
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectro-organic green synthesis of dicyano-2-(2-oxoindolin-3-ylidene) malononitriles using molecular iodine as catalyst†
Neetu Vermaa,
Vinay Kumar Mishrab,
Sundaram Singha,
Manisha Malviya*a and
Ram Sagar *bc
*bc
aDepartment of Chemistry, IIT (Banaras Hindu University), Varanasi 221005, India. E-mail: manisha.apc@itbhu.ac.in
bDepartment of Chemistry, Institute of Science, Banaras Hindu University, Varanasi 221005, India. E-mail: ram.sagar@jnu.ac.in
cGlycochemistry Laboratory, School of Physical Sciences, Jawaharlal Nehru University, New Delhi 110067, India
First published on 16th May 2023
Abstract
The first electrochemical molecular iodine promoted, domino reactions for the green synthesis of biologically relevant dicyano 2-(2-oxoindolin-3-ylidene) malononitriles (11 examples, up to 94% yield) from readily available isatin derivatives, malononitrile, and iodine at room temperature have been presented. This synthesis method showed tolerance towards various EDG and EWG and was completed in a short reaction time at the constant low current density of 5 mA cm−2 in the low redox potential range of −0.14 to 0.07 V. The present study exhibited by-product-free formation, easy operation, and product isolation. In particular the formation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond was observed at room temperature with a high atom economy. Furthermore, in the present study, the electrochemical behavior of dicyano 2-(2-oxoindolin-3-ylidene) malononitrile derivatives using a cyclic voltammetry (CV) technique in 0.1 M NaClO4 in acetonitrile solution was studied. All the chosen substituted isatin exhibited well-defined diffusion-controlled quasi-reversible redox peaks except 5-substituted derivatives. This synthesis could serve as an alternative strategy to synthesize other biologically important oxoindolin-3-ylidene malononitrile derivatives.
C bond was observed at room temperature with a high atom economy. Furthermore, in the present study, the electrochemical behavior of dicyano 2-(2-oxoindolin-3-ylidene) malononitrile derivatives using a cyclic voltammetry (CV) technique in 0.1 M NaClO4 in acetonitrile solution was studied. All the chosen substituted isatin exhibited well-defined diffusion-controlled quasi-reversible redox peaks except 5-substituted derivatives. This synthesis could serve as an alternative strategy to synthesize other biologically important oxoindolin-3-ylidene malononitrile derivatives.
The dicyano derivative of 2-(2-oxoindolin-3-ylidene) malononitrile, or isatylidene malononitrile, is an interesting Michael acceptor used to construct potential bio-active molecules. Because of the unique structural features and interesting biological properties of 2-(2-oxoindolin-3-ylidene) malononitrile, the design of new strategies for constructing this scaffold has attracted considerable interest. General interest in synthesizing isatylidene malononitrile-based structures comes not only from their structural properties and biological applications1–4 but also from their electrochemical properties. Many researchers have reported a two or three-component reaction to synthesize isatylidene malononitriles as an intermediate. Isatylidene malononitriles have been prepared by many researchers5,6 using different techniques. Among these, the most common methods for the synthesis of 2-(2-oxoindolin-3-ylidene) malononitriles are the condensation of isatins with malononitriles in the presence of a catalyst, such as a piperidine acetate,7 DBU, Al2O3, N(CH2CH2OH)3, chitosan.6,8,9 Recently, microwave irradiation10,11 and iodine12 at elevated temperature have also been applied for the synthesis of isatylidene malononitriles.
In the last few years, molecular iodine has emerged as a powerful catalyst in various organic transformations as it is mild, soluble in common organic solvents, non-toxic, cost-effective, non-hazardous, and environmentally benign catalyst for the synthesis of a variety of heterocyclic compounds.13 It also constitutes an alternative to transition metal catalysis. Its mild Lewis acidity and halogen bond activation favor the catalytic properties. Generally, organic synthesis reactions reported in the presence of molecular iodine occurs at room temperatures (25 °C).14 However, most of these procedures have significant drawbacks such as long reaction times, low yields, harsh reaction conditions, tedious workup, and environmentally toxic or expensive reagents etc. Thus, there is still a need to develop a simple and general protocol for condensing isatin with malononitrile. Until now, molecular iodine catalyzed transformations were carried out in conventional as well as under microwave and ultrasound irradiation only for such types of transformations. We are disclosing here the first report, to the best of my knowledge, on use of molecular iodine in the electro-organic synthesis of the dicyano derivative of 2-(2-oxoindolin-3-ylidene) malononitrile. Electro-organic synthesis is considered a clean and efficient synthetic methodology.15,16 Molecular iodine is the exciting and essential catalyst of the present electro-organic synthesis. Using electron as a reagent, the following benefits were observed: the number of steps reduced, cleaner reaction mixtures, easier isolation of product, and pollution caused by the use of chemicals decreased significantly. Summary of previous strategies for the synthesis of dicyano 2(2-oxoindolin-3-ylidene) malononitrile derivatives using various methods with shortcomings of the methods viz. use of metallic catalyst,17 temperature12 and access of only few derivatives18,19 their comparison with present green electro-organic synthesis catalysed with iodine are presented in Scheme 1.
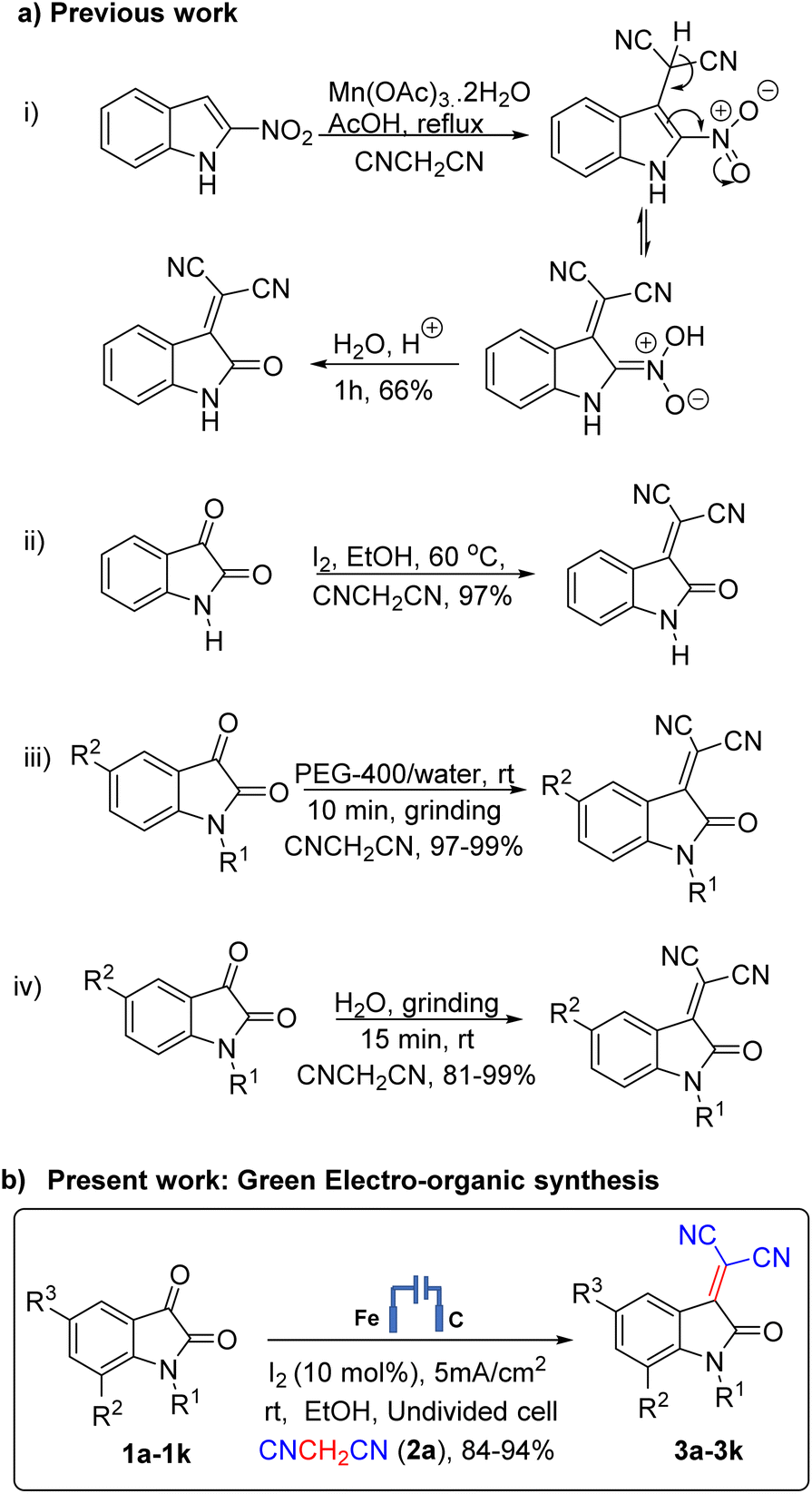 | ||
| Scheme 1 Strategies for the synthesis of dicyano 2-(2-oxoindolin-3-ylidene) malononitrile derivatives. | ||
In the present work, after screening the various reaction conditions (Tables S1–S3, ESI†), we herein reporting a three-component reaction using isatin 1a (1.0 mmol), malononitrile 2a (1.0 mmol), and iodine (10 mol%) in ethanol (20 mL) to furnish the desired isatylidene malononitrile 3a in 94% isolated yield. The electrochemical synthesis was carried out in an undivided cell equipped with carbon cloth (2 cm2) as an anode and iron (2 cm2) as cathode at room temperature under a constant current density of 5 mA cm−2. The progress of the reaction was monitored by TLC using a hexane/ethyl acetate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) solvent mixture. Initially, we selected isatin 1a and malononitrile 2a as the model starting materials to optimize the reaction. The use of 1.0 equivalent of 1a, 2a and I2 (0.1 mmol) was found to be optimal reaction conditions (Table S3, ESI†). The yield got decreased with no change in product was observed when their equivalents were increased or decreased. Subsequently, the influence of solvents, time of reaction, and applied current density were studied (Tables S1 and S2, ESI†). We have performed electrolysis in various LR grade solvents such as EtOH, MeOH, MeCN, DCM, DMF and H2O and found that reaction proceeded in all these solvents and was observed to be best in EtOH in terms of yield and reaction time (Table S1, ESI†). The experiment manifested that no product was formed without any solvent, while a low conversion was observed in acetonitrile and dichloromethane (may be due to poor transfer of e−) while ethanol enhanced the yield. The reaction with changes in equivalents ratio of substrates 1a and 2a along with iodine was also carried out which showed no change in product expect very little variation in yields (Table S3, ESI†). As shown in Table S2 (entries 4, ESI†), applied current density of 5 mA cm−2 is optimal to synthesize isatylidene malononitrile derivatives owing to maximum yield. At the same time, lower or higher current densities resulted in decreased yield.
:1, v/v) solvent mixture. Initially, we selected isatin 1a and malononitrile 2a as the model starting materials to optimize the reaction. The use of 1.0 equivalent of 1a, 2a and I2 (0.1 mmol) was found to be optimal reaction conditions (Table S3, ESI†). The yield got decreased with no change in product was observed when their equivalents were increased or decreased. Subsequently, the influence of solvents, time of reaction, and applied current density were studied (Tables S1 and S2, ESI†). We have performed electrolysis in various LR grade solvents such as EtOH, MeOH, MeCN, DCM, DMF and H2O and found that reaction proceeded in all these solvents and was observed to be best in EtOH in terms of yield and reaction time (Table S1, ESI†). The experiment manifested that no product was formed without any solvent, while a low conversion was observed in acetonitrile and dichloromethane (may be due to poor transfer of e−) while ethanol enhanced the yield. The reaction with changes in equivalents ratio of substrates 1a and 2a along with iodine was also carried out which showed no change in product expect very little variation in yields (Table S3, ESI†). As shown in Table S2 (entries 4, ESI†), applied current density of 5 mA cm−2 is optimal to synthesize isatylidene malononitrile derivatives owing to maximum yield. At the same time, lower or higher current densities resulted in decreased yield.
With the optimized electrolysis parameters in hand, we applied the electro-organic synthesis method to a range of substrates to elucidate the reaction's scope (Scheme 2). We were contented to find that reaction works with a wide range of isatin derivatives. We carried out the reaction at room temperature under an air atmosphere and prepared isatylidene malononitrile with electron-donating or electron-neutral, or electron-withdrawing substituents, such as 5-methyl, 5-chloro, 5- bromo, 5-fluoro, 7-chloro, 7-bromo, 7-fluoro, N-methyl and N-phenyl by Knoevenagel condensation of isatin and malononitrile using I2 successfully, leading to the corresponding products in good to very good yields (Scheme 2). To our delight, this transformation could be performed under an air atmosphere, affording the targeted products in excellent yields. These results demonstrated that this condensation reaction is relatively easy to handle and practically applicable under electro-organic synthesis conditions. Different substituents on the isatin core were well tolerated and furnished the respective products in very good to excellent yields. Expectedly, an electrical current is necessary for this reaction (Table S2, ESI†).
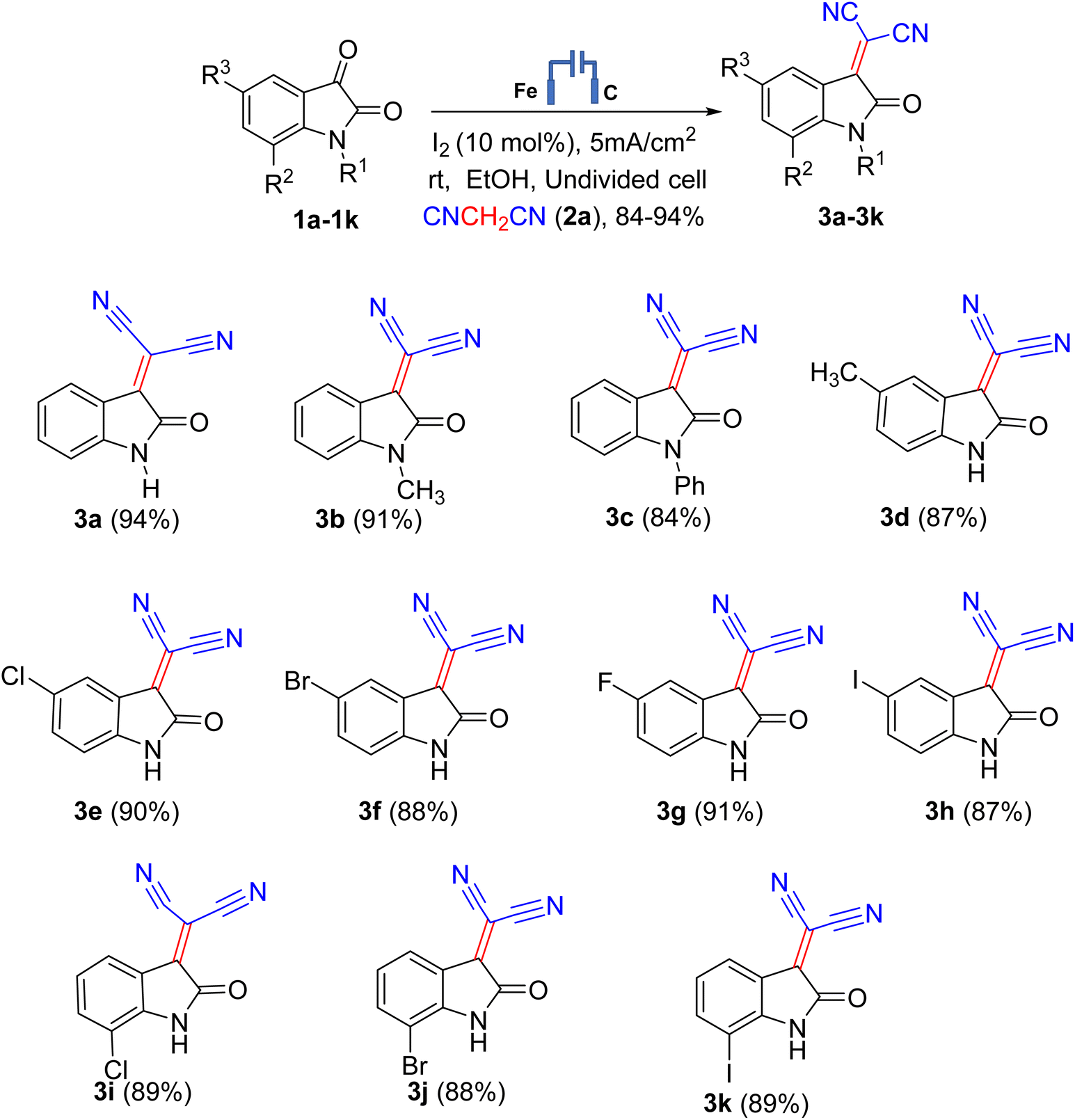 | ||
| Scheme 2 Electro-organic synthesis of dicyano 2-(2-oxoindolin-3-ylidene) malononitriles. | ||
Open circuit potential (OCP) was measured before applying fixed current density. An open circuit potential is equal to the electrochemical potential of electrode with respect to the electrolyte of the reaction mixture. OCP was observed for different isatin derivatives, as shown in Table 1, in the electro organic synthesis reaction mixture in a potential region of +1 V to −1 V for 6.67 min. In almost all cases, OCP was constant and varying with ±0.001 V in the reaction mixture containing 1b, 1c, and 1f. OCP was recorded for different electron-withdrawing and electron donating substituents at 1, 5, and 7 positions of isatin substrates and shown in Table 1.
| Entry | Electrochemical reaction mixture (substrate + I2 + 2a) | Open circuit potential OCP (in volts) |
|---|---|---|
| 1 | 1a | −0.056 |
| 2 | 1b | −0.008 |
| 3 | 1c | 0.073 |
| 4 | 1d | −0.011 |
| 5 | 1e | −0.026 |
| 6 | 1f | −0.029 |
| 7 | 1g | 0.0053 |
| 8 | 1h | −0.030 |
| 9 | 1i | 0.074 |
| 10 | 1j | 0.068 |
| 11 | 1k | 0.060 |
| 12 | 1a + I2 + 2a +TEMPO | 0.041 |
Comparative OCP for electro-organic synthesis of a reaction mixture containing (5- and 7-)substituted isatins (1.0 mmol) + iodine (10 mol%) + malononitrile (1.0 mmol) was studied prior to application of applied current density (Fig. 1). The result showed that the 5-methyl derivative has lower OCP as an electron donating group while 5-substituted –Br, –Cl, –F, and 7-Cl have higher OCP due to electron-withdrawing behaviour. Similar open circuit potential data were presented by Assary et al.20 for a broader set of aromatic nitrogen-containing molecules, especially quinoxaline derivatives in ethanol.
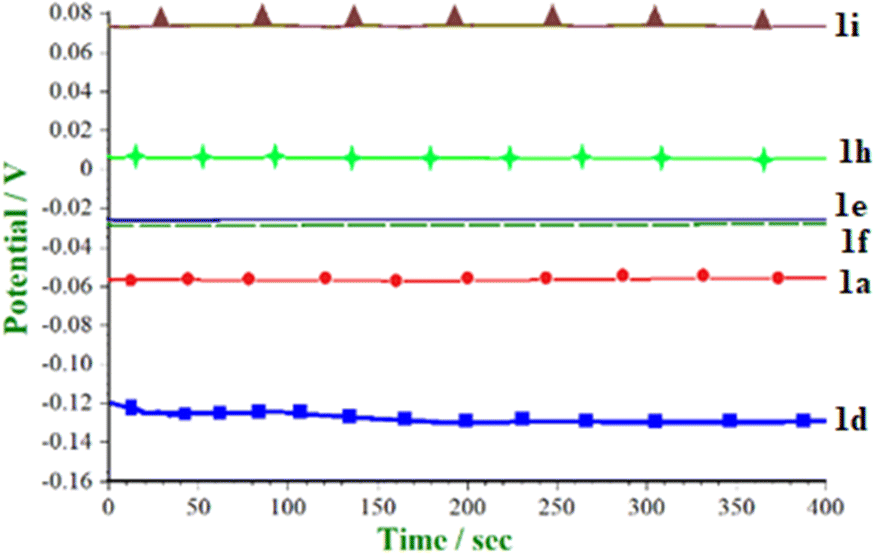 | ||
| Fig. 1 OCP electro-organic synthesis reaction mixture. | ||
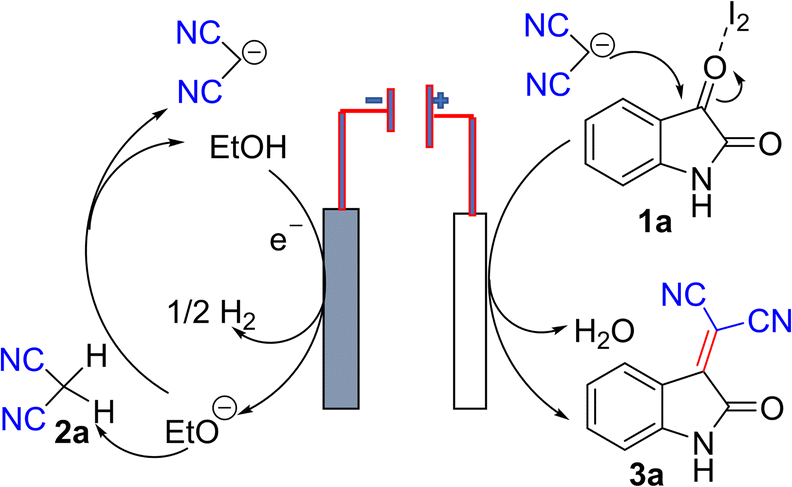 | ||
| Fig. 2 The postulated reactions at each electrode in the reaction mixture. | ||
OCP of 5-halo substituted isatin was found to be in the following order: 1g > 1e > 1f > 1h, due to the −I effect, fluoro being highly electron withdrawing. Among alkyl/aryl substituents, the OCP values are in the following order: 1c > 1b > 1d, where there is a +I effect from the conjugation of 5-methyl group.
Thus, electro-organic synthesis was carried out at a constant current density of 5 mA cm−2 for different times (∼1.5 h to ∼3 h), and the progress of the reaction was monitored by TLC using hexane and ethyl acetate (1:1) using pre-coated TLC plates (silica gel 60 F254). Loaded TLC plates were visualized in an UV chamber under a wavelength of 254 λ. It is worth mentioning here that, the electron-withdrawing and electron-donating groups are compatible with the electro-organic synthesis, and the desired product was obtained in 91% ± 3% yields. From the experimental data, it can be inferred that the strong −I effect raised the redox potential for the electro organic synthesis. For example, 5-halo substituted isatin derivatives (–Cl, –Br, –I) show electron releasing behavior and thus have low redox potential. However –F behaves differently because of the strong −I effect and has high redox potential for electro-organic synthesis.21 Therefore, the redox potential for synthesis of 3g is comparatively higher than the electro-organic synthesis of 3e, 3f, and 3h (Table 2).
| Entry | (Substrate + I2 + 2a) | Potential (in volts) | Time (seconds) | Products | Yield (%) |
|---|---|---|---|---|---|
| 1 | 1a | (−0.058 to −0.0560) | 8.78 × 103 | 3a | 94 |
| 2 | 1b | (−0.11 to −0.09) | 6.31 × 103 | 3b | 91 |
| 3 | 1c | (0.058 to 0.050) | 1.06 ×104 | 3c | 84 |
| 4 | 1d | (−0.14 to −0.13) | 1.06 ×104 | 3d | 87 |
| 5 | 1e | (−0.028 to −0.026) | 6.0 × 103 | 3e | 90 |
| 6 | 1f | (−0.030 to −0.026) | 1.06 × 104 | 3f | 88 |
| 7 | 1g | (0.004 to 0.087) | 7.5 × 103 | 3g | 91 |
| 8 | 1h | (−0.031 to −0.030) | 1.05 × 104 | 3h | 87 |
| 9 | 1i | (0.065 to 0.050) | 1.00 ×104 | 3i | 89 |
| 10 | 1j | (0.071 to 0.045) | 1.04 × 104 | 3j | 88 |
| 11 | 1k | (0.058 to 0.035) | 1.05 × 104 | 3k | 89 |
| 12 | 1a + I2 + 2a +TEMPO | (−0.0315 to 0.0310) | 1.01 × 104 | 3a | 88 |
We were interested to see whether reaction proceeded through free radical intermediate formation or not, therefore a reaction was performed using 1a and 2a by adding TEMPO (free radical scavenger) adopting optimized standard reaction conditions. It was observed that, formation of desired product 3a (88%) took place (Table 2, entry 12), which confirms there is no radical formation taking place under used reaction conditions. Prior to synthesis, a constant value of OCP was noted as 0.041 V. However, synthesis has initiated at −0.03 V relatively higher reduction potential than that of the standard reaction mixture and furnished the final product in ∼2 h 45 min. This rise in reaction time may be due to the presence of TEMPO, which hindered the mass transfer of the reaction mixture at the electrode surface.
Effect of substituents in the synthesis of isatylidene derivatives: the effect of a methyl group can be seen on 3b and 3d synthesis in comparison to 3a synthesis, the methyl group being electron donating, lowers the reduction potential as well as the time of product formation by ∼50 mV and ∼25 min during 3b synthesis. On the other hand, for the synthesis of 3d, the reduction potential was lowered by 80 mV, and it took ∼15 min longer to attain the maximum yield. However, synthesis of 3c, having a −I effect group, was found to raise the potential and reaction time by 100 mV and 32 min longer than 3a. This may be due to a decrease in electron density owing to the electron-withdrawing tendency of the phenyl group to a greater extent. At the same time, the N-methyl group showed decreased redox potential than 3a since it is present on a non-conjugated, non-aromatic ring and pushes its electron density towards C2, which affects the electrophilicity of C3 and thus lowers the reduction potential and is observed to have enhanced rate of reaction for the synthesis of 3b.
The introduction of electron donating groups (EDG) at the ring nitrogen increases the electron density and, hence, decreases the electron affinity and reduction potential. It is important to note that, in this electro-organic synthesis, explicit effects of electrolyte, solvent, reagents, concentration, and electrode materials play a significant role in deciding the fate of the reaction. Electron withdrawing groups (EWG) like fluoride, chloride, and phenyl groups have increased the electron affinity and hence increased redox potential for synthesis. For instance, under the present experimental conditions, the OCP of 5-fluoro isatin for the synthesis of 3g was 0.005 V, which was higher than that of unsubstituted isatin. Similarly, substitution at ring nitrogen with methyl and phenyl groups increases the reduction potential by ∼1.2 V for the phenyl group. All 7-substituted products showed higher redox potential compared to their 5-substituted derivatives. For example, the synthesis of 7-chloro isatylidene malononitrile was started at ∼0.07 V, while synthesis of 5-chloro isatylidene malononitrile started at −0.03 V. The reason for the higher redox potential for 7-substituted derivatives can be considered that it is adjacent to the –CONH– group, which is an electron-withdrawing, meta-directing group and deactivated the ring more significantly as compared to 5-substituted derivatives. Generally, isatin bearing EWG gave a better yield within a short reaction time than EDG-substituted isatin. The formation of the 3b product was faster than the 3c, keeping 3a reactivity in between, which may be owed to the +I effect of the methyl group and the steric effect of the phenyl group.22 It was found that when the electrochemical reaction is controlled by the mass transfer step, the electrolysis current density relates to the magnitude of the concentration gradient of the substrate molecule or the intermediate species at the electrode surface and interface.23
Overall, it was observed that chronopotentiographs of (3a–h) have followed similar pattern (Fig. S1, ESI†) and their potential region lies in 0.058 V, 0.004 V, −0.028 V, −0.030 V, −0.031 V, −0.058 V, −0.09 V and −0.14 V in the order as follows: N-phenyl > –F > –Cl > –Br > –I > IS > N-methyl > –CH3 with the high yield of 3a (94%) and 3g (91%) and others in good yield in the range of 84–90%. The better yield of 3g can be conferred to the −I effect, which eventually affects the electrophilicity of the C3O. It was observed that the synthesis of 3a, 3b, 3e, and 3g took relatively lesser time to furnish the final products. Importantly, it was observed that their synthesis got initiated at redox potentials or nearby OCP value.
Electro organic synthesis of 3a initiated with −0.055 V and raised to −0.059 V within 13 s. This shows that a reductive step follows initial oxidation. Due to the halogen bond activation mode of catalysis by molecular I2,24 wherein I2 was reduced and further showed a plateau at the constant potential for 30 seconds, representing the transition time for the reaction. It is followed by a continuous reaction reduction process up to −0.061 V for about an initial 1000 s and followed by oxidation at −0.056 V and remains constant throughout the synthesis process. It indicates a more significant reaction and diffusion at the electrode surface.25 Precipitate started to form after 16.67 min and continued to form until the reaction completed. The progress of the reaction was observed by TLC. After completion of the reaction, the precipitate was washed with ethanol, filtered, and vacuum dried, followed by column chromatography to get pure products. Remarkably, we did not observe any by-products. The crude product can be also recrystallized using ethanol to get pure product. The details of electro-organic synthesis of other compounds 3b–k are discussed in ESI.†
Cyclic voltammetry (CV) is a powerful and important electrochemical tool commonly used to investigate the reduction and oxidation processes of molecular species. Therefore, CV measurements were carried out (Fig. 3) using a glassy carbon (GCE) (d = 3 mm) working electrode, a Pt counter electrode (1 cm2), and a non-aqueous Ag/AgNO3 (0.1 M AgNO3 in acetonitrile) as reference electrode, respectively and NaClO4 (0.1 M) as supporting electrolytes was used. Mechanical treatment of GCE was carried out as follows: polished using Alumina powder (particle size 0.01 μm) before each experiment, rinsed thoroughly, and cleaned with isopropyl alcohol (IPA). Then GCE was placed in an electrolyte, and various cyclic voltammograms were recorded until a steady state baseline voltammogram was obtained. This procedure ensured very reproducible experimental results. The cyclic voltammogram consists of two segments, i.e., anodic and cathodic (Table S4, ESI†).
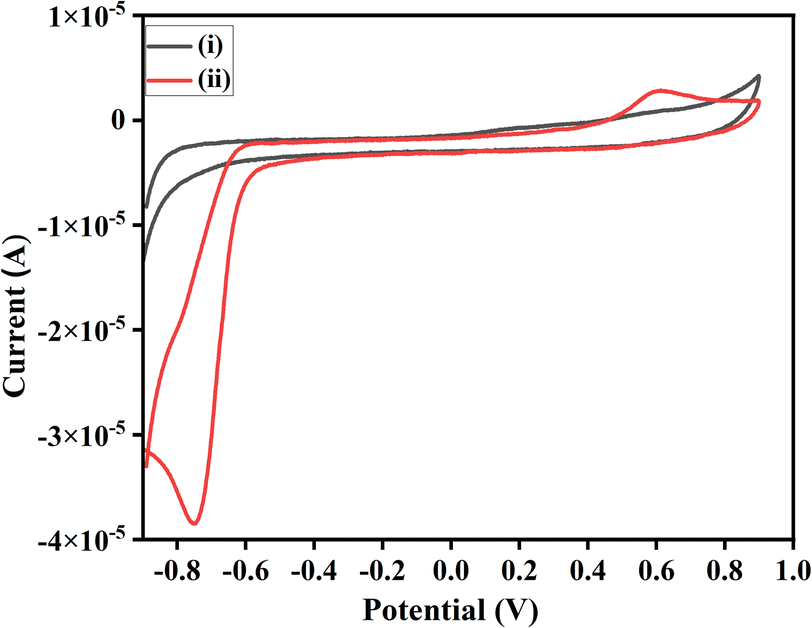 | ||
| Fig. 3 Cyclic voltammograms: (i) 1a (ii) 3a. | ||
All the chosen substituted isatins (3a–k) exhibited well-defined diffusion-controlled quasi-reversible redox peaks except 5-substituted derivatives. 5-Substituted derivatives showed a typical irreversible peak with an insignificant reduction in all cases while an oxidation peak for 3g.26 A study of the effect of scan rate is made to evaluate the mechanism and the feasibility of all product's electrochemical reactions involved at GCE in the electrolytic condition. Fig. 4 shows an exemplary cyclic voltammogram of 3c where the oxidation and reduction current peaks increase with the increasing scan rate; however, in the case of oxidation, the increase of current is not as significant as for the reduction. For instance, in the case of 3c, the linear dependence of peak current on the inverse of the square root of the sweep rate. Fig. 4 indicates that the migration of 3c is proportional to the concentration of the species at the interface and hence it was a diffusion-controlled electrode process.27 The inset shows the Randles–Sevcik plots for product 3c, where the linear slope signifies the diffusion-controlled reaction. The linearity of the data points with a regression coefficient of ca. 0.98 indicates that the relationship is promising.
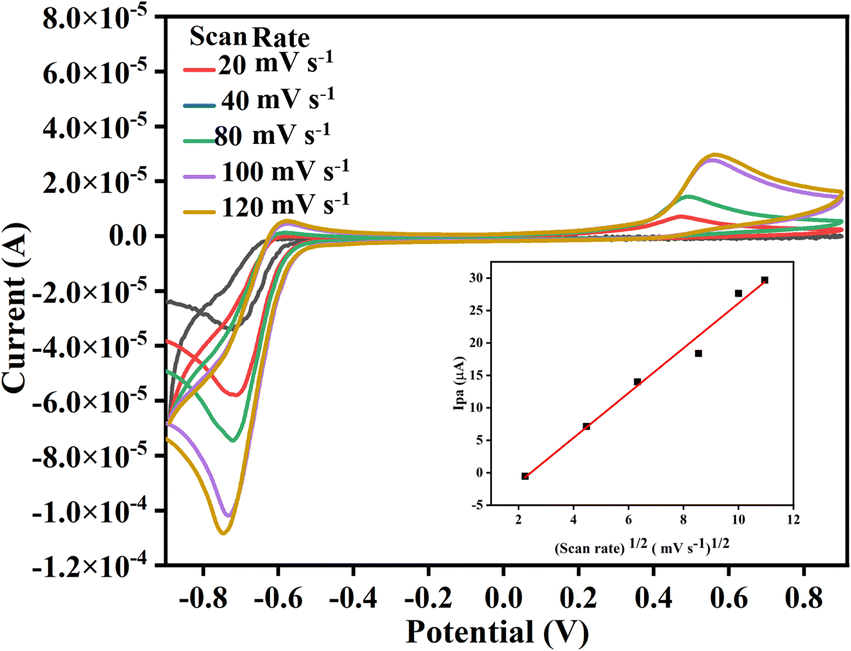 | ||
| Fig. 4 Cyclic voltammograms of 3c at different scan rates. Inset shows the plot for Ipa vs. square root of scan rate. | ||
Cyclic voltammetry of isatin and malononitrile were not showing any peak, and hence redox inactive under the given reaction condition. However, iodine is quite reactive and exhibits quasi-reversible redox peaks. The interaction of I2 with isatin showed a high current density compared to its interaction with malononitrile. This indicates that I2 preferably interacts with the C3 position of isatin, being highly electrophilic rather than malononitrile. Moreover, the onset potential of catalysis by I2 via halogen bond activation and with 1a is lower than that of 2a, the difference of 0.022 V, which shows that 1a reacts with I2 at a faster rate than 2a. We monitored the anodic and cathodic peak currents, and peak potentials achieved during the interaction of I2 reacted with 1a and 2a. CV was recorded in the potential range of +0.90 V and −0.90 V in an electrolytic solution (Fig. 5), as mentioned in experimental details (ESI†), both 1a and 2a do not exhibit any redox activity. Whereas, when the CV was recorded in the standard electrolytic solution with dissolved I2 + 1a and I2 + 2a, in the potential range of +0.90 V and −0.90 V, quasi-reversible redox processes were observed for both. However, the peak current for I2 + 1a was higher than I2 + 2a because with 1a the exchange of electrons is swift and interaction is more effective due to the involvement of partial negative charge of O-atom and lone pair of I2. Hence, I2 is bound with the carbonyl group at the C3-positions of isatin and exhibits a higher peak current. Whereas in the latter case, the exchange of electrons is slower due to subsided electronegativity of the CN group due to the presence of methylene groups; hence interaction is weaker. This concept directs us towards the inference that I2 comes in contact with isatin at the anode and simultaneously malononitrile generates anion in the electrolytic solution, as shown in the given plausible mechanism (Scheme 3 and Fig. 2).
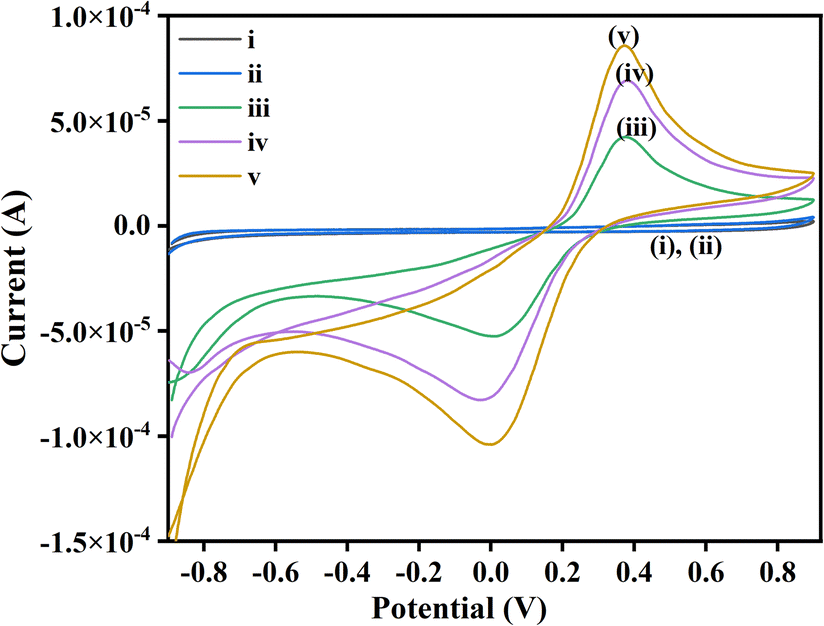 | ||
| Fig. 5 Cyclic voltammograms in the standard electrolyte: (i)1a, (ii) 2a, (iii) I2 + 2a, (iv) I2 + 1a, (v) I2. | ||
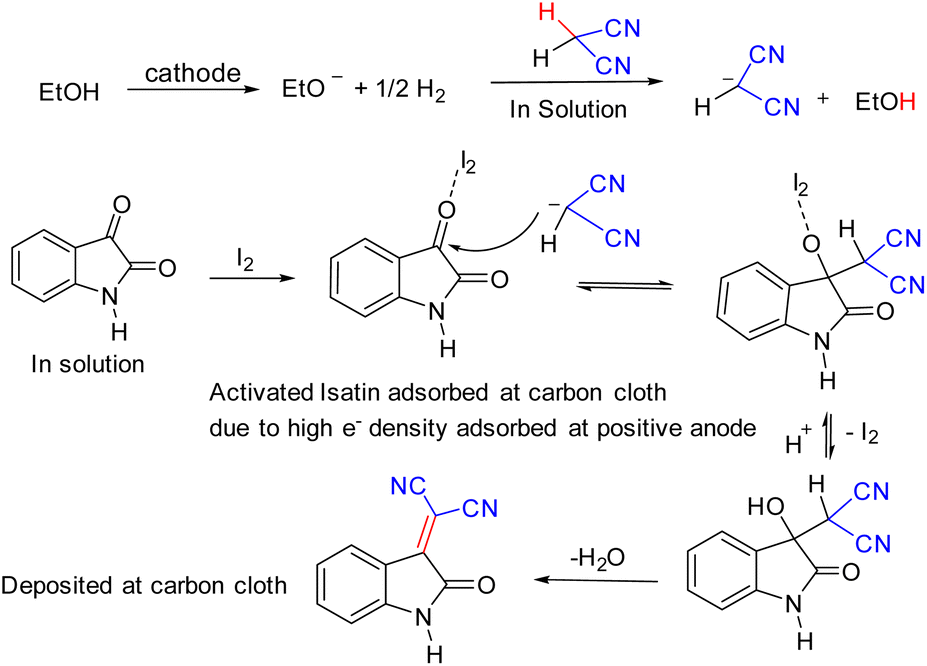 | ||
| Scheme 3 Plausible mechanism. | ||
Based on the above observations and a recent study,24 which supported that I2 exhibits halogen bonding activation, which is a strong catalytic event, we can propose that, during synthesis, at the cathode, ethoxide ion generated and releases one electron to the cathode and former helps to generate malononitrile anion in solution. At the anode, isatin preferably undergoes a charge transfer reaction with I2, which helps lower the activation energy for nucleophilic attack and thus facilitates the nucleophilic substitution at C3 position to furnish the product 3a in excellent isolated yield.
Conclusions
In conclusion we have demonstrated that, molecular iodine catalyzed electro-organic synthesis of dicyano-isatylidene malononitrile derivatives can be achieved in greener way at ambient temperature and cost-effective manner. This methodology can be used for the gram-scale synthesis of dicyano-isatylidene malononitrile derivatives. This finding will be helpful in the electro-organic synthesis of other isatin and indole derivatives which are biologically active compounds, and their synthesis and isolation are tedious. We are also in the process of elucidate further mechanistic details of these types of reactions in order to understand their scope and limitations, which will be disclosed in due course of time.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
NV gratefully acknowledges the IIT BHU forTeaching Assistant fellowship and providing funds to carry out this work. M. Malviya acknowledges the central instrumentation facility centre (CIFC)-IIT BHU for analytical facilities. VKM is thankful to UGC for junior research fellowship. RS thankful to Banaras Hindu University (BHU) and Jawaharlal Nehru University (JNU) for providing Lab Facilities. RS is grateful to Ultra International and Sanganeria Foundation for supporting lab furniture to Glycochemistry laboratory at JNU.Notes and references
- T. H. Yang, C. I. Lee, W. H. Huang and A. R. Lee, Molecules, 2017, 22, 1 Search PubMed.
- Z. Liang, D. Zhang, J. Ai, L. Chen, H. Wang, X. Kong, M. Zheng, H. Liu, C. Luo, M. Geng, H. Jiang and K. Chen, Bioorg. Med. Chem. Lett., 2011, 21, 3749 CrossRef CAS PubMed.
- A.-R. Lee, J. Cancer Treat. Diagn., 2018, 2, 24 CrossRef.
- S. Rossiter, Tetrahedron Lett., 2002, 43, 4671 CrossRef CAS.
- S. M. Khalil and A. M. A. Hassaan, Acta Phys. Pol. A, 1993, 83, 477 CrossRef CAS.
- I. A. Abdelhamid, Synlett, 2009, 4, 625–627 CrossRef.
- Y. S. Kayukov, O. V. Kayukova, E. S. Kalyagina, I. N. Bardasov, O. V. Ershov, O. E. Nasakin and V. A. Tafeenko, Russ. J. Org. Chem., 2011, 47, 392 CrossRef CAS.
- I. A. Abdelhamid, M. H. Mohamed, A. M. Abdelmoniem and S. A. S. Ghozlan, Tetrahedron, 2009, 65, 10069 CrossRef CAS.
- A. Dandia, H. Taneja, R. Gupta and S. Paul, Synth. Commun., 1999, 29, 2323 CrossRef CAS.
- R. Pandey, D. Singh, N. Thakur and K. K. Raj, ACS Omega, 2021, 6, 13240 CrossRef CAS PubMed.
- V. P. Mehtaab and B. Punji, RSC Adv., 2013, 3, 11957 RSC.
- X. J. Yang and Y. S. Zhang, J. Chem., 2013, 543219 Search PubMed.
- P. M. Jadhav, A. B. Rode, L. Kótai, R. P. Pawar and S. U. Tekale, New J. Chem., 2021, 45, 16389 RSC.
- C. Monika, S. Ram and P. K. Sharma, Asian J. Org. Chem., 2023, 12, 111–138 CrossRef.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484 CAS.
- D. A. Androsov, T. L. S. Kishbaugh and G. W. Gribble, Tetrahedron Lett., 2008, 49, 6621 CrossRef CAS.
- I. Azad, T. Khan, R. Ahmad, A. Kamal, A. R. Khan and M. Nasibullah, J. Mol. Struct., 2021, 1228, 129451 CrossRef CAS.
- D. V. Demchuk, M. N. Elinson and G. I. Nikishin, Mendeleev Commun., 2011, 21, 224 CrossRef CAS.
- R. S. Assary, F. R. Brushett and L. A. Curtiss, RSC Adv., 2014, 4, 57442 RSC.
- A. M. Ismail, R. F. Radman and N. A. Al-Jallal, Prog. React. Kinet. Mech., 2010, 35, 281 CrossRef CAS.
- S. R. Waldvogel, Angew. Chem., Int. Ed., 2015, 54, 9751 CrossRef CAS.
- D. Von Der Heiden, S. Bozkus, M. Klussmann and M. Breugst, J. Org. Chem., 2017, 82, 4037 CrossRef CAS PubMed.
- H. Gerischer and P. Delahay, Anal. Chim. Acta, 1958, 18, 12 Search PubMed.
- A. K. Gupta and R. S. Sindal, J. Chem. Sci., 2009, 121, 347–351 CrossRef CAS.
- E. M. Espinoza, J. A. Clark, J. Soliman, J. B. Derr, M. Morales and V. I. Vullev, J. Electrochem. Soc., 2019, 166, H3175 CrossRef CAS.
- A. Dandia, K. Arya, M. Sati and R. Sharma, Heterocycl. Commun., 2003, 9, 415 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra02152a |
| This journal is © The Royal Society of Chemistry 2023 |