Derawi, J. Cleaner Prod., 2020, 274, 122850 CrossRef CAS.
- X. Lei, H. Xin, X. Du, H. Yang, Y. Zeng, L. Zhou, C. Juan, H. Zhang, D. Li and C. Hu, Fuel, 2022, 324, 124548 CrossRef CAS.
- W. Nor Adira Wan Khalit, N. Asikin-Mijan, T. Shafirah Marliza, M. Safa-Gamal, M. Razali Shamsuddin, I. Nur Azreena, M. Izham Saiman and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2022, 164, 105505 CrossRef.
- L. E. Oi, M.-Y. Choo, H. V. Lee, Y. H. Taufiq-Yap, C. K. Cheng and J. C. Juan, Int. J. Hydrogen Energy, 2020, 45, 11605–11614 CrossRef CAS.
- X. Y. Ooi, L. E. Oi, M.-Y. Choo, H. C. Ong, H. V. Lee, P. L. Show, Y.-C. Lin and J. C. Juan, Fuel Process. Technol., 2019, 194, 106120 CrossRef CAS.
- M. Žula, M. Grilc and B. Likozar, Chem. Eng. J., 2022, 444, 136564 CrossRef.
- K. W. Cheah, M. J. Taylor, A. Osatiashtiani, S. K. Beaumont, D. J. Nowakowski, S. Yusup, A. V. Bridgwater and G. Kyriakou, Catal. Today, 2020, 355, 882–892 CrossRef CAS.
- J. E. Lam, A. R. Mohamed, A. N. Kay Lup and M. K. Koh, Fuel Process. Technol., 2022, 236, 107394 CrossRef CAS.
- E. Santillan-Jimenez, T. Morgan, J. Shoup, A. E. Harman-Ware and M. Crocker, Catal. Today, 2014, 237, 136–144 CrossRef CAS.
- N. Arun, R. V. Sharma and A. K. Dalai, Renewable Sustainable Energy Rev., 2015, 48, 240–255 CrossRef CAS.
- M.-Y. Choo, L. E. Oi, T. C. Ling, E.-P. Ng, Y.-C. Lin, G. Centi and J. C. Juan, J. Anal. Appl. Pyrolysis, 2020, 147, 104797 CrossRef CAS.
- N. Asikin-Mijan, N. A. Rosman, G. AbdulKareem-Alsultan, M. S. Mastuli, H. V. Lee, N. Nabihah-Fauzi, I. M. Lokman, F. A. Alharthi, A. A. Alghamdi, A. A. Aisyahi and Y. H. Taufiq-Yap, Process Saf. Environ. Prot., 2020, 142, 336–349 CrossRef CAS.
- A. Yıldız, J. L. Goldfarb and S. Ceylan, Fuel, 2020, 263, 116750 CrossRef.
- D.-P. Phan and E. Y. Lee, J. Catal., 2020, 386, 19–29 CrossRef CAS.
- P. Šimáček and D. Kubička, Fuel, 2010, 89, 1508–1513 CrossRef.
- B. Veriansyah, J. Y. Han, S. K. Kim, S.-A. Hong, Y. J. Kim, J. S. Lim, Y.-W. Shu, S.-G. Oh and J. Kim, Fuel, 2012, 94, 578–585 CrossRef CAS.
- A. Srifa, N. Viriya-empikul, S. Assabumrungrat and K. Faungnawakij, Catal. Sci. Technol., 2015, 5, 3693 RSC.
- C. Hu, D. Creaser, S. Siahrostami, H. Grönbeck, H. Ojagh and M. Skoglundh, Catal. Sci. Technol., 2014, 4, 2427–2444 RSC.
- X. Cao, J. Zhao, F. Long, P. Liu, X. Jiang, X. Zhang, J. Xu and J. Jiang, Appl. Catal., B, 2022, 312, 121437 CrossRef CAS.
- M. de Oliveira Camargo, J. L. Castagnari Willimann Pimenta, M. de Oliveira Camargo and P. A. Arroyo, Fuel, 2020, 281, 118719 CrossRef CAS.
- J. L. C. W. Pimenta, M. de Oliveira Camargo, R. Belo Duarte, O. A. A. dos Santos and L. M. de Matos Jorge, Fuel, 2021, 290, 119979 CrossRef CAS.
- M. F. Wagenhofer, E. Baráth, O. Y. Gutiérrez and J. A. Lercher, ACS Catal., 2017, 7, 1068–1076 CrossRef CAS.
- F. Deng, J. Huang, E. E. Ember, K. Achterhold, M. Dierolf, A. Jentys, Y. Liu, F. Pfeiffer and J. A. Lercher, ACS Catal., 2021, 11, 14625–14634 CrossRef CAS.
- S. Fu, D. Li, T. Liu, L. Liu, H. Yang and C. Hu, Catalysts, 2022, 12, 569 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, J. C. Juan, A. R. Noorsaadah and Y. H. Taufiq-Yap, RSC Adv., 2017, 7, 46445–46460 RSC.
- H. Yang, Y. Zeng, Y. Zhou, X. Du, D. Li and C. Hu, J. Catal., 2022, 413, 297–310 CrossRef CAS.
- Y. Bie, J. Lehtonen and J. Kanervo, Appl. Catal., A, 2016, 526, 183–190 CrossRef CAS.
- J. Chen and Q. Xu, Catal. Sci. Technol., 2016, 6, 7239–7251 RSC.
- S.-U. Lee, E. S. Kim, T.-W. Kim, J.-R. Kim, K.-E. Jeong, S. Lee and C.-U. Kim, J. Ind. Eng. Chem., 2020, 83, 366–374 CrossRef CAS.
- Y. Zhou, X. Liu, P. Yu and C. Hu, Fuel, 2020, 278, 118295 CrossRef CAS.
- K. N. Papageridis, N. D. Charisiou, S. Douvartzides, V. Sebastian, S. J. Hinder, M. A. Baker, S. AlKhoori, K. Polychronopoulou and M. A. Goula, Renewable Energy, 2020, 162, 1793–1810 CrossRef CAS.
- R. S. R. M. Hafriz, N. A. Arifin, A. Salmiaton, R. Yunus, Y. H. Taufiq-Yap, N. M. Saifuddin and A. H. Shamsuddin, Fuel, 2022, 308, 122041 CrossRef CAS.
- B. Rozmysłowicz, P. Mäki-Arvela, S. Lestari, O. A. Simakova, K. Eränen, I. L. Simakova, D. Yu. Murzin and T. O. Salmi, Top. Catal., 2010, 53, 1274–1277 CrossRef.
- P. Šimáček, D. Kubička, G. Šebor and M. Pospíšil, Fuel, 2009, 88, 456–460 CrossRef.
- H. Yan, S. Yao, T. Zhang, D. Li, X. Tang, M. Chen, Y. Zhou, M. Zhang, Y. Liu, X. Zhou, X. Feng, X. Chen and C. Yang, Appl. Catal., B, 2022, 306, 121138 CrossRef CAS.
- S. T. Mohammed, K. I. Hamad, S. A. Gheni, D. Y. Aqar, S. M. R. Ahmed, M. A. Mahmood, S. Ceylan and G. H. Abdullah, Chem. Eng. Sci., 2022, 251, 117489 CrossRef CAS.
- F. Pinto, S. Martins, M. Gonçalves, P. Costa, I. Gulyurtlu, A. Alves and B. Mendes, Appl. Energy, 2013, 102, 272–282 CrossRef CAS.
- M. Snåre, I. Kubičková, P. Mäki-Arvela, K. Eränen, J. Wärnå and D. Y. Murzin, Chem. Eng. J., 2007, 134, 29–34 CrossRef.
- M. Arend, T. Nonnen, W. F. Hoelderich, J. Fischer and J. Groos, Appl. Catal., A, 2011, 399, 198–204 CrossRef CAS.
- H. Bernas, K. Eränen, I. Simakova, A. R. Leino, K. Kordás, J. Myllyoja, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, Fuel, 2010, 89, 2033–2039 CrossRef CAS.
- M. Jin and M. Choi, Mol. Catal., 2019, 474, 110419 CrossRef CAS.
- J. L. F. Oliveira, L. M. B. Batista, N. Alburquerque dos Santos, A. M. M. Araújo, V. J. Fernandes, A. S. Araujo, A. P. M. Alves and A. D. Gondim, Renewable Energy, 2021, 168, 1377–1387 CrossRef CAS.
- K. Praserttaweeporn, T. Vitidsant and W. Charusiri, Results Eng., 2022, 14, 100461 CrossRef CAS.
- M. Safa-Gamal, N. Asikin-Mijan, M. Arumugam, W. N. A. W. Khalit, I. Nur Azreena, F. S. Hafez and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2021, 160, 105334 CrossRef CAS.
- B. H. Susanto, M. Nasikin, Sukirno and A. Wiyo, Procedia Chem., 2014, 9, 139–150 CrossRef CAS.
- P. Šimáček, D. Kubička, I. Kubičková, F. Homola, M. Pospíšil and J. Chudoba, Fuel, 2011, 90, 2473–2479 CrossRef.
- C. Miao, O. Marin-Flores, S. D. Davidson, T. Li, T. Dong, D. Gao, Y. Wang, M. Garcia-Pérez and S. Chen, Fuel, 2016, 166, 302–308 CrossRef CAS.
- J. A. Capunitan and S. C. Capareda, Fuel Process. Technol., 2014, 125, 190–199 CrossRef CAS.
- J. Horáček, Z. Tišler, V. Rubáš and D. Kubička, Fuel, 2014, 121, 57–64 CrossRef.
- Q. Liu, H. Zuo, Q. Zhang, T. Wang and L. Ma, Chin. J. Catal., 2014, 35, 748–756 CrossRef CAS.
- S. Kovács, T. Kasza, A. Thernesz, I. W. Horváth and J. Hancsók, Chem. Eng. J., 2011, 176–177, 237–243 CrossRef.
- I. Nur Azreena, H. L. N. Lau, N. Asikin-Mijan, M. A. Hassan, S. M. Izham, M. Safa Gamal, W. Nor Adira Wan Khalit, M. Arumugam, E. Kennedy, M. Stockenhuber and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2022, 161, 105406 CrossRef CAS.
- S. Ding, Y. Guo, M. J. Hülsey, B. Zhang, H. Asakura, L. Liu, Y. Han, M. Gao, J. Hasegawa, B. Qiao, T. Zhang and N. Yan, Chem, 2019, 5, 3207–3219 CAS.
- D. Kubička, J. Horáček, M. Setnička, R. Bulánek, A. Zukal and I. Kubičková, Appl. Catal., B, 2014, 145, 101–107 CrossRef.
- C. Detoni, F. Bertella, M. M. V. M. Souza, S. B. C. Pergher and D. A. G. Aranda, Appl. Clay Sci., 2014, 95, 388–395 CrossRef CAS.
- P. Mäki-Arvela, I. Kubickova, M. Snåre, K. Eränen and D. Y. Murzin, Energy Fuels, 2007, 21, 30–41 CrossRef.
- F. Wang, H. Xu, S. Yu, H. Zhu, Y. Du, Z. Zhang, C. You, X. Jiang and J. Jiang, Renewable Energy, 2022, 197, 40–49 CrossRef CAS.
- X. Liu, M. Yang, Z. Deng, A. Dasgupta and Y. Guo, Chem. Eng. J., 2021, 407, 126332 CrossRef CAS.
- K. A. Rogers, J. Fu, Y. Xu and Y. Zheng, Catal. Today, 2022, S0920586122001249 Search PubMed.
- P. Arora, E. L. Grennfelt, L. Olsson and D. Creaser, Chem. Eng. J., 2019, 364, 376–389 CrossRef CAS.
- L. Fu, Y. Li, H. Cui, W. Ba and Y. Liu, Appl. Catal., A, 2021, 623, 118258 CrossRef CAS.
- G. C. R. Silva and M. H. C. de Andrade, Biomass Convers. Biorefin., 2021, 13, 1843–1857 CrossRef.
- J. G. Immer and H. H. Lamb, Energy Fuels, 2010, 24, 5291–5299 CrossRef CAS.
- D. Kubička and L. Kaluža, Appl. Catal., A, 2010, 372, 199–208 CrossRef.
- J. Wang, X. Chen, X. Chen, C. Zhao, Y. Ling and C. Liang, Sustainable Energy Fuels, 2022, 6, 3025–3034 RSC.
- K. Jeništová, I. Hachemi, P. Mäki-Arvela, N. Kumar, M. Peurla, L. Čapek, J. Wärnå and D. Yu. Murzin, Chem. Eng. J., 2017, 316, 401–409 CrossRef.
- S. Janampelli and S. Darbha, Energy Fuels, 2018, 32, 12630–12643 CrossRef CAS.
- N. Asikin-Mijan, G. AbdulKareem-Alsultan, M. S. Mastuli, A. Salmiaton, M. Azuwa Mohamed, H. V. Lee and Y. H. Taufiq-Yap, Fuel, 2022, 325, 124917 CrossRef CAS.
- H. Du, Q. Yu, G. Liu, J. Li, J. Zhang, W. Wang, G. Duan, Y. Meng and H. Xie, Fuel, 2022, 317, 123367 CrossRef CAS.
- Z. Ding, T. Zhao, Q. Zhu, S. Liao, L. Ning, Y. Bi and H. Chen, Biomass Bioenergy, 2020, 143, 105879 CrossRef CAS.
- W. Zhou, H. Xin, H. Yang, X. Du, R. Yang, D. Li and C. Hu, Catalysts, 2018, 8, 153 CrossRef.
- K.-W. Jeon, H.-R. Park, Y.-L. Lee, J.-E. Kim, W.-J. Jang, J.-O. Shim and H.-S. Roh, Fuel, 2022, 311, 122488 CrossRef CAS.
- A. Galadima and O. Muraza, J. Ind. Eng. Chem., 2015, 29, 12–23 CrossRef CAS.
- S. Lestari, P. Mäki-Arvela, I. Simakova, J. Beltramini, G. Q. M. Lu and D. Yu. Murzin, Catal. Lett., 2009, 130, 48–51 CrossRef CAS.
- K. Hengst, M. Arend, R. Pfützenreuter and W. F. Hoelderich, Appl. Catal., B, 2015, 174–175, 383–394 CrossRef CAS.
- J. Lu, M. Faheem, S. Behtash and A. Heyden, J. Catal., 2015, 324, 14–24 CrossRef CAS.
- T. Morgan, D. Grubb, E. Santillan-Jimenez and M. Crocker, Top. Catal., 2010, 53, 820–829 CrossRef CAS.
- E. S. K. Why, H. C. Ong, H. V. Lee, W.-H. Chen, N. Asikin-Mijan, M. Varman and W. J. Loh, Energy, 2022, 239, 122017 CrossRef CAS.
- L. J. Konwar and J.-P. Mikkola, Appl. Catal., A, 2022, 638, 118611 CrossRef CAS.
- R. Loe, K. Huff, M. Walli, T. Morgan, D. Qian, R. Pace, Y. Song, M. Isaacs, E. Santillan-Jimenez and M. Crocker, Catalysts, 2019, 9, 200 CrossRef.
- M. Oh, M. Jin, K. Lee, J.-C. Kim, R. Ryoo and M. Choi, Chem. Eng. J., 2022, 439, 135530 CrossRef CAS.
- A. Ali and C. Zhao, Chin. J. Catal., 2020, 41, 1174–1185 CrossRef CAS.
- L. Zhou, W. Lin, K. Liu, Z. Wang, Q. Liu, H. Cheng, C. Zhang, M. Arai and F. Zhao, Catal. Sci. Technol., 2020, 10, 222–230 RSC.
- M. S. Gamal, N. Asikin-Mijan, W. N. A. W. Khalit, M. Arumugam, M. Izham Saiman and Y. H. Taufiq-Yap, Fuel Process. Technol., 2020, 208, 106519 CrossRef CAS.
- S. Janampelli and S. Darbha, Catal. Today, 2021, 375, 174–180 CrossRef CAS.
- S. Thongkumkoon, W. Kiatkittipong, U. W. Hartley, N. Laosiripojana and P. Daorattanachai, Renewable Energy, 2019, 140, 111–123 CrossRef CAS.
- C. Sarkar, S. Chandra Shit, D. Quang Dong, J. Lee, N. Han Tran, R. Singuru, K. An, N. Dang Nam, Q. Van Le, P. Nana Amaniampong, A. Drif, F. Jerome, P. Thanh Huyen, T. To Nga Phan, D.-V. N. Vo, N. Thanh Binh, Q. Thang Trinh, M. P. Sherburne and J. Mondal, Green Chem., 2020, 22, 2049–2068 RSC.
- S. Lestari, P. Mäki-Arvela, K. Eränen, J. Beltramini, G. Q. Max Lu and D. Yu. Murzin, Catal. Lett., 2010, 134, 250–257 CrossRef CAS.
- J. G. Immer, M. J. Kelly and H. H. Lamb, Appl. Catal., A, 2010, 375, 134–139 CrossRef CAS.
- B. Rozmysłowicz, P. Mäki-Arvela, A. Tokarev, A.-R. Leino, K. Eränen and D. Yu. Murzin, Ind. Eng. Chem. Res., 2012, 51, 8922–8927 CrossRef.
- E. Meller, U. Green, Z. Aizenshtat and Y. Sasson, Fuel, 2014, 133, 89–95 CrossRef CAS.
- S. K. Kim, J. Y. Han, H. Lee, T. Yum, Y. Kim and J. Kim, Appl. Energy, 2014, 116, 199–205 CrossRef CAS.
- A. Srifa, K. Faungnawakij, V. Itthibenchapong and S. Assabumrungrat, Chem. Eng. J., 2015, 278, 249–258 CrossRef CAS.
- L. Fu, Z. Liu, X. Li, Y. Li, H. Yang and Y. Liu, Fuel, 2022, 322, 124027 CrossRef CAS.
- H. Wang, S. Yan, S. O. Salley and K. Y. Simon Ng, Fuel, 2013, 111, 81–87 CrossRef CAS.
- A. A. Stepacheva, A. I. Sidorov, V. G. Matveeva, M. G. Sulman and E. M. Sulman, Chem. Eng. Technol., 2019, 42, 780–787 CrossRef CAS.
- Y. Zhou, L. Liu, G. Li and C. Hu, ACS Catal., 2021, 11, 7099–7113 CrossRef CAS.
- V. K. Soni, S. Dhara, R. Krishnapriya, G. Choudhary, P. R. Sharma and R. K. Sharma, Fuel, 2020, 266, 117065 CrossRef CAS.
- M. Lin, X. Zhang, L. Zhan, X. Li, X. Song and Y. Wu, Fuel, 2022, 318, 123605 CrossRef CAS.
- A. S. Berenblyum, R. S. Shamsiev, T. A. Podoplelova and V. Y. Danyushevsky, Russ. J. Phys. Chem., 2012, 86, 1199–1203 CrossRef CAS.
- A. S. Berenblyum, V. Y. Danyushevsky, E. A. Katsman, R. S. Shamsiev and V. R. Flid, Pet. Chem., 2013, 53, 362–366 CrossRef CAS.
- J. Liang, Z. Zhang, K. Wu, Y. Shi, W. Pu, M. Yang and Y. Wu, Fuel Process. Technol., 2019, 188, 153–163 CrossRef CAS.
- Z. Zhang, H. Chen, C. Wang, K. Chen, X. Lu, P. Ouyang and J. Fu, Fuel, 2018, 230, 211–217 CrossRef CAS.
- S. Zulkepli, H. V. Lee, N. Abd. Rahman, L. T. Chuan, P. L. Show, W.-H. Chen and J. C. Juan, Fuel, 2022, 308, 121860 CrossRef CAS.
- T. Thangadurai and C. T. Tye, Period. Polytech., Chem. Eng., 2021, 65, 350–360 CrossRef CAS.
- J. Bian, Y. Wang, Q. Zhang, X. Fang, L. Feng and C. Li, RSC Adv., 2017, 7, 47279–47287 RSC.
- S. N. Zdainal Abidin, H. V. Lee, N. Asikin-Mijan, J. C. Juan, N. A. Rahman, M. S. Mastuli, Y. H. Taufiq-Yap and P. S. Kong, Pure Appl. Chem., 2020, 92, 587–600 CrossRef CAS.
- R. S. R. M. Hafriz, I. Nor Shafizah, A. Salmiaton, N. A. Arifin, R. Yunus, Y. H. Taufiq Yap and S. Abd Halim, Arabian J. Chem., 2020, 13, 8146–8159 CrossRef CAS.
- J. Zhong, Q. Deng, T. Cai, X. Li, R. Gao, J. Wang, Z. Zeng, G. Dai and S. Deng, Fuel, 2021, 292, 120248 CrossRef CAS.
- G. C. R. Silva, D. Qian, R. Pace, O. Heintz, G. Caboche, E. Santillan-Jimenez and M. Crocker, Catalysts, 2020, 10, 91 CrossRef CAS.
- N. A. Rashidi, E. Mustapha, Y. Y. Theng, N. A. A. Razak, N. A. Bar, K. B. Baharudin and D. Derawi, Fuel, 2022, 313, 122695 CrossRef CAS.
- M. V. Tsodikov, A. V. Chistyakov, M. A. Gubanov, P. A. Zharova, S. S. Shapovalov, A. A. Pasynskii, V. V. Kriventsov and I. I. Moiseev, Russ. Chem. Bull., 2015, 64, 2062–2068 CrossRef CAS.
- N. Chen, Y. Ren and E. W. Qian, J. Catal., 2016, 334, 79–88 CrossRef CAS.
- X. Li, Q. Wang, J. Chen, S. Li, D. Wang and Z. Zheng, J. Anal. Appl. Pyrolysis, 2021, 156, 105121 CrossRef CAS.
- P. A. Zharova, A. V. Chistyakov, S. S. Shapovalov, A. A. Pasynskii and M. V. Tsodikov, Energy, 2019, 172, 18–25 CrossRef CAS.
- H. Shi, X. Gu, Y. Shi, D. Wang, S. Shu, Z. Wang and J. Chen, Front. Chem. Sci. Eng., 2022, 17, 139–155 CrossRef.
- R. Loe, E. Santillan-Jimenez, T. Morgan, L. Sewell, Y. Ji, S. Jones, M. A. Isaacs, A. F. Lee and M. Crocker, Appl. Catal., B, 2016, 191, 147–156 CrossRef CAS.
- A. V. Chistyakov, V. V. Kriventsov, A. V. Naumkin, A. Y. Pereyaslavtsev, P. A. Zharova and M. V. Tsodikov, Pet. Chem., 2016, 56, 607–615 CrossRef CAS.
- L. Zhao, B. Li and C. Zhao, ACS Sustainable Chem. Eng., 2021, 9, 12970–12977 CrossRef CAS.
- L. Chen, H. Li, J. Fu, C. Miao, P. Lv and Z. Yuan, Catal. Today, 2016, 259, 266–276 CrossRef CAS.
- K. C. Kwon, H. Mayfield, T. Marolla, B. Nichols and M. Mashburn, Renewable Energy, 2011, 36, 907–915 CrossRef CAS.
- C. H. Bartholomew and R. J. Farrauto, Fundamentals of Industrial Catalytic Processes, John Wiley & Sons, Inc., 2nd edn, 2006 Search PubMed.
- J. Cheng, Z. Zhang, X. Zhang, Z. Fan, J. Liu and J. Zhou, Int. J. Hydrogen Energy, 2019, 44, 11765–11773 CrossRef CAS.
- F. P. Sousa, L. N. Silva, D. B. de Rezende, L. C. A. de Oliveira and V. M. D. Pasa, Fuel, 2018, 223, 149–156 CrossRef CAS.
- B. F. Honorato de Oliveira, L. F. de França, N. C. Fernandes Corrêa, N. F. d. P. Ribeiro and M. Velasquez, Catalysts, 2021, 11, 1088 CrossRef CAS.
- J. M. Crawford, C. S. Smoljan, J. Lucero and M. A. Carreon, Catalysts, 2019, 9, 42 CrossRef.
- M. Arroyo, L. Briones, H. Hernando, J. M. Escola and D. P. Serrano, Energy Fuels, 2021, 35, 17167–17181 CrossRef CAS.
- P. Aiamsiri, D. Tumnantong, B. Yoosuk, C. Ngamcharussrivichai and P. Prasassarakich, Energy Fuels, 2021, 35, 14793–14804 CrossRef CAS.
- H. Xin, H. Yang, X. Lei, X. Du, K. Zhou, D. Li and C. Hu, Ind. Eng. Chem. Res., 2020, 59, 17373–17386 CrossRef CAS.
- Z. Zhang, G. Bi, H. Zhang, A. Zhang, X. Li and J. Xie, Fuel, 2019, 247, 26–35 CrossRef CAS.
- M. Liu, Y. Shi, K. Wu, J. Liang, Y. Wu, S. Huang and M. Yang, Catal. Commun., 2019, 129, 105726 CrossRef CAS.
- F. Wang, R. Pace, Y. Ji, J. Jiang, X. Jiang, A. Krystianiak, O. Heintz, G. Caboche, E. Santillan-Jimenez and M. Crocker, Renewable Energy, 2022, 195, 1468–1479 CrossRef CAS.
- K. N. Papageridis, N. D. Charisiou, S. L. Douvartzides, V. Sebastian, S. J. Hinder, M. A. Baker, S. AlKhoori, K. Polychronopoulou and M. A. Goula, Fuel Process. Technol., 2020, 209, 106547 CrossRef CAS.
- T. Hengsawad, T. Jindarat, D. E. Resasco and S. Jongpatiwut, Appl. Catal., A, 2018, 566, 74–86 CrossRef CAS.
- I.-H. Choi, J.-S. Lee, C.-U. Kim, T.-W. Kim, K.-Y. Lee and K.-R. Hwang, Fuel, 2018, 215, 675–685 CrossRef CAS.
- X. Li, X. Yang, Q. Wang, S. Li, Y. Ye, D. Wang and Z. Zheng, J. Cleaner Prod., 2022, 350, 131520 CrossRef CAS.
- W. N. A. W. Khalit, N. Asikin-Mijan, T. S. Marliza, M. S. Gamal, M. R. Shamsuddin, M. I. Saiman and Y. H. Taufiq-Yap, Biomass Bioenergy, 2021, 154, 106248 CrossRef CAS.
- N. Aliana-Nasharuddin, N. Asikin-Mijan, G. Abdulkareem-Alsultan, M. I. Saiman, F. A. Alharthi, A. A. Alghamdi and Y. H. Taufiq-Yap, RSC Adv., 2020, 10, 626–642 RSC.
- G. Abdulkareem-Alsultan, N. Asikin-Mijan, L. K. Obeas, R. Yunus, S. Z. Razali, A. Islam and Y. Hin Taufiq-Yap, Chem. Eng. J., 2022, 429, 132206 CrossRef CAS.
- M. S. Gamal, N. Asikin-Mijan, W. N. A. W. Khalit, M. Arumugam, S. M. Izham and Y. H. Taufiq-Yap, Fuel Process. Technol., 2020, 208, 106519 CrossRef CAS.
- IPCC, Climate Change 2022: Mitigation of Climate Change. Contribution of Working Group III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2022 Search PubMed.
- IRENA, World Energy Transitions Outlook 2022: 1.5 °C Pathway – Executive Summary, International Renewable Energy Agency, Abu Dhabi, 2022 Search PubMed.
- ICAO, Climate Change Mitigation: Overview, The International Civil Association, 2022 Search PubMed.
- IEA, Renewables 2022: Analysis and forecast to 2027, International Energy Agency, 2022 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |