
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC-Glycopyranosyl aldehydes: emerging chiral synthons in organic synthesis
Sandeep Kumar
 a,
Vinod Khatri
ab,
Priyanka Mangla
a,
Rajni Johar Chhatwal
c,
Virinder S. Parmar
*adef and
Ashok K. Prasad
*a
a,
Vinod Khatri
ab,
Priyanka Mangla
a,
Rajni Johar Chhatwal
c,
Virinder S. Parmar
*adef and
Ashok K. Prasad
*a
aBioorganic Laboratory, Department of Chemistry, University of Delhi, Delhi, India. E-mail: ashokenzyme@gmail.com; virparmar@gmail.com
bT. D. L. Govt College for Women, Murthal-131027, Haryana, India
cMaitreyi College, Department of Chemistry, University of Delhi, Delhi, India
dThe City University of New York-Medgar Evers College, Department of Chemistry and Environmental Science, USA
eNanoscience Program, CUNY-Graduate Center and City College, Departments of Chemistry and Biochemistry, USA
fInstitute of Click Chemistry Research and Studies, Amity University, Noida 201303, India
First published on 3rd July 2023
Abstract
Herein, we have summarized the vast array of synthetic processes that have been developed for the synthesis of C-glycopyranosyl aldehydes and diverse C-glycoconjugates derived from them by covering the literature reported from 1979 to 2023. Notwithstanding its challenging chemistry, C-glycosides are considered stable pharmacophores and are used as important bioactive molecules. The discussed synthetic methodologies to access C-glycopyranosyl aldehydes take advantage of seven key intermediates, viz. allene, thiazole, dithiane, cyanide, alkene, and nitromethane. Furthermore, the integration of complex C-glycoconjugates derived from varied C-glycopyranosyl aldehydes involves nucleophilic addition/substitution, reduction, condensation, oxidation, cyclo condensation, coupling, and Wittig reactions. In this review, we have categorized the synthesis of C-glycopyranosyl aldehydes and C-glycoconjugates on the basis of the methodology used for their synthesis and on types of C-glycoconjugates, respectively.
 Sandeep Kumar | Dr Sandeep Kumar obtained his PhD from the Department of Chemistry, University of Delhi, India in February 2022 under the supervision of Prof. Ashok Kumar Prasad in the field of nucleosides and carbohydrate chemistry. His research interest lies in the synthesis of modified nucleosides and sugar based heterocyclic molecules for therapeutic applications. Dr Kumar secured All India Rank 14 in CSIR-JRF and thereafter awarded SPM (Shyama Prasad Mukherjee) fellowship during his PhD. Dr Kumar has been DST-INSPIRE Fellow during graduation and post-graduation. Also, Dr Kumar is recipient of many meritorious fellowships from Haryana Government and Kurukshetra University. He has published around twenty research papers in internationally reputed journals such as New Journal of Chemistry, Carbohydrate Research, Beilstein Journal of Organic Chemistry, Journal of Organic Chemistry etc. Currently, Dr Kumar is an Assistant Professor in Department of Chemistry, Acharya Narendra Dev College, University of Delhi. |
 Vinod Khatri | Dr Vinod Khatri studied Chemistry at Kurukshetra University, Kurukshetra Haryana and received his PhD from University of Delhi under the supervision of Professor Ashok K. Prasad. After PhD in 2017, he joined IISER Mohali as National Postdoctoral Fellow (DST-SERB) and worked on metal-catalyzed reactions on carbohydrates. In 2018, he joined as an Assistant Professor in the Department of Higher Education Haryana. He worked as postdoctoral fellow at Freie University Berlin Germany for one year with Dr Sumati Bhatia in 2021. His research interests are C-glycosides, sugar based copolymers and multivalent glycoconjugates. |
 Priyanka Mangla | Dr Priynka received her PhD in Organic and Medicinal Chemistry in 2019 from University of Delhi (India), where she developed modified nucleosides using diastereoselective biocatalytic transformations of sugars and catalytic C–H functionalization of nucleobases. To continue her passion in the field of nucleic acid chemistry, she joined the Oligonucleotide Chemistry team at AstraZeneca, Sweden in 2021 as a post-doctoral research scientist. Her research focuses on the development of novel pH-sensitive endosomolytic agents to enhance the endosomal escape of oligonucleotides and improve gene-silencing. |
 Rajni Johar Chhatwal | Dr Chhatwal obtained her BSc and MSc degree from Kurukshetra University, Kurukshetra in 1993 and 1995, respectively. Then Dr Chhatwal did PhD from Guru Jambeshwar University in 2005. Her area of interest is “Spectral, Structural Elucidation and Co-ordination Abilities of Tin and Aluminium with Triphenyl Oxo Propoxide to Yield Starting Material Triphenyl Tin(iv) Aluminium(III) Oxo Popoxide Compound”. Currently, Dr Chhatwal is an Assistant Professor in Maitreyi College-University of Delhi. |
 Virinder S. Parmar | Professor Virinder S. Parmar is a Professor of Chemistry at Medgar Evers College-City University of New York, USA. He has also been a faculty member at St Stephen's College and the University of Delhi for 44 years, he recently retired as Professor of Chemistry and has served as Head of the Department of Chemistry and as Chairman of the Board of Research Studies, and Provost of Gwyer Hall at Delhi university. Professor Parmar's research interests include: green/sustainable Chemistry, nanotechnology, organic synthesis, nucleic acid chemistry, advanced materials, medicinal chemistry, biocatalysis and the chemistry of natural products. He has mentored 85 PhD and postdoctoral scientists and has published over 500 research papers. He was awarded the Dean's Research Excellence Award of the School of Science, Health and Technology, MEC-CUNY, New York, USA for the years 2019 & 2020. |
 Ashok K. Prasad | Late Professor Ashok K. Prasad (1961–2023): Professor Prasad obtained his PhD from the Department of Chemistry, University of Delhi, in 1990 in the area of synthesis of bioactive polyphenolic natural products. After spending about a decade as a post-doctoral fellow/visiting scientist at the University of Southern Denmark, the Max Planck-Institute for Molecular Physiology (Germany), Sapienza University Rome (Italy), and the University of Massachusetts Lowell (USA), Professor Prasad joined the Department of Chemistry, University of Delhi, as Reader in 2001 and subsequently became Professor in 2009. He was the Head of the Chemistry Department and Dean of Science Faculty, University of Delhi. He had published more than 300 research papers in journals of international repute. His research interests were in the areas of nucleic acid chemistry, biotransformations, natural product chemistry, and synthesis of bioactive heterocyclic compounds. We dedicate this review to the fond memory of Late Professor Ashok K. Prasad. |
1. Introduction
C-Glycosides constitute an important and useful class of organic molecules, in which anomeric carbon of a sugar is attached to an aglycon through C–C bonds. Molecules of this class are mimics of O-glycosides, where the sugar moiety is attached with aglycon through C-atom instead of O-atom at the anomeric position.1 Glycosides have made their presence felt in foodstuffs to the components of nucleic acids and cell surface glycoconjugates. The structural alteration resulting from the transformation of the anomeric centre from C–O acetal to a strong C–C bond in C-glycosides stimulates the resistance towards chemical/enzymatic hydrolysis and metabolic processes.2 As a consequence, the dominance of C-glycosides to personate native O-glycosides as potential therapeutic candidates has become undoubtedly apparent. Moreover, the occurrence of C-glycosidic linkage is common in natural products obtained from plants and microorganisms, such as saptomycin B,3 mangiferin,4 aspalathin,5 tunicamycin V,6 and aureonuclemycin,7 which have shown significant biological activities8 (Fig. 1).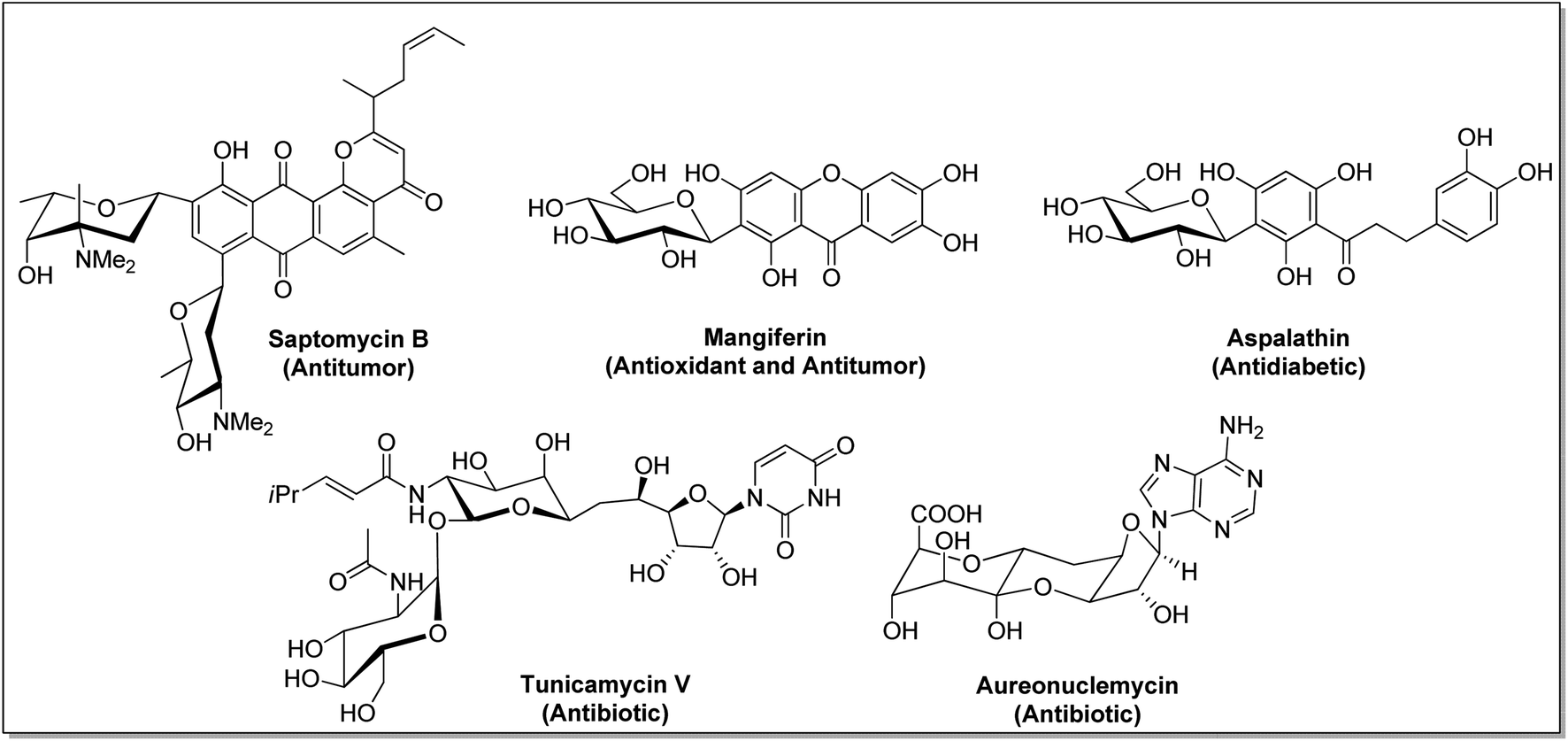 | ||
| Fig. 1 Structures of some selected naturally occurring bioactive C-glycosides. | ||
Remarkable structural diversity in naturally occurring C-glycosides has drawn considerable attention to chemically synthesized complex C-glycosides. The successful examples of the chemical evolution of C-glycosides include pro-xylene as anti-aging cosmetic agent,9 dapagliflozin, canagliflozin and empagliflozin as SGLT2 inhibitors for the treatment of type II diabetes,10–12 metabolically stable C-analogue of KRN7000 as anticancer agent,13 C-mannosyl-Trp for post-transitional modification affecting folding, stability and other functions of proteins,14 etc. (Fig. 2).
 | ||
| Fig. 2 Structures of some bioactive synthetic C-glycosides. | ||
A literature survey on C-glycosides illustrates that earlier reported review articles have mainly been on exo-glycals,15 C-mannopyranosides,16 C-nucleosides,17 C-glycoconjugates,18 C-arylglycosides,19,20 and on chemical synthesis of C-glycosides.21,22 Although α- and β-C-glycopyranosyl aldehydes constitute an important class of precursor molecules for the formation of complex C-glycosides, it has been overlooked to make their explicit presence in any review article. About two decades ago, Dondoni presented a review that mainly referred to sugar thiazole as a key synthetic intermediate for the synthesis of C-glycopyranosyl aldehyde and explained its role as a precursor for the formation of C-disaccharide and C-amino acids.23 Consequently, these facts encouraged us to compile the literature on the synthesis of α- and β-C-glycopyranosyl aldehydes and their applications in the synthesis of diverse biologically relevant C-glycoconjugates in this review article.
2. Synthesis of C-glycopyranosyl aldehydes
Generally, syntheses of C-glycosides involve the nucleophilic addition of aglycon part on sugar moiety followed by treatments using dehydration, oxidation, reduction, and reductive hydrolysis reactions to obtain targeted C-glycopyranosyl aldehydes. In comparison to the syntheses of α-C-glycopyranosyl aldehydes, more methodologies have been reported on the synthesis of β-C-glycopyranosyl aldehydes. Further, it has been observed that α-C-glycopyranosyl aldehydes can be easily converted into β-C-glycopyranosyl aldehydes in the presence of organic bases. Seven key intermediates have been used for the preparation of α- and β-C-glycopyranosyl aldehydes, which have been described below (Fig. 3).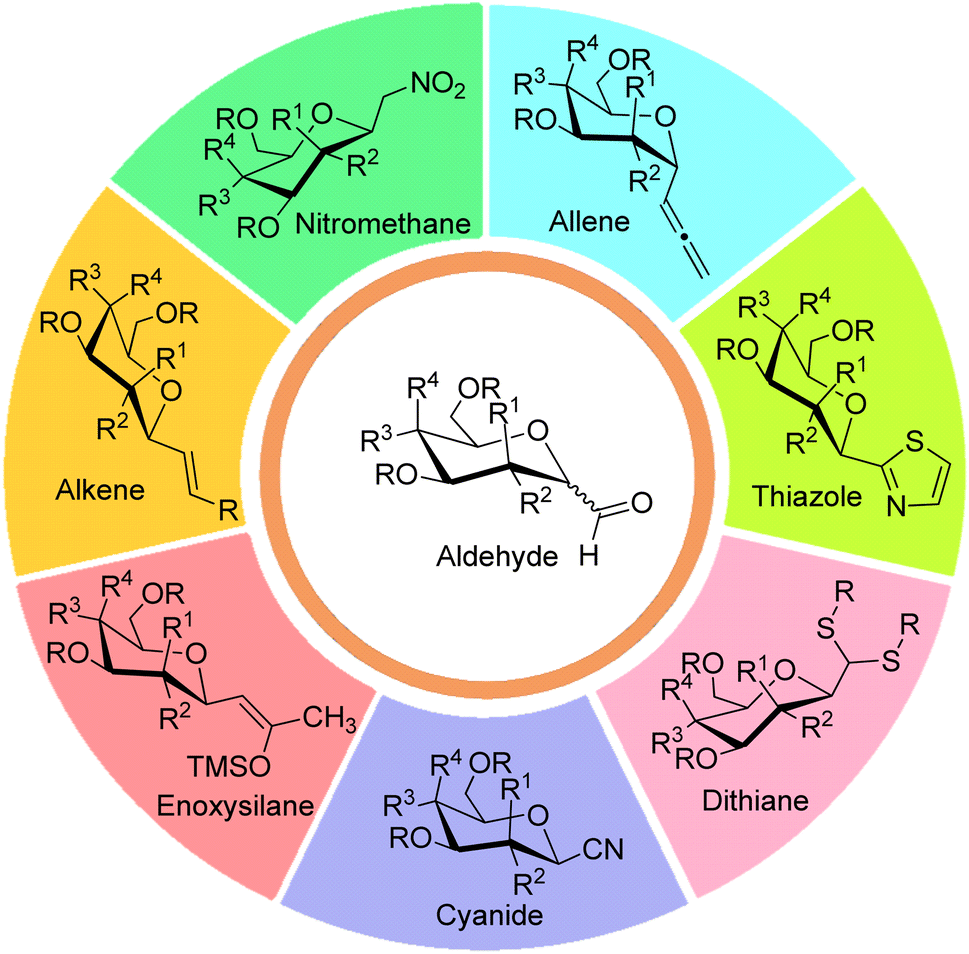 | ||
| Fig. 3 Key intermediates for the synthesis of C-glycopyranosyl aldehydes. | ||
2.1 Allene approach
Kobertz et al.24 reported the use of α-C-glycopyranosyl allenes 2a–c as key synthetic intermediates for the synthesis of C-glycopyranosyl aldehydes, which in turn can be synthesized from methyl tetra-O-benzyl-α-D-glycopyranosides 1a–c with high diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) via Lewis acid catalysed nucleophilic substitution reaction using propargyl trimethylsilane in the presence of trimethylsilyl triflate (TMSOTf) in acetonitrile. Furthermore, ozonolysis of allenes 2a–c in dichloromethane at −78 °C provided α-C-glycopyranosyl aldehydes 3a–c, which on treatment with 10% triethylamine in isopropanol:DCM (1:1) at 25 °C for 24 h yielded β-C-glycopyranosyl aldehydes 4a–c (Scheme 1). The overall yields and equilibrium ratio of β/α for the conversion of C-glycopyranosyl allenes 2a–c into β-linked C-glycopyranosyl aldehydes 4a–c are shown in Table 1.
:1) via Lewis acid catalysed nucleophilic substitution reaction using propargyl trimethylsilane in the presence of trimethylsilyl triflate (TMSOTf) in acetonitrile. Furthermore, ozonolysis of allenes 2a–c in dichloromethane at −78 °C provided α-C-glycopyranosyl aldehydes 3a–c, which on treatment with 10% triethylamine in isopropanol:DCM (1:1) at 25 °C for 24 h yielded β-C-glycopyranosyl aldehydes 4a–c (Scheme 1). The overall yields and equilibrium ratio of β/α for the conversion of C-glycopyranosyl allenes 2a–c into β-linked C-glycopyranosyl aldehydes 4a–c are shown in Table 1.
 | ||
| Scheme 1 Synthesis of β-C-glycopyranosyl aldehydes 4a–c. | ||
| Reactant | β-linked products | β:α |
Yield (%) |
|---|---|---|---|
| 2a | 4a | >20:1 |
41 |
| 2b | 4b | 8:1 |
42 |
| 2c | 4c | 10:1 |
49 |
In another synthetic approach reported by Kroger et al.25 peracetylated D-galactose 5 was used as a starting material to access the sugar allene precursor (Scheme 2). 1,2,3,4,6-Penta-O-acetyl-β-D-galactopyranose (5) was converted into the corresponding α-C-glycosidic allene 6 using propargyl trimethylsilane in the presence of Lewis acid (BF3·Et2O) in acetonitrile at 0 °C in 48% yield. Further, compound 6 on ozonolysis afforded acetylated α-C-galactopyranosyl aldehyde 7, which was found to be very labile and further used without isolation for the synthesis of O-glycosyl amino acid mimetics. The details of synthetic methods for these O-glycosyl amino acid mimetics are described in Section 3.4 (Scheme 50).
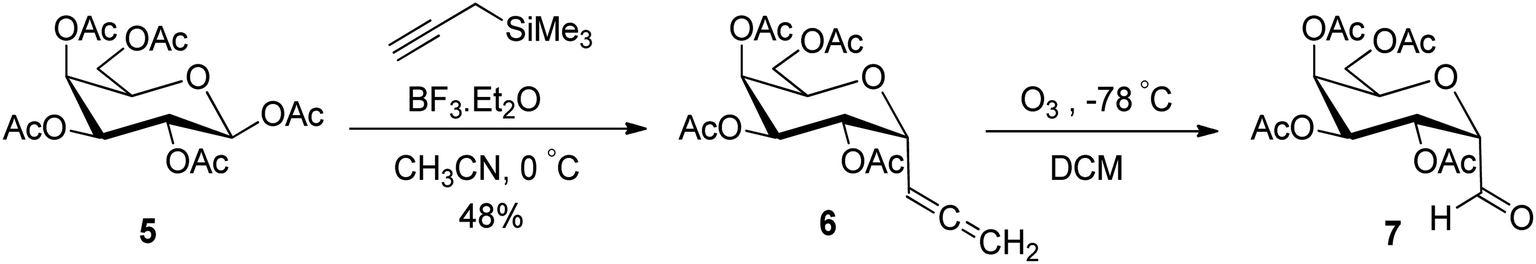 | ||
| Scheme 2 Synthesis of α-C-galactopyranosyl aldehyde 7. | ||
Alternatively, Kolympadi et al.26 synthesized both α- and β-linked glycopyranosyl aldehydes using allene as the key precursor, which was obtained from peracetylated D-galactose 5 following the method of Kroger et al.25 (Scheme 3). However, here combination of Lewis acids BF3–Et2O–TMSOTf produced allene 6 in 80% yield. Since Kroger observed that the acetyl-protected glycopyranosyl aldehyde 7 was very labile, here, acetylated allene 6 was converted into the perbenzylated α-C-glycosyl allene 2c by deprotection of acetyl group with sodium methoxide (NaOMe)/methanol followed by the protection of generated hydroxyl groups with benzyl bromide in the presence of sodium hydride in DMF. Further, compound 2c was converted into β-C-galactopyranosyl aldehyde 4c by following the methodology developed by Kobertz et al.24
 | ||
| Scheme 3 Synthesis of β-C-galactopyranosyl aldehyde 4c. | ||
Guillaume et al.27 designed a route for the synthesis of C-glycopyranosyl aldehyde 11 initiating from perbenzylated α-methyl galactoside 1c. The aim for the synthesis of orthogonally protected C-glycopyranosyl aldehyde 11 was to be utilized for the synthesis of C6-modified α-C-GalCer analogues (Section 3.1, Scheme 32a). Thus, treatment of galactoside 1c with propargyl trimethylsilane and trimethylsilyl triflate in acetonitrile at 0 °C for 48 hours followed by acetylation using acetic anhydride yielded sugar allene 8 in 66% yield. Deprotection of the C6-OBn group in 1c under condition propargyl trimethylsilane and trimethylsilyl triflate in acetonitrile at 0 °C for 48 was due to the excess use of Lewis acid, i.e. trimethylsilyl triflate and longer reaction time. Acetyl protection of compound 8 was replaced with PMB on treatment with ammonia in methanol followed by protection of alcohol with PMBCl resulting in allene 9 in 78% yield, which upon ozonolysis and in situ reduction with sodium borohydride afforded primary alcohol 10 in 73% yield. The α-C-galactopyranosyl aldehyde 11 was obtained by re-oxidation of alcohol 10 via the Dess–Martin reaction. α-C-Galactopyranosyl aldehyde 11 was also found labile and further used without isolation (Scheme 4).
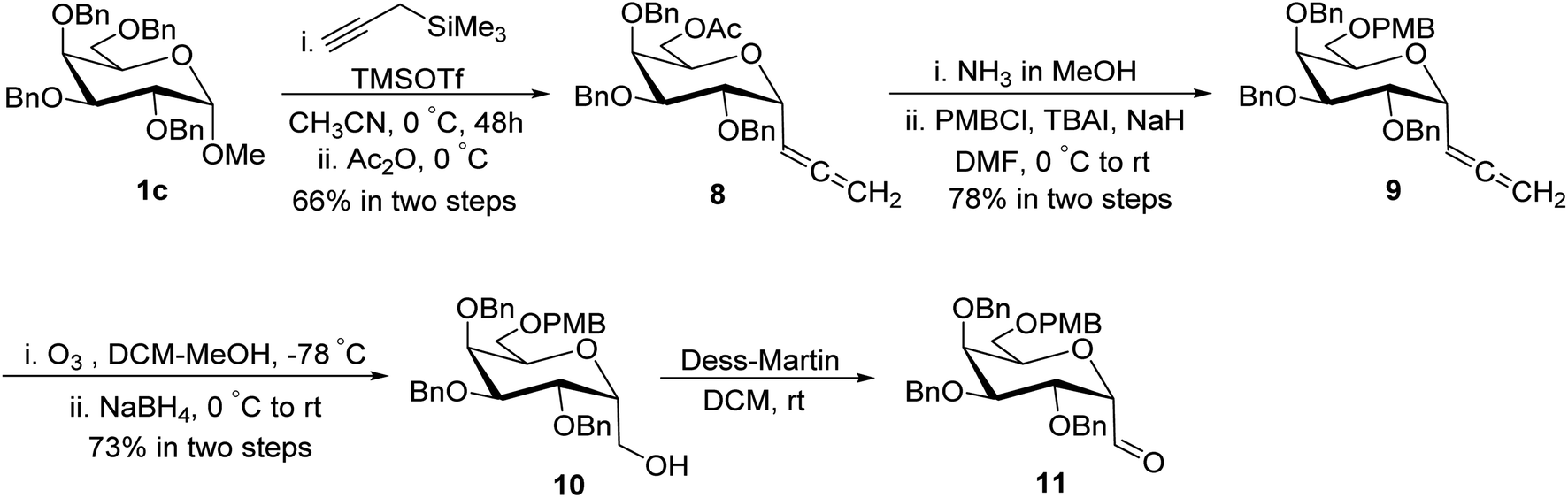 | ||
| Scheme 4 Synthesis of α-C-galactopyranosyl aldehyde 11. | ||
2.2 Thiazole approach
Thus, the allene approach carried out by these researchers concluded that the use of both Lewis acids BF3–Et2O–TMSOTf produced allene in good yield from sugar precursors. Excess use of Lewis acids furnished deprotection of the C6-OBn group and it was also observed that α-C-glycopyranosyl aldehydes are less stable than β-C-glycopyranosyl aldehydes and the earlier one could be easily converted into later on treatment with base triethylamine in iPrOH-DCM. However, the synthesis of C-glycopyranosyl aldehydes via the allene approach has posed the limitation of using expensive reagents such as propargyl trimethylsilane, troublesome cleavage by ozonolysis and a long reaction time. So, a more convenient approach was developed by Dondoni et al.28 in which C-glycopyranosyl aldehyde was synthesized using sugar thiazole as a key synthetic intermediate.In this approach, the sugar thiazole was synthesized via the nucleophilic addition reaction followed by reductive dehydroxylation. The nucleophilic addition of 2-lithiothiazole (2-LTT, prepared in situ by the reaction of 2-bromothiazole and butyl lithium) on 2,3,4,6-tetra-O-benzyl-D-glucono-1,5-lactone 12 at −78 °C in THF yielded 2,3,4,6-tetra-O-benzyl-β-1-C-(2-thiazolyl)-D-glucopyranose (13) in 80% yield. On acetylation, compound 13 yielded anomerically activated compound 14, which on further reduction with triethylsilane in trimethylsilyl triflate (TMSOTf) at room temperature for 15 minutes gave an anomeric mixture of α- and β-linked 2-thiazolyl-C-glucopyranosides 15a–b in 1:1 ratio. The anomeric mixture 15a–b when subjected to N-methylation followed by reduction and then hydrolysis in the presence of corresponding reagents produced α- and β-linked C-glucopyranosyl aldehydes 16a–b in the ratio of 1:1. Treatment of 16a–b with 10% triethylamine afforded β-C-glucopyranosyl aldehyde 4a in 60% yield (Scheme 5).
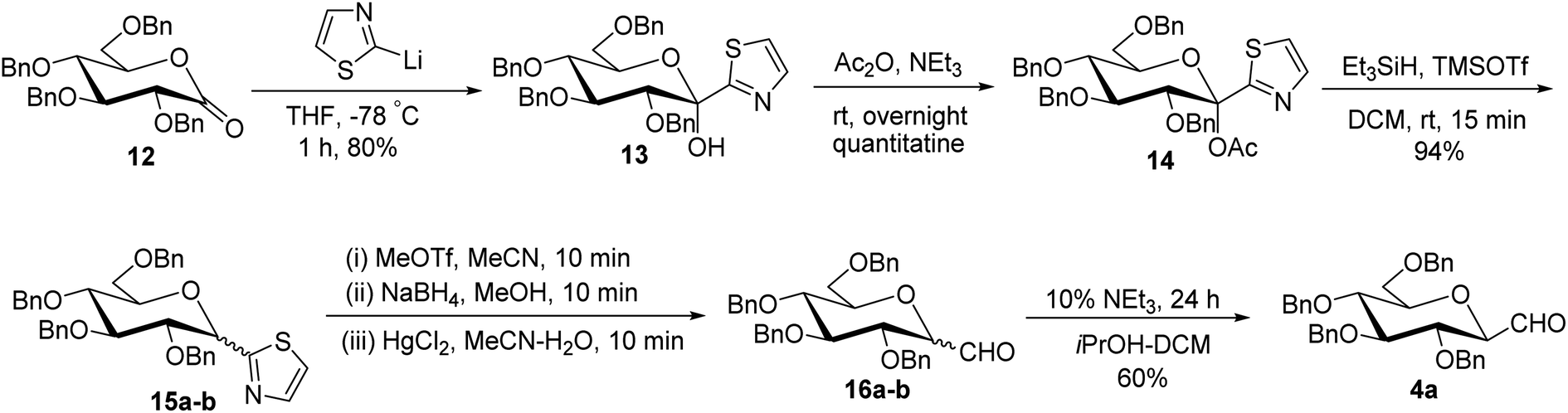 | ||
| Scheme 5 Synthesis of β-C-glucopyranosyl aldehyde 4a. | ||
Further, Dondoni et al.29 applied the same approach to other sugar precursors to study the kinetic and thermodynamic aspects of improving the stereochemical control and chemical efficacy (Scheme 6). Different sugar precursors such as 2,3,4,6-tetra-O-benzyl-glucose/-mannose/-galactose and 2-azido-3,4,6-tri-O-benzyl-2-deoxy-galactose 17a–d were examined for the given approach where thiazole was used as a formyl group equivalent to synthesise C-glycopyranosyl aldehyde 4a–d. A mixture of α- and β-isomers of C-glucopyranosyl aldehyde 4a was obtained in a 1:1 ratio, while, on the other hand, remaining C-glycopyranosyl aldehydes 4b–d were formed preferentially in β-configuration.
 | ||
| Scheme 6 Synthesis of C-glycopyranosyl aldehydes 4a–d. | ||
Ketol acetates 19a–d were synthesised by stereoselective addition of 2-lithiothiazole to sugar lactones 17a–d, resulting in ketol molecules and further acetylation of the corresponding hydroxy group in situ or after isolation. Two pathways have been studied for the conversion of 17a–d into 19a–d (Scheme 7). These two pathways produced an anomeric mixture in different yields along with different ratios as shown in Table 2.
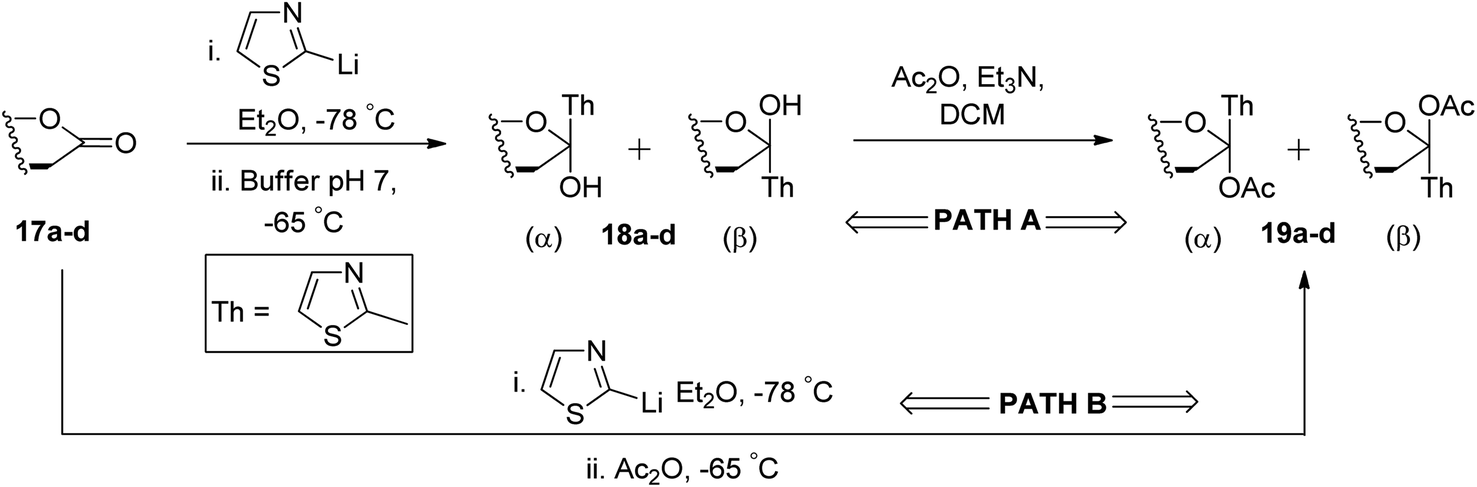 | ||
| Scheme 7 The conversion process of 17a–d into 19a–d via two pathways, A and B. | ||
| Lactone | Ketol acetate | Condition (path) | α:β |
Yield (%) |
|---|---|---|---|---|
| 17a | 19a | A | 1:0 |
80 |
| B | 1:7 |
87 | ||
| 17b | 19b | A | 1:0 |
73 |
| B | 0:1 |
78 | ||
| 17c | 19c | A | 1:0 |
78 |
| B | 1:10 |
75 | ||
| 17d | 19d | A | 1:0 |
77 |
| B | 0:1 |
80 |
Thus, ketols 18a–d were found as a mixture of kinetically and thermodynamically controlled products where the kinetic products were attributed to the steric effect of substituent and the thermodynamic products to the electronic effect of ring oxygen. On the further reaction of compounds 19a–d with triethylsilane and trimethylsilyl triflate (TMSOTf), the acetate group was reduced yielding thiazolyl C-glycoside 20a–d in excellent yield, which is independent of stereochemistry at C-1 position of glycosyls 19a–d. Further, a reaction sequence of N-methylation followed by reduction of hydride and hydrolysis gave β-linked C-glycopyranosyl aldehydes 4a–d in 72–80% yields. The use of thiazole ring as a masked formyl group has been proven to be very efficient and flexible as it worked well on four different substrates, which is further supported by good overall yields (52–65%) of the isolated products. Also, the thiazolyl-masked precursor was indefinitely stable and tolerated the synthetic elaboration of the product to yield delicate sugar aldehydes.
Later, Dondoni et al.30 reported a multigram scale synthesis of C-glucopyranosyl aldehyde, where benzothiazole was used in place of thiazole as a formyl group equivalent. The precursor ketose 21 was prepared as a single anomer by the reaction of 2-lithiobenzothiazole (prepared in situ by the reaction of butyl lithium and benzothiazole) with 2,3,4,6-tetra-O-benzyl-D-glucopyrano-1,5-lactone 12 in 78% yield. The anomeric position was activated by O-acetylation with acetic anhydride in triethylamine to afford compound 22 in 88% yield. Next, compound 22 was reacted with triethylsilane in the presence of trimethylsilyl triflate (TMSOTf) underwent silane-based deoxygenation, and afforded benzothiazolyl α- and β-C-glucosides 23a–b in 4:6 ratio in 80% yield. After the recovery of β-C-glucoside, the benzothiazole ring was converted to a formyl group by a multistep sequence starting with N-methylation, followed by hydride reduction and then hydrolysis to give hydrated gem-diol compound 24 along with formyl C-glucopyranosyl aldehyde 4a in 82% yield. A mixture of compounds 24 and 4a, when heated with DMSO-d6 at 160 °C produced almost pure β-linked C-glucopyranosyl aldehyde 4a (Scheme 8).
 | ||
| Scheme 8 Synthesis of C-glucopyranosyl aldehyde 4a. | ||
2.3 Dithiane approach
Although the above-described thiazole approach proved efficient in respect of the overall yield obtained, the lack of diastereofacial selectivity arises during the addition of 2-lithiothiazole to sugar lactones and the formation of the anomeric mixture of C-glycopyranosyl aldehyde has been the major limitation. However, it has been observed that kinetic and thermodynamic conditions control the diastereoselectivity of thiazole attack on sugar lactones. Hence, for stereoselective formylation, Sanchez et al.31 introduced the sugar dithiane as the key intermediate and synthesized single β-anomer of C-glycopyranosyl aldehyde using anucleophilic addition reaction followed by Lewis acid-catalysed stereospecific reductive dehydroxylation. 2,3,4,6-Tetra-O-benzyl-D-glucopyrano-1,5-lactone (12) on reaction with 2-lithio-1,3-dithiane gave a single diastereoisomer, sugar lactol 25 in 73% yield. This transformation indicated an equatorial attack on carbonyl to generate corresponding equatorial dithiane substituted sugar and hence compound 25 was assumed to be β-C-glucopyranosyl dithiane. Further, in the presence of boron trifluoride diethyl etherate (BF3·Et2O) at −40 °C, the hydroxyl group was stereoselectively removed by triethylsilane yielding 2,3,4,6-tetra-O-benzyl-β-1-C-(2-dithianyl)-D-glucopyranose (26) in 89% yield. Subsequently, compound 26 was hydrolysed to give β-C-glucopyranosyl aldehyde 4a in the presence of sodium carbonate (Na2CO3) and methyl iodide (MeI) in acetone at 35 °C in 61% yield along-with 20% of unreacted starting material (Scheme 9). | ||
| Scheme 9 Synthesis of β-C-glucopyranosyl aldehyde 4a. | ||
Later on, Labeguere et al.32 reported another route for the highly diastereoselective synthesis of β-C-glucopyranosyl aldehyde with improved overall yield. The commercially available precursor 2,3,4,6-tetra-O-benzyl-D-glucopyranose (27) underwent oxidation in the presence of dimethyl sulfoxide (DMSO) in acetic anhydride to produce2,3,4,6-tetra-O-benzyl-D-glucono-1,5-lactone 12 in 99% yield. Under Umpolung Seebach reaction condition, compound 12 was transformed into 1-C-[bis(methylthio)methyl]-α-D-glucopyranose (28) in 74% yield via in situ formation of bis(methylthio)methyl carbanion. In the presence of BF3·Et2O and triethylsilane (Et3SiH) in DCM at −78 °C, the anomeric hydroxyl group of 28 was reduced to obtain the single β-anomer, i.e. β-1-bis(methylthio)methyl-tetra-O-benzyl-D-glucopyranoside (29) with 99% yield. Finally, treatment with methyl iodide and calcium carbonate in a solvent mixture of acetonitrile-water afforded single β-linked glucopyranosyl aldehyde 4a (Scheme 10).
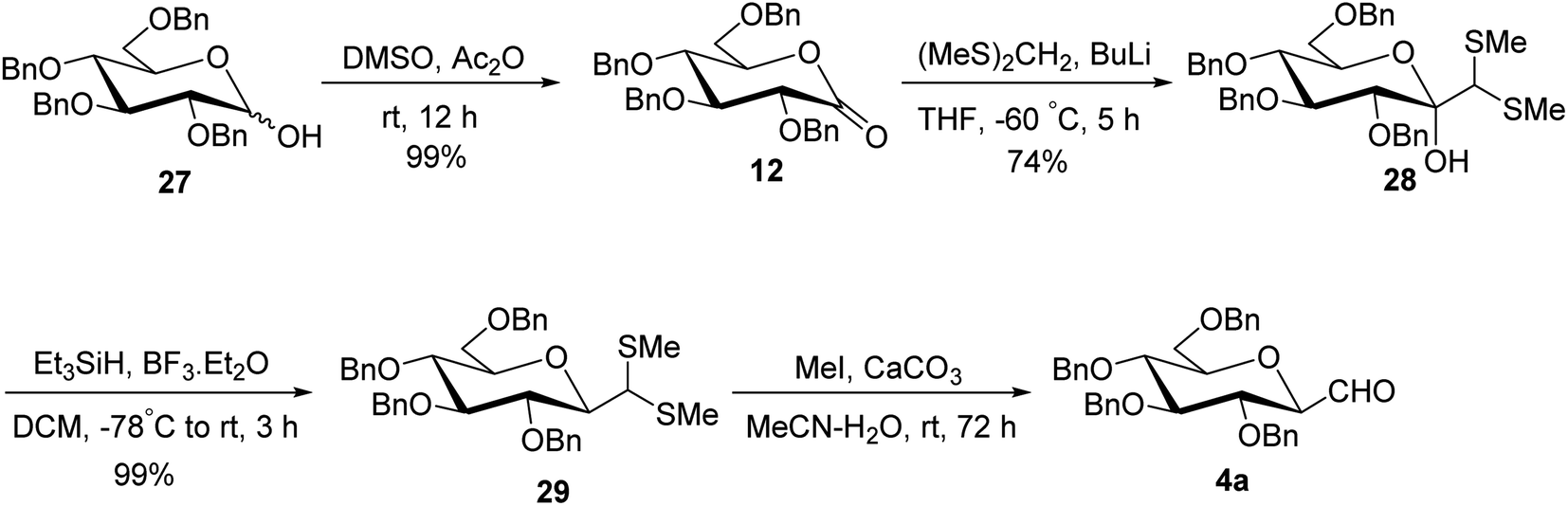 | ||
| Scheme 10 Synthesis of β-C-glucopyranosyl aldehyde 4a. | ||
Gerber et al.33 developed a unique protocol for the stereoselective synthesis of β-C-manno-pyranosyl aldehyde (Scheme 11). Here, 7-oxabicyclo[2.2.1]hept-5-en-2-one (30) was converted into (−)-6-endo-(benzyloxy)-5-exo-hydroxy-7-oxabicyclo[2.2.1]heptane-2-one (31) in 68% yield using epoxidation by mCPBA followed by nucleophilic attack on the epoxide ring by BnOH and hydroxy group protection using tert-butyldimethylsilyl chloride. Further, treatment with triethylsilyl chloride followed by ClCH2I/Et2Zn in DCE and subsequent oxidation with FeCl3/pyridine afforded enone 32 in 45% yield. Epoxidation using t-BuOOH, DBU followed by reduction with sodium borohydride after that protection with benzoyl chloride and deprotection of the silyl group using fluoride donor Bu4NF afforded 33 in 60% yield. Dess–Martin Periodinane oxidation and epoxide ring opening using CF3COOH–H2O and acetyl protection produced 34 in 79% yield, which on Baeyer–Villiger treatment using mCPBA furnished lactone 35 in 83% yield. Further, treatment with ethanethiol in acidic media (EtSH–TfOH) and then with methanol produced dithioacetal 36, which on hydrolysis using Hg(ClO4)2·xH2O and then Ag2CO3 liberated unstable C-glycopyranosyl aldehyde 37.
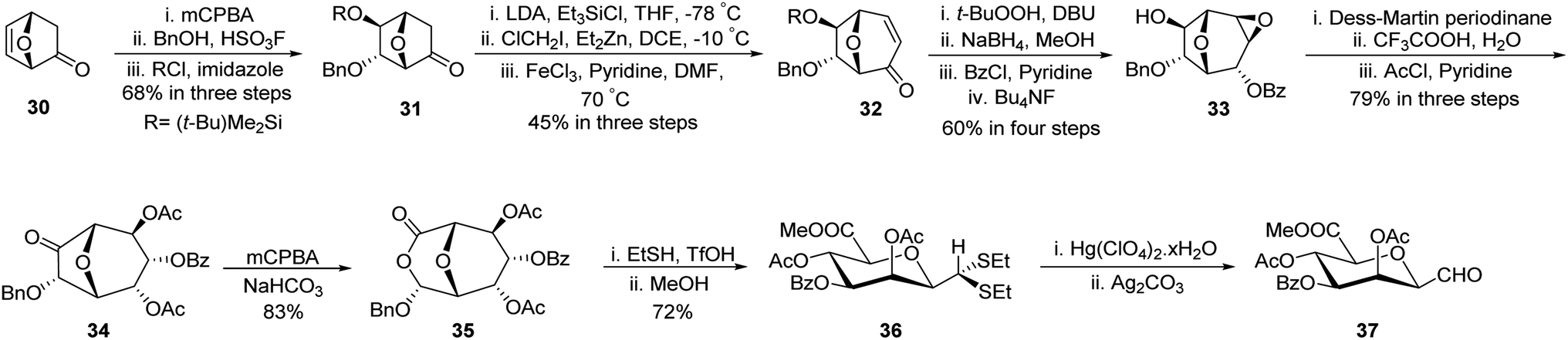 | ||
| Scheme 11 Synthesis of β-C-glycopyranosyl aldehyde 37. | ||
2.4 Cyanide approach
In the dithiane approach, both cyclic and acyclic 1,3-dithiane were used, and it was observed that the acyclic dithiane could be easily removed in the last step for the synthesis of desired C-glycopyranosyl aldehydes. The cyclic 1,3-dithiane removal consists of a lower yield and has a longer reaction time along-with the recovery of the starting material. Therefore, Sipos et al.34 introduced a new approach to synthesise β-C-glycopyranosyl aldehyde by reductive hydrolysis of C-glycosyl cyanides. All glycosyl cyanides (38a–f) except 38c(β) were transformed into the expected aldehydes (39a–f) on direct treatment with DIBAL-H in ammonium chloride at −78 °C. On the other hand, 1-C-formyl glycal product 40c was formed from galactosyl cyanide (38c(β)) (Scheme 12a). Similarly, the benzylated glycalnitriles 41a–b were reduced using DIBAL-H under the same reaction conditions to afford 2-deoxy glycopyranosyl aldehyde derivatives 40a–b, which were observed to be very stable (Scheme 12b). The corresponding reaction was carried out by taking the same precursors 41a–b where reduction using Pd/C in ethanol resulted in the formation of 2-deoxy-β-C-glycopyranosyl cyanides 42a–b, which on further reduction with DIBAL-H in ammonium chloride at −78 °C afforded β-C-glycopyranosyl aldehydes 43a–b (Scheme 12c). While compounds 40a–c and 43a–b were found to be very stable and could be stored, compounds 39a–f in the case of gluco-, manno- and galacto-derivatives were very sensitive to the elimination of 2-alkoxy group and a one pot strategy was developed to store them (Scheme 12d). In this process, 38a(β) was treated with DIBAL-H at −78 °C and excess hydride was quenched with acetic acid after 30 min followed by the addition of Wanzlick's reagent to afford 44a(β) in 57% yield.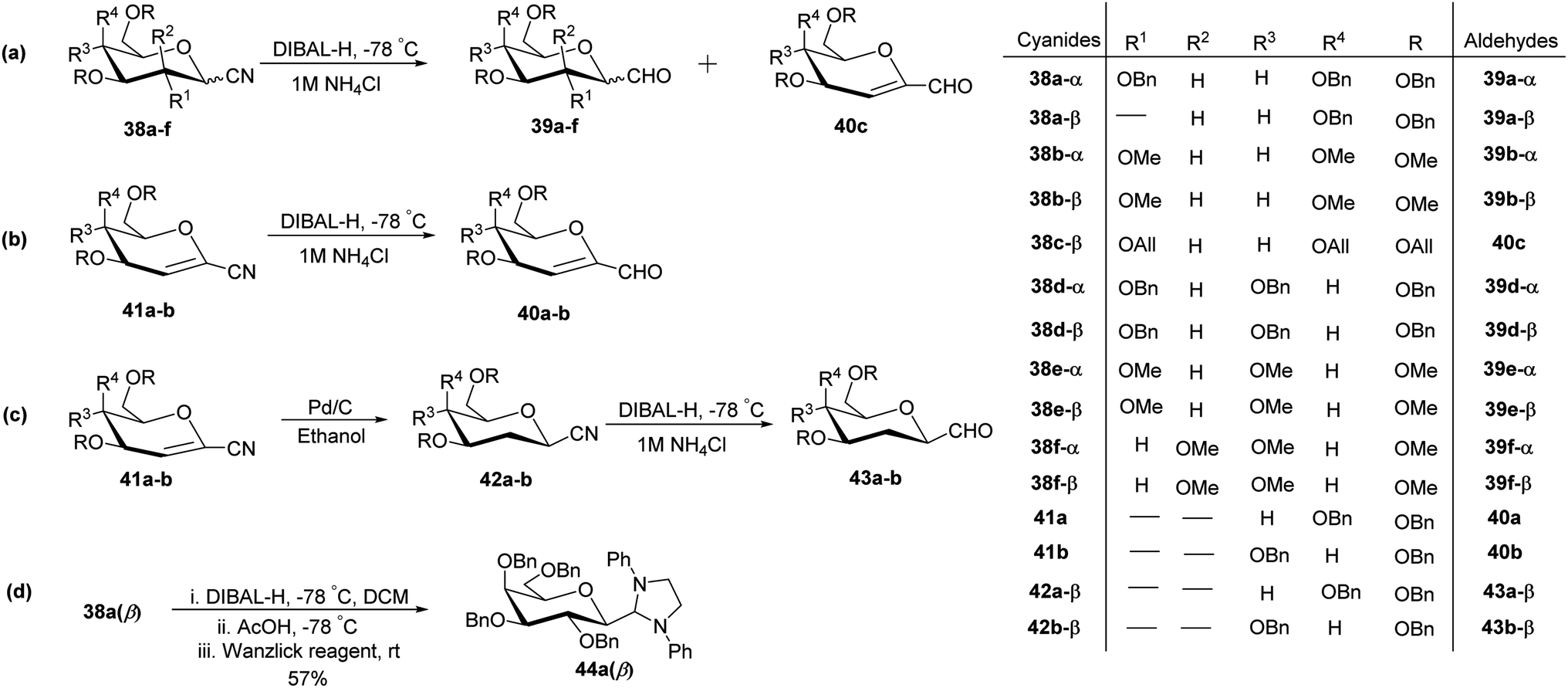 | ||
| Scheme 12 Synthesis of C-glycopyranosyl aldehydes 39a–f, 40a–c, 43a–b. | ||
Lopez et al.35 reported the synthesis of α-C-glucopyranosyl aldehyde via sugar cyanide as a key intermediate. Here, 1-O-acetyl-2,3,4,6-tetra-O-benzyl-D-glycopyranose 45a–b on reaction with trimethylsilyl cyanide and boron trifluoride etherate in acetonitrile afforded anomeric mixture of β- and α-glycopyranosyl cyanide 46a–b and 47a–b in 80–90% yield, which could be separated by column chromatography. Further, the reaction of glucopyranosyl cyanide 46a and 47a with lithium aluminium hydride in THF produced 1-amino-2,6-anhydro-3,4,5,7-tetra-O-benzyl-1-deoxy-D-glycero-D-gulo-heptitol 48a and 1-amino-2,6-anhydro-3,4,5,7-tetra-O-benzyl-1-deoxy-D-glycero-D-ido-heptitol 49a (Scheme 13a). C-Glucopyranosyl aldehyde 3a was obtained by direct reduction of α-glucopyranosyl cyanide 47a with lithium aluminium hydride in THF followed by hydrolysis with ammonium hydroxide (Scheme 13b).
 | ||
| Scheme 13 Synthesis of α-C-glucopyranosyl aldehyde 3a. | ||
Dettinger et al.36 designed a protocol for the synthesis of β-C-galactopyranosyl aldehyde 50 by a series of reactions starting from 2,3,4,6-tetra-O-acetyl-D-galactopyranosyl cyanide 48. Acetyl-protected cyanide 48 underwent a reduction in the presence of RANEY®-Nickel followed by protection with N,N-diphenylethylenediaminein pyridine to yield 49 in 72% yield. Subsequently, compound 49 on treatment with PTSA in DCM–acetone afforded β-C-galactopyranosyl aldehyde 50 in just 37% yield (Scheme 14).
 | ||
| Scheme 14 Synthesis of β-C-galactopyranosyl aldehyde 50. | ||
Fujiwara et al.37 synthesised β-C-glycopyranosyl aldehyde starting from tri-O-acetyl-D-glucal (Scheme 15). Acetyl-protected D-glucal 51 was converted into 52 by treatment with TMSCN, SnCl4 in DCM at −78 °C in 61% yield, which transformed into benzylidene-protected compound 53 by using solid support acid (Amberlyst 15) in methanol followed by benzaldehyde in chloroform, which was refluxed using Dean–Stark apparatus to afford benzylidene protection. The yield obtained in two steps was 85%. Alkene 53 underwent dihydroxylation by treatment with osmium tetraoxide, NMO in dioxane–water to furnish diol 54 in quantitative yield. Silyl protection was carried out using TBSOTf in the presence of 2,6-lutidine in DCM to afford protected cyanide 55 quantitatively, which was further subjected to DIBAL-treatment to furnish benzylidene-protected pyranosyl aldehyde 56 in 95% yield.
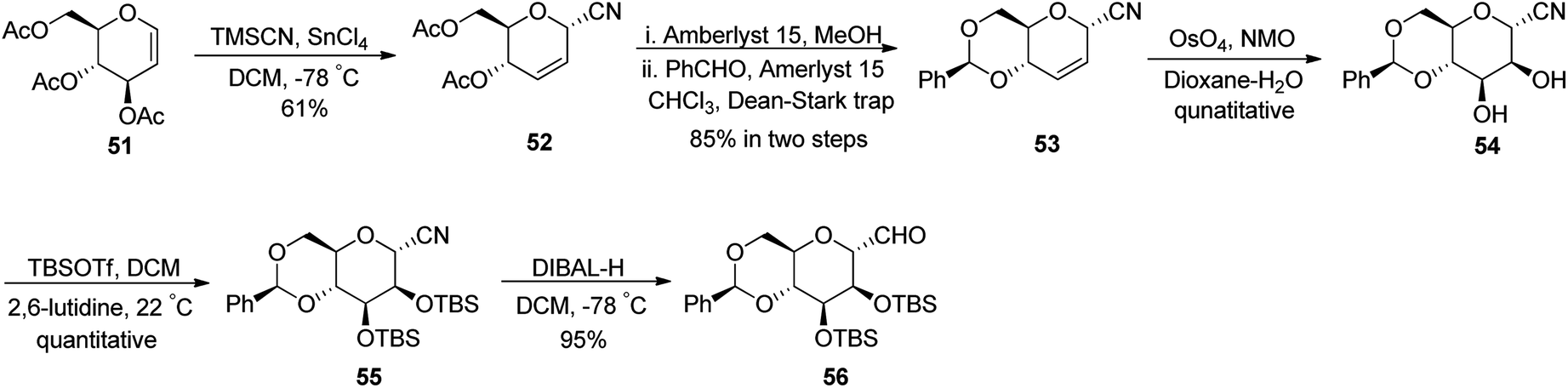 | ||
| Scheme 15 Synthesis of α-C-glycopyranosyl aldehyde 56. | ||
C-Glycopyranosyl aldehydes from cyanide precursors were also obtained by Toth et al.38 using Dettinger et al.36 protocol where reduction of glycosyl cyanides 57a–f was achieved by RANEY® nickel and sodium hypophosphite in pyridine-aqueous acetic acid in the presence of N,N-diphenylethylenediamine to obtain imidazolidine derivatives 58a–f. Further, by using PTSA in DCM–acetone under acidic reaction conditions C-glycopyranosyl aldehydes 59a–f were obtained (Scheme 16). Further, the synthesized C-glycopyranosyl aldehydes 59a–f were used for the synthesis of exo-glycals.
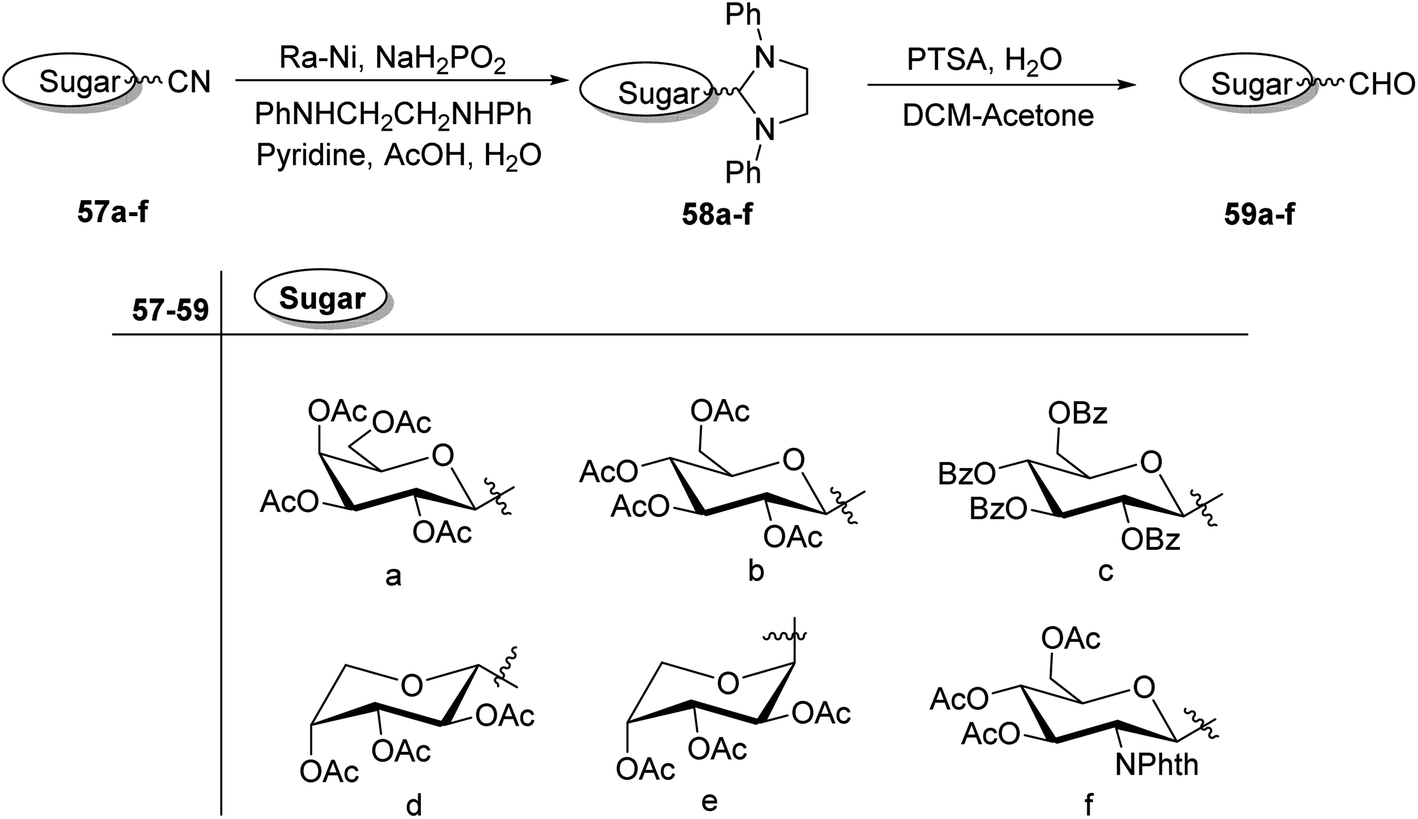 | ||
| Scheme 16 Synthesis of C-glycopyranosyl aldehydes 59a–f. | ||
2.5 Enoxysilane approach
The cyanide approach furnished the desired C-glycopyranosyl aldehyde in just one step, i.e. protection of the cyanide group with N,N-diphenylethylenediaminein pyridine followed by treatment with PTSA and direct reduction of cyanide using reducing agents DIBAL-H or LiAlH4. However, the increased interest in the synthesis of β-C-glycopyranosyl aldehydes due to their demanding application as intermediates in the formation of various complex C-glycosides led Zeitouni et al.39 to introduce a new synthetic approach using enoxysilanes as a key intermediate. In this approach, the β-D-glucosidic ketone 60 was synthesised from D-glucose in 96% yield using pentane-2,4-dione. Next, compound 60 was subjected to benzylation (reagents: Ag2O, BnBr in DMF) and acetylation (reagents: Ac2O in pyridine) individually yielding 61a and 61b with a yield of 30–45% and 91%, respectively, which on enolization using TMSCl in pyridine formed structural isomers 62a and 63a (from 61a) and similarly, compounds 62b from 61b. However, better regioselectivity was observed for compound 61b as compared to compound 61a. It has been observed that it was difficult to perform benzylation with the classical benzylation process (NaH, benzyl bromide, DMF) as compounds had ketone functional group present in the molecule, which further resulted in non-regioselective enolisation of compound 61a. Subsequently, the scheme further proceeded with the reaction of enoxysilane 62b with a freshly prepared solution of dimethyldioxirane (DMDO) to yield α-hydroxy ketone 64, which on further treatment with sodium metaperiodate in THF–water produced desired β-C-glycopyranosyl aldehyde 65a. Compound 65a turned out to be unstable and less pure, so it was stored as aminal 66 using N,N-dibenzylethylene diamine in toluene. Compound 66 obtained in a yield of 68% was in turn benzylated to give 67 in a yield of 90%. On the deprotection of compound 67 with Dowex-H+ resin quantitatively led to the formation of extremely pure β-C-glucopyranosyl aldehyde 4a with a yield of 94% (Scheme 17). | ||
| Scheme 17 Synthesis of β-C-glucopyranosyl aldehydes 65a and 4a. | ||
Later, Norsikian et al.40 applied this effective approach to introduce the formyl group at the anomeric position of other sugar precursors and reported it to be very efficient for all sugars except D-mannose. β-C-Glycosyl ketones 68a–g were treated with trimethylsilyl chloride in pyridine followed by sodium iodide in acetonitrile to afford enolized products 69a–g and 70a–g. The enoxysilanes thus produced were subjected to oxidation with DMDO, which produced α-hydroxy ketones 71a–g as major products. Further, α-hydroxy ketones 71a–g on treatment with sodium metaperiodate in THF–water gave the desired β-C-glycopyranosyl aldehydes 73a–g. Due to the less stability of β-C-glycopyranosyl aldehydes and with an aim to achieve a more pure form of it, they were stored as aminal 74a–g, which on deprotection with Dowex-H+ resin yielded the purest forms of β-C-glycopyranosyl aldehydes 73a–g (Scheme 18).
 | ||
| Scheme 18 Synthesis of β-C-glycopyranosyl aldehydes 73a–g. | ||
Xia et al.41 reported synthesis of L-glucose and L-galactose starting from D-glucose and D-mannose, respectively. In this synthesis, C-glycopyranosyl aldehyde was achieved as an intermediate, which was further converted into L-glucose and L-galactose sugars (Section 3.8, Scheme 81). Thus, both sugars, i.e. D-glucose and D-mannose were treated with pentan-2,4-dione, sodium bicarbonate in water to afford ketones 60 and 75, respectively. The primary hydroxyl group was selectively protected with trityl chloride followed by acetylation of the secondary hydroxyl group to afford 76a–b in 80–85% yield. Further, the Norsikian40 condition was applied on ketone 76a–b to produce silyl enol ether 77a–b, which upon ozonolysis furnished C-glycopyranosyl aldehyde 78a–b (Scheme 19).
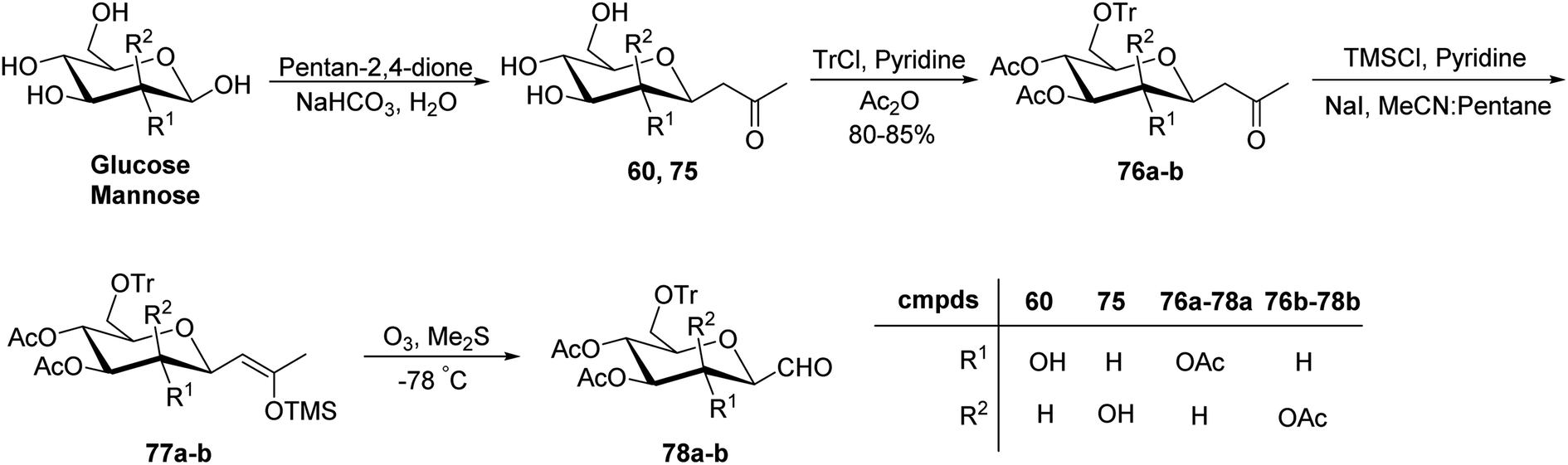 | ||
| Scheme 19 Synthesis of C-glycopyranosyl aldehydes 78a–b. | ||
2.6. Alkene approach
The approaches using allene and thiazole ring as a masked formyl group were low yielding and in addition, the thiazole deprotection involved a lengthy process. Besides, enoxysilane approach incriminated difficult synthesis of benzylated glycopyranosyl aldehydes due to by-product formation during DMDO treatment and the use of DMDO, which made this synthetic procedure cumbersome. Dietrich et al.42 utilized Wittig reaction conditions to carry out a reaction between commercially available 2,3,4,6-tetra-O-benzyl-D-glucopyranose (27) and methylenetriphenylphosphorane to afford alkene 79 following the reported procedure.43 Further, mercury-mediated cyclisation of compound 79 afforded oxymercuration product 80 with the known procedure.44 Then, oxidative demercuration of 80 using O2–NaBH4 produced alcohol 81,45 which was subjected to Swern oxidation to afford α-C-glucopyranosyl aldehyde 3a (Scheme 20). The obtained compound 3a was found to be very labile and immediately used for the synthesis of benzyl-α-C-glucosides and anilinomethyl-α-C-glucosides, which act as α-glucosidase inhibitors (Section 3.5, Scheme 62). | ||
| Scheme 20 Synthesis of α-C-glycopyranosyl aldehyde 3a. | ||
Sanchez et al.31 introduced the sugar alkene as a key synthetic intermediate, which was obtained via nucleophilic addition and reductive dehydroxylation. This approach provided the stereochemical control during reductive dehydroxylation, before unmasking of the formyl group, unlike allene and thiazole approaches where both α- and β-isomer were formed. Sugar lactone 12 on treatment with phenylacetylyllithium at −78 °C gave a diastereomeric mixture (1:1) of lactol 82 in 84% yield, which furnished single isomer β-C-glycoside 83 in 88% yield after stereoselective removal of the hydroxyl group by triethylsilane in the presence of Lewis acid. After the stereoselectivity was achieved, Lindlar's catalyst was used in the presence of quinoline for hydrogenation, resulting in cis-alkene 84 in 97% yield. Further, alkene 84 was subjected to ozonolysis to afford exclusively β-C-glucopyranosyl aldehyde 4a in 81% yield (Scheme 21).
 | ||
| Scheme 21 Synthesis of α-C-glycopyranosyl aldehyde 4a. | ||
Leclere et al.46 prepared the α-C-glycopyranosyl aldehyde 90 from easily available galactosyl bromide 85. Galactosyl bromide 85 undergoes allylation in the presence of allyltributylstannane and Et3B/air and yielded α-C-allyl galactose derivative 86 in 70% yield and a trace amount, i.e. 6% of compound 87 resulting from in situ acyl migration. Terminal alkene 86 in the presence of a catalyst (Ph2MeP)2Ir(COD)PF6 isomerised into internal alkene 2-propenyl derivative 88 using the designed protocol.47 Initial attempts to produce desired C-glycopyranosyl aldehyde from acetyl-protected alkene 88 failed, so deprotection of acetate of compound 88 with K2CO3 in methanol followed by protection with iso-propylidenes using 2-methoxypropane, TsOH in DMF afforded compound 89 in 75% yield, which on ozonolysis yielded α-C-glycopyranosyl aldehyde 90 in good yield (Scheme 22).
 | ||
| Scheme 22 Synthesis of α-C-glycopyranosyl aldehyde 90. | ||
Chen et al.48 incorporated easily and readily available starting material galactosyl pentaacetate 5. The galactosyl pentaacetate 5 was subjected to allylation at the anomeric position in the presence of allyl trimethylsilane in BF3·Et2O to afford compound 86 in 77% yield, which next underwent de-protection in the presence of sodium methoxide in methanol followed by benzyl protection in the presence of BnBr led to the formation of compound 91 in 93% yield. Palladium-mediated isomerisation of the terminal alkene of compound 91 was performed into internal sugar alkene 92 with a 90% yield. Subsequently, compound 92 on ozonolysis followed by in situ reduction and further treatment with Swern oxidation resulted in the formation of α-C-galactopyranosyl aldehyde (Scheme 23).
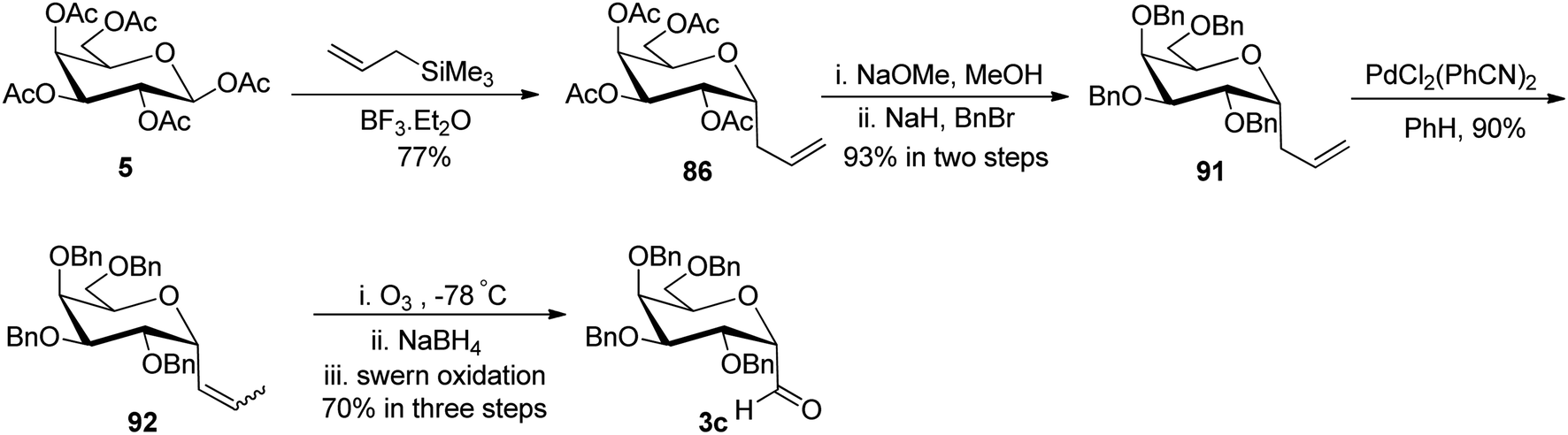 | ||
| Scheme 23 Synthesis of α-C-galactopyranosyl aldehyde 3c. | ||
Guillaume et al.27 designed a strategy to synthesise α-C-galactopyranosyl aldehyde 96 using galactosyl pentaacetate 5, which is a readily available starting material. The C-glycosylation of compound 5 was performed with allyl trimethylsilane using conditions of Chen et al.48 to afford terminal sugar alkene 86 in 44% yield, which on palladium catalysed double bond isomerisation resulted in propene 93 in 63% yield. Compound 93 upon Zemplen deacetylation with sodium methoxide in methanol followed by selective protection of primary alcohol with tri-isopropylsilyl ether and secondary alcohol were masked in the presence of PMBCl to obtain compound 94 in 75% yield. Alkene 94 on osmium-catalysed dihydroxylation resulted in vicinal diol 95, which underwent further reaction with sodium-periodate resulting α-C-galactopyranosyl aldehyde 96 (Scheme 24), which was further utilized for the synthesis of C6-modified α-C-GalCer analogues.
 | ||
| Scheme 24 Synthesis of α-C-galactopyranosyl aldehyde 96. | ||
Desire et al.49 reported synthesis of α-C-glucopyranosyl aldehyde via alkene as a key intermediate (Scheme 25). 6-O-Acetyl-2,3,4-tri-O-benzyl-α-D-glucopyranosyl chloride (99) was synthesised by ring opening of 1,6-anhydro-2,3,4-tri-O-benzyl-β-D-glucopyranose (97) or from 6-O-acetyl-2,3,4-tri-O-benzyl-D-glucopyranose (98) by following reported literature.50 Further treatment of compound 99 with tributyl(phenylethynyl)tin in the presence of silver tetrafluoroborate in 1,2-dichloroethane at 0 °C produced selectively (α-D-glucopyranosyl) phenylacetylene 100 in 73% yield. Hydrogenation of compound 100 was carried out by using powdered zinc in AcOH–MeOH for a long time of 2–5 days to give Z-alkene 101 in 80% yield, which underwent ozonolysis to afford α-C-glucopyranosyl aldehyde 102, which was found to be prone to decomposition; therefore, it was masked by N,N′-diphenylethtylenediamine as stable 1,3-diphenylimidazolidines 103 with 61% yield and could be regenerated by mild acidic treatment. Deacetylation was achieved using triethylamine in methanol–water to afford 104.
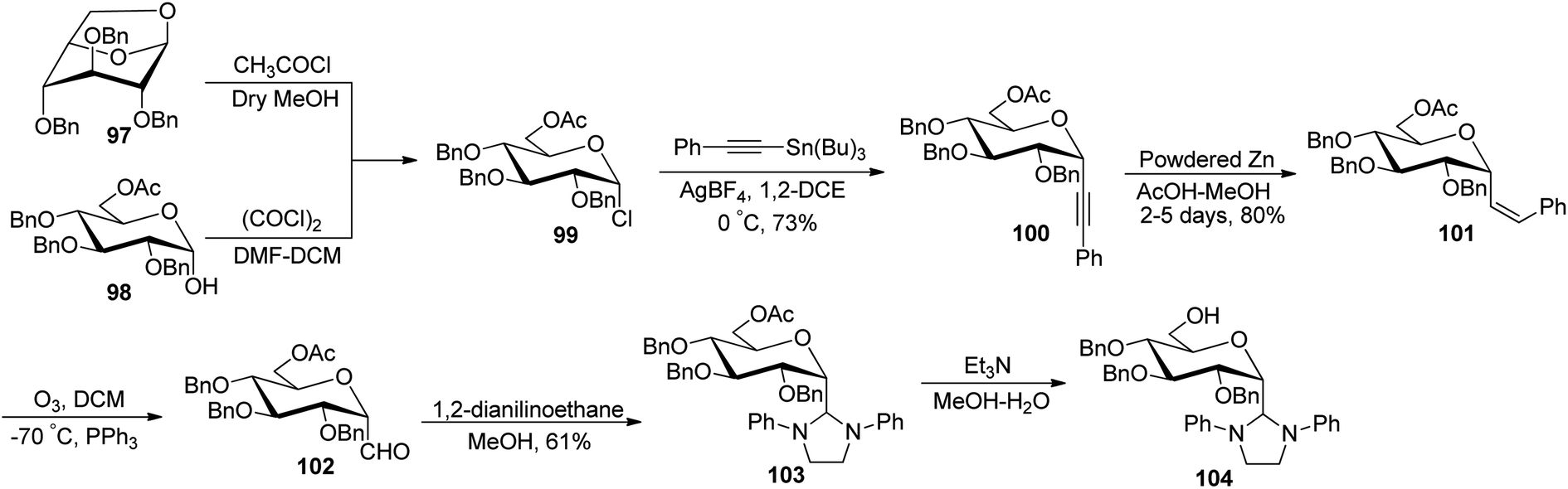 | ||
| Scheme 25 Synthesis of α-C-glucopyranosyl aldehyde 102. | ||
Among the other alkene approaches, the metal-catalysed isomerisation of allyl species was also achieved by McGarvey et al.51 where the benzyl protected β-isomer, 105 and the α-isomer, 106 on exposure to iridium catalyst, (Ph2Me)2IrCOD·PF6 (10 mol%) yielded isomeric vinyl glycosides 107a and 107b in 80% yield. These isomeric vinyl glycosides further gave β- and α-linked C-glycopyranosyl aldehydes, where the β-linked isomer was the major product. Substrate 107b upon ozonolysis gave the desired 108b isomer with >90% yield. Similarly, compound 107a underwent oxidative cleavage using Lemieux reaction conditions yielding 108a analogue. The aldehydes 108a and 108b thus obtained acted as intermediates for various further syntheses (Scheme 26).
 | ||
| Scheme 26 Synthesis of C-glycopyranosyl aldehydes 108a–b. | ||
Khatri et al.52 reported another synthesis of stereoselective β-linked C-glycopyranosyl aldehyde via an alkene approach using easily available precursors i.e., D-galactose, D-glucose, and D-mannose. The native sugars were treated under a developed procedure,53 i.e. reaction with dibenzoylmethane–sodium bicarbonate in aqueous alcoholic solution gave β-C-glycosyl benzoylmethane 109a–c, which further reacted with acetic anhydride–DMAP in DMF yielding the peracetylated derivatives of β-C-glycosyl benzoylmethane 110a–c in 94–97% yield. Compounds 110a–c upon reduction with NaBH4 gave corresponding alcohols 111a–c in 93–96% yield, which was followed by dehydration reaction with dehydrating reagent P2O5 in dichloromethane to obtain alkene derivatives 112a–c in 91–95% yield. The peracetylated C-glycosides 112a–c were converted to the corresponding perbenzylated sugar alkene 113a–c since the former gave very unstable products on oxidation. Hence, 112a–c were firstly deacetylated using sodium methoxide followed by perbenzylation using benzyl bromide in NaH to obtain the perbenzylated sugar alkene 113a–c in 80–92% yield. These perbenzylated analogues 113a–c upon oxidation with OsO4–NaIO4 resulted in β-C-glycopyranosyl aldehydes 4a–c in 78–81% yield (Scheme 27).
 | ||
| Scheme 27 Synthesis of β-C-glycopyranosyl aldehydes 4a–c. | ||
2.7 Nitromethane approach
In the above-discussed approaches, the alkene approach was found to be most efficient in respect of overall yield obtained and selectivity due to the formation of only one anomeric isomer of C-glycopyranosyl aldehydes. Moreover, the synthetic routes consisted of cheap and easily available starting materials. Now we are moving towards the end of approaches developed for the synthesis of C-glycopyranosyl aldehydes and at last we are discussing nitromethane as a key synthetic intermediate. So, Martin et al.54 developed a synthetic route for the synthesis of the β-C-glucopyranosyl aldehyde 65a starting from β-D-glucosylnitromethane 114, which in turn can be synthesized from readily available D-glucose following two steps.55 The β-D-glucosylnitromethane 114 on reaction with tert-butyldimethylsilyl chloride, DBU in DCM, yielded the silyl nitronate derivative 115 in 95% yield, which on ozonolysis afforded the 2,3,4,6-tetra-O-acetyl-β-C-glucopyranosyl aldehyde 65a (Scheme 28).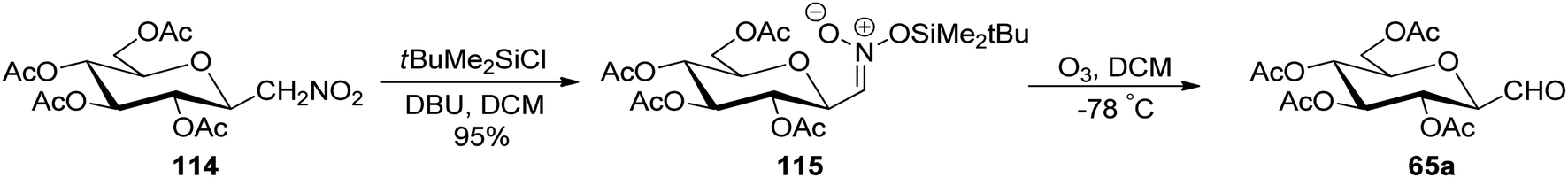 | ||
| Scheme 28 Synthesis of β-C-glucopyranosyl aldehyde 65a. | ||
Simo et al.56 achieved the synthesis of C-glycopyranosyl aldehyde starting from nitromethane sugar precursor (Scheme 29). Here, C-(4,6-O-benzylidene-β-D-glucopyranosyl) nitromethane (116) was reduced to hydroxylamino derivative 117 using H2, Lindlar catalyst in methanol which was difficult to isolate and directly oxidised in the air under basic condition (NH4OH) to obtain cis–trans mixture of oxime 118 in 70% yield. The oxime 118 reacted with H2 in the presence of RANEY® nickel affording the dimeric aminal 119 in 79%, which on reaction with 1,2-dianilinoethane (Wanzlick base) produced imidazolidine 120 in 90% yield. Acetylation was carried out using acetic anhydride in pyridine to afford 121, which was hydrolysed by p-toluenesulfonic acid monohydrate in dry THF to furnish C-glycopyranosyl aldehyde 122 in 57% yield.
 | ||
| Scheme 29 Synthesis of C-glycopyranosyl aldehyde 122. | ||
Petrusova et al.57 designed a one-step protocol for the synthesis of β-C-glycopyranosyl aldehydes from β-D-glucosylnitromethane (Scheme 30). 2,6-Anhydro-7-deoxy-7-nitro-L-glycero-L-galacto-heptitol 123a–d treated with an aqueous alkaline solution followed by ozonolysis at room temperature to give β-C-glycopyranosyl aldehydes 124a–d. It was observed that in an aqueous solution, the compound 124a–d exists in its hydrate form predominantly. Thus, the resulting native C-glycopyranosyl aldehydes 124a–d were found very much labile and were used further for the synthesis of 2-β-D-glycopyranosyl-nitroethenes and -nitroethanes (Section 3.8, Scheme 82).
 | ||
| Scheme 30 Synthesis of β-C-glycopyranosyl aldehydes 2a–d. | ||
3. Synthesis of C-glycoconjugates
The availability of numerous methods to bring diversity in the formation of aldehyde at the anomeric position of sugar benefited the development of C-galactosphingo lipid analogues, C-glycopyranosyl disaccharides, C-glycopyranosyl amino acid precursors, C-glycopyranosyl-amino acids and dipeptides, glycopyranosyl phenyl methane and its fluoro derivatives, C-glycopyranosyl heterocycles, glycopyranosyl bis-amides, several natural product fragments, and many other C-linked glycoconjugates. Some of them play important roles in biological systems, for instance, glycoconjugate 125 is a potent inhibitor of ice recrystallization and could protect embryonic liver cells from cryo-injury;46 the acyl- and benzyl-C-β-D-glucosides (126 and 127) work as glucose uptake promotor;58 C-mannosides analogues (128a–b) block uropathogenic Escherichia coli from colonizing the lower urinary tract;59 C-glycosides 129a–c were determined by ELISA of a blood sample obtained from mice stimulated by a 1 μg injection of glycolipid in the buffer. It was found that E-isomer 129b is superior to Z-isomer 129c as a ligand for CD1d/NKT immunity pathway;48 C-linked galactosphingo lipid analogue (130) blocks the interaction of HIV-1 gp120 with GalCer;60 C-linked disaccharide analogue (131a) of TF epitope induces a strong immune response in mice, which imparts it a possibility to be developed as a therapeutic vaccine;61 C-linked disaccharide (132a) mimics O-β-D-galactopyranosyl-(1–3)-D-galactopyranosidesand proved to be a suitable agent for O-glycosidation and construction of glycoconjugates;62 carbohydrate amino acids mimetic (133) shows in vivo stability towards α-galactosidase enzyme and might function as glycosidase inhibitor(Fig. 4).25 Henceforth, we describe the following synthetic processes for the development of structurally diversified and complex C-glycoconjugates.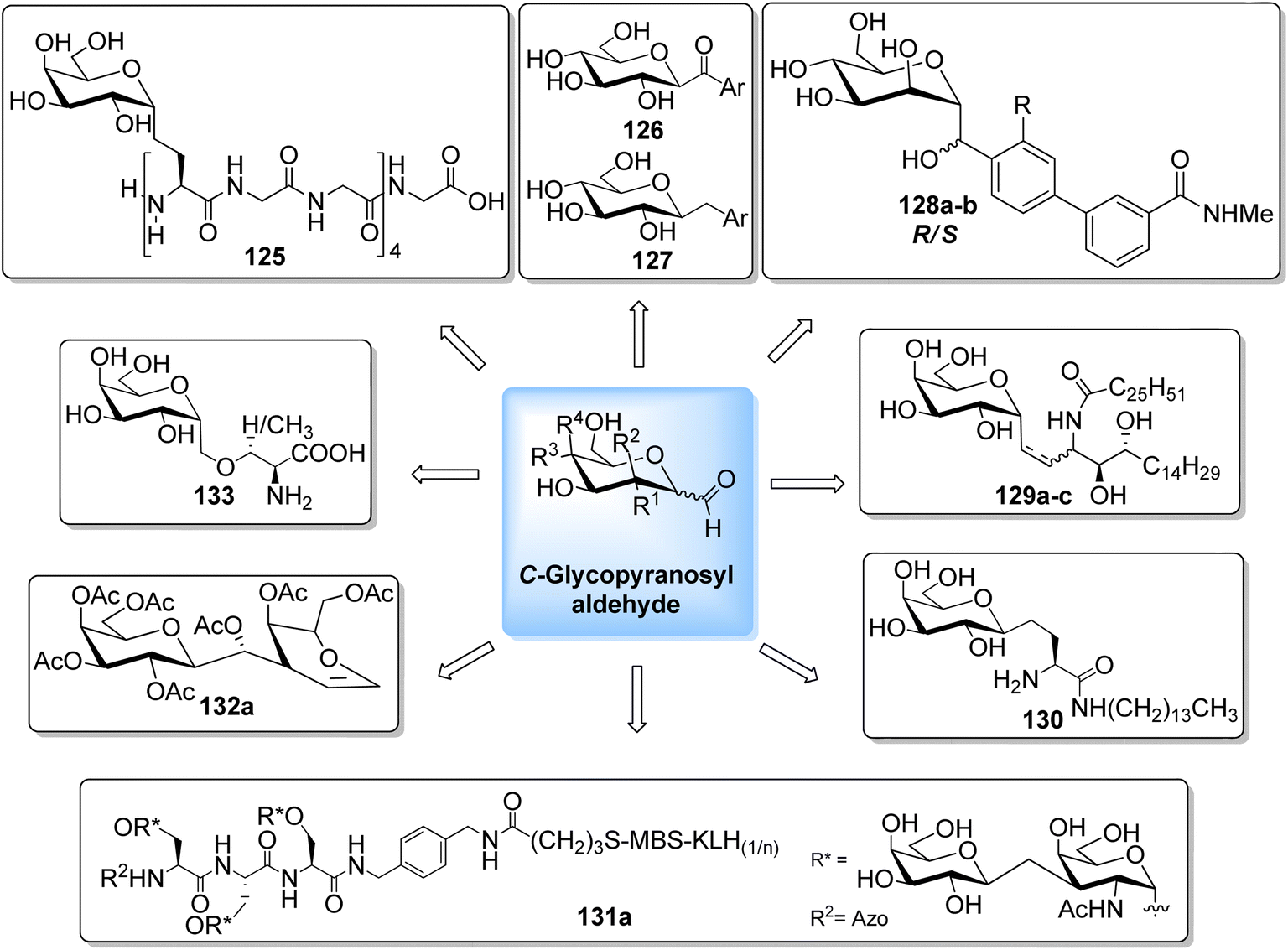 | ||
| Fig. 4 Structures of various biologically important C-glycoconjugates derived from C-glycopyranosyl aldehyde. | ||
3.1 C-Glycolipid analogues
Bertozzi et al.60 designed a range of water-soluble, C-linked galactosphingo lipid derivatives, which bind specifically to HIV-1 gp120, inhibiting its interaction with CD4 of host cells. The designed compounds have β-linked galactose mimicking galactosyl ceramide (GalCer) antibodies, which inhibit the infection of two CD4-negative neural cell lines. The synthesis of these C-linked galactosphingo lipids was initiated from β-C-galactopyranosyl aldehyde 4c, which upon condensation with Wittig reagent 134 furnished oxazolidinone 135 in 34% yield. Next, alkene was reduced by tosyl hydrazine and sodium acetate in DME-H2O producing compound 136 in 92% yield, which was transformed into compound 137 using the reaction condition of Boc-anhydride/triethylamine in DMF, and further calcium carbonate in methanol. Upon oxidation with Jones reagent, compound 138 was achieved in 95% yield, which was transformed to its amide derivative 139 by reaction with tetradecylamine in the presence of coupling reagent (EDC and HOBt). The deprotection of Boc and benzyloxy group was achieved on treatment with TFA and H2 in Pd/C to afford C-linked galactosphingo lipid 130 quantitatively (Scheme 31). Similarly, other derivatives 140, 141, 142, and 143 were also synthesized from the condensation of acid derivative 138 and different varieties of amines (Table 3). The introduction of different amines displayed different inhibition range against gp120 GalCer binding at 1 mg mL−1 as given in Table 3. | ||
| Scheme 31 Synthesis of galactosphingo lipid 130. | ||
| Entry | Compounds | % inhibition of 1 mg mL−1 |
|---|---|---|
| 1 |  |
86 (IC50 = 120 μM) |
| 2 |  |
34 |
| 3 |  |
0 |
| 4 |  |
0 |
| 5 |  |
96 (IC50 = 160 μM) |
It was observed that compound 130 (entry 1) showed the highest affinity for recombinant gp120 at IC50 = 120 μM and slightly better than their O-linked glycolipid analogue 143 (entry 5). A decrease in inhibitory activity was observed with a decrease in the length of hydrocarbon.
Guillaume et al.27 designed a strategy to synthesise immunogenic glycolipids due to their importance in the medical field. These have the potential to act as vaccine adjuvants to fight cancer and other microbial infections. One such glycolipid synthesised is α-GalCer analogues starting from C-galactopyranosyl aldehyde 96 (Scheme 32a). Aldehyde 96 upon Julia–Kocienski olefination with the known benzothiazole 144 yielding inseparable α–β anomers of olefin 145a–b in just 44% yield. Olefin 145a–b underwent deprotection of silyl ethers on treatment with tetrabutylammonium fluoride, leading to the separation of both anomers and α-anomer alcohol 146 in 65% yield was subjected to Mitsunobu reaction with phthalimide to afford compound 147 in 88% yield. This compound 147 on treatment with methylhydrazine did not reduce the double bond, while the same compound on treatment with hydrazine monohydrate reduced the alkene and liberated free ammonia. Further treatment of both analogues with 1-napthyl isocyanate furnished corresponding urea derivatives 148 and 150, in 66% and 77% yields, respectively. Further, deprotection of compounds 148 and 150 was achieved by treatment of 4 M HCl in dioxane after that acylation with cerotoyl chloride in a biphasic mixture of THF and 1 M KOH solution gave α-C-GalCer derivatives 149 and 151 in 29% yield, respectively.
 | ||
| Scheme 32 (a) Synthesis of α-GalCer analogues 149 and 151. (b) Synthesis of α-GalCer analogues 153 and 156. | ||
Further, for the synthesis of α-GalCer analogues 153 and 156, the alcohol 146 on treatment with p-nitrophenyl chloroformate followed by the addition of 4-aminopyridine resulted in the 4-pyridylcarbamate intermediate 152 in 62% yield, while the same sequence when followed after the reaction with hydrazine and 1-napthyl isocyanate afforded 4-pyridylcarbamate intermediate 155. Both compounds 152 and 155 when treated with HCl followed by acetylation afforded the corresponding pyridinylcarbamate analogues 153 and 156 in 17% and 44% yields, respectively (Scheme 32b).
Chen et al.48 reported the synthesis of E and Z α-C-galactosylceramides using Julia–Lythgoe–Kocienski reaction as the key step between C-glycopyranosyl aldehyde 3c and sulfones 157 or 158 using lithium hexamethyldisilazide (LiHMDS) at −78 °C to afford olefin 159 along with a trace amount of 1-C-formyl glycal (Scheme 33a). Only an E-isomer was formed in the case of BT-sulfone while in the case of PYR-sulfone E/Z ratio (39:61) was obtained. Further, the installation of fatty amide side chain was achieved by treatment of olefin 159 with trifluoro acetic acid (TFA), triethylsilane (Et3SiH) in DCM followed by di-isopropanyl carbodiimide (DIC), DMAP in butyl alcohol to obtain the precursor of targeted compound 160 in 80% yield. The trans isomer in amide 160 could be easily separated from its cis counterpart by flash chromatography. Finally, amide 160 was elaborated by either the retention or removal of double bonds. Hydrogenation of the double bond together with debenzylation was achieved using H2, Pd/C to afford C-glycoside 129a in 80% yield, while unsaturated compounds 129b and 129c were prepared in 80% and 84% yield, respectively, by Birch reduction (Na–NH3).
 | ||
| Scheme 33 Synthesis of α-C-galactosylceramides 129a–c. | ||
IL-12, IFN-γ and IL-4 levels of these synthetic C-glycosides 129a, 129b, and 129c were determined by ELISA of blood samples obtained from mice stimulated by a 1 μg injection of glycolipid in the buffer. It was found that E-isomer 129b is superior to Z-isomer 129c as a ligand for the CD1d/NKT immunity pathway.
Both sulfones used in the above Julia–Lythgoe–Kocienski reaction were prepared from commercially available phytosphingosine (Scheme 33b). BOC protection was achieved from phytosphingosine to afford butyl carbamate 161 in 93% yield. Further, primary alcohol was temporarily blocked by the silyl protecting group (TBSCl) followed by isopropylidene protection of vicinal diol and deprotection of primary alcohol, producing precursor 162 in 87% yield. Mitsunobu transformation to thioether using PPh3, BtSH, and DIAD followed by oxidation with m-CPBA afforded BT-sulfone 157 in 95% yield. Similarly, the mesylation of the hydroxyl group in 162 followed by substitution with PYRSH afforded 164, which on oxidation with m-CPBA produced PYR-sulfone 158 in 92% yield.
Dondoni et al.63 developed a strategy for the synthesis of β-D-galactosyl ceramide methylene isostere starting from C-galactopyranosyl aldehyde (Scheme 34). First, C-galactopyranosyl aldehyde 4c was transformed into alkyloxazolidine 164 by a sequence of reactions.64 Acetonide protective group removal was achieved by AcOH–H2O to afford N-Boc amino alcohol, which on further oxidation under Swern conditions obtained aldehyde 165 in 55% yield. Addition of lithium 1-pentadecyne in anhydrous THF produced alcohol 166 as a mixture of S/R (syn/anti) isomer in a 70:30 ratio. Due to the isomer formation, oxidation–reduction steps were carried out with 166. First, Swern oxidation afforded ketone 167 followed by reduction with L-selectride in THF afforded anti isomer 168 as a major product with S/R (syn/anti) isomer in a 5:95 ratio. BOC deprotection was carried out with 4.8 M HCl in dioxane-afforded amino alcohol 169 in 90% yield. Further, stereoselective hydrogenation of triple bond using lithium aluminium hydride produced alkene 170 where installation of N-palmitoyl group produced benzyl protected β-D-galactosyl ceramide methylene isostere 171 in 80% yield. Switching of the protective group produced another β-D-galactosyl ceramide methylene isostere 172 in 55% yield. Isostere 172 with a 45% yield was also obtained from 169 via another sequence of reactions (Scheme 34).
 | ||
| Scheme 34 Synthesis of β-D-galactosyl ceramide methylene isostere 171 and 172. | ||
3.2 C-Glycopyranosyl di- and polysaccharides
Due to the presence of ubiquitous glycosidases and their ability to carry out hydrolysis, a short lifetime of the usual O-linked disaccharide conjugates was observed in the bloodstream whereas C-linked disaccharides are stable enough towards hydrolysis so that they could be utilised as a disaccharide-based vaccine. Due to the immense potential of C-linked disaccharides, Kobertz et al.65 described the synthesis of C-linked disaccharide using nitro aldol condensation between C-galactopyranosyl aldehyde 4c and nitro sugar 175 in the presence of KF in acetonitrile, which afforded compound 176 as a diastereomeric mixture in 52% yield (Scheme 35). Nitro sugar derivative 175 used herein was prepared from compound 1a by acid catalysed selective debenzylation followed by substitution with the azide group to obtain 174. Next, compound 174 was subjected to reduction followed by oxidation to afford compound 175 in 61% yield. Compound 176 was converted into completely protected compound 177 using n-butyl lithium followed by phenyl chlorothionocarbonate (PTC-Cl), which was used in the next step without isolation. Further, a radical-assisted elimination reaction using Bu3SnH-AIBN was carried out to afford alkene 178 in 10% yield calculated from compound 176. The reduction of olefin 178 was carried out by diimide (generated in situ from tosyl hydrazine and sodium acetate) to obtain disaccharides 179 in 98% yield. The debenzylation of 179 was carried out by dissolving metal reduction in liquid ammonia to obtain compound 180 in quantitative yield. | ||
| Scheme 35 Synthesis of disaccharide 180. | ||
Demange et al.66 designed a synthesis of C-(1→3)-linked disaccharides via the Oshima–Nozaki condensation of β-C-glucopyranosyl aldehyde with isolevoglucosenone and levoglucosenone (Scheme 36). In both condensations, an exclusively (1′R)-hydroxymethano linker was formed. β-C-Glucopyranosyl aldehyde 4a was condensed with isolevoglucosenone 181 using Et2AlI in DCM at −95 °C to generate enone 182 in 80% yield. Further, the addition of thiophenol was taken using base Et3N followed by direct reduction with Me4N[B(OAc)3]H in MeCN–AcOH to give 183 in 50% yield. Treatment with RANEY®-Nickel produced 184 in 69% yield, where debenzylation followed by acetylation was achieved to obtain compound 185 in 56% yield. Further, treatment with Ac2O–CF3COOH afforded an anomeric mixture 186 where ammonolysis was carried out to give pure C-disaccharide 187 in 76% yield (Scheme 36a).
 | ||
| Scheme 36 Synthesis of C-linked disaccharides 187, 192, 193, and 200. | ||
The addition of thiophenol to enone 182 was followed by a reduction with NaBH4 instead of Me4N[B(OAc)3]H to obtain 188 in 82% yield, which on reduction with RANEY®-Nickel afforded compound 189 in 85% yield. C-Disaccharides 192 and 193 were obtained by applying the same sequence of reactions as carried out for the synthesis of C-disaccharide 187. Exchange of the benzyl protective group with acetyl produced 190, which on acetolysis afforded anomer mixture 191 in 90% yield. Further, deacetylation was achieved by NH3–MeOH to produce the mixture of 192 and 193 in 73% yield (Scheme 36b).
C-Glucopyranosyl aldehyde 4a and levoglucosenone 194 were subjected to Oshima–Nozaki condensation, which produced enone 195 in just 30% yield. The addition of thiophenol followed by reduction with DIBAL-H afforded 196 in 74% yield, which was further treated with RANEY®-Nickel to obtain 197 in 72% yield. The debenzylation of compound 197 followed by acetylation was carried out to obtain 198 in 83% yield. Further, acetolysis was carried out using triethylsilyltrifluoromethanesulfonate in acetic anhydride to obtain an anomeric mixture 199 in 98% yield, which was deacetylated using NH3–MeOH to give C-disaccharide 200 in 71% yield (Scheme 36c).
Demange et al.62 reported the synthesis of C-linked disaccharides, which mimics O-β-D-galactopyranosyl-(1→3)-D-galactopyranosides starting from C-galactopyranosyl aldehyde. Three C-linked disaccharides 132a, 132b, and 132c were synthesized, out of which 132a was found to be a suitable agent for O-glycosidation and for the construction of conjugates (Scheme 37). C-galatopyranosyl aldehyde 4c and 201 reacted with 181 to prepare 202a and 202b in 73% and 95% yields, respectively. Michael addition of thiophenol to 202a afforded 203, which was directly converted into 204 with 81% yield by reduction using sodium borohydride in methanol. Treatment with RANEY®-nickel converted compound 204 into compound 205 in 87% yield, which on debenzylation following the acetylation furnished peracetylated 206 in 88% yield. Treatment of 206 with Ac2O-TFA gave 207a–b as a mixture of anomers (α:β, 1:0.3) in 81% yield, which on refluxing in toluene afforded 132a in 82% yield. Further replacement of acetyl protection with the benzyl group produced 132b in 76% yield (Scheme 37a).
 | ||
| Scheme 37 (a) Synthesis of disaccharides 132a–b. (b) Synthesis of β-D-galactopyranosyl-D-galactal 132c. | ||
C-Galactoside 202b on reduction with RANEY®-nickel produced a mixture of aldols 208–211 in quantitative yield. From this mixture, compounds 208 and 209 were isolated by flash chromatography in 57% and 43% yield, respectively. This mixture of aldols 208 and 209 when treated with CH3SO2Cl/pyridine and DMAP formed unstable enone 212, which when reduced with Ra–Ni in aq. THF produced a mixture of ketones 210 and 211 in 43% and 44% yield, respectively. The ketone 210 thus produced when reduced with lithium borohydride in THF at low temperature formed alcohol 213 in 79% yield, which on treatment with triethylsilyltrifluoromethanesulfonate(Et3SiOTf) and acetic anhydride underwent ring opening of 1,6-anhydro moiety followed by acylation to give a mixture of α- and β-pyranosides 214a–b. Anomeric mixture 214a–b was directly treated with silica gel in benzene and heated under refluxing conditions, which resulted in a C-linked analogue of β-D-galactopyranosyl-D-galactal 132c in 60% yield (Scheme 37b).
In 2001, Zhu et al.67 and in 2005, Awad et al.68 reported the synthesis of a C-linked disaccharide analogue of epitope T. First, Oshima–Nozaki condensation69 was carried out between C-glycopyranosyl aldehyde 201 and commercially available isolevoglucosenone 181 using Et2AlI in DCM to afford 202b in 96% yield. Further conjugate addition of MeONHBn to enone 202b produced a stereoisomeric mixture of 215 and 216, which were isomerised during column chromatography, and adduct 215 was recovered in 82% yield. Reduction of 215 was carried out with lithium borohydride in THF at −78 °C to afford compound 217, which was directly treated with TBAF in THF following acetylation to furnish 218 quantitatively. Treatment of peracetylated compound 218 with TMSSPh/ZnI2-DCM followed by TBAF and acetylation furnished 219 in 93% yield, which was reacted with protected serine to afford β-C-glycopyranosyl disaccharide 220 in 92% yield (Scheme 38a).
 | ||
| Scheme 38 (a) Synthesis of β-C-galactopyranosyl disaccharide 220. (b) Synthesis of β-C-glycopyranosyl disaccharide 225. | ||
The bulky 2-MeONBn group present on compound 219 enforced the reaction to undergo β-glycosylation, which afforded compound 220 (Scheme 38a). To achieve α-glycosylation, 2-MeONBn group was removed initially from compound 217 using Na/NH3 reduction conditions followed by hydrogenation with H2–Pd/C to afford compound 221 (Scheme 38b). Further, compound 221 was treated without isolation with TfN3-CuSO4 following treatment with TBAF in THF and acetic anhydride in pyridine to furnish 222 in 81% yield. The peracetylated compound 222 on treatment with Ac2O-TMSOTf afforded 223, which reacted with protected serine to afford α-glycosylated product 224 in 92% yield. The treatment of compound 224 with CH3COSH-Ac2O followed by further deacetylation afforded targeted compound 225 in 55% yield.
In 2012, Awad et al.61 reported another interesting application of C-glycopyranosyl aldehyde by synthesizing C-linked disaccharide, which was found to induce a strong immune response in mice. The TBS-protected C-galactopyranosyl aldehyde 201 and isolevoglucosenone 181 were condensed followed by a reaction with MeONHBn to afford 216 in 60% yield. Compound 216 on treatment with TFA in pyridine following DBU furnished dehydration, further reduction of the double bond with RANEY®-nickel, and then by LiBH4 converted the keto group to afford compound 226 in 61% yield. Compound 227 was achieved in 72% yield by reduction of the 2-MeONBn group, its conversion into azide by diazo transfer, and desilylation following acetylation of 226. α-O-Galactosidation was achieved with protected serine to afford compound 228, which on reaction with Zn–AcOH followed by treatment with TFA in DCM afforded compound 229 in 88% yield, which was coupled with trityl polymer to give 230. Compound 231 was obtained first by removal of Fmoc protection from 230 and then by coupling it with the –COOH group of 229.
The deprotection of Fmoc from compound 231 followed by capping with 4-(dimethylamino)-azobenzene-4′-carboxylic acid (Azo-OH) gave compound 232. Coupling with pentaflurophenylacetylthioacetate after the liberation of protected triglycopeptides from the polymer furnished 233, which was treated with sodium methoxide to give peptide 234. Finally, the reaction of peptide 234 with MBS-KLH under the Michael addition reaction condition furnished antigen 131a (Scheme 39a).
 | ||
| Scheme 39 (a) Synthesis of antigen 131a. (b) Synthesis of antigen 131b. | ||
A similar synthetic protocol was developed for TBS protected α-C-galactppyranosyl aldehyde 235 (Scheme 39b). In this strategy, trityl resin-monoprotected diamine 1,4-di(aminomethyl)benzene was coupled with compound 242. So, following the same sequence of reactions, antigen 131b was prepared.
Zeitouni et al.70 introduced a new class of C-linked disaccharides 253a–c, which were obtained by the Mukaiyama aldol71 reaction between β-C-glucopyranosyl aldehyde 4a and trimethylsilyl enol ether 250. This enol ether was synthesised from readily available methyl-2,6-di-O-benzyl-β-D-galactopyranoside 246 (Scheme 40a). The diol 246 was reacted with SOCl2–Et3N followed by reaction with RuCl3·H2O–NaIO4 to afford 3,4-cyclic sulfate 247 in 84% yield, which on treatment with 10 M NaOH gave unstable compound 248 and subsequent acidic hydrolysis furnished ketone 249 in 67% yield. Final enol ether 250 was obtained quantitatively by treatment with LDA-TMSCl in THF.
 | ||
| Scheme 40 Synthesis of C-linked disaccharide 253a–c. | ||
Condensation of 4a and 250 in the presence of Yb(OTf)3 in THF–H2O furnished diastereomeric mixture 251a and 251b in a 59:41 ratio out of four possible diastereomers in 95% yield. The diastereomers could not be separated by column chromatography so they were further reduced with sodium borohydride to furnish three diol products 252a, 252b, and 252c in 94% yield (Scheme 40b). Here, only 252c could be separated by column chromatography which was reacted with benzaldehyde dimethyl acetal in acetonitrile in the presence of PTSA to give 253c in 23% yield (Scheme 40c). The same reaction was also carried out on the 253a/253b mixture to give 253a and 253b in 71% yield, they could be separated easily (Scheme 40d).
Gerber et al.33 designed a stereoselective synthesis of C-linked disaccharides and oligosaccharides due to their advantage of being resistant to acidic and enzymatic hydrolysis. The enolate 254, which in turn was synthesised from enone 32 using Me2AlSPh was reacted with C-glycopyranosyl aldehyde 37 in tetrahydrofuran to furnish aldol 255, which turned out to be unstable and hence was reduced with NaBH4 in methanol to afford diol 256 with 56% yield. Diol 256 on acetylation with Ac2O in pyridine, followed by desilylation with HF in H2O and MeCN, and then undergoing Dess–Martin periodinane oxidation furnished a ketone, which upon Baeyer-Villiger oxidation afforded compound 257. Compound 257 on treatment with EtSH/CF3SO3H/CH2Cl2 and with CH2N2 in DCM afforded β-D-C-manno-pyranoside 258 in 61% yield (Scheme 41).
 | ||
| Scheme 41 Synthesis of β-D-C-manno-pyranoside 258. | ||
Levoirier et al.72 developed a strategy for the synthesis of β-(1→4)-C-disaccharides using Barbier-type allylation catalysed by indium in aqueous media (Scheme 42). C-Glucopyranosyl aldehyde was reacted with 6-bromo-4,6-dideoxy-α-D-threo-4-enopyranoside 259 using indium in THF–phosphate buffer to obtain an inseparable mixture of C-disaccharide 260 and 261 in 45% yield, which was acetylated using acetic anhydride in pyridine affording 262 and 263, respectively which could be separated easily. Hydroboration with diborane-tetrahydrofuran followed by oxidation with hydrogen peroxide in phosphate buffer and then acetylation was carried out on 262 and 263 to obtain protected C-disaccharides 264 and 265 in 91% and 83% yields, respectively (Scheme 42a).
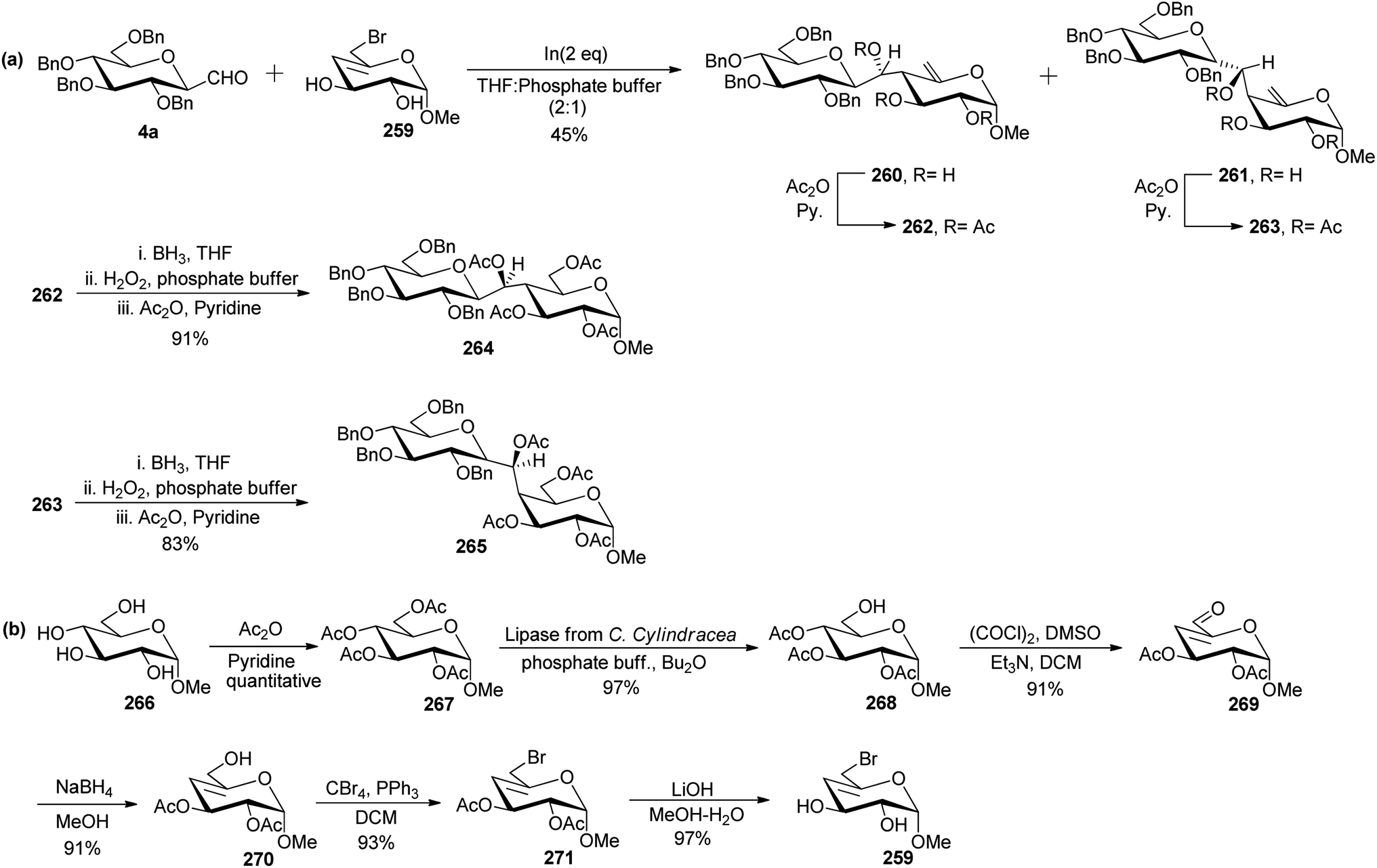 | ||
| Scheme 42 Synthesis of β-(1→4)-C-disaccharides 264 and 265. | ||
6-Bromo-4,6-dideoxy-α-D-threo-4-enopyranoside 259 used in the above Barbier-type allylation was synthesised from methyl α-D-glucopyranoside 266, which on acetylation produced peracetylated derivative 267 quantitatively. Selective deacetylation of peracetylated derivative was performed after treatment with lipase from C. Cylindracea in a mixture of phosphate buffer and di-n-butyl ether produced methyl 2,3,4-tri-O-acetyl-α-D-glucopyranoside 268 in 97% yield. Compound 268 underwent Swern oxidation to afford unsaturated aldehyde 269 in 91% yield, which on further reduction with NaBH4 in MeOH produced allyl alcohol 270 in 91% yield. Treatment of allyl alcohol 270 with carbon tetrabromide and triphenylphosphine in DCM followed by deacetylation using LiOH in MeOH–H2O afforded 259 in 97% yield (Scheme 42b).
Canac et al.73 designed a protocol for the synthesis of C-disaccharides under indium-promoted Barbier-type allylation (Scheme 43). Here, C-glucopyranosyl aldehyde 4a was reacted with 4-bromo-2-enopyranoside 272 using indium in H2O–THF. A mixture of diastereomers 273a–b with a ratio (1:1) in 55% yield was obtained.
 | ||
| Scheme 43 Synthesis of C-disaccharide 273a–b. | ||
The key compound 272 used in the above Barbier-type allylation was synthesised from 2-enopyranoside 274 using a sequence of deprotection of the acetyl group, selective protection of the primary hydroxyl group, bromination followed by deprotection reaction (Scheme 43b).
Dondoni et al.74 achieved the synthesis of (1→6)-C-disaccharides by Wittig olefination reaction between C-glycopyranosyl aldehyde and pyranose 6-phosphoranes (Scheme 44). First, C-glycosyl aldehydes 4a–d were reacted with galactose 6-phosphorane 275 using n-butyl lithium, THF–HMPA at −30 °C to afford alkenes 276a–d in 42–80% yield. In the case of mannopyranosyl aldehyde 4b low yield (42%) was observed due to the formation of unexpected side products (formed by the elimination of BnOH from C1 and C2 of sugar). Further, treatment of alkenes 276a–d with H2, Pd(OH)2 led to the debenzylation and reduction of the double bond, which on deprotection with amberlite IR-120 in water furnished β-D-(1→6)-C-disaccharides 277a–d in 40–85% yield.
 | ||
| Scheme 44 Synthesis of (1→6)-C-disaccharides 277 and 280. | ||
Similarly, C-glycopyranosyl aldehydes 4a–d were reacted with glucose 6-phosphorane 278 under Wittig conditions to furnish alkenes 279a–d with 53–77% yield, which on treatment with H2, Pd(OH) 2 followed by Amberlite IR-120 resin produced disaccharides 280a–d. Further, disaccharides 280a–d were acetylated using acetic anhydride in pyridine to afford 281a–d (Scheme 44b).
Dondoni et al.75–77 worked on designing synthetic routes for oligosaccharides and glycoconjugates both being mediators in inflammation, fertilization, cancer metastasis, and viral and bacterial infections in human beings. For the synthesis of these oligosaccharides, two complementary routes were designed taking C-galactopyranosyl aldehyde 282 in the first route while in the second, α-linked galactosylmethylenephosphorane 10 was taken as the starting material. The formyl group of C-galactospyranosyl aldehyde 282 on reaction with NaBH4 reduces to alcohol followed by iodination with I2 and PPh3 finally phosphination in the presence of PPh3 yielding galactosylmethylenephosphonium iodide 283 in 65% yield, which reacted with galactose derived aldehyde 284 in the presence of BuLi. The reaction afforded olefin 285 in a good yield, which further undergoes removal of the silyl protecting group followed by oxidation to primary alcohol to yield aldehyde 286 in 70% yield. The same sequence of addition of galactosylmethylenephosphonium iodide 283 followed by oxidation of alcohol to aldehyde with Bu4NF and PCC resulted in the formation of the desired oligosaccharide chain 290 in very low overall yield (Scheme 45a). In the other route, galactosylmethylenephosphorane 292 was reacted with C-glycopyranoside 282 in the presence of BuLi in THF-HMPA, affording biglycosylated olefin 293 in excellent yield. Further, olefin 293 upon subsequent installation of the phosphonium group via desilylation (Bu4NF), followed by iodination (I2, PPh3), and phosphanation (PPh3) yielded sugar phosphonium iodide 294 in 88% yield, which on further reaction with C-glactopyranosyl aldehyde 282 followed by the same sequence of steps yielded bis-olefin 295 in 87% yield and same steps were followed to obtain oligosaccharide 299, which on debenzylation and hydrogenation produced oligosaccharide 300 with good overall yield (Scheme 45b).
 | ||
| Scheme 45 (a) Synthesis of oligosaccharide 290. (b) Synthesis of oligosaccharide 300. | ||
3.3 C-Glycopyranosyl amino acid precursors
Sipos et al.78 designed the synthesis of C-glycosyl-aminoacetonitriles 301a–b, 302a–b, and 303 under Strecker reaction conditions from various C-glycopyranosyl aldehydes. C-glycosyl aminoacetonitriles 301a–b were formed in 67–70% yield by Strecker reaction upon 43a–b with DCM for 43a and THF for 43b. The diastereomeric ratios for 301a–b were 8.25 and 5.17, respectively. Similarly, 302a–b were formed in 48–78% yield by Strecker reaction upon 40a–b by reacting them with chiral amines such as S- or R-PEA or achiral amines such as benzylamine (BA), benzhydrylamine (BHA) in the presence of cyanide donor HCN. The diastereomeric ratio and yield obtained for C-glycosyl-aminoacetonitriles are shown in Scheme 46.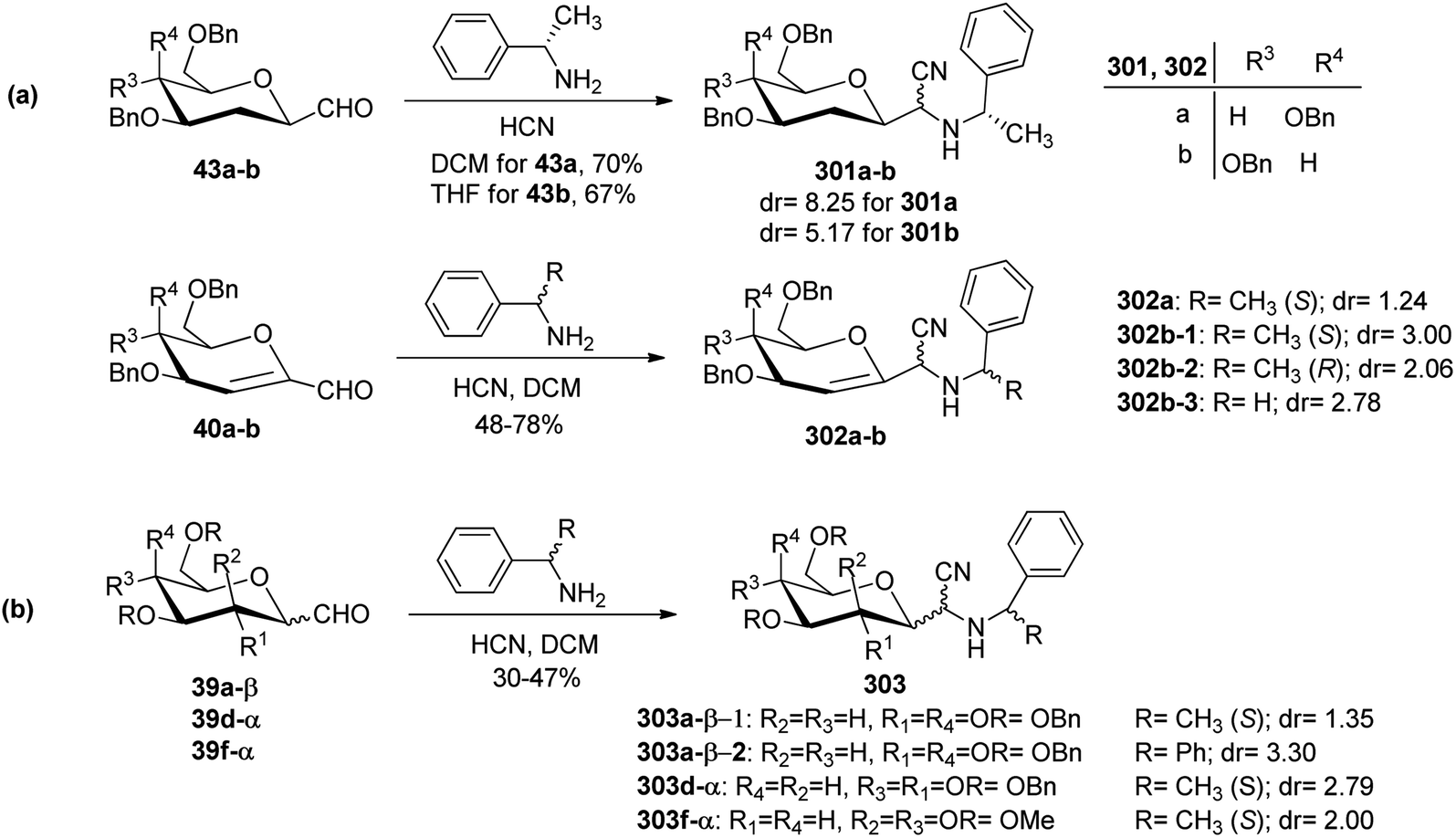 | ||
| Scheme 46 Synthesis of 2-amino-2-C-D-glycosyl-acetonitriles 301a–b, 302a–b, 303. | ||
In addition, glycosyl-formaldehydes 39a, 39d, and 39f also underwent Strecker reaction leading to the formation of C-glycosyl aminoacetonitriles 303a, 303d, and 303f (Scheme 46b). The hydrolysis of these C-glycosyl-α-aminoacetonitrile led to the formation of C-glycosyl glycine, which can be used for the development of biologically-active glycol-peptidomimetics.
Charfedinne et al.79 described the synthesis of some alkynes and iodoalkynes bearing protected amino alcohol moieties as amino acid precursors, which were used for the alkynylation reaction of sugar derivatives. The alkynyl iodide 306 was obtained in a multi-step reaction process (Scheme 47). Garner's aldehyde 304 was synthesised from L-serine in a five-step procedure.80 This aldehyde was then homologated into terminal alkyne 305 with 67% yield using Ohira–Bestman reagents (Seyferth–Colvin–Gilbert reaction). The alkyne derivative 305 when treated with iodine and morpholine in benzene at 45 °C resulted in iodinated tert-butyl-(R)-4-iodoethynyl-2,2-dimethylexazelidine-3-carboxylate (306) in 90% yield.
 | ||
| Scheme 47 Synthesis of C-glycopyranosyl propargylic alcohol 307. | ||
The reaction of alkynyl-iodide 306 with 2,3,4,5-tetra-O-benzyl-1-formylglucopyranose (4a) in the presence of metallic indium in dichloromethane resulted in a coupling reaction producing the corresponding propargylic alcohol 307 in 66% yield with a diastereomeric ratio of 2/1 (Scheme 47).
3.4 C-Glycopyranosylamino acids and dipeptides
Dondoni et al.81,82 developed complementary one-pot Mannich- and Reformatsky-type multicomponent approaches to synthesise a library of C-glycosyl β-amino acids (Scheme 48). Both methods involved C-glycopyranosyl aldehydes 4a–c as starting compounds. Mannich route coupled C-glycopyranosyl aldehyde 4a–c, p-methoxy benzylamine 308, and commercially available ketene silyl acetal 309 in the presence of indium chloride in methanol to afford C-glycosyl β-amino ester 310a–c as single diastereomer observed by 1H NMR analysis with 80% yields. In the case of 4b (manno sugar), DCM was chosen as the solvent because of the insolubility of the intermediate in methanol and also reaction was performed at 0 °C to avoid the formation of glycal amino ester (resulting from the elimination of BnOH from C-1 and C-2 of manno sugar). Further, for the reagent diversity ketene silyl acetal 312 was also chosen (Scheme 48b). Unfortunately, this process suffered from low yields of C-glycosyl β-amino esters 313a and 313c in 58–60% yield as well as undesired product formation 313b and 314b in case of 4b (manno sugar).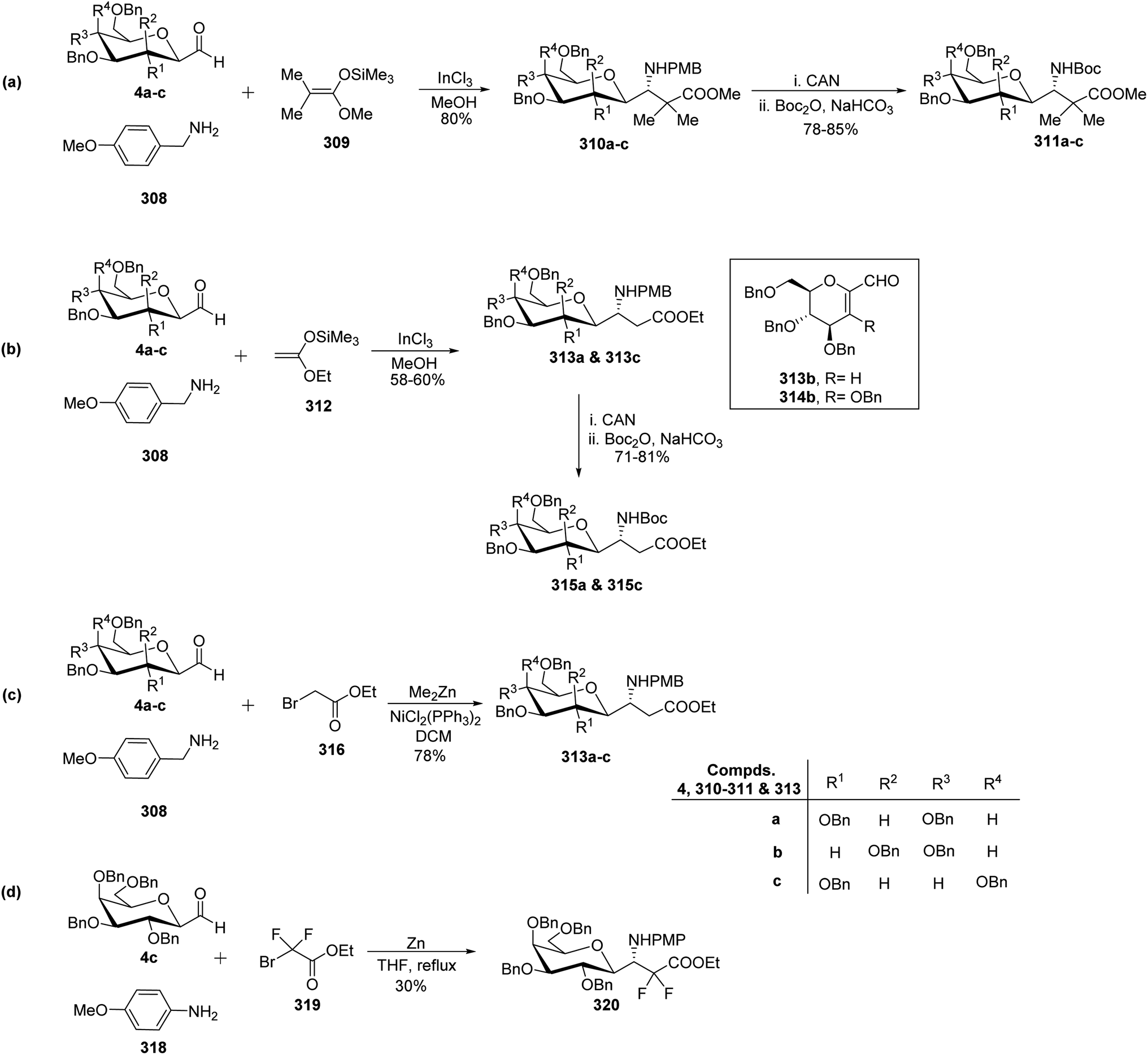 | ||
| Scheme 48 Synthesis of C-glycopyranosyl β-amino acids 311, 313, 315, 320. | ||
In view of Boc-based peptide synthesis, all amino esters, 310a–c, 313a, and 313c, were transformed into their corresponding N-Boc derivatives 311a–c, 315a, and 315c by treatment of ceric ammonium nitrate (CAN) followed by Boc2O and NaHCO3 in 78–85% and 71–81% yields, respectively (Scheme 48a–b).
Due to some limitations in the above Mannich-type approach another route for the synthesis of C-glycosyl β-amino esters was performed. Here, ethyl bromoacetate 316 was taken in place of ketene silyl acetate (Scheme 48c). C-glycopyranosyl aldehyde 4a–c, p-methoxy benzylamine 308, and ethyl bromoacetate 316 were reacted in the presence of zinc donor Me2Zn, NiCl2(PPh3)2 as a catalyst in DCM to produce C-glycosyl β-amino esters 313a–c in 78% yield. Thus, the Reformatsky route demonstrated over the Mannich route in terms of overall yield and desired products.
Due to the application of fluorinated amino acids in biological and medicinal studies, synthesis of α,α-difluoro C-glycosyl β-amino acid 320 was also achieved by the Refromatsky-type reaction in 30% yield (Scheme 48d).
Dondoni et al.83,84 employed threonine-derived silyl enol ethers 321 to serve as a synthetic equivalent of the homoalanine carbanion to introduce α-amino side chains at the anomeric carbon position of sugar moieties. This functionalized silyl enol ether 321 was prepared from methyl N-Boc-L-threoninate in six steps with a 48% yield. Tetra-O-benzylated formyl C-glycopyranoside 4a–c upon condensation with 321 in the presence of Lewis acid boron trifluoride etherate underwent Mukaiyama coupling in CH2Cl2 at −30 °C, which afforded aldols 322a–c in 70–78% yield. The α,β-enones 323a–c were obtained with 80–81% yield from dehydration of 322a–c using DCC-Cu(OTf)2 for α,β-elimination reaction. The carbonyl and carbon–carbon double bonds were then reduced by following a sequence of reactions on 323a–c to yield 324a–c with 49–50% yield in four steps. Next, when compounds 324a–c were subjected to Jones' reagent, they underwent oxidative cleavage of oxazolidine ring, which afforded C-glycosyl amino acids β-D-galactose (CH2)2-asparagine (β-Gal-(CH2)2-Asn, 325c), and the corresponding gluco- and manno-isomers 325a and 325b, respectively in 95% yield (Scheme 49).
 | ||
| Scheme 49 Synthesis of C-glycosyl amino acids 325a–c. | ||
Kroger et al.25 synthesised O-glycosyl amino acid mimetics 3′-O-(2,6-anhydro-D-glycero-L-gluco-heptitol-1-yl)-L-serine (328a) and -L-threonine (328b), which acted as building blocks for O-glycopeptides. The synthesis proceeded via α-C-galactopyranosyl aldehyde 7, which in turn was prepared from 1,2,3,4,6-penta-O-acetyl-β-D-galactopyranose 5 via an allene approach. Aldehyde 7 on reduction with sodium borohydride gave C-glycosidic methylene galactitol 326 in 62% yield. Compound 326 in the presence of Lewis acid BF3·Et2O in toluene, reacted with the activated aziridine ring containing compounds 327a–b to form compounds 328a–b in 52–55% yield. The C-glycosyl amino acids 133a–b in 53–87% yields were obtained by hydrogenolytic deprotection followed by Zemplen deacetylation of compounds 328a–b (Scheme 50). C-Glycosyl amino acids 133a–b were found to be a competitive inhibitor of α-glycosidase from Aspergillus niger.
 | ||
| Scheme 50 Synthesis of C-glycosyl amino acids 133a–b. | ||
Risseeuw et al.85 worked to design two synthetic pathways to obtain δ-substituted pyranoid sugar amino acids (SAAs). This synthesis was initiated from easily and readily available C-glucopyranosyl aldehyde 4a. Condensation of formyl tetra-O-benzyl-β-D-C-glucopyranoside 4a with R-tert-butanesulfinyl amide yielded R-tert-butanesulfamide 329 in 70% yield, which on alkylation in the presence of MeMgBr in DCM afforded R-methyl adduct 330 in excess over the corresponding Z-diastereoisomer 331. Adduct 330 on further acid-mediated hydrolysis and installation of Fmoc protecting group resulted in 332 in 70% yield, which on selective acidolysis of primary benzyl ether, ester hydrolysis, and oxidation afforded the carboxylate 333 in 64% yield. The same stereoisomer 332 was formed in 75% yield when the whole reaction sequence was repeated with S-tert-butanesulfinamide, and the final product obtained was carboxylate 333 (Scheme 51a).
 | ||
| Scheme 51 (a) Synthesis of δ-substituted pyranoid sugar amino acid 333. (b) Synthesis of L,L-dipeptides isosters 340a–d. | ||
Treatment of C-glucopyranosyl aldehyde 4a with different Grignard reagents afforded single isolated diastereomers 337a–d in 36–68% yield. Further, Mitsunobu displacement using HN3, DEAD, and PPh3 in toluene afforded azide 338a–d in 48–92% yield, with inverted stereochemistry. The reduction of azide 338b using Me3P, THF–H2O followed by Fmoc protection produced 341b in 73% yield. Selective acidolysis of primary benzyl protection of compounds 338a–d with ZnCl2–AcOH yielded compounds 339a–d in 68–81% yield, which were further oxidized with TEMPO to afford L,L dipeptide isosters 340a–d in 76–92% yield (Scheme 51b).
Three-component reaction on C-glycopyranosyl aldehyde was achieved by Dondoni et al.86 to synthesize C-glycopyranosyl-β-lactams and further elaborated synthesis of C-glycosyl isoserines and dipeptide precursors. C-Galactopyranosyl aldehyde 4c was reacted with easily available primary amines and substituted with acetyl chlorides to give a small library of C-galactopyranosyl-β-lactams 342a–g with 65–94% yield (Scheme 52a).
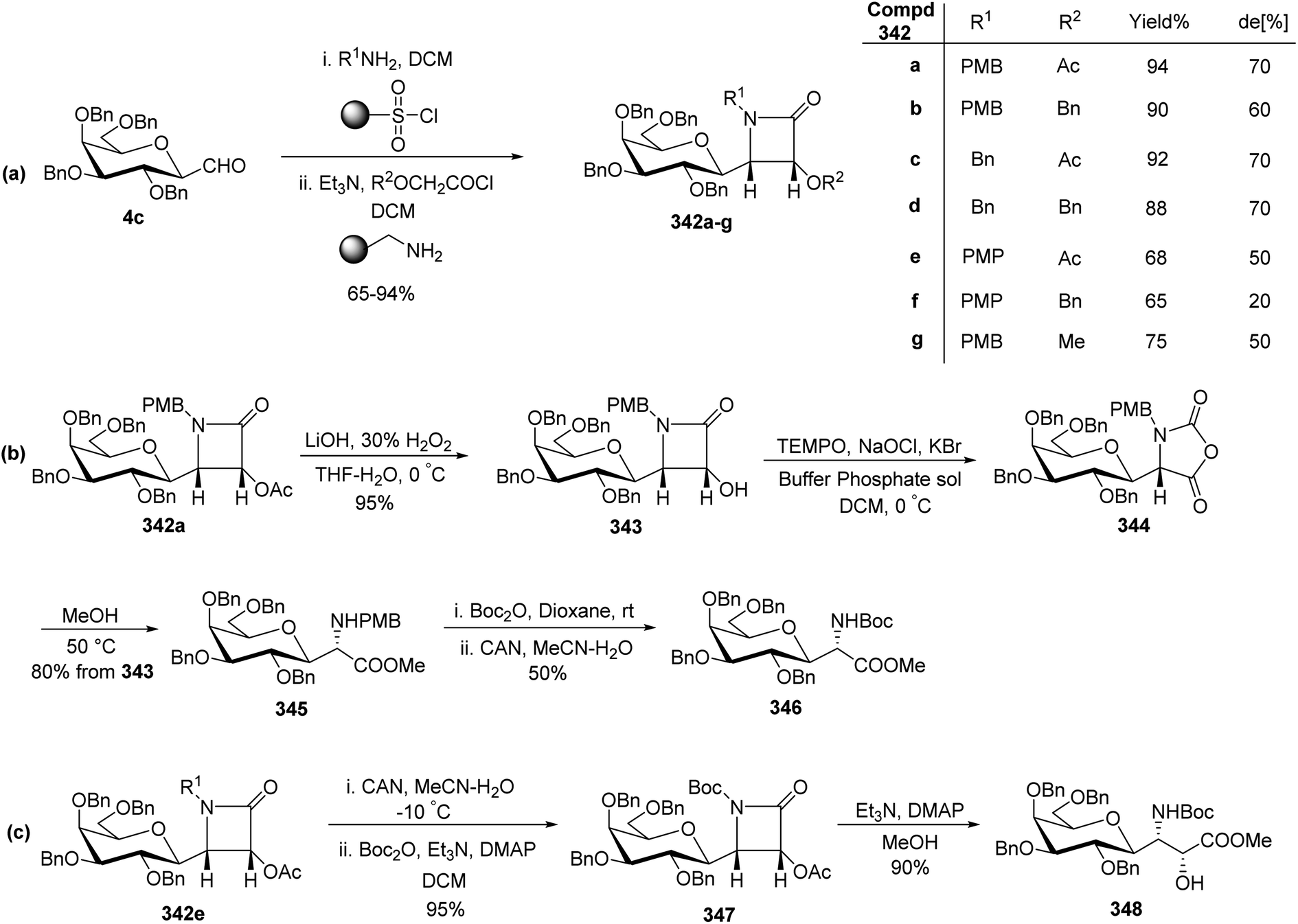 | ||
| Scheme 52 Synthesis of C-galactopyranosyl-β-lactams 342a–g, α-amino ester 346, C-galactosyl isoserine methyl ester 348. | ||
First, C-glycosylimine was generated by the reaction between C-galactopyranosyl aldehyde and an excess amount of primary amine derivatives in DCM, and thereafter resin supported sulfonyl chloride removed the excess of unreacted alkylamine from the solution. The resulting mixture was treated with substituted acetyl chloride in the presence of triethylamine to afford C-galactopyranosyl-β-lactam. The amino-methylated resin was used to remove excess ketene and unreacted acetyl chloride from the reaction mixture. These β-lactams could be utilised to synthesise other classes of biologically active compounds, i.e. β-amino acids.
Deacetylation of C-glycosyl-β-lactam 342a was achieved using LiOH-30% H2O2 to afford 343 in 95% yield, which on oxidation with TEMPO-NaOCl furnished N-carboxy anhydride 344 (Scheme 52). Ring-opening of 344 using methanol followed by protection of –NH with Boc group and deprotection of PMB functional group with CAN produced compound 346 in 50% yield (Scheme 52b).
Synthesis of C-galactopyranosyliso-serine ester 348 was achieved with 90% yield by conversion of C-glycosyl-β-lactam 342e into 347 with 95% yield via switching of N-protective group using Et3N–DMAP in MeOH (Scheme 52c).
In 1998, Dondoni et al.64 reported the synthesis of C-galactopyranosyl-α-amino acids i.e. C-analogue of β-D-galactopyranosyl-L-serine. First of all, β-C-galactopyranosyl aldehyde 4c was reduced using NaBH4 in MeOH followed by iodination with iodine–PPh3–imidazole furnished compound 349 in 71% yield, which was efficiently converted into sugar-based Wittig reagent 350 in 92% yield by reaction with PPh3. Subsequently, Wittig coupling between 350 and 351 in the presence of butyl lithium–THF–HMPA afforded 352 with as a mixture of stereoisomers (Z-54%, E-8%). Reduction with diimide (generated in situ) gave 164 in 84% yield, which was treated with Jones' reagent and then methylation with diazomethane afforded C-linked analogue of β-D-galactopyranosyl-L-serine 353 in 94% yield (Scheme 53).
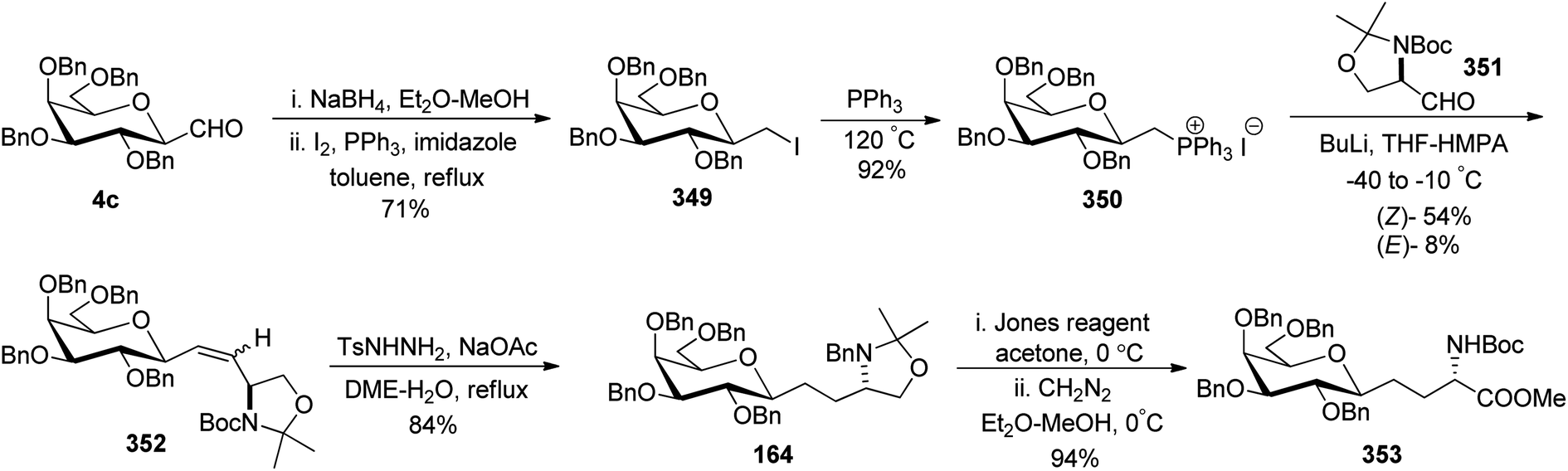 | ||
| Scheme 53 Synthesis of β-D-galactopyranosyl-L-serine 353. | ||
Boutard et al.87 synthesized C-glucopyranosyl-α-amino acids 355 in 38% yield with 50% diastereomeric excess where the reaction between C-glucopyranosyl aldehyde 4a and organozinc reagent of amino acid 354 was carried out in presence of Lewis acid BF3·Et2O in acetonitrile (Scheme 54a). Compound 354 was synthesized from α-bromo ester in three steps as shown in (Scheme 54b).
 | ||
| Scheme 54 Synthesis of C-glucopyranosyl-α-amino acid 355. | ||
Guerrini et al.88 designed one-pot synthesis of constrained N,O-orthogonally protected C-glycosyl norstatines (α-hydroxy-β-amino acids) 365e–h, which was formed by the reaction of enolates of dioxolanones 361a–b with (S)-N-sulfinyl azomethines 362c–d following Seebach's SRS (Self-Regenerating Stereocenters) principle (Scheme 55). In the compounds 363e–h, the N and O were orthogonally protected with the sulfinyl group at the nitrogen atom and the acetal moiety of the dioxolanone ring. The sulfinyl group present in the compounds 363e–h was removed by using 2 N HCl acidic condition in a solvent mixture of methanol and diethyl ether (1:1) leading to the formation of N-deprotected 1-C-glycosyl amino-dioxolan-4-ones 364e–h in 94–95% yield. The acetal groups of 363e–h were removed by methoxide-induced methanolysis forming, corresponding methyl C-glycosyl-sulfinylamino-isoserinates 365e–h with 86–96% yield. Compounds 364e and 364g–h were converted into corresponding β-lactams 366e and 366g–h, respectively, with 35–83% yield using LHMDS in THF (Scheme 55).
 | ||
| Scheme 55 Synthesis of C-glycosyl norstatines (α-hydroxy-β-amino acids) 365e–h. | ||
Kumar et al.89 recently designed a protocol based on the Passerini90 multi-component reaction to synthesize a library of β-C-glycopyranosyl-α-acyloxy amides (Scheme 56). The component C-glycopyranosyl aldehyde 4a–b, aryl/alkyl acid derivatives 367a–p, and isocyanides 368a–c were reacted by using DCM as the solvent at room temperature to afford glucopyranosyl α-acyloxy amides 369a–t in good to excellent yield (60–72%). These glucopyranosyl α-acyloxy amides are commonly known as depsipeptides and are biologically significant. Further, 369a was debenzylated using H2, Pd/C in methanol to obtain 370a in 90% yield.
 | ||
| Scheme 56 Synthesis of depsipeptides 369a–t and 370a. | ||
Raunkjaer et al.91 synthesised highly functional, novel dipeptide isosters via diastereoselective alkylation/arylation of sugar amino acids. This novel sugar amino acid (SAA) when applied in peptide synthesis protocol afforded a cyclic tetramer with excellent overall yield. The synthesis started with perbenzylated β-C-glucopyranosyl aldehyde 4a, which on condensation with (R)-tert-butylsulfinamide in the presence of anhydrous CuSO4 in DCM-derived sulfinimine 329 in 70% yield, which was treated with PhMgBr in toluene leading to the formation of sulfinamide 371 in >95% diastereomeric excess. Compound 371 was desulfinylated by HCl in methanol giving HCl salt of 372, which was Boc protected using Boc2O, DIPEA in DCM to produce corresponding tert-butoxycarbonylate, which underwent hydrogenolysis of benzyl ether group in the presence of Pd/C followed by removal of protecting group leading to the synthesis of compound 375 in 62% yield (Scheme 57a). The HCl salt of compound 372 was Fmoc protected to derive 373 with 63% yield in the presence of Fmoc-OSu, DIPEA, and DCM in dioxane. Compound 373 underwent selective debenzylation at the primary hydroxyl group on treatment with anhydrous ZnCl2 in AcOH and Ac2O, followed by acid-catalyzed deacetylation (HCl in MeOH) and oxidation using TEMPO and BAIB, leading to sugar amino acid 374 in 80% yield (Scheme 57b).
 | ||
| Scheme 57 Synthesis of benzylated cyclic tetramer 381. | ||
The sulfinimine derivative 329 was employed to synthesize a series of substituted SAAs 377a–c. Compound 329 was treated with RMgBr in toluene and HCl in methanol followed by the removal of the sulfoxyl auxiliary and Fmoc protection, which led to the production of compounds 376a–c in 50–71% yield. The primary benzyl-protected hydroxyl functionalities were converted into corresponding carboxylic acid groups following the reaction sequence leading to the formation of compounds 377a–c in 64–74% yield (Scheme 57c). The alkylated SAAs were synthesized for their incorporation into oligomeric sequences. SAA 377a was condensed with glycine-functionalized Wang resin 378 (Scheme 57d), thus producing immobilized product 379, which upon removal of the Fmoc protecting group, and repetition of coupling steps followed by TFA-mediated cleavage from the solid support transformed into the linear tetramer 380. This linear tetramer 380 was cyclized under high dilution to afford benzylated cyclic tetramer 381 in 74% yield.
Singh et al.92 reported the synthesis of sugar–amino acid hybrid macrocycle starting from 2,6-anhydro heptitol 383a, which was synthesized from C-glycopyranosyl aldehyde 4a. Synthesis of 2,6-anhydro heptitol 383a–c was achieved by Khatri et al.52 from the reduction of aldehyde 4a–c using NaBH4 in methanol afforded 382a–c in 90–95% yield, followed by treatment with trifluoro acetic acid, acetic anhydride, and sodium methoxide in methanol (Scheme 58a). 2,6-Anhydroheptitol 383a was condensed with N-boc-glycinate using EDC·HCl, DMAP in dichloromethane produced 2,6-anhydro heptitolyl bis-glycinate 384 in 99% yield. Treatment with trifluoro acetic acid afforded deprotection of the Boc-group to produce 385 in 99% yield, which was reacted with succinic/pyridine dicarboxylic acid to obtain sugar–amino acid hybrid macrocycle 386 and 388 in 40% yield (Scheme 58b). The deprotection of the benzyl group using Pd/C in methanol was achieved on macrocycle 386 to produce macrocycle 387 in 95% yield. Further host–guest anion interaction studies were carried out on macrocycles 386 and 388 with TBA salt of Boc-glycinate. Based on 1H NMR titration experiments, it was found that the carboxylate anion of Boc-glycinate salt exhibits interaction with the host macrocycle through hydrogen bonding. The macrocyclic compounds 386 and 388 have binding constants (Ka) of 9.201 × 103 M−1 and 1.437 × 104 M−1, respectively.
 | ||
| Scheme 58 Synthesis of 2,6-anhydro heptitol 383a–c and sugar-amino acid hybrid macrocycles 386–388. | ||
Leclere et al.46 reported a highly selective synthesis of C-AFGP analogue 125 (Scheme 59). Julia–Kocienski–Lythgoe (JKL) olefination reaction was carried out between C-glycopyranosyl aldehyde 90 and sulfone 389 to obtain glycopyranosyl alkene 390 in 65% yield. The sulfone 389 used here could be synthesized from orthogonally protected L-serine methyl ester 394 (Scheme 59b). Thus, the Mitsunbou reaction of 389 with 5-phenyl tetrazolyl thiol furnished 395 in 82% yield. Subsequently, reduction with lithium borohydride followed by N,O-isopropylidene formation and oxidation using sharpless conditions furnished 396 in 65% yield.
 | ||
| Scheme 59 Synthesis of C-AFGP analogue 125. | ||
The glycopyranosyl alkene 390 was reduced with concomitant deprotection of the Cbz group afforded amino alcohol 391 in 95% yield using H2, Pd(OH)2 in ethanol. Protection with Fmoc carbamate furnished 392 in 75% yield, which was oxidised to carboxylic acid 393 with 80% yield using TEMPO, PhI(OAc)2, and NaClO2. Finally, synthesis of glycoconjugate 125 was achieved using standard Fmoc-based solid phase synthesis with Wang resin in 20% yield. Glycoconjugate 125 is a potent inhibitor of ice recrystallization and could protect embryonic liver cells from cryo-injury at millimolar concentration. Thus C-AFGP analogue 125 is an effective cryoprotectant for human embryonic liver cells and inhibition of ice recrystallization in the course of cryopreservation is an important function for a cryoprotectant.
3.5 C-Glycopyranosyl phenyl methane and its derivatives
Kolympadi et al.26 synthesized (α-D-galactosyl)phenylmethane (402) and α- and β-D-galactosyl)-(difluoro)phenylmethane, i.e. anomers 401 and 398, respectively. α- and β-Glycopyranosyl aldehydes 4c and 3c on the addition of phenylmagnesium bromide followed by oxidation with PCC in DCM afforded corresponding ketones 396 and 399 in 50% and 45% yields, respectively. Ketones 396 and 399 upon difluorination with Deoxo-Fluor reagent in the presence of catalytic HF in pyridine afforded β- and α-CF2 protected sugar analogues 397 and 400 in 23% and 77% yields, respectively. Finally, benzyl protection of compounds 397 and 400 was deprotected by hydrogenolysis to give (β- and α-D-alactosyl)-(difluoro)phenylmethane 398 and 401 in 67% and 70% yield, respectively. (α-D-Galactosyl)phenylmethane 402 was obtained from glycopyranosyl ketone 399 by reduction with catalytic hydrogenolysis followed by deprotection of benzyl ether (Scheme 60). | ||
| Scheme 60 Synthesis of compounds 398, 401, and 402. | ||
McGrane et al.93 worked on designing a synthetic route for C-mannosides analogues, which specifically block uropathogenic Escherichia coli from colonizing the lower urinary tract. C-Mannosides were synthesised via a new pathway starting from C-linked glycopyranosyl aldehyde 3b, which on reaction with organolithium reagent 403a–b formed in situ by lithiation of 1,4-dibromobenzene with butyl lithium afforded R/S mixture of alcohol 404a–b. The palladium-mediated reaction of alcohol 404a–b with 3-(N-methylaminocarbonyl)phenylboronate furnished the cross-coupled carboxamide 405a–b, which on further hydrogenation with H2, Pd/C afforded mannosides 128a–b and 406 (Scheme 61). Methylene C-mannoside can be synthesised only by differing the organolithium reagent (4-bromo-2-methyliodobenzene).
 | ||
| Scheme 61 Synthesis of C-mannosides analogues 128a–b and 406. | ||
Dietrich et al.42 utilised the α-C-glycopyranosyl aldehyde for the synthesis of benzyl-α-C-glucosides and anilinomethyl-α-C-glucosides 412a–d, which act as α-glucosidase inhibitors. α-C-glucopyranosyl aldehyde 3a was treated with Grignard reagent phenylmagnesium bromide to afford diastereoselectively one isomer of compound 407 in 80% yield. Phenylcarbinol derivative 407 was converted into mesylated product 409, which on treatment with tetramethylguanidine azide in DMF afforded 410 together with elimination product 408. Azido compound 410 was reduced with LiAlH4 to furnish 411 in 56% yield, which was converted into one of the desired compounds 412a via a sequence of reactions (Scheme 62).
 | ||
| Scheme 62 Synthesis of benzyl-α-C-glucosides and anilinomethyl-α-C-glucosides 412a–d. | ||
Due to the low yield of the product, another route for the synthesis of 412a was investigated. Swern oxidation of 407 afforded compound 413 in 92% yield, which on treatment with hydroxylammonium chloride produced diastereomeric oximes 414a–b in a 1:1 ratio. Reduction of an isomeric mixture of 414a–b with LiAlH4 furnished diastereomeric amines 411, 415 and 416 in a ratio of 1:1:1 with 20%, 29%, and 24% yield, respectively. Finally, desired 412a, 412c, and 412d were obtained in 64–68% yield in four steps from 411, 415, and 416 following the reaction sequence as shown in Scheme 62.
Direct hydrogenolysis of compound 413 with palladium on charcoal following O-acetylation of crude product and treatment with sodium methoxide was carried out to obtain desired 412b in 70% yield.
Geng et al.94 synthesized C-(α-D-glucopyranosyl)-phenyldiazomethane using α-C-glucopyranosyl aldehyde 3a as the starting precursor (Scheme 63). A library of compounds with different substituent groups at the para position of the phenyl ring was prepared and further investigated for their stability. C-Glucopyranosyl aldehyde 3a was reacted with different substituted arylmagnesium bromide in THF to afford α-C-glucopyranosyl benzylic alcohols 417a–d in 65–81% yield with different substituents at the para position of the phenyl ring. Further oxidation with PCC in DCM led to the formation of 418a–d with 71–85% yield, which on reaction with hydroxylammonium chloride furnished compounds 419a–d with 92–98% yield. Reduction of oximes 419a–d was carried out to furnish a diastereomeric mixture of amines 420a–d and 421a–d in 86–93% yield. Reaction with methylchloroformate and triethylamine with each of these diastereomers 420a–d and 421a–d afforded 422a–d and 423a–d, respectively in 88–95% yield. Hydrogenolytic debenzylation with H2–Pd/C followed by acetylation with acetic anhydride, further treatment with NaNO2 in Ac2O–AcOH mixture afforded 424a–d and 425a–d, respectively in 80–88% yield in three steps. Both diastereomers on treatment with sodium methoxide in methanol furnished C-(α-D-glucopyranosyl)-phenyldiazomethanes 426a–d in 92–95% yield.
 | ||
| Scheme 63 Synthesis of C-(α-D-glucopyranosyl)-phenyldiazomethanes 426a–d. | ||
Investigation of the stability of these diazo compounds showed that substituent groups on the phenyl ring had a strong influence. The presence of an electron-withdrawing cyano group at the phenyl ring as in the case of compound 426a was found to be stable enough to resist the solvolysis. Compound 426a was further tested for inhibiting activity and type of inhibition towards α-glucosidase from Saccharomyces cerevisiae and found that compound was an irreversible inhibitor of α-glucosidase.
Picard et al.95 introduced the indium-mediated alkynylation of β-C-galactopyranosyl aldehydes in order to synthesize various β-C-glycosides. Galactopyranosyl aldehydes 4c and 59a were reacted with phenylacetylene iodide in the presence of indium in DCM, which resulted in the formation of corresponding propargylic alcohols 427a–b and 428a–b, respectively in 71–87% yield, as a diastereomeric mixture (65:35). Propargylic alcohols 427a–b upon oxidation with 2-iodoxybenzoicacid (IBX) in DMSO led to the formation of propargylic ketone 429 with 97% yield. Ketone 429 gave difluorinated product 430 on treatment with neat diethylaminosulfur trifluoride DAST (Scheme 64).
 | ||
| Scheme 64 Synthesis of β-C-glycosides 430–433. | ||
Also, propargylic alcohols 427a–b were mono-fluorinated by treatment with DAST, forming corresponding fluorinated compound 431 in 96% yield (Scheme 64). The propargylic alcohol 428a–b underwent palladium-catalysed hydrogenation leading to the formation of alcohol 432 in 95% yield, which upon treatment with thiocarbonyldiimidazole and tributyltin hydride afforded dehydroxylated compound 433 in 54% yield (Scheme 64).
Reddy et al.58 developed a scalable synthetic approach to synthesise acyl-C-β-D-glucosides and benzyl-C-β-D-glucosides, which proved to promote glucose-uptake activity in C2C12 mytotubes. The synthesis of these acyls and benzyl-C-β-D-glucosides was executed from β-C-glucopyranosyl aldehyde 4a, which was oxidised under Pinnick oxidation reaction conditions using NaClO4 and H2O2 affording carboxylic acid 434 in 97% yield. Carboxylic acid 434 on reaction with pivolyl chloride in Et3N followed by HCl·NH(Me)(Ome) in DCM afforded amide 435 in 93% yield. The amide 435 was easily converted to a range of ketone derivatives 436a–p in 60–94% yields with differently substituted aryl groups by Grignard reagent ArMgX in dry THF. The ketone derivatives 436a–p were converted to corresponding acyl-C-β-D-glucoside derivatives by first reacting with TMSOTf in acetic anhydride yielding peracetylated compounds 437a–p in 46–97% yield, followed by its deprotection using NaOMe in methanol afforded acyl-C-β-D-glucosides 126a–p in 60–85% yield (Scheme 65a).
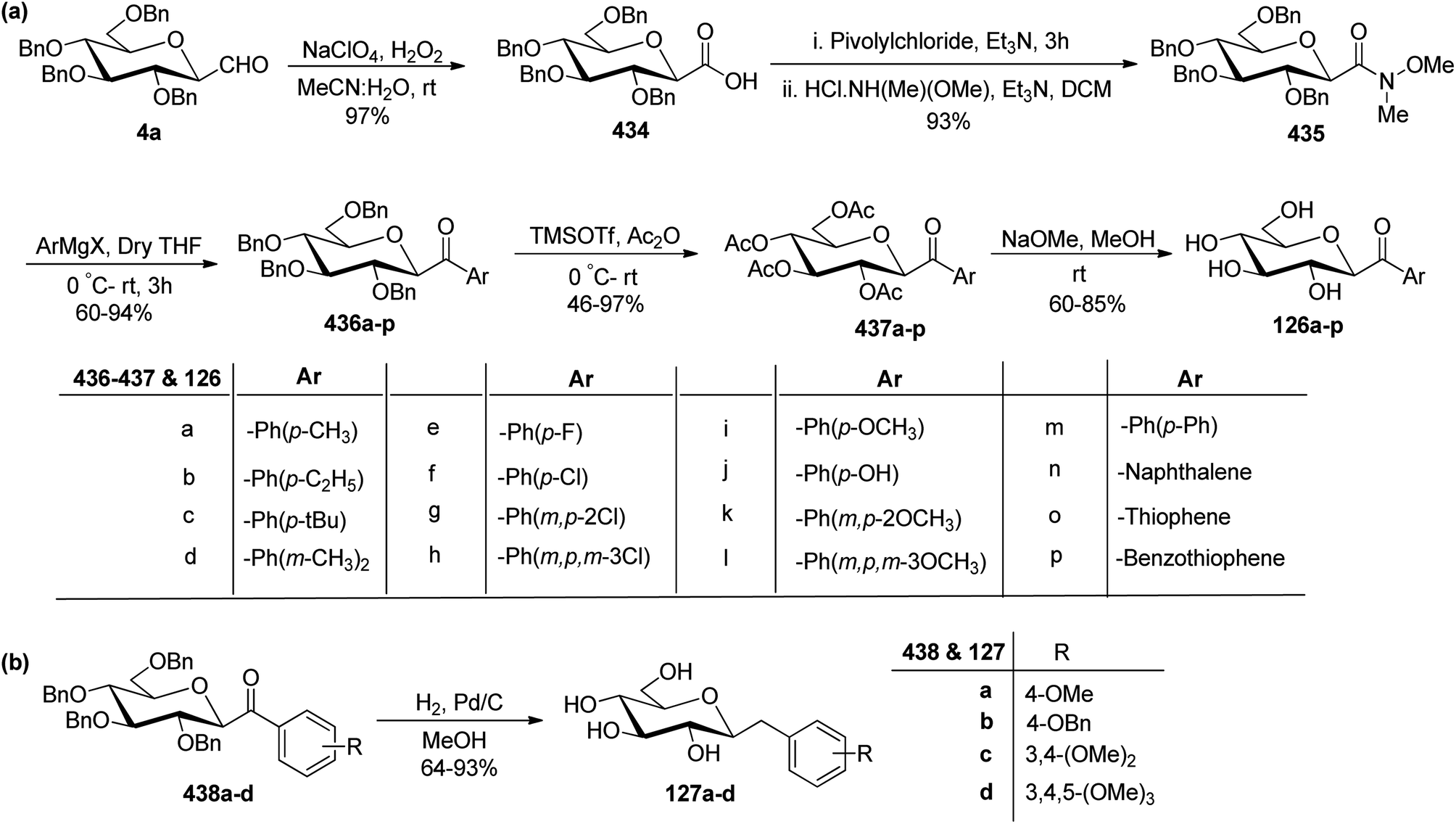 | ||
| Scheme 65 Synthesis of C-β-D-glucosides 126a–p and 127a–d. | ||
The C-β-D-glucosides 438a–d were also synthesised by one-pot reduction of the carbonyl group and benzyl ether deprotection from the corresponding perbenzylated-C-β-D-glucosides 127a–d using H2, Pd/C in methanol in 64–93% yield (Scheme 65b).
Zeitouni et al.70 reported a new strategy for the formation of C-glycosides by using the Mukaiyama aldol reaction. C-Glucopyranosyl aldehyde 4a was reacted with trimethylsilyl enol ether 439 using Mukaiyama aldol reaction in the presence of Yb(Otf)3 to furnish 440 and 441 as a diastereomeric mixture in ratio 59:41 with 95% yield (Scheme 66).
 | ||
| Scheme 66 Synthesis of C-glycosides 440 and 441. | ||
3.6 C-Glycopyranosyl heterocycles
The three-component Biginelli cyclo condensation reaction involving aldehyde–ketoester–urea was explored by Dondoni et al.96 using C-glycopyranosyl aldehyde to design a potential library of dihydropyrimidinone glycoconjugates. Under suitable acid additive and optimal reaction conditions, perbenzylated formyl C-galactopyranosyl aldehyde 4c, ethyl acetoacetate 442, and urea 443 were combined to form the cyclocondensed product 444 in 65% yield (Scheme 67). | ||
| Scheme 67 Synthesis of various sugar-based dihydropyrimidinones. | ||
Aiming to prepare a collection of mono-glycosylated and multiple-glycosylated DHPM derivatives, various substituted sugar residues were employed as substrates, which were in turn prepared from sugar aldehydes (Scheme 67). The C-glycosyl-β-ketoesters 446a–c were synthesised in 60–75% yield starting from sugar aldehydes 445a–c via coupling with ethyl diazoacetate in the presence of BF3·Et2O. Similarly, the sugar aldehydes 445a–c were reduced in the presence of NaBH4 followed by DPPA (diphenylphosphorylazide), DBU, and sodium azide to synthesize glycosylmethylazides 447a–c in 95–98% yield via the formation of corresponding alcohol molecules as intermediate. Azides 447a–c upon catalytic hydrogenation formed the corresponding amine, which on condensation with nitrourea afforded ureido sugars 448a–c with good overall yields (80–86%). Thus, various cyclocondensations were studied taking differently substituted substrates via the Biginelli reaction and the results were found to adhere to the standard method, giving a collection of glycosylated DHPM derivatives (Scheme 67).
Sudarshan et al.97 developed a route for the synthesis of 3-glycosylated isocoumarins by using modified Julia-olefination for initial C–C bond formation between C-glycopyranosyl aldehyde and benzylic-sulfones (Scheme 68). C-Glucopyranosyl aldehyde 4a and benzyl-sulfones 449, 454, and 459 were subjected to Julia-olefination reaction to afford alkenes 450, 455, and 460 in 62–80%, 62–84%, and 60% yields, respectively, with E-isomer as the major product (Scheme 67a–c). These olefins further oxidised to ketone intermediates 451, 456, and 461 in 44–74%, 54–84%, and 65% yields, respectively, using palladium-catalysed Meinwald rearrangement i.e. m-CPBA in DCM-H2O followed by Pd(OAc)2, PBu3 in tert-butanol with good yields. The ketone intermediates 451, 456, and 461 were transformed into isocoumarins 452, 457, and 462 by base-promoted intramolecular cyclisation using DBU in dry DCM. Finally, 3-glycosylated isocoumarin 453 and 458 were obtained in 42% and 51% yield by debenzylation of 452 and 457 using boron tribromide in dry DCM.
 | ||
| Scheme 68 Synthesis of 3-glycosylated isocoumarins 453, 458, and 462. | ||
Mukkamala et al.98 discovered a pathway to synthesise C-aryl glucoside analogues of Dapagliflozin, which act as SGLT inhibitors for the treatment of type-2 diabetes. Key products benzyl C-glucosyl aryl ketone 464 was synthesised from glucopyranosyl aldehyde 4a and functionalized Grignard reagent 463 generated in situ from anthranilic acid 470. Iodinated anthranilic acid 471 was obtained with 87% yield by the reaction of 470 with KI/NaIO4/NaCl in acetic acid. Acid 471 on diazotisation reaction followed by the replacement of the diazonium group by chloro-group afforded carboxylic acid 472 in 93% yield, which afforded Weinerb-amide 473 in 95% yield on the reaction with SOCl2 in DCM–DMF followed by Me(MeO)NHHCl. Functionalized arylmagnesium bromide 463 was obtained by reacting acid 473 with iPrMgBr under Paul Knochel's condition (Scheme 69b). Synthesised Grignard reagent 463 was reacted directly with aldehyde 4a to afford addition product 464 in 68% yield, which undergoes subsequent deoxygenation on reaction with phenyl chlorothionocarbonate in 4-dimethylaminopyridine (DMAP) and acetonitrile yielding thionocarbonate derivative 465. Compound 465 on radical de-oxygenation in tri-n-butyl tin hydride and AIBN in toluene afforded functionalized benzyl C-glucoside 466 in 75% yield, which upon addition of arylmagnesium bromides afforded benzyl C-glucosyl diaryl ketones 467a–f in 67–97% yield. Ketone 467a–f when subjected to simple hydrogenation with H2, Pd/C in dichlorobenzene afforded de-protected C-benzylglucosyl ketones 469a, 469c, and 469f in 81–90% yield while for other derivatives the hydrogenation was performed with 40 psi pressure affording C-benzyl analogues of dapagliflozin 468a–f in 80–84% yield (Scheme 69a).
 | ||
| Scheme 69 Synthesis of C-aryl glucoside 467–469. | ||
Chanteau et al.99 designed a stereo-controlled synthetic pathway for the synthesis of carbohydrate-derived acylsilanes starting from C-glucopyranosyl aldehyde 4a. The aldehyde 4a undergoes nucleophilic addition on reaction with bis(trimethylsilyl)methoxymethane (474) in n-butyl lithium–THF via Peterson reaction to furnish enol ether 475 in 49% yield. Acidic hydrolysis of ether 475 with HCl in THF–H2O afforded acylsilane 476 in 85% yield, which on treatment with perfluorobutyl iodide with methyllithium furnished the corresponding hemifluorinatedenone 477 and α-hydroperfluorobutyl ketone 478 in a ratio of 75:25 in 84% yield. The mixture of 477 and 478 when reacted with acetamide leads to cyclisation affording the pyridine derivative 479 with a 79% yield. Also, acylsilane 476 when reacted with perfluorobutyl iodide with methyllithium followed by methylhydrazine at room temperature afforded pyrazole derivative 480 with 83% yield (Scheme 70).
 | ||
| Scheme 70 Synthesis of glycopyranosyl pyridine and pyrazole derivatives 479 and 480. | ||
Dondoni et al.100,101 synthesized C-glycosylated dihydropyridine by Hantzsch condensation reaction using C-glycopyranosyl aldehyde (Scheme 71). One pot three-component reaction was performed by taking C-glycopyranosyl aldehyde 4b–c, ethyl acetoacetate 442, and ethyl 3-aminobut-2-enoate 481 in the presence of Yb(OTf)3 in THF or EtOH to furnish C(4)-glycosylated dihydropyridine 482b–c. The overall yield with respect to the solvent taken is given in Scheme 71.
 | ||
| Scheme 71 Synthesis of C-glycosylated dihydropyridine 482b–c. | ||
Verma et al.102 reported a very simple methodology for the synthesis of hexopyranosyl pyrimidine homonucleosides and hexopyranosyl double-headed pyrimidine homonucleosides using C-glycopyranosyl aldehyde as a precursor. 2,6-anhydro heptitols 382a–b and 383a–b were converted into their tosylated form using tosyl chloride in pyridine-DCM to afford 483a–b and 486a–b in 92–93% yield, respectively. Further nucleophilic substitution reactions on 483a–b and 486a–b were carried out using thymine/uracil as nucleobase, K2CO3, 18-crown-6 in DMF to obtain hexopyranosyl homonucleosides 484a–d, and 487a–c, respectively. Treatment with H2, Pd/C in methanol afforded debenzylated form of homonucleosides 485a–d and 488a–c in 78–80% and 72–80% yield, respectively (Scheme 72).
 | ||
| Scheme 72 Synthesis of homonucleosides 485a–d and 488a–c. | ||
Dondoni et al.103 used β-C-glycopyranosyl aldehydes for the synthesis of glycosyl nitrile oxides and nitrones, which in turn could be used for the synthesis of more complex C-glycoconjugates. C-Glycopyranosyl aldehydes 4a–d on treatment with hydroxylamine and sodium carbonate in THF–H2O at room temperature afforded corresponding oximes 489a–d in 78–88% yield. Oximes 489a–d on treatment with N-bromosuccimimide (NBS) followed by triethylamine, led to the formation of nitrile oxide 490a–d. Upon heating, these nitrile oxides dimerised to give furoxans 491a–d in 60–80% yield. Compound 490a reacted with dimethyl acetylenedicarboxylate, which underwent the cycloaddition reaction and afforded corresponding C-glycosylated isoxazole 493a with 75% yield. Also, compounds 4a–d afforded the corresponding N-benzyl nitrones 492a–d in 75–95% yield on treatment with N-benzylhydroxyl-amine (Scheme 73).
 | ||
| Scheme 73 Synthesis of glycosyl nitrile oxide 490a and glycosyl nitrones 492a–d. | ||
Reddy et al.104 reported the synthesis of 2-β-D-glucopyranosyl pyridines by using Bohlmann–Rahtz hetero-annulation starting from β-C-glucopyranosyl aldehyde (Scheme 74). Nucleophilic addition of TMS–ethynyl magnesium bromide was carried out on C-glucopyranosyl aldehyde 4a to afford a diastereomeric mixture of 494a and 494b in 67% yield, which on further treatment with K2CO3 in methanol removed the TMS group and furnished 495a and 495b, respectively, in 97% yield. Oxidation of the diastereomeric mixture of 495a–b with IBX in DMSO produced 496 in 94% yield, which was followed by Bohlmann–Rahtz hetero-annulation reaction conditions with β-aminocrotonate 497 in toluene–AcOH at 70 °C to afford trisubstituted pyridine 498 in 90% yield.
 | ||
| Scheme 74 Synthesis of 2-β-D-glucopyranosyl pyridines 502a–f. | ||
Further, reduction of glycoside 498 with LiAlH4 produced 499 in 89% yield, which on oxidation with IBX in DMSO afforded 500 in 95% yield. Compound 500 was treated with substituted arylmagnesium bromide to afford a diastereomeric mixture of compounds 501a–f in 72–90% yield, which on direct hydrogenation with palladium on charcoal afforded targeted compounds 502a–f in 45–90% yield. These compounds were found to be the potential analogue of Dapagaliflozin, an approved drug that lowers blood glucose levels by inhibiting the sodium-glucose transport in the kidney.
Very recently our research group has synthesized a library of C-glucosides of 1-azaindolizines from C-glucopyranosyl aldehyde using the Groebke–Blackburn–Bienayame105–107 (GBB) reaction protocol (Scheme 75).108 The multicomponent reaction of C-glucopyranosyl aldehyde (4a) with 2-aminopyridine derivatives 503a–g and isocyanides 504a–b was carried out in the presence of the catalytic amount of InCl3 in MeOH at 70 °C to afford C-glucosides of 1-azaindolizines 505a–n in 56–88% yield. Deprotection was carried out for 505h using H2, Pd/C in methanol to obtain 506 in 92% yield.
 | ||
| Scheme 75 Synthesis of C-glucosides of 1-azaindolizines 505a–n and 506. | ||
3.7 C-Glycopyranosyl based natural product fragments
A new approach to the synthesis of ambruticin fragment by employing β-C-glycoside aldehyde as starting material was generated by Michelet et al.109 The key phenomena introduced was stereoselective chain-elongating hydroxy alkylation of β-C-glucopyranosyl aldehyde 4a by using various alkynyl derivatives 507a–d in the presence of methyllithium and magnesium dibromide leading to formation of diastereomeric alcohols 508a–d and 510a–d with a diastereomeric ratio of 75:25 in 75% yield. The major alcohol derivative 508b on treatment with a series of reagents led to the formation of alcohol 510 in 66% yield, which was hydrogenated in the presence of Pd/C followed by alkylation with NaCH(COOMe)2, palladium acetate, and DPPE in THF forming compound 511 as a single isomer in just 30% yield. Alcohol 511 on activation with 2,4-dichlorobenzoyl chloride followed by cyclization afforded tetra-O-benzylated cyclopropane 512 in 48% yield (Scheme 76a), which in turn is the western part of ambruticin.
 | ||
| Scheme 76 (a) Synthesis of tetra-O-benzylated cyclopropane 512. (b) Synthesis of the western part of Ambruticin 519. | ||
Synthesis of the western part of ambruticin was achieved from C-glycosyl aldehyde 513 (Scheme 76b). C-Glycosyl aldehyde 513 was reacted with alkyne 507b in the presence of MeLi–MgBr2 to afford alcohol mixtures in 12:88 diastereomeric ratio (R,R)-513 and (S,R)-514 in 52% yield. Mitsunobu reaction was applied on (S,R)-514 to invert the stereochemistry of alcohol, which produced 516 in 66% yield. Further treatment of potassium carbonate in 2 mol% methanol followed by H2, Pd/C in ethyl acetate produced 517 in 49% yield. Compound 518 was achieved using Pd(OAc)2, dimethyl malonate sodium salt in THF. Finally, the western part of Ambruticin 519 was obtained in 48% yield using Pd-catalysed cyclization.
3.8 Miscellaneous
The Ugi reaction is a multicomponent reaction where aldehyde, amine, carboxylic acid, and isocyanide components are combined to form bis-amide (Scheme 77a). Lockhoff et al.110 used this reaction to synthesise glycoconjugates libraries by using carbohydrate building blocks for the Ugi condensation. C-Glycosyl aldehyde 4a which also serves as an aldehyde building block could be easily oxidised into carboxylic acid 520 in 81% yield by using KMnO4–NaH2PO4. Similarly, amine and isocyanide building blocks were synthesised starting from C-glycosyl aldehyde (Scheme 77b). Compound 4a was reduced with LiBH4 to afford glycosylmethyl alcohol 521 in 95% yield, which was converted into amine 522 in 71% yield by following the reaction sequence, i.e. tosylation, azide substitution followed by reduction. Further, N-formylation of 522 and dehydration afforded isocyanide 523 in 52% yield. All these C-glycosides are the desired building blocks for Ugi condensation and were used to synthesise glycoconjugates (Table 4). | ||
| Scheme 77 Synthesis of sugar-based building blocks for the Ugi reaction, i.e. acid 520, amine 522, and isocyanide 523. | ||
| Ugi building blocks | Ugi product | ||||||
|---|---|---|---|---|---|---|---|
| Aldehyde | Amine | Acid | Isocyanide | R1 | R2 | R3 | R4 |
| a For R1, R2, R3, R4 see Scheme 77a, PMB = 4-methoxybenzyl, A = 2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl. | |||||||
| 4a | PMB | Acetic acid | Cyclohexyl isocyanide | A | PMB | CH3 | C6H11 |
| Benzaldehyde | 522 | Acetic acid | Cyclohexyl isocyanide | C6H5 | A-CH2 | CH3 | C6H11 |
| Benzaldehyde | PMB | 520 | Cyclohexyl isocyanide | C6H5 | PMB | A | C6H11 |
| Benzaldehyde | PMB | Acetic acid | 523 | C6H5 | PMB | CH3 | A-CH2 |
| 4a | 522 | Acetic acid | Cyclohexyl isocyanide | A | A-CH2 | CH3 | C6H11 |
| 4a | PMB | 520 | Cyclohexyl isocyanide | A | PMB | A | C6H11 |
| 4a | PMB | Acetic acid | 523 | A | PMB | CH3 | A-CH2 |
| Benzaldehyde | 522 | 520 | Cyclohexyl isocyanide | C6H5 | A-CH2 | A | C6H11 |
| Benzaldehyde | 522 | Acetic acid | 523 | C6H5 | A-CH2 | CH3 | A-CH2 |
| Benzaldehyde | PMB | 520 | 523 | C6H5 | PMB | A | A-CH2 |
| 4a | 522 | 520 | Cyclohexyl isocyanide | A | A-CH2 | A | C6H11 |
| 4a | 522 | Acetic acid | 523 | A | A-CH2 | CH3 | A-CH2 |
| 4a | PMB | 520 | 523 | A | PMB | A | A-CH2 |
| Benzaldehyde | 522 | 520 | 523 | C6H5 | A-CH2 | A | A-CH2 |
| 4a | 522 | 520 | 523 | A | A-CH2 | A | A-CH2 |
Dondoni et al.111 synthesized [60]fulleropyrrolidine glycoconjugates by using C-glycosyl aldehyde 4c and 524 (Scheme 78). In this protocol, C-glycosyl aldehydes 4c and 524, [60]fullerene, and N-methylglycine (sarcosine) 525 were refluxed in toluene to afford a diastereomeric mixture of [60]fulleropyrrolidine glycoconjugates 527a–b in 10–14% yield. The reaction proceeds through the formation of sugar azomethine ylide intermediate 526. It was found that an attack of azomethine ylide 526 occurred on a 6,6-ring junction of C60 through 1,3-dipolar cycloaddition reaction. Deprotection of the benzoyl group was carried out to obtain 528b using sodium methoxide in methanol-toluene.
 | ||
| Scheme 78 Synthesis of [60]fulleropyrrolidine glycoconjugates 527a–b and 528b. | ||
Auge et al.112 while studying vinyl halides realised that these are the building blocks of olefin synthesis and hence, they designed various vinyl halide synthesis. One of them involved the C-glycopyranosyl aldehydes as the starting product 4a, which upon treatment with CHI3 and CrCl3 in the presence of zinc in sodium iodide yielded the corresponding vinyl iodide 529 in an isomeric mixture in 63% yield (Scheme 79).
 | ||
| Scheme 79 Synthesis of C-glucopyranosyl vinyl iodide 529. | ||
Janes et al.113 and team explored the scope of organometallic C-glycosides containing chromium carbene and used β-C-glucopyranosyl aldehyde 4a for the synthesis. Glucopyranosyl aldehyde 4a on reaction with pentacarbonyl[(methoxy)-methylcarbene] chromium 530 underwent aldol condensation in the presence of TiCl4 in iPr2NEt to afford the novel C-glycoside compound, 1-deoxy-2,3,4,6-tetra-O-benzyl-β-D-glucopyranose 531 in 54% yield (Scheme 80).
 | ||
| Scheme 80 Synthesis of organometallic C-glycosides 531. | ||
C-glycopyranosyl aldehydes 78a–b synthesized from D-glucose and D-mannose by Xia et al.41 were utilised for the synthesis of L-sugar derivatives also. Reduction of 78a–b with sodium triacetoxyborohydride (NaBH(OAc)3) afforded 532a–b, which on further acetylation produced 533a–b. The hydroxyl group at C5 was deprotected using TMSCl, NaI in acetonitrile to produce 534a–b in 36–54% yield, which was oxidised to carboxylic acid 535a–b in 70–85% yield using TEMPO, iodosobenzene diacetate (DIB) in MeCN–H2O. Oxidative decarboxylation mediated by lead(iv)tetraacetate in THF-AcOH afforded L-sugar derivatives 536a–b in 76–80% yield (Scheme 81).
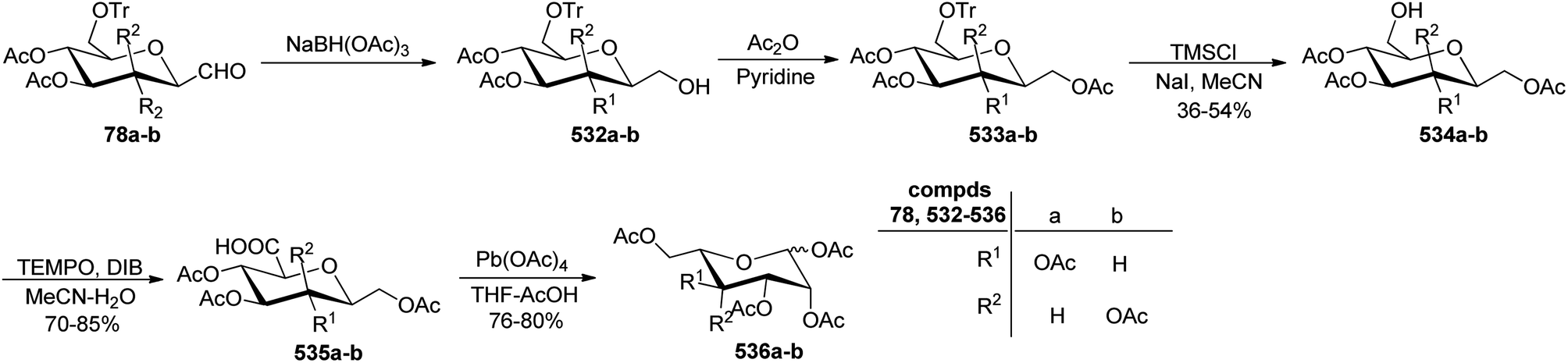 | ||
| Scheme 81 Synthesis of L-sugar derivatives 536a–b. | ||
C-Glycopyranosyl aldehydes synthesised by Petrusova et al.57 via the nitromethane route were further utilised for the synthesis of 2-(β-D-glycopyranosyl)nitroethenes and -nitroethanes (Scheme 82). The addition of nitromethane in the presence of NaOMe afforded epimericnitroalcohols 537a–d with 57% yield, which on further acetylation using acetic anhydride in the presence of sulfuric acid produced epimeric mixtures of 538a–c in 85% yield. The treatment of compounds 538a–c with sodium bicarbonate in benzene at 80 °C afforded 2-(β-D-glycopyranosyl)nitroethenes 539a–d in 90–94% yield. Hydrogenolysis was carried out using H2, Pd/C in ethyl acetate to give 2-(β-D-glycopyranosyl)nitroethanes 540a–d in 80–86% yield. Finally, deacetylation was achieved using sodium methoxide in methanol to afford 541a–d in 85–94% yield.
 | ||
| Scheme 82 Synthesis of 2-(β-D-glycopyranosyl)nitroethenes and -nitroethanes 539a–d & 541a–d. | ||
Lehmann et al.114 explored the synthetic route for the synthesis of 4,8-anhydro-2,3-dideoxy-D-galacto- and -D-gluco-non-3-enose dimethyl acetals which can be used for the determination of the steric course of glycoside hydrolases-initiated protonation. C-Linked galacto- and glucopyranosyl aldehyde 59a–b were treated with formylmethylenetriphenylphosphorane 542 in benzene to furnish β-D-galacto- and gluco-non-2-enose 543a–b in 75% yield, respectively, which on addition of HBr followed by acetylation afforded diastereomeric 3-bromides 544a–b in 42% yield. Elimination of HBr from 544a–b in the presence of AgF in pyridine furnished dimethyl acetal 545a–b with 80% yield, which upon treatment with sodium methoxide in methanol afforded acetylated product 546a–b in 89% yield. Enzymatic deuteriohydration of 546b was performed in the presence of α-D-galactosidase in the sodium/potassium phosphate–D2O buffer to give 2,3-dideoxy-α-D-galacto-(3-2H)nonos-4-ulose dimethyl acetal 547b in 94% yield. The reduction was carried out using sodium borohydride in water to afford epimers 548 and 549 in 44% yield, which could be separated by HPLC technique and were treated separately with 0.5 M CF3COOH in methanol to afford 1,6-anhdro-2,3-dideoxy-3(S)-D-glycero-L-gluco-(3-2H)nonopyranose 550 and 551 in 41% yield, respectively (Scheme 83).
 | ||
| Scheme 83 Synthesis of 1,6-anhydro-2,3-dideoxy-3(S)-D-glycero-L-gluco-(3-2H)nonopyranose 550 and 551. | ||
Khatri et al.115 recently synthesized a sugar–PEG copolymer starting from 2,6-anhydro heptitol 383a–b, which was obtained from β-C-glycopyranosyl aldehydes 4a–b by reduction with NaBH4 followed by selective debenzylation of primary benzyl protection. The synthesized two diastereomeric sugar alcohol derived from D-glucose and D-mannose were employed for the synthesis of sugar PEG-based copolymers using PEG-1000 diethyl ester 552 and 553 in 80% and 76% yields, respectively (Scheme 84). This synthesis of copolymers was achieved by Novozym®-435 catalyzed transesterification reaction. The configurational change at one of the carbons of diastereomeric 2,6-anhydro-heptitols has a significant effect on the degree of polymerization of the synthesized polymers. The aqueous solution of these copolymers forms small spherical micelles, which have the capability to encapsulate Nile red dye.
 | ||
| Scheme 84 Synthesis of gluco- and manno-heptitol-PEG copolymers 552 and 553. | ||
Very recently our group developed an efficient methodology for the synthesis of (2R,3S,4R)-chromanes 558a–i from C-glucopyranosyl aldehyde 4a (Scheme 85).116 Thus, C-glucopyranosyl aldehyde 4a was submitted to Claisen–Schmidt condensation using various acetophenones 553a–j in the presence of 5% aq. sodium hydroxide in ethanol to obtain 1-(E-1-arylpropenon-3-yl)-3,4,6-tri-O-benzyl-D-glucals 554a–j in 68–88% yields. Further, a cross dehydrogenative reaction was carried out on these C-1 substituted glucalpropenones using alkenes 555a–g in the presence of Pd(OAc)2, CuI, AgOTf in DMF-DMSO solvent system at 80 °C to afford 1,2-disubstituted glucals 556a–i in 78–90% yields. The CDC reaction afforded products with (E) stereoselectivity in good to excellent yield. Thus, E,Z,E-triene 556a–i heated at 160 °C in xylene, which on 6π-electrocyclization followed by in situ dehydrogenative aromatization produced chromanes 557a–i in 66–73% yields. The cyclization of trienes 557e and 557f was not achieved under optimised conditions. The benzyl protection was removed using 1 M BCl3 in DCM at −78 °C to furnish desired (2R,3S,4R)-chromanes derivatives 558a–i in 82–92% yields.
 | ||
| Scheme 85 Synthesis of (2R,3S,4R)-chromanes 558a–i. | ||
4. Conclusion
In this review, we have described various approaches for the synthesis of C-glycopyranosyl aldehydes and their applications in the synthesis of diverse biologically relevant C-glycoconjugates. A total of seven approaches have been designed based on the key intermediates involved in the synthesis of C-glycopyranosyl aldehydes, which refer to facile and efficient methods developed including multistep synthetic protocols. In addition, the purpose of this review is to scrutinize the importance of C-glycopyranosyl aldehydes to the vast field of organic synthesis. This review will serve as a collection of literature reports, where researchers will find both the synthetic methodologies to access C-glycopyranosyl aldehydes as well as their applications to synthesize various complex C-glycoconjugates as key precursor molecules.5. Future prospects
This review describes a wide scope of syntheses of C-glycopyranosyl aldehydes via different key intermediates along with a vast library of C-glycoconjugates derived from them. Although many synthetic routes are found to be very interesting even though some methods are economically unfavourable due to very expensive reagents used in the synthetic routes, also in some procedures harsh conditions make the syntheses cumbersome. Therefore, there is a need to develop more synthetic protocols that must be simple, economical, and environment friendly. We believe that the upcoming decades will bring us many exciting reports on the syntheses of C-glycopyranosyl aldehydes and glycoconjugates. The synthesis of glycoconjugates must be explored in the desire of many biologically important sugar-based natural products and the total synthesis of target molecules. Since the last decade, we have had our own interest in these C-glycopyranosyl aldehydes and to further utilise these compounds for the syntheses of glycoconjugates. At present, we are working on the cost-effective, greener, and easy synthetic routes for the synthesis of C-glycopyranosyl aldehydes and glycoconjugates and will discuss them in the coming years.Abbreviations
| AIBN | Azobisisobutyronitrile |
| BtOH | Butanol |
| BAIB | Bis(acetoxy)iodobenzene |
| BCl3 | Boron trichloride |
| BF3·Et2O | Boron trifluoride diethyl etherate |
| Bu3SnH | Tributyltin hydride |
| CH3COSH | Thioacetic acid |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCC | N,N-Dicyclohexylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| DIBAL-H | Diisobutylaluminiumhydride |
| DAST | Diethylaminosulfur trifluoride |
| DPPE | 1,2-Bis(diphenylphosphino)ethane |
| DMDO | Dimethyldioxirane |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| Et3SiH | Triethylsilane |
| HOBt | Hydroxybenzotriazole |
| 2-LTT | 2-Lithiothiazole |
| PTSA | para-Toluenesulfonic acid |
| P2O5 | Phosphorous pentoxide |
| Pd/C | Palladium on charcoal |
| PMB | para-Methoxybenzyl |
| PEG | Polyethylene glycol |
| TMSOTf | Trimethylsilyl triflate |
| TMSCl | Trimethylsilyl chloride |
Author contributions
Sandeep Kumar and Vinod Khatri wrote the review. Priyanka Mangla and Rajni Johar checked the review thoroughly and made suitable changes. The review was written under the supervision of Virinder S. Parmar and Ashok K. Prasad.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Sandeep Kumar thanks CSIR, New Delhi for the award of the SPM Research Fellowship [File No. 09/045(0269)/2018-EMR-1]. V. K. and P. M. thank CSIR-UGC, New Delhi for the award of Junior/Senior Research Fellowship.References
- D. E. Levy and C. Tang, The Chemistry of C-glycosides, Pergamon Publishers, 1995 Search PubMed.
- M. Brito-Arias, Synthesis and Characterization of Glycosides, Springer, Boston, MA, 2007 Search PubMed.
- K. Kitamura, Y. Maezawa, Y. Ando, T. Kusumi, T. Matsumoto and K. Suzuki, Angew. Chem., Int. Ed., 2014, 53, 1262–1265 CrossRef CAS PubMed.
- Z. Wu, G. Wei, G. Lian and B. Yu, J. Org. Chem., 2010, 75, 5725–5728 CrossRef CAS PubMed.
- Z. Han, M. Achilonu, P. S. Kendrekar, E. Joubert, D. Ferreira, S. L. Bonnet and J. H. van der Westhuizen, J. Nat. Prod., 2014, 77, 583–588 CrossRef CAS PubMed.
- J. Li and B. Yu, Angew. Chem., Int. Ed., 2015, 54, 6618–6621 CrossRef CAS PubMed.
- D. Hager, P. Mayer, C. Paulitz, J. Tiebes and D. Trauner, Angew. Chem., Int. Ed., 2012, 51, 6525–6528 CrossRef CAS PubMed.
- (a) P. G. Hultin, Curr. Top. Med. Chem., 2005, 5, 1299–1331 CrossRef CAS PubMed; (b) K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Chem. Rev., 2018, 118, 1495–1598 CrossRef CAS PubMed.
- (a) A. Cavezza, C. Boulle, A. Guéguiniat, P. Pichaud, S. Trouille, L. Ricard and M. Dalko-Csiba, Bioorg. Med. Chem. Lett., 2009, 19, 845–849 CrossRef CAS PubMed; (b) L. Leseurre, C. Merea, S. Duprat de Paule and A. Pinchart, Green Chem., 2014, 16, 1139–1148 RSC.
- X.-J. Wang, L. Zhang, D. Byrne, L. Nummy, D. Weber, D. Krishnamurthy, N. Yee and C. H. Senanayake, Org. Lett., 2014, 16, 4090–4093 CrossRef CAS PubMed.
- J. P. Henchke, C.-W. Lin, P.-Y. Wu, W.-S. Tsao, J.-H. Liao and P.-C. Chiang, J. Org. Chem., 2015, 80, 5189–5195 CrossRef PubMed.
- C. Guo, M. Hu, R. J. DeOrazio, A. Usyatinsky, K. Fitzpatrick, Z. Zhang, J.-H. Maeng, D. B. Kitchen, S. Tom, M. Luche, Y. Khmelnitsky, A. J. Mhyre, P. R. Guzzo and S. Liu, Bioorg. Med. Chem., 2014, 22, 3414–3422 CrossRef CAS PubMed.
- G. Yang, J. Schmieg, M. Tsuji and R. W. Franck, Angew. Chem., Int. Ed., 2004, 43, 3818–3822 CrossRef CAS PubMed.
- A. Shcherbakova, M. Preller, M. H. Taft, J. Pujols, S. Ventura, B. Tiemann, F. F. Buettner and H. Bakker, Elife, 2019, 8, e52978 CrossRef CAS PubMed.
- C. Taillefumier and Y. Chapleur, Chem. Rev., 2004, 104, 263–292 CrossRef CAS PubMed.
- M. Choumane, A. Banchet, N. Probst, S. Gerard, K. Ple and A. C. R. Haudrechy, Chimie, 2011, 14, 235–273 CrossRef CAS.
- J. Stambasky, M. Hocek and P. Kocovsky, Chem. Rev., 2009, 109, 6729–6764 CrossRef CAS PubMed.
- E. Von Moos and R. N. Ben, Curr. Top. Med. Chem., 2005, 5, 1351–1361 CrossRef CAS PubMed.
- D. Y. W. Lee and M. He, Curr. Top. Med. Chem., 2005, 5, 1333–1350 CrossRef CAS PubMed.
- E. Bokor, S. Kun, D. Goyard, M. Toth, J. P. Praly, S. Vidal and L. Somsak, Chem. Rev., 2017, 117, 1687–1764 CrossRef CAS PubMed.
- K. Lalitha, K. Muthusamy, Y. S. Prasad, P. K. Vemula and S. Nagarajan, Carbohydr. Res., 2015, 402, 158–171 CrossRef CAS PubMed.
- Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS PubMed.
- A. Dondoni, Pure Appl. Chem., 2000, 72, 1577–1588 CrossRef CAS.
- W. R. Kobertz, C. R. Bertozzi and M. D. Bednarski, Tetrahedron Lett., 1992, 33, 737–740 CrossRef CAS.
- L. Kroger, D. Henkensmeier, A. Schafer and J. Thiem, Bioorg. Med. Chem. Lett., 2004, 14, 73–75 CrossRef CAS PubMed.
- M. Kolympadi, M. Fontanella, C. Venturi, S. Andre, H. J. Gabius, J. J. Barbero and P. Vogel, Chem. - Eur. J., 2009, 15, 2861–2873 CrossRef CAS PubMed.
- J. Guillaume, T. Seki, T. Decruy, K. Venken, D. Elewaut, M. Tsuji and S. V. Calenbergh, Org. Biomol. Chem., 2017, 15, 2217–2225 RSC.
- A. Dondoni and M. C. Scherrmann, Tetrahedron Lett., 1993, 34, 7319–7322 CrossRef CAS.
- A. Dondoni and M. C. Scherrmann, J. Org. Chem., 1994, 59, 6404–6412 CrossRef CAS.
- A. Dondoni and A. Marra, Tetrahedron Lett., 2003, 44, 13–16 CrossRef CAS.
- M. E. L. Sanchez, V. Michelet, I. Besnier and J. P. Genet, Synlett, 1994, 705–708 CrossRef CAS.
- F. Labeguere, J. P. Lavergne and J. Martinez, Tetrahedron Lett., 2002, 43, 7271–7272 CrossRef CAS.
- P. Gerber and P. Vogel, Tetrahedron Lett., 1999, 40, 3165–3168 CrossRef CAS.
- S. Sipos and I. Jablonkai, Carbohydr. Res., 2011, 346, 1503–1510 CrossRef CAS PubMed.
- M. T. G. Lopez, F. G. D. Las Heras and A. S. Felix, Carbohydr. Chem., 1987, 6, 273–279 CrossRef.
- H. M. Dettinger, G. Kurz and J. Lehmann, Carbohydr. Res., 1979, 74, 301–307 CrossRef CAS PubMed.
- K. Fujiwara, Y. Koyama, E. Doi, K. Shimawaki, Y. Ohtaniuchi, A. Takemura, S. I. Souma and A. Murai, Synlett, 2002, 9, 1496–1499 CrossRef.
- M. Toth, K. E. Kover, A. Benyei and L. Somsak, Org. Biomol. Chem., 2003, 1, 4039–4046 RSC.
- J. Zeitouni, S. Norsikian and A. Lubineau, Tetrahedron Lett., 2004, 45, 7761–7763 CrossRef CAS.
- S. Norsikian, J. Zeitouni, S. Rat, S. Gerard and A. Lubineau, Carbohydr. Res., 2007, 342, 2716–2728 CrossRef CAS PubMed.
- T. Y. Xia, Y. B. Li, Z. J. Yin, X. B. Meng, S. C. Li and Z. J. Li, Chin. Chem. Lett., 2014, 25, 1220–1224 CrossRef CAS.
- H. Dietrich and R. R. Schmidt, Carbohydr. Res., 1993, 250, 161–176 CrossRef CAS.
- (a) J. R. Pougny, M. A. M. Nassr and P. Sinaj, J. Chem. Soc., Chem. Commun., 1981, 375–376 RSC; (b) J. M. Lancelin, J. R. Pougny and P. Sinay, Carbohydr. Res., 1985, 136, 369–374 CrossRef CAS.
- F. Nicotra, R. Perego, F. Ronchetti, G. Russo and L. Toma, Carbohydr. Res., 1984, 131, 180–184 CrossRef CAS.
- T. V. RajanBabu and G. S. Reddy, J. Org. Chem., 1986, 51, 5458–5461 CrossRef CAS.
- M. Leclere, B. K. Kwok, L. K. Wu, D. S. Allan and R. N. Ben, Bioconjugate Chem., 2011, 22, 1804–1810 CrossRef CAS PubMed.
- R. Patnam, J. M. Juarez-Ruiz and R. Roy, Org. Lett., 2006, 8, 2961–2964 CrossRef PubMed.
- G. Chen, M. Chien, M. Tsuji and R. W. Franck, ChemBioChem, 2006, 7, 1017–1022 CrossRef CAS PubMed.
- J. Desire and A. Veyrieres, Carbohydr. Res., 1995, 268, 177–186 CrossRef CAS.
- (a) R. Gigg, A. A. E. Penglis and R. Conant, J. Chem. Soc., Perkin Trans. 1, 1977, 1, 2014–2017 RSC; (b) T. Iversen and D. R. Bundle, Carbohydr. Res., 1982, 103, 29–40 CrossRef CAS PubMed.
- G. J. McGarvey, F. W. Schmidtmann, T. E. Benedum and D. E. Kizer, Tetrahedron Lett., 2003, 44, 3775–3779 CrossRef CAS.
- V. Khatri, A. Kumar, B. Singh, S. Malhotra and A. K. Prasad, J. Org. Chem., 2015, 80, 11169–11174 CrossRef CAS PubMed.
- W. Feng, Z. Fang, J. Yang, B. Zheng and Y. Jiang, Carbohydr. Res., 2011, 346, 352–356 CrossRef CAS PubMed.
- O. R. Martin, F. E. Khamis and S. P. Rao, Tetrahedron Lett., 1989, 30, 6143–6146 CrossRef CAS.
- (a) L. Petrus, S. Bystricky, T. Sticzay and V. Bilik, Chem. Zvesti, 1982, 36, 103 CAS; (b) A. Fortsch, H. Kogelberg and P. Koll, Carbohydr. Res., 1987, 164, 391 CrossRef.
- O. Simo, K. Michael, W. Yoshida, V. A. Chertkov and P. H. Gross, Synth. Commun., 2005, 35, 1589–1599 CrossRef CAS.
- M. Petrusova, J. N. BeMiller, A. Krihova and L. Petrus, Carbohydr. Res., 1996, 295, 57–67 CAS.
- M. R. Reddy, S. Hemaiswarya, H. Kommidi, I. S. Aidhen and M. Doble, Eur. J. Org. Chem., 2019, 6053–6070 CrossRef CAS.
- L. M. McGrane, Z. Cusumano, Z. Han, J. Binkley, M. Kostakioti, T. Hannan, J. S. Pinkner, R. Klein, V. Kalas, J. Crowley, N. P. Rath, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2016, 59, 9390–9408 CrossRef PubMed.
- C. R. Bertozzi, D. G. Cook, W. R. Kobertz, F. G. Scarano and M. D. Bednarski, J. Am. Chem. Soc., 1992, 114, 10639–10641 CrossRef CAS.
- L. Awad, R. Madani, A. Gillig, M. Kolympadi, M. Philgren, A. Muhs, C. Gerand and P. Vogel, Chem. - Eur. J., 2012, 18, 8578–8582 CrossRef CAS PubMed.
- R. Demange, L. Awad and P. Vogel, Tetrahedron: Asymmetry, 2004, 15, 3573–3585 CrossRef CAS.
- A. Dondoni, D. Perrone and E. Turturici, J. Org. Chem., 1999, 64, 5557–5564 CrossRef CAS PubMed.
- A. Dondoni, A. Marra and A. Massi, Tetrahedron, 1998, 54, 2827–2832 CrossRef CAS.
- W. R. Kobertz, C. R. Bertozzi and M. D. Bednarski, Org. Chem., 1996, 61, 1894–1897 CrossRef CAS PubMed.
- R. Demange, C. Buhlmann and P. Vogel, Helv. Chim. Acta, 2003, 86, 361–376 CrossRef CAS.
- Y. H. Zhu and P. Vogel, Synlett, 2001, 79–81 CrossRef CAS.
- L. Awad, J. Riedner and P. Vogel, Chem. - Eur. J., 2005, 11, 3565–3573 CrossRef CAS PubMed.
- (a) A. Itoh, S. Ozawa, K. Oshima and H. Nozaki, Tetrahedron Lett., 1980, 21, 361–364 CrossRef; (b) A. Itoh, S. Ozawa, K. Oshima and H. Nozaki, Bull. Chem. Soc. Jpn., 1981, 54, 274–278 CrossRef CAS.
- J. Zeitouni, S. Norsikian, D. Merlet and A. Lubineau, Adv. Synth. Catal., 2006, 348, 1662–1670 CrossRef CAS.
- T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 2, 1011–1014 CrossRef.
- E. Levoirier, Y. Canac, S. Norsikian and A. Lubineau, Carbohydr. Res., 2004, 339, 2737–2747 CrossRef CAS PubMed.
- Y. Canac, E. Levoirier and A. Lubineau, J. Org. Chem., 2001, 66, 3206–3210 CrossRef CAS PubMed.
- A. Dondoni, H. M. Zuurmond and A. Boscarato, J. Org. Chem., 1997, 62, 8114–8124 CrossRef CAS PubMed.
- A. Dondoni, M. Kleban, H. Zuurmond and A. Marra, Tetrahedron Lett., 1998, 39, 7991–7994 CrossRef CAS.
- A. Dondoni, M. Mizuno and A. Marra, Tetrahedron Lett., 2000, 41, 6657–6660 CrossRef CAS.
- A. Dondoni, A. Marra, M. Mizuno and P. P. Giovannini, J. Org. Chem., 2002, 67, 4186–4199 CrossRef CAS PubMed.
- S. Sipos, I. Jablonkai, O. Egyed and M. Czugler, Carbohydr. Res., 2011, 346, 2862–2871 CrossRef CAS PubMed.
- A. Charfedinne, P. Julien, L. G. Nadege, U. Jacques and A. Jacques, Sci. China: Chem., 2010, 53, 1921–1926 CrossRef.
- A. Dondoni and D. Perrone, Org. Synth., 2000, 77, 64–77 CrossRef CAS.
- A. Dondoni, A. Massi, S. Sabbatini and V. Bertolasi, Tetrahedron Lett., 2004, 45, 2381–2384 CrossRef CAS.
- A. Dondoni, A. Massi and S. Sabbatini, Chem. - Eur. J., 2005, 11, 7110–7125 CrossRef CAS PubMed.
- A. Dondoni, A. Massi and A. Marra, Tetrahedron Lett., 1998, 39, 6601–6604 CrossRef CAS.
- A. Dondoni and A. M. A. Massi, J. Org. Chem., 1999, 64, 933–944 CrossRef CAS PubMed.
- M. D. P. Risseeuw, J. Mazurek, A. V. Langenvelde, G. A. V. D. Marel, H. S. Overkleeft and M. Overhand, Org. Biomol. Chem., 2007, 5, 2311–2314 RSC.
- A. Dondoni, A. Massi, S. Sabbatini and V. Bertolasi, Adv. Synth. Catal., 2004, 346, 1355–1360 CrossRef CAS.
- N. Boutard, F. Labeguere, Y. Vidal, J. P. Lavergne and J. Martinez, Synth. Commun., 2012, 42, 1461–1471 CrossRef CAS.
- A. Guerrini, G. Varchi and A. Battaglia, J. Org. Chem., 2006, 71, 6785–6795 CrossRef CAS PubMed.
- B. Kumar, J. Maity, B. Shankar, S. Kumar, Kavita and A. K. Prasad, Carbohydr. Res., 2021, 500, 108236 CrossRef CAS PubMed.
- (a) M. Passerini and L. Simone, Gazz. Chim. Ital., 1921, 51, 126 CAS; (b) M. Passerini, Gazz. Chim. Ital., 1921, 51, 181 CAS.
- M. Raunkjaer, F. E. Oualid, G. A. Van der Marel, H. S. Overkleeft and M. Overhand, Org. Lett., 2004, 6, 3167–3170 CrossRef PubMed.
- A. Singh, V. K. Maikhuri, V. Verma, R. J. Chhatwal, D. Sharma and A. K. Prasad, Synth. Commun., 2020, 50, 2787–2795 CrossRef CAS.
- L. M. McGrane, Z. Cusumano, Z. Han, J. Binkley, M. Kostakioti, T. Hannan, J. S. Pinkner, R. Klein, V. Kalas, J. Crowley, N. P. Rath, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2016, 59, 9390–9408 CrossRef PubMed.
- Y. Geng, A. Kumar, H. M. Faidallah, H. A. Albar, I. A. Mhkalid and R. R. Schmidt, Bioorg. Med. Chem., 2013, 21, 4793–4802 CrossRef CAS PubMed.
- J. Picard, N. L. Germain, J. Uziel and J. Auge, Synthesis, 2006, 6, 979–982 Search PubMed.
- A. Dandoni, A. Massi, S. Sabbatini and V. Bertolasi, J. Org. Chem., 2002, 67, 6979–6994 CrossRef PubMed.
- K. Sudarshan and I. S. Aidhen, Eur. J. Org. Chem., 2017, 2017, 34–38 CrossRef CAS.
- R. Mukkamala, R. Kumar, S. K. Banerjee and I. S. Aidhen, Eur. J. Org. Chem., 2020, 2020, 1828–1839 CrossRef CAS.
- F. Chanteau, R. P. Royon and C. Portella, Synlett, 2004, 3, 512–516 Search PubMed.
- A. Dondoni, A. Massi and E. Minghini, Synlett, 2002, 1, 89–92 CrossRef.
- A. Dondoni, A. Massi and E. Minghini, Helv. Chim. Acta, 2002, 85, 3331–3348 CrossRef CAS.
- V. Verma, V. K. Maikhuri, V. Khatri, A. Singh and A. K. Prasad, Synth. Commun., 2021, 51, 446–452 CrossRef CAS.
- A. Dondoni and P. P. Giovannini, Synthesis, 2002, 12, 1701–1706 CrossRef.
- M. R. Reddy and I. S. Aidhen, Eur. J. Org. Chem., 2018, 5744–5753 CrossRef CAS.
- K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 661–663 CrossRef CAS.
- C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635–3638 CrossRef CAS.
- H. Bienayme and K. Bouzid, Angew. Chem. Int. Ed., 1998, 37, 2234–2237 (Angew. Chem. Int. Ed., 1998, 110, 2349) CrossRef CAS PubMed.
- B. Kumar, B. Shankar, S. Kumar, J. Maity and A. K. Prasad, Synth. Commun., 2020, 50, 2853–2859 CrossRef CAS.
- V. Michelet, K. Adiey, S. Tanier, G. Dujardin and J. P. Genet, Eur. J. Org. Chem., 2003, 2947–2958 CrossRef CAS.
- O. Lockhoff, Angew. Chem., Int. Ed., 1998, 37, 3436–3439 CrossRef CAS PubMed.
- A. Dondoni and A. Marra, Tetrahedron Lett., 2002, 43, 1649–1652 CrossRef CAS.
- J. Auge, V. Boucard, R. Gil, N. L. Germain, J. Picard and J. Uziel, Synth. Commun., 2003, 33, 3733–3739 CrossRef CAS.
- E. Janes and K. H. Dotz, J. Organomet. Chem., 2003, 669, 1–5 CrossRef CAS.
- H. F. J. Lehmann, M. S. Schuchardt and W. Weiser, Carbohydr. Res., 1991, 218, 129–141 CrossRef.
- V. Khatri, S. Bhatia, S. Deep, E. Kohli, R. Haag, N. N. Senapati and A. K. Prasad, New J. Chem., 2020, 44, 15369–15375 RSC.
- B. Shankar, V. Khatri, B. Kumar, V. K. Maikhuri, A. Kumar, R. Tomar and A. K. Prasad, ACS Omega, 2021, 6, 11248–11259 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |