
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated pipeline for ultrasensitive protein detection in cancer nanomedicine†
Chi-An Cheng *,
Li-Chiao Chiang and
Yu-Syuan Chu
*,
Li-Chiao Chiang and
Yu-Syuan Chu
School of Pharmacy, College of Medicine, National Taiwan University, Taipei, 10050, Taiwan. E-mail: cacheng123@ntu.edu.tw
First published on 15th May 2023
Abstract
Although nanotechnologies have attractive attributes in cancer therapy, their full potential has yet to be realized due to challenges in their translation to clinical settings. The evaluation of cancer nanomedicine efficacy in preclinical in vivo studies is limited to tumor size and animal survival metrics, which do not provide adequate understanding of the nanomedicine's mechanism of action. To address this, we have developed an integrated pipeline called nanoSimoa that combines an ultrasensitive protein detection technique (Simoa) with cancer nanomedicine. As a proof-of concept, we assessed the therapeutic efficacy of an ultrasound-responsive mesoporous silica nanoparticle (MSN) drug delivery system on OVCAR-3 ovarian cancer cells using CCK-8 assays to evaluate cell viability and Simoa assays to measure IL-6 protein levels. The results demonstrated significant reductions in both IL-6 levels and cell viability following nanomedicine treatment. In addition, a Ras Simoa assay (limit of detection: 0.12 pM) was developed to detect and quantify Ras protein levels in OVCAR-3 cells, which are undetectable by commercial enzyme-linked immunosorbent assays (ELISA). These results suggest that nanoSimoa has the potential to guide the development of cancer nanomedicines and predict their behavior in vivo, making it a valuable tool for preclinical testing and accelerating the development of precision medicine if its generalizability is confirmed.
Cancer nanomedicine has experienced significant growth in recent years, with over 300 trials listed on http://www.Clinicaltrials.gov for cancer and nanoparticles as of 2022. However, the low success rate of cancer nanomedicine in clinical trials highlights the need for more rigorous preclinical evaluation.1 Currently, preclinical in vivo studies primarily rely on tumor size and animal survival metrics to evaluate the efficacy of cancer nanomedicines, which may not provide sufficient insight into the mechanism of action of these nanomedicines.2–4 In vitro testing should be conducted to evaluate nanomedicines before proceeding to animal studies. The development of in vitro and in vivo data correlation (IVIVC) tools may improve the clinical success rate of nanomedicines.5,6
Liquid biopsy is a non-invasive method that has revolutionized oncology in the past decade by offering ease in tumor sampling and continuous monitoring of cancer by repeated sampling. The analysis of tumor-derived biomarkers such as proteins,7 small molecules, circulating tumor cells,8,9 circulating tumor DNA (ctDNA),10,11 and tumor extracellular vesicles (tEVs)12–14 present in biofluids provides insight into endpoint biology that genes alone cannot provide. Proteins have proven to be valuable biomarkers as they often play crucial roles in the development and progression of diseases and are frequently targeted by therapeutic treatments. Targeted proteomics in combination with genome and transcriptome sequencing allows clinicians to develop personalized treatment plans tailored to an individual's specific medical condition, leading to more effective treatments.15,16 Identification of protein biomarkers associated with tumor drug resistance or sensitivity to chemotherapy or immunotherapy is now being used to predict the most appropriate treatment options.17 However, developing predictive tests for the in vivo efficacies of nanomaterials remains a challenge, due to a limited understanding of biomolecular interactions with these materials, as well as individual differences among patients.2
The enzyme-linked immunosorbent assay (ELISA)18 is the most commonly used protein detection method, with a sensitivity range of approximately picomolar.19 However, because protein baseline levels can vary among patients, a more precise method for assessing immune responses and physiological changes at an early stage would be to monitor changes in protein levels relative to each individual's baseline.20 Unfortunately, many proteins circulate in biofluids at concentrations as low as femtomolar and even attomolar, which cannot be detected using ELISA.19 While mass spectrometry is another common protein detection method, it has limitations such as not being inherently quantitative, requiring extensive sample preparation, and only providing limited information about the intact protein.21 Additionally, due to the low amounts of secreted proteins by tumor cells, a large number of cells, ranging from 108 to 109, are necessary to obtain adequate material for successful protein analysis using mass spectrometry.19 Therefore, there is an urgent need for a highly sensitive and high-resolution protein detection method that can accurately detect absolute changes in protein levels. This method would be critical for assessing cancer treatment efficacy, monitoring disease progression, and personalizing therapeutic regimens.
The Single-molecule array (Simoa) platform, also known as digital ELISA, is a highly advanced technology that can detect low abundance proteins at extremely low concentrations (sub-femtomolar range, and in some cases, 10−19 M).22,23 It uses a sandwich ELISA approach and is based on the selectivity of a pair of antibodies to quantify analytes. This reaction takes place on beads so that the signal from a single binding event can be imaged.24 Simoa offers unparalleled analytical sensitivity, precision, and accuracy, enabling reliable measurements of biomarkers at both low and high concentrations.22 Compared to ELISA, Simoa can detect cytokines at much lower concentrations, spanning four orders of magnitude with a wide dynamic range for serological characterization from early to late disease stages. Simoa requires only small sample volumes, facilitating widespread testing with finger prick or dried blood spot sample collection.25 Additionally, it is highly versatile, cost-effective, and reproducible, making it suitable for use in academic, industrial, and medical laboratories. Furthermore, Simoa is high-throughput, rapid, and multiplexed, allowing for efficient evaluation of multiple analytes.23,26
Mesoporous silica nanoparticles (MSN) possess unique properties, including a large pore volume and high surface area for efficient cargo loading and delivery, biocompatibility, high cellular internalization efficiency, and ease of surface functionalization, making them a promising drug delivery system.27–31 In a previous study, we developed an ultrasound-responsive drug delivery system based on MSN that enabled controlled release of drugs with precise dose, timing, and location.29 The system incorporated gadopentetate dimeglumine (Gd(DTPA)2−), a gadolinium-based magnetic resonance imaging (MRI) contrast agent, as an imageable cargo, and delivered the cargo at predetermined conditions.29 Ultrasound stimulation induced drug release, while mild hyperthermia induced by ultrasound enhanced chemotherapy efficacy by improving drug delivery to tumor tissue.32–34 This drug delivery system holds great potential for precision medicine and cancer therapy.
In this study, we used the MSN-based drug delivery system developed previously,29 but modified it with an ultrasound-responsive moiety to release topotecan (TOP), a water-soluble drug used to treat various cancers,35–38 including ovarian cancer cells. To accurately quantify and profile protein biomarkers from cancer cells, we developed a novel pipeline called nanoSimoa, which integrates the Simoa protein detection platform with cancer nanomedicine (Scheme 1). This pipeline enables the precise detection of low-abundance biomarkers and high-resolution profiling of biomarkers without extensive sample enrichment and purification. This allows for the detection of even small changes in biological responses, making it possible to monitor early-stage responses of tumor cells to anti-tumor drugs and detect disease early. Additionally, multiplexed Simoa assays allow for efficient evaluation of multiple cancer nanomedicines,26 with potential applications in vaccine development, adjuvant screening, and immunotherapy. To our knowledge, nanoSimoa is the first integrated pipeline of cancer nanomedicine and Simoa, representing a significant advancement in analyzing the biological interfaces of nanoparticles. It also offers the potential for predictive tests to guide nanomaterial design and enable precision nanomedicine.
 | ||
| Scheme 1 Scheme of the nanoSimoa pipeline, which integrates cancer nanomedicine to treat cancer cells and single molecule-array (Simoa) to profile protein level changes in response to the nanomedicine. Specifically, PEGylated mesoporous silica nanoparticles (MSN-PEG) carrying topotecan (TOP), a water-soluble drug used to treat various cancers, are utilized in this study. The nanoparticles are ultrasound-responsive, and their release of TOP molecules is triggered by ultrasound stimulation inside cancer cells. To evaluate the cellular responses to the nanomedicine, cell viability is measured, and Simoa assays are used to profile protein biomarkers. If the results are unsatisfactory, we will redesign the nanomedicine and repeat the nanoSimoa pipeline. Moreover, nanoSimoa can assess multiple nanomedicines in parallel. | ||
Results and discussion
Design of ultrasound-responsive drug delivery system
To demonstrate the feasibility of the nanoSimoa pipeline, we employed a pre-existing mesoporous silica nanoparticle (MSN)-based drug delivery system.29 The MSN served as drug carriers and were synthesized through a sol–gel reaction, as previously described in our publication.29 The obtained nanoparticles had a diameter of 95.4 ± 10.1 nm (Fig. S1†) and possessed an MCM-41 type structure (Fig. 1a). To enable the loading of topotecan (TOP) in the pores of MSN and conjugation of dicarboxylic acid-terminated PEG (HOOC-PEG-COOH) (average Mw = 2000 Da) on the surface of MSN, the inner and outer surfaces of MSN were functionalized with amine groups by post-grafting the silica surface with (3-aminopropyl)triethoxysilane (APTS) (designated as MSN-APTS) (Scheme S1†). The TOP molecule becomes negatively charged at physiological pH, as its lactone moiety undergoes a reversible pH-dependent conversion to a carboxylated open-ring form. Those positively charged amine groups on the inner surfaces of MSN would increase the loading amount of TOP molecules through electrostatic interactions. On the outer surface, HOOC-PEG-COOH was conjugated to the amine groups by a standard 1-ethyl-3-(3-diethylaminopropyl) carbodiimide hydrochloride (EDC-HCl) and N-hydroxysulfosuccinimide sodium salt (sulfo-NHS) coupling reaction through amide bond formation (designated as MSN-PEG) (Scheme S1†). From transmission electron microscopy (TEM) images, it was observed that amine functionalization and PEGylation did not change the morphology of mesoporous structures of the nanoparticles (Fig. 1a). Confirmation of amine functionalization and PEGylation of MSN was achieved through zeta potential (Fig. 1b) and dynamic light scattering (DLS) measurements (Fig. 1c) after each functionalization step. MSNs exhibited a characteristic zeta potential of −22.1 mV in deionized H2O at pH 7, which shifted to +25.5 mV after APTS modification (Fig. 1b). Upon PEGylation, the zeta potential decreased to +9.7 mV due to the screening of charges by the newly formed amide bonds. The observed increase in DLS size of MSN (191.4 ± 7.1 nm), MSN-APTS (221.1 ± 4.5 nm), and MSN-PEG (310.2 ± 5.8 nm) in deionized H2O provided further evidence of successful amine functionalization and PEGylation (Fig. 1c). Additionally, the effectiveness of PEGylation in improving the colloidal stability of the nanoparticles was examined in phosphate-buffered saline (PBS) (pH 7.4). MSN-APTS showed significant aggregation in PBS, resulting in a much larger particle size (812.1 ± 12.8 nm) than that observed in H2O. However, PEGylation led to similar particle sizes in both solvents, indicating improved colloidal stability (Fig. 1c). This improvement may be due to the steric repulsion provided by PEG, which blocks surface contact between nanoparticles and prevents their aggregation. The total grafting amounts of APTS and PEG on the surface of MSN were found to be 22% of the total mass based on thermogravimetric analysis (TGA) (Fig. 1d). The functional groups on the surface of MSNs were characterized by Fourier transform-infrared spectroscopy (FT-IR) within the range of 4000–400 cm−1 (Fig. 1e). The appearance of new absorption peaks at ν = 1546 cm−1 (amide II) and ν = 2922 cm−1 (C–H stretching) in the FT-IR spectra of MSNs-PEG confirmed the successful PEGylation of the MSNs. From the N2 adsorption–desorption isotherms of MSN and MSN-PEG (Fig. 1f), compared with as-synthesized MSN, which exhibited a Brunauer–Emmett–Teller (BET) surface area of 1123 m2 g−1, a pore volume of 1.21 cc g−1 in pore volume, and an average pore diameter of 2.4 nm, MSN-PEG demonstrated a reduction in BET surface area (142 m2 g−1) and total pore volume (0.32 cc g−1) (Table S1†). These reductions can be attributed to the surface coverage of the nanoparticles and the blockage of the pore openings that occurred after the sequential grafting of APTS and PEG. | ||
| Fig. 1 Characterizations of MSN, MSN-APTS, and MSN-PEG. (a) TEM images of MSN (left) and MSN-PEG (right). (b) Zeta potential values and (c) DLS size of MSN, MSN-APTS, and MSN-PEG in deionized H2O, and MSN-APTS and MSN-PEG in PBS, respectively. (d) TGA results, (e) FT-IR spectra, and (f) N2 adsorption–desorption isotherms of MSN and MSN-PEG. | ||
The loading of TOP was accomplished by immersing 3 mg of MSN-APTS in 0.6 mL of TOP solution (2.5 mg mL−1) and stirring the mixture overnight. The TOP-loaded MSN-APTS were then conjugated with HOOC-PEG-COOH (Mw = 2000 Da) using an EDC/NHS coupling reaction to seal the pores. The mixture was reacted overnight and washed thoroughly with deionized H2O and PBS to remove excess TOP, EDC-HCl, sulfo-NHS, and PEG. Finally, TOP-MSN-PEG was stored in 3 mL of PBS for further studies. The amount of TOP loaded in MSN was determined by measuring the difference in absorbance (at 267 nm) of the initial and the recovered solutions using UV-vis spectroscopy. The loading capacity of TOP, defined as (mass of TOP loaded in pores/mass of MSN) × 100%, was calculated to be approximately 8%.
Validation of ultrasound-responsive drug release in tube
The ultrasound-stimulated TOP release was evaluated in a vial triggered by a probe sonicator (VCX 130, Sonics & Materials, Inc., Newtown, CT). The TOP-loaded MSN-PEG (designated as TOP-MSN-PEG) was suspended in physiologically relevant PBS (pH 7.4) and stimulated with the probe sonicator (20 kHz, power density: 75 W cm−2) for 2, 5, 10, 20, and 60 min. The TOP release efficiency was defined as (mass of released TOP/mass of TOP loaded in pores) × 100%. The mass of released TOP was quantified by UV-vis spectroscopy. Increasing the sonication time led to a higher amount of released TOP, with a release efficiency of 41% achieved after 60 min of sonication (Fig. 2).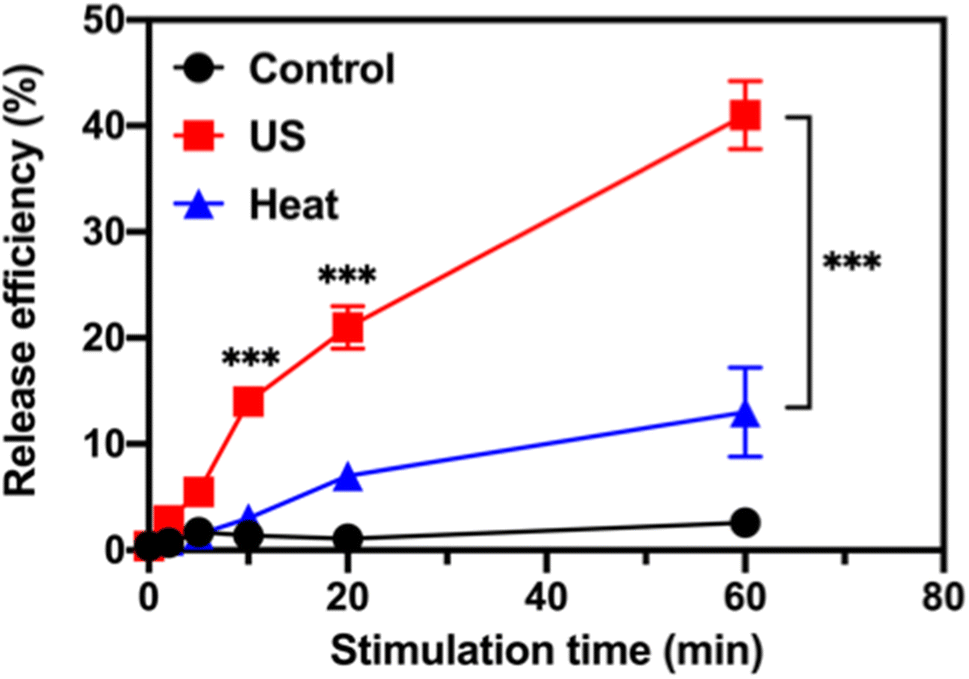 | ||
| Fig. 2 Ultrasound-stimulated topotecan (TOP) release using a probe sonicator. Time-dependent release profile of TOP from MSN-PEG after probe sonication (red), or immersed in a 50 °C (blue) water bath. The control (black) was also included. Data are means ± SD of three independent experiments (***P < 0.001). | ||
After incubating the drug-loaded nanoparticles in a hot water bath at 50 °C, a significant decrease in TOP release efficiency was observed. Only 13% of the TOP was released after 60 min of sonication (Fig. 2), which confirms that ultrasound waves were responsible for uncapping the PEG-covered pores of MSN, and that the amount of released cargo was controllable based on the triggering time. Our previous research showed that besides heat, the mechanical effects of ultrasound (such as cavitation) could rupture the PEG layer and facilitate the efficient release of the entrapped cargo.29 At a molecular level, cavitation causes small molecules (e.g., solvent molecules and TOP) to move rapidly, generating friction, increasing strain, and eventually leading to bond rupture.39 TEM image of MSN-PEG after ultrasound treatment and heating at 50 °C for 30 min (Fig. S2†) showed that the mesoporous structure was still intact, suggesting that the release of TOP was primarily controlled by the PEG gatekeeper and not the destruction of the mesoporous silica structure.
In vitro evaluation of ultrasound-responsive drug release and therapeutic efficacy
After fully characterizing the MSN-PEG drug delivery system and evaluating its ultrasound-responsive TOP release capability in a tube, the next step was to investigate its in vitro ultrasound-responsive behavior, resulting therapeutic efficacy, and biological responses using human ovarian carcinoma OVCAR-3 cells as a cell model.To assess the cytotoxicity of MSN-PEG on OVCAR-3 cells, different nanoparticle concentrations were used to treat the cells for 4 h, followed by overnight incubation for cell proliferation. It was observed that MSN-PEG did not exhibit any inherent cytotoxicity to OVCAR-3 cells at both time points, even at a high particle concentrations of up to 100 μg mL−1, as analyzed by a CCK-8 assay (Fig. 3a). The tightness of the MSN-PEG drug delivery system prior to ultrasound stimulation was also examined. The drug-loaded nanoparticles exhibited minimal cytotoxicity after 4 h of incubation, even at high particle concentration of 100 μg mL−1 (Fig. 3b). Additionally, cell proliferation after 20 h of growth was not significantly affected at this nanoparticle concentration, suggesting minimal drug leakage in the biological environment at 37 °C over the test period (Fig. 3b and S3†).
 | ||
Fig. 3 (a) Optimization of ultrasound-responsive drug delivery system for cell study. OVCAR-3 cells were treated with (a) MSN-PEG or (b) TOP-MSN-PEG at different nanoparticle concentrations for 4 h with and without an additional 20 h of incubation. Cell viability was measured by a CCK-8 assay and normalized to the control without MSN-PEG treatment. Data are means ± SD of three independent experiments. (c) Fluorescence microscope images of OVCAR-3 cells after 4 h treatment with TOP-MSN-PEG. The control groups include cells treated with MSN-PEG or TOP alone, and cells without any treatment. The panels from top to bottom show the blue-emitting nucleus stained by Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342, green emission from TOP, and the merged images. (d) Optimization of ultrasound stimulation parameters for cells. The attached OVCAR-3 cells were exposed to ultrasound at different power densities and stimulation times by a probe sonicator. The viability of the treated cells was determined by the CCK-8 assay and normalized to the control group without ultrasound treatment. Data are means ± SD of three independent experiments. The scale bar is 20 μm. 342, green emission from TOP, and the merged images. (d) Optimization of ultrasound stimulation parameters for cells. The attached OVCAR-3 cells were exposed to ultrasound at different power densities and stimulation times by a probe sonicator. The viability of the treated cells was determined by the CCK-8 assay and normalized to the control group without ultrasound treatment. Data are means ± SD of three independent experiments. The scale bar is 20 μm. | ||
To confirm the uptake and delivery of TOP-MSN-PEG in cells, a concentration of 75 μg mL−1 was employed for subsequent cellular experiments. Fluorescence microscope images were analyzed, revealing that OVCAR-3 cells treated with TOP-MSN-PEG for 4 h exhibited significantly higher green fluorescence, derived from TOP. This suggests that the cells had taken up the TOP-MSN-PEG and exhibited a higher cellular uptake of TOP compared to the control groups of cells without treatment, cells treated with nanoparticles only, and cells treated with the drug alone (Fig. 3c).
To ensure cell safety, we optimized the ultrasound stimulation parameters in vitro. Initially, cells were allowed to attach to the bottom of an 8-well plate overnight without exposure to nanoparticles. We then exposed the cells to different power densities and stimulation durations using the probe sonicator. Our results revealed that cell viability gradually decreased with longer exposure times at a power density of 75 W cm−2 (Fig. 3d). We also monitored the temperature increases of the cell supernatant post-ultrasound treatments. No significant temperature increases were observed at a power density of 75 W cm−2 and 15 s of ultrasound stimulation duration, or a power density lower than 56.3 W cm−2 and duration shorter than 60 s (Table S2†). However, with ultrasound stimulation duration at 75 W cm−2, gradual temperature increases occurred. Based on these findings, we selected a power density of 75 W cm−2 and a stimulation time of 30 s as the ultrasound conditions for our in vitro drug release study.
Prior to conducting the cell study, the behavior of TOP-MSN-PEG in RPMI medium was investigated. After subjecting the nanoparticles to ultrasound at a power density of 75 W cm−2, they were divided into two groups, one incubated overnight and the other not. Results showed that although drug release was slower and significantly lower with brief ultrasound stimulation, the release efficiency could match that of longer ultrasound treatments after an overnight incubation period (Fig. 4a).
 | ||
| Fig. 4 Ultrasound-stimulated TOP release in RPMI medium using a probe sonicator. (a) Time-dependent release profile of TOP from MSN-PEG immediately (black) or after a 20 h incubation (red) post probe sonication. (b) Cell viability of OVCAR-3 cells treated with TOP-MSN-PEG, the equivalent amount of TOP alone (positive control), MSN-PEG, and cells only (negative control), with or without ultrasound stimulation (power density: 75 W cm2; stimulation time: 30 s). Cell viability was determined by the CCK-8 assay and normalized to the control group without any treatment. Data are means ± SD of three independent experiments (***P < 0.001). | ||
Once the ultrasound conditions were optimized, the cells were treated with the appropriate dose of TOP-MSN-PEG and subjected to ultrasound to release the drug. The group that received the TOP-MSN-PEG and ultrasound treatment displayed the highest therapeutic efficacy, as evidenced by the lowest cell viability compared to the control groups (Fig. 4b). The presence of the drug carrier facilitated the delivery of a larger amount of TOP to the cells, resulting in higher therapeutic efficacy than the drug-only group (Fig. 4b). To confirm the importance of the drug carrier, the cells were treated with supernatants obtained from the TOP-MSN-PEG solution after ultrasound treatment. Cells treated with the supernatant from the TOP-MSN-PEG solution that underwent longer ultrasound stimulation displayed a slight reduction in cell viability, highlighting the crucial role of the MSN-PEG drug carrier (Fig. S3†).
Simoa-based quantification of IL-6 secretion from OVCAR-3 cells and correlation with therapeutic efficacy
Studies have recently emphasized the significant role of IL-6 in tumor progression,40–42 particularly in ovarian carcinoma, where elevated serum levels of IL-6 are associated with poor clinical outcomes.43–45 This study represents the first integration of cancer nanomedicine and Simoa in a pipeline (nanoSimoa). The aim of this research is to investigate the impact of the ultrasound-responsive drug delivery system on IL-6 secretion under different treatment conditions, evaluate the ability of Simoa to quantify changes in IL-6 concentrations, and assess the correlation between IL-6 levels quantified by Simoa and therapeutic efficacy measured by the CCK-8 assay.To establish a baseline level of IL-6 secretion by human ovarian cancer cell lines, OVCAR-3 cells were seeded at varying cell densities (103, 104, 105, and 106 cells per well) in growth medium (RPMI containing 20% fetal bovine serum (FBS) and 0.01 mg mL−1 bovine insulin) in individual wells of a 6-well plate. After 24 hours of incubation, the cells were washed with DPBS, and a fresh growth medium was added. Following a 48 hour incubation, the supernatants were collected and analyzed for the presence of IL-6 using a commercially available ELISA kit (R&D Systems). Fig. 5a illustrates that OVCAR-3 cells exhibited a baseline level of secreted IL-6 that correlated with cell density. However, the lowest cell seeding density did not produce detectable levels of IL-6, possibly due to the suboptimal sensitivity (about 1–10 pg mL−1) of the ELISA kit used.
 | ||
| Fig. 5 Simoa-based quantification of IL-6 secretion from OVCAR-3 cells. (a) Correlation between IL-6 baseline levels of OVCAR-3 cells measured by Simoa (solid red) or ELISA kit (dotted black) at different cell-seeded density. (b) Simoa-based quantification of IL-6 secretion from OVCAR-3 cells treated with TOP-MSN-PEG, the equivalent amount of TOP alone (positive control), MSN-PEG, and cells only (negative control), with or without ultrasound stimulation (power density: 75 W cm2; stimulation time: 30 s). IL-6 concentrations were measured by IL-6 Simoa assay. Data are means ± SD of two independent experiments (*P < 0.05; **P < 0.01; ***P < 0.001). | ||
To evaluate the Simoa platform's ability to measure the baseline level of secreted IL-6, an IL-6 Simoa assay with a three-step format was developed using the HD-X analyzer (Quanterix). The cell culture supernatant was diluted in Homebrew Detector/Sample Diluent, and a biotinylated anti-IL-6 antibody and streptavidin-β-galactosidase (SβG) concentrate were used in the assay. System Wash Buffer 1, System Wash Buffer 2, resorufin β-D-galactopyranoside (RGP), and Simoa Sealing oil were used according to the manufacturer's instructions.
In the first step of the assay, 25 μL of the anti-IL-6 antibody-coupled beads were incubated with 100 μL of the diluted cell supernatant for 15 min. The total number of beads used per reaction was 500000, comprising 125000 IL-6 capture beads and 375000 helper beads. After incubation, the beads were washed six times with System Wash Buffer 1. In the second step, the beads were resuspended in 100 μL of biotinylated anti-IL-6 antibody and incubated for 5.25 min, followed by six wash steps with System Wash Buffer 1. In the third step, the beads were resuspended in 100 μL of SβG, incubated for 5.25 min, and washed six times. Finally, the beads were resuspended in 25 μL of RGP and loaded into the microwell array for analysis. The microwell array was sealed with oil and imaged in five optical channels, and the HD-X analyzer software calculated the Average Enzyme per Bead (AEB) values. All samples were measured in duplicates.
The IL-6 Simoa assay was able to detect and quantify IL-6 secretion from as few as 103 seeded cells, which was not possible with the commercial ELISA kit (Fig. 5a). The limit of detection (LOD) of the IL-6 Simoa assay was calculated to be 0.013 pg mL−1 (0.6 fM), which was two to three orders of magnitude more sensitive than the conventional IL-6 ELISA assay (1–10 pg mL−1). Furthermore, a strong correlation was observed between cell density and IL-6 baseline levels, and the IL-6 concentrations measured by both ELISA and Simoa were comparable, confirming the reliability of the Simoa platform.
To investigate the impact of the ultrasound-responsive drug delivery system on IL-6 secretion levels under various treatment conditions, OVCAR-3 cells were seeded at a density of 105 cells per well and treated with varying conditions. The cell culture supernatants were then analyzed using the IL-6 Simoa assay. Remarkably, only the group treated with TOP-MSN-PEG and ultrasound stimulation showed a significant reduction in IL-6 secretion, indicating that the combination of drug delivery and ultrasound may have a synergistic effect in reducing IL-6 levels (Fig. 5b). The lack of significant changes in IL-6 levels in the other treatment groups suggests that these treatments did not significantly impact the IL-6 signaling pathway. However, further experiments may be necessary to fully understand the effects of these treatments on IL-6 secretion and their potential therapeutic implications.
Simoa-based quantification of Ras baseline levels of OVCAR-3 cells
For years, Ras proteins encoded by HRAS, KRAS, and NRAS genes expressed throughout the body have been recognized for their crucial role in human cancer, with frequent mutations occurring in cancerous cells. These proteins have been the target of significant research efforts to develop inhibitors.46–48 Ras proteins not only promote cell growth and differentiation but also induce IL-6 secretion in various cell types, facilitating angiogenesis and tumor growth.49,50 Inhibiting IL-6 production through knockdown, genetic ablation, or neutralizing antibodies has been shown to impede Ras-driven tumorigenesis, indicating that targeting IL-6 could be therapeutically beneficial in cancers with oncogenic Ras mutations.49 Despite the availability of Western blotting using Ras-specific antibodies for detecting Ras proteins, limited Ras immunoassays have been established to date.51,52 Additionally, since Ras is an intracellular protein that has not been investigated as a biomarker, its concentration in the blood remains unknown. Thus, developing a sensitive tool capable of accurately quantifying Ras protein levels is crucial.To assess the potential of the Simoa platform for quantifying Ras protein levels in cells, we developed a Ras Simoa assay to determine Ras protein levels in OVCAR-3 cells. Given the connection between Ras and IL-6, we theorized that measuring Ras protein levels using the Simoa assay could serve as a useful indicator of IL-6 production and its potential role in Ras-driven cancer. The cells were lysed in cell lysis buffer (Thermo Fisher) at a concentration of 1000 cells per μL and evaluated for Ras protein levels using the HD-X analyzer. The assay followed a similar protocol to the IL-6 Simoa assay, using biotinylated anti-Ras antibodies (ab209974, Abcam) as the detector antibody and anti-Ras antibodies (ab55391, Abcam) as the capture antibody.
Our study successfully detected and quantified Ras protein levels using the Ras Simoa assay with a LOD of 0.12 pM (Fig. 6a). We further tested the assay in OVCAR-3 cell lysate and found that it could detect Ras proteins at cell densities as low as 14 cells per mL (Fig. 6b). By correlating Ras protein levels with cell density, we estimated that OVCAR-3 cells have approximately one million Ras protein molecules per cell (Fig. 6c). These results suggest that Ras Simoa assay may be a valuable tool for measuring Ras protein levels in the bloodstream, and identifying and monitoring Ras-mediated diseases. Additionally, the assay's high sensitivity may facilitate the development of targeted therapies for Ras-driven tumors. By integrating Ras and IL-6 Simoa assays, it may be possible to gain a better understanding of how these two molecules interact in the context of cancer progression. This knowledge could be used to develop targeted therapies for Ras-driven tumors.49
 | ||
| Fig. 6 Simoa-based quantification of Ras baseline levels of OVCAR-3 cells. Calibration curves of Ras Simoa assay at different (a) Ras concentrations or (b) OVCAR-3 cell densities. The correlation between Ras concentrations and cell densities. Ras concentrations from Ras recombinant proteins or cell lysates were measured by Ras Simoa assay. Data are means ± SD of two independent experiments. | ||
Overall, the integrated nanoSimoa pipeline has the potential to enable the rational design of nanotherapeutics and provide insights into how those nanotherapeutics may behave in the context of Ras-mediated cancer. If this approach proves to be effective and applicable to other cancer types, it could serve as a valuable tool for preclinical testing of cancer nanomedicine and accelerate the development of more effective cancer treatments.
Conclusions
In summary, this study introduced a new pipeline, nanoSimoa, that integrates cancer nanomedicine with a high-throughput protein detection tool to analyze cellular responses to nanomedicine with high sensitivity and resolution. The nanoSimoa pipeline has the potential to facilitate the rational development of nanotherapeutics and predict their in vivo behaviors, thereby reducing the use of animals in preclinical cancer nanomedicine studies. Our approach aligns with the 3 Rs principle of replacement, reduction, and refinement,53 which guides animal research and has the potential to advance the field of cancer nanomedicine significantly. The success of the nanoSimoa pipeline underscores the importance of combining advanced analytical tools with a rigorous approach to accelerate the success rate of cancer nanomedicine.Materials and methods
Materials and chemicals
Hexadecyltrimethylammonium bromide (CTAB, 99+%), tetraethyl orthosilicate (TEOS, 98%), (3-aminopropyl)triethoxysilane (APTS, 99%), ammonium nitrate (NH4NO3, 98+%), 2-(N-morpholino)ethanesulfonic acid hydrate (MES hydrate, 99.5+%), 1-ethyl-3-(-3-dimethylami-nopropyl) carbodiimide hydrochloride (EDC-HCl, 99+%), and N-hydroxysulfosuccinimide sodium salt (sulfo-NHS, 99+ %), topotecan hydrochloride hydrate (TOP, ≥98%), bovine insulin, trypsin–EDTA, phosphate-buffered saline (PBS, 10×), and bisBenzimide H 33342 trihydrochloride (Hoechst 33342, 98+%) were purchased from Sigma-Aldrich. EDC (No-Weigh™ Format) and sodium hydroxide (NaOH, 97+%) was purchased from ThermoFisher Scientific. α,ω-Bis-carboxy polyethylene glycol (HOOC-PEG-COOH, Mw = 2000 Da) was purchased from Iris Biotech GmbH. RPMI-1640 and fetal bovine serum (FBS) were purchased from Gibco. fetal bovine serum (FBS), antibiotics (10000 U per mL penicillin, 10000 μg per mL streptomycin, and 29.2 mg per mL L-glutamine), Dulbecco's phosphate-buffered saline (DPBS), and trypsin–ethylenediaminetetraacetic acid (trypsin–EDTA) (0.05%) were purchased from Gibco. Cell counting kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc. Paraformaldehyde solution (4% in PBS) was purchased from USB Corporation. Ethanol was purchased from Echo Chemical. Human epithelial ovarian adenocarcinoma cell line NIH-OVCAR-3 was purchased from ATCC. 750 nm dye-encoded carboxylated paramagnetic beads (Quanterix), Bead Wash Buffer, Bead Conjugation Buffer, Bead Blocking Buffer, Bead Diluent, Homebrew Detector/Sample Diluent, Streptavidin-β-galactosidase (SβG) concentrate, SβG Diluent, System Wash Buffer 1, System Wash Buffer 2, resorufin β-D-galactopyranoside (RGP) and Simoa Sealing Oil were purchase from Quanterix. Anti-IL-6 antibody and biotinylated anti-IL-6 antibody were purchased from R&D Systems. Anti-Ras antibody and biotinylated anti-Ras antibody were purchased from Abcam. All chemicals were used without further purification.
Characterization
The characterization of nanoparticle morphology and diameter was performed using transmission electron microscopy (TEM, Tecnai T12) with an operating voltage of 120 kV. MSN and MSN-APTS were dispersed in ethanol at a concentration of 0.1 mg mL−1, and 10 μL of the resulting nanoparticle suspension was dropped onto a carbon-coated copper grid and dried at room temperature. The surface area, pore diameter, and pore volume of MSN and MSN-PEG were determined by measuring N2 adsorption–desorption isotherms at 77 K (Autosorb-iQ, Quantachrome Instruments). Prior to measurement, the nanoparticles were degassed at 120 °C for 20 h. The surface area and pore diameter distribution of the nanoparticles were analyzed using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods. Thermogravimetric analysis (TGA) was conducted on a PerkinElmer Pyris Diamond TG/DTA machine under air flow. MSN and MSN-PEG (5–10 mg) were loaded in aluminum pans, and the data were recorded from 30 °C to 550 °C at a scan rate of 10 °C min−1. An empty aluminum pan served as the reference. The functional groups on the surface of MSNs were characterized by Fourier transform infrared spectroscopy (FT-IR, JASCO FT/IR-420) spectrometer in the range of 4000–400 cm−1. The loading capacity, release capacity, and release efficiency of topotecan (TOP) were determined by UV-vis spectroscopy (Cary 5000). The absorbance of the peaks was used for quantification by the Beer–Lambert law.Synthesis of mesoporous silica nanoparticles (MSN)
The synthesis of mesoporous silica nanoparticles involved a sol–gel reaction with the use of cationic surfactant templates. In general, CTAB (250 mg) was dissolved with vigorous stirring in a 250 mL round bottom flask containing 120 mL of deionized H2O and 875 μL of NaOH solution (2 M). The resulting solution was then heated to 80 °C in an oil bath and maintained at this temperature for 30 min before the dropwise addition of 1.25 mL of TEOS. The reaction mixture was further stirred for 2 h to facilitate the formation of MSN. Following this, the solution was cooled to room temperature, centrifuged, and washed three times with ethanol to eliminate any unreacted precursors and free surfactants.Synthesis of amine functionalized MSN (MSN-APTS)
In order to add amine groups to the surface of MSN, a solution of MSN (180 mg) in a mixture of 50 mL of anhydrous toluene and 150 μL of APTS was heated to 110 °C and refluxed for 12 h under vigorous stirring. The resulting APTS-modified MSN (MSN-APTS) was washed twice with ethanol. To remove the surfactant template, MSN-APTS were dispersed in a solution of 100 mL of ethanol and 2 g of NH4NO3 in a 250 mL round bottom flask, heated to 78 °C, and refluxed for 1 h. This extraction process was repeated twice. After extraction, the MSN-APTS were centrifuged and washed twice with deionized H2O and ethanol, respectively. The final product was stored in ethanol for future use.Topotecan (TOP) loading in MSN-APTS and PEG capping (MSN-PEG)
To prepare TOP-loaded MSN-APTS, 3 mg of MSN-APTS were washed once with MES solution (100 mM, pH = 6.0) and then soaked in 0.6 mL of TOP solution (2.5 mg mL−1). After stirring overnight to allow TOP molecules to diffuse into the pores of MSN-APTS, the pores were sealed by conjugating with HOOC-PEG-COOH (Mw = 2000 Da). EDC-HCl and sulfo-NHS coupling reagents were dissolved in MES solution, and HOOC-PEG-COOH was activated by stepwise addition to the solution every 10 min. Activated PEG was then stepwise added into the TOP-loaded MSN-APTS solution every 10 min to react with the primary amines on the surface of MSN-APTS. The resulting MSN-PEG was washed three times with deionized H2O to remove excess reactants and unloaded TOP. The TOP-loaded MSN-PEG (designated as TOP-MSN-PEG) was stored in deionized H2O for further studies on ultrasound-stimulated release.Ultrasound-stimulated release of TOP
A solution of TOP-MSN-PEG (1 mg mL−1) was prepared in deionized H2O in an Eppendorf tube. The tip of the probe sonicator (VCX 130, Sonics & Materials, Inc., Newtown, CT) was positioned in the center of the solution, and the ultrasound parameter was set with a frequency of 20 kHz and an output power of 21 W (power density: 75 W cm−2). Following various ultrasound stimulation time durations (2, 5, 8, 10, or 30 min) with the probe sonicator, the solution was centrifuged (14000 rpm, 10 min) to separate the particles and the supernatant containing released TOP. The supernatant and pellet were collected separately to further quantify TOP loading capacity and release efficiency by UV-vis spectroscopy.
TOP loading capacity analysis
In general, 3 mg of MSN was dispersed in MES with 0.6 mL of TOP solution (2.5 mg mL−1) and left to stir for 24 h. Subsequently, the TOP-MSN-PEG were centrifuged at 14000 rpm for 10 min, and the pellets were washed three times with deionized H2O while being sonicated to remove any excess TOP. Then, the TOP-MSN-PEG was washed twice with PBS and centrifuged to obtain a pellet. This washing procedure was repeated multiple times until the supernatant was clear. The resulting supernatants were collected and analyzed using UV-vis spectroscopy. The loading capacity of TOP was determined using Beer's law, a calibration curve, and the formula: (mass of loaded TOP/mass of MSN) × 100%, based on the maximum absorbance at 267 nm.
TOP release efficiency analysis after ultrasound stimulation
Following ultrasound stimulation, the TOP-MSN-PEG was centrifuged to separate the pellet and the supernatant. The collected supernatants, containing the released TOP, were analyzed using UV-vis spectroscopy with a maximum absorbance at 267 nm. The TOP released after being heated at 80 °C for 30 min was used as the reference for 100% release. The release efficiency of TOP was calculated using the definition: (mass of released TOP/mass of TOP loaded in pores) × 100%.OVCAR-3 cell culture
Human ovarian cancer cells (OVCAR-3) (HTB-161, ATCC) were cultured in T-75 flasks (Corning) with vented caps in RPMI-1640 supplemented with 20% fetal bovine serum (FBS) and 0.01 mg mL−1 bovine insulin at 37 °C and 5% CO2 in a humidity-controlled incubator. The culture media were refreshed daily, and the cells were harvested using 0.05% trypsin–EDTA for passaging every 2–3 days.OVCAR-3 cell viability and proliferation
The cell counting kit-8 (CCK-8) assay was used to evaluate the impact of MSN-PEG and TOP-MSN-PEG on the viability and proliferation of OVCAR-3 cells. The cells were initially seeded in 96-well plates at a density of 5 × 103 cells per well in 200 μL of RPMI-1640 supplemented with 20% fetal bovine serum (FBS) and 0.01 mg mL−1 bovine insulin and incubated for 24 h. After the attachment, the medium was removed and the cells were treated with varying concentrations (0, 10, 25, 50, 75, 100, and 200 μg mL−1) of MSN-PEG or TOP-MSN-PEG for 4 h in fresh RPMI at 37 °C. After treatment, the medium was removed and the cells were washed three times with DPBS and incubated with RPMI (100 μL) and CCK-8 reagent (10 μL) for 2 h at 37 °C. The viability of the cells was determined by measuring the absorbance at 450 nm and 650 nm (as the reference) using a plate reader (Tecan M1000), with RPMI containing CCK-8 reagent as a background. For the cell proliferation study, the cells were treated with MSN-PEG or TOP-MSN-PEG for 4 h, washed twice with DPBS, and then allowed to grow in a fresh culture medium for 20 h before conducting the CCK-8 assay.Fluorescence microscope images of OVCAR-3 cells
OVCAR-3 cells were seeded in a 24-well chamber slide at a density of 6 × 104 cells per well in 300 μL of RPMI supplemented with 20% FBS and 0.01 mg mL−1 bovine insulin at 37 °C for 48 h. After attachment, the cells were treated with 22.5 μg of TOP-MSN-PEG (75 μg mL−1) in 300 μL of fresh RPMI for 4 h at 37 °C, washed three times with DPBS, then fixed with 4% paraformaldehyde in DPBS for 20 min. The fixed cells were then washed with DPBS three times. Afterwards, the cell nuclei were stained with Hoechst 33342 (500 μL, 5 μg mL−1) for 20 min and washed with DPBS five times. The stained cells were covered with a mounting medium cover glass before being observed under a Zeiss fluorescence microscope. Control groups, including cells only (negative control), cells treated with an equivalent amount of free TOP (positive control) to the TOP-MSN-PEG, and cells treated with MSN-PEG (75 μg mL−1), were also evaluated.
Ultrasound-stimulated TOP release and resulting cell viability
OVCAR-3 cells were cultured in a 24-well chamber slide at a density of 6 × 104 cells per well in 300 μL of RPMI supplemented with 10% FBS and 1% antibiotics for 48 h at 37 °C. Following attachment, the medium was removed and the cells were treated with TOP-MSN-PEG (75 μg mL−1) in 300 μL of fresh RPMI and incubated at 37 °C. Control groups, including cells only (negative control), cells treated with an equivalent amount of free TOP (positive control) to the TOP-MSN-PEG, and cells treated with MSN-PEG (75 μg mL−1), were also evaluated. After 4 h of incubation, the medium was removed, and the cells were washed twice with DPBS. The probe sonicator was positioned in the center of the well to stimulate the cells with ultrasound. The ultrasound parameter was set at a power density of 75 W cm−2 and a stimulation time of 30 s. The cell viability following ultrasound stimulation was measured using the CCK-8 assay.Preparation of IL-6 and Ras capture beads for single-molecule array (Simoa) assays
The IL-6 Simoa assay capture beads were prepared as follows. Prior to bead conjugation, 2.8 × 108 of 750 nm dye-encoded carboxylated paramagnetic beads (Quanterix) were washed three times with 200 μL of Bead Wash Buffer (Quanterix), followed by three washes with 200 μL of Bead Conjugation Buffer (Quanterix), and resuspended in 300 μL of Bead Conjugation Buffer. Freshly prepared 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (ThermoFisher Scientific 77149) (10 mg mL−1 in Bead Conjugation Buffer) was immediately added to activate the beads. The beads were then agitated on a HulaMixer (ThermoFisher Scientific) for 30 min at 4 °C and washed once with a Bead Conjugation Buffer. The beads were resuspended in a Bead Conjugation Buffer containing anti-IL-6 antibodies (MAB206, R&D Systems) and agitated on the HulaMixer for 2 h at 4 °C. Following bead conjugation, the anti-IL-6 antibody-coupled beads were washed twice with Bead Wash Buffer and blocked with Bead Blocking Buffer (Quanterix) for 30 min at room temperature. After blocking, the beads were washed once with Bead Wash Buffer and twice with Bead Diluent (Quanterix) before resuspending in 200 μL of Bead Diluent. Finally, the beads were counted using a Beckman Coulter Z1 Particle Counter and stored at 4 °C for future use.The preparation of the capture beads for the Ras Simoa assay was similar to the one described earlier, except that the capture antibody used was anti-Ras antibodies (ab55391, Abcam).
IL-6 and Ras Simoa assays
The IL-6 Simoa assays were performed in an automated three-step assay format onboard the HD-X Analyzer (Quanterix). The supernatant from the cell culture or the cell lysate was collected and diluted in Homebrew Detector/Sample Diluent (Quanterix). Biotinylated anti-IL-6 antibodies (BAF206, R&D Systems) were diluted in Homebrew Detector/Sample Diluent to final concentrations of 0.3 μg mL−1. Streptavidin-β-galactosidase (SβG) concentrate (Quanterix) was diluted to 150 pM in SβG Diluent (Quanterix). System Wash Buffer 1, System Wash Buffer 2, resorufin β-D-galactopyranoside (RGP), and Simoa Sealing Oil were purchased from Quanterix and loaded onto the HD-X Analyzer per the manufacturer's instructions. In the first step of the assay, 25 μL of the anti-IL-6 antibodies-coupled beads were incubated with 100 μL of diluted cell supernatant for 15 min. The total number of beads used per reaction was 500000 (125000 of the IL-6 capture beads and 375000 of helper beads). After incubation, six wash steps were performed with System Wash Buffer 1. In the second step, the beads were resuspended in 100 μL of the biotinylated anti-IL-6 antibody and incubated for 5.25 min, then washed six times with System Wash Buffer 1. In the third step, the beads were resuspended in 100 μL of SβG, incubated for 5.25 min and washed six times. The beads were mixed with 25 μL of RGP and loaded into the microwell array for analysis. The microwell array was then sealed with oil and imaged. The software in the HD-X analyzer was used to calculate the average number of enzymes per bead (AEB) values. A four-parameter logistic weighted regression was used to fit the calibration curve and determine protein concentrations in the samples based on their measured AEB values. The limit of detection (LOD) value was calculated by adding 3 times the standard deviation to the mean AEB of the background.
The Ras Simoa assay was conducted using a procedure similar to the one described above, except that the detector antibody used was biotinylated anti-Ras antibodies (ab209974, Abcam).
Author contributions
C.-A. C. is the first and corresponding author. C.-A. C.: conceptualization (lead); data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); project administration (lead); resources (lead); validation (lead); visualization (lead); writing – original draft (lead); writing – review and editing (lead). L.-C. C. and Y.-S.C.: data curation (supporting); investigation (supporting); methodology (supporting); validation (supporting); visualization (supporting). All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge all the financial supports received from NSTC, MOE, NTU, NTU College of Medicine, and NTU School of Pharmacy in Taiwan. This work was largely funded through a grant from the National Science and Technology Council, Taiwan (NSTC 111-2628-B-002-051) from the 2030 Cross-Generation Young Scholars Program awarded to C.-A.C. In addition, C.-A.C. was supported by the grant (MOE 111V1025-1) from Yushan Fellow Program provided by the Ministry of Education (MOE), Taiwan; National Taiwan University (NTU) Academic Research-Career Development Project (Sprout Research Project 112L7847); New Faculty Founding Research Grants and Match Funds provided from NTU, NTU College of Medicine, and School of Pharmacy.References
- H. He, L. Liu, E. E. Morin, M. Liu and A. Schwendeman, Survey of Clinical Translation of Cancer Nanomedicines—Lessons Learned from Successes and Failures, Acc. Chem. Res., 2019, 52(9), 2445–2461 CrossRef CAS PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Cancer nanomedicine: progress, challenges and opportunities, Nat. Rev. Cancer, 2017, 17(1), 20–37 CrossRef CAS PubMed.
- S. Y. Choi, D. Lin, P. W. Gout, C. C. Collins, Y. Xu and Y. Wang, Lessons from patient-derived xenografts for better in vitro modeling of human cancer, Adv. Drug Delivery Rev., 2014, 79–80, 222–237 CrossRef CAS PubMed.
- N. E. Sharpless and R. A. Depinho, The mighty mouse: genetically engineered mouse models in cancer drug development, Nat. Rev. Drug Discovery, 2006, 5(9), 741–754, DOI:10.1038/nrd2110.
- Y. Lu, S. Kim and K. Park, In vitro–in vivo correlation: perspectives on model development, Int. J. Pharm., 2011, 418(1), 142–148 CrossRef CAS PubMed.
- S. H. Shin, M. Yu, D. C. Hammell, P. Ghosh, S. G. Raney, H. E. Hassan and A. L. Stinchcomb, Evaluation of in vitro/in vivo correlations for three fentanyl transdermal delivery systems using in vitro skin permeation testing and human pharmacokinetic studies under the influence of transient heat application, J. Controlled Release, 2022, 342, 134–147 CrossRef CAS PubMed.
- J. D. Cohen, L. Li, Y. Wang, C. Thoburn, B. Afsari, L. Danilova, C. Douville, A. A. Javed, F. Wong and A. Mattox, et al., Detection and localization of surgically resectable cancers with a multi-analyte blood test, Science, 2018, 359(6378), 926–930 CrossRef CAS PubMed.
- W. Zhang, W. Xia, Z. Lv, C. Ni, Y. Xin and L. Yang, Liquid Biopsy for Cancer: Circulating Tumor Cells, Circulating Free DNA or Exosomes?, Cell. Physiol. Biochem., 2017, 41(2), 755–768 CrossRef CAS PubMed.
- D. Lin, L. Shen, M. Luo, K. Zhang, J. Li, Q. Yang, F. Zhu, D. Zhou, S. Zheng and Y. Chen, et al., Circulating tumor cells: biology and clinical significance, Signal Transduction Targeted Ther., 2021, 6(1), 404 CrossRef CAS PubMed.
- J. C. M. Wan, C. Massie, J. Garcia-Corbacho, F. Mouliere, J. D. Brenton, C. Caldas, S. Pacey, R. Baird and N. Rosenfeld, Liquid biopsies come of age: towards implementation of circulating tumour DNA, Nat. Rev. Cancer, 2017, 17(4), 223–238 CrossRef CAS PubMed.
- E. Crowley, F. Di Nicolantonio, F. Loupakis and A. Bardelli, Liquid biopsy: monitoring cancer-genetics in the blood, Nat. Rev. Clin. Oncol., 2013, 10(8), 472–484 CrossRef CAS PubMed.
- X. Zhang, X. Yuan, H. Shi, L. Wu, H. Qian and W. Xu, Exosomes in cancer: small particle, big player, J. Hematol. Oncol., 2015, 8(1), 83 CrossRef PubMed.
- R. Rahbarghazi, N. Jabbari, N. A. Sani, R. Asghari, L. Salimi, S. A. Kalashani, M. Feghhi, T. Etemadi, E. Akbariazar and M. Mahmoudi, et al., Tumor-derived extracellular vesicles: reliable tools for Cancer diagnosis and clinical applications, Cell Commun. Signaling, 2019, 17(1), 73 CrossRef PubMed.
- J. Wu, Z. Lin, Z. Zou, S. Liang, M. Wu, T. Y. Hu and Y. Zhang, Identifying the Phenotypes of Tumor-Derived Extracellular Vesicles Using Size-Coded Affinity Microbeads, J. Am. Chem. Soc., 2022, 144(51), 23483–23491 CrossRef CAS PubMed.
- Y. C. Song, G. X. Lu, H. W. Zhang, X. M. Zhong, X. L. Cong, S. B. Xue, R. Kong, D. Li, Z. Y. Chang and X. F. Wang, et al., Proteogenomic characterization and integrative analysis of glioblastoma multiforme, Oncotarget, 2017, 8(57), 97304–97312 CrossRef PubMed.
- A. Akcakanat, X. Zheng, C. X. Cruz Pico, T.-B. Kim, K. Chen, A. Korkut, A. Sahin, V. Holla, E. Tarco and G. Singh, et al., Genomic, Transcriptomic, and Proteomic Profiling of Metastatic Breast Cancer, Clin. Cancer Res., 2021, 27(11), 3243–3252 CrossRef CAS PubMed.
- D. Ren, Y. Hua, B. Yu, X. Ye, Z. He, C. Li, J. Wang, Y. Mo, X. Wei and Y. Chen, et al., Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy, Mol. Cancer, 2020, 19(1), 19 CrossRef PubMed.
- E. Engvall and P. Perlmann, Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G, Immunochemistry, 1971, 8(9), 871–874 CrossRef CAS PubMed.
- L. Cohen and D. R. Walt, Highly Sensitive and Multiplexed Protein Measurements, Chem. Rev., 2019, 119(1), 293–321 CrossRef CAS PubMed.
- D. Wu, T. L. Dinh, B. P. Bausk and D. R. Walt, Long-Term Measurements of Human Inflammatory Cytokines Reveal Complex Baseline Variations between Individuals, Am. J. Pathol., 2017, 187(12), 2620–2626 CrossRef CAS PubMed.
- Y. W. Kwon, H. S. Jo, S. Bae, Y. Seo, P. Song, M. Song and J. H. Yoon, Application of Proteomics in Cancer: Recent Trends and Approaches for Biomarkers Discovery, Front. Med., 2021, 8, 747333 CrossRef PubMed.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang and A. J. Rivnak, et al., Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations, Nat. Biotechnol., 2010, 28(6), 595–599 CrossRef CAS PubMed.
- D. M. Rissin, D. H. Wilson and D. C. Duffy, Chapter 2.13 – Measurement of Single Protein Molecules Using Digital ELISA, in The Immunoassay Handbook, ed. D. Wild, Elsevier, 4th edn, 2013, pp. 223–242 Search PubMed.
- D. M. Rissin and D. R. Walt, Digital Concentration Readout of Single Enzyme Molecules Using Femtoliter Arrays and Poisson Statistics, Nano Lett., 2006, 6(3), 520–523 CrossRef CAS PubMed.
- D. M. Rissin, D. R. Fournier, T. Piech, C. W. Kan, T. G. Campbell, L. Song, L. Chang, A. J. Rivnak, P. P. Patel and G. K. Provuncher, et al., Simultaneous detection of single molecules and singulated ensembles of molecules enables immunoassays with broad dynamic range, Anal. Chem., 2011, 83(6), 2279–2285 CrossRef CAS PubMed.
- A. J. Rivnak, D. M. Rissin, C. W. Kan, L. Song, M. W. Fishburn, T. Piech, T. G. Campbell, D. R. DuPont, M. Gardel and S. Sullivan, et al., A fully-automated, six-plex single molecule immunoassay for measuring cytokines in blood, J. Immunol. Methods, 2015, 424, 20–27 CrossRef CAS PubMed.
- C.-A. Cheng, W. Chen, L. Zhang, H. H. Wu and J. I. Zink, Magnetic resonance imaging of high-intensity focused ultrasound-stimulated drug release from a self-reporting core@shell nanoparticle platform, Chem. Commun., 2020, 56(71), 10297–10300 RSC.
- C. A. Cheng, T. Deng, F. C. Lin, Y. Cai and J. I. Zink, Supramolecular Nanomachines as Stimuli-Responsive Gatekeepers on Mesoporous Silica Nanoparticles for Antibiotic and Cancer Drug Delivery, Theranostics, 2019, 9(11), 3341–3364 CrossRef CAS PubMed.
- C.-A. Cheng, W. Chen, L. Zhang, H. H. Wu and J. I. Zink, A Responsive Mesoporous Silica Nanoparticle Platform for Magnetic Resonance Imaging-Guided High-Intensity Focused Ultrasound-Stimulated Cargo Delivery with Controllable Location, Time, and Dose, J. Am. Chem. Soc., 2019, 141(44), 17670–17684 CrossRef CAS PubMed.
- W. Chen, C.-A. Cheng and J. I. Zink, Spatial, Temporal, and Dose Control of Drug Delivery using Noninvasive Magnetic Stimulation, ACS Nano, 2019, 13(2), 1292–1308 CAS.
- W. Chen, C.-A. Cheng, B.-Y. Lee, D. L. Clemens, W.-Y. Huang, M. A. Horwitz and J. I. Zink, Facile Strategy Enabling Both High Loading and High Release Amounts of the Water-Insoluble Drug Clofazimine Using Mesoporous Silica Nanoparticles, ACS Appl. Mater. Interfaces, 2018, 10(38), 31870–31881 CrossRef CAS PubMed.
- M. Ma, H. Xu, H. Chen, X. Jia, K. Zhang, Q. Wang, S. Zheng, R. Wu, M. Yao and X. Cai, et al., A drug-perfluorocarbon nanoemulsion with an ultrathin silica coating for the synergistic effect of chemotherapy and ablation by high-intensity focused ultrasound, Adv. Mater., 2014, 26(43), 7378–7385 CrossRef CAS PubMed.
- H. Tang, Y. Guo, L. Peng, H. Fang, Z. Wang, Y. Zheng, H. Ran and Y. Chen, In Vivo Targeted, Responsive, and Synergistic Cancer Nanotheranostics by Magnetic Resonance Imaging-Guided Synergistic High-Intensity Focused Ultrasound Ablation and Chemotherapy, ACS Appl. Mater. Interfaces, 2018, 10(18), 15428–15441 CrossRef CAS PubMed.
- M. Souri, M. Soltani and F. Moradi Kashkooli, Computational modeling of thermal combination therapies by magneto-ultrasonic heating to enhance drug delivery to solid tumors, Sci. Rep., 2021, 11(1), 19539 CrossRef CAS PubMed.
- S. De Placido, G. Scambia, G. Di Vagno, E. Naglieri, A. V. Lombardi, R. Biamonte, M. Marinaccio, G. Cartenì, L. Manzione and A. Febbraro, et al., Topotecan Compared With No Therapy After Response to Surgery and Carboplatin/Paclitaxel in Patients With Ovarian Cancer: Multicenter Italian Trials in Ovarian Cancer (MITO-1) Randomized Study, J. Clin. Oncol., 2004, 22(13), 2635–2642 CrossRef CAS PubMed.
- J. Topotecan Garst, An evolving option in the treatment of relapsed small cell lung cancer, Ther. Clin. Risk Manage., 2007, 3(6), 1087–1095 CAS.
- M. Martínez-Carmona, D. Lozano, M. Colilla and M. Vallet-Regí, Selective topotecan delivery to cancer cells by targeted pH-sensitive mesoporous silica nanoparticles, RSC Adv., 2016, 6(56), 50923–50932, 10.1039/C6RA07763C.
- C. Kollmannsberger, K. Mross, A. Jakob, L. Kanz and C. Bokemeyer, Topotecan - A novel topoisomerase I inhibitor: pharmacology and clinical experience, Oncology, 1999, 56(1), 1–12 CrossRef CAS PubMed.
- H. Kawasaki, Y. Takeda and R. Arakawa, Mass Spectrometric Analysis for High Molecular Weight Synthetic Polymers Using Ultrasonic Degradation and the Mechanism of Degradation, Anal. Chem., 2007, 79(11), 4182–4187 CrossRef CAS PubMed.
- T. Hirano, IL-6 in inflammation, autoimmunity and cancer, Int. Immunol., 2021, 33(3), 127–148 CrossRef CAS PubMed.
- A. Masjedi, V. Hashemi, M. Hojjat-Farsangi, G. Ghalamfarsa, G. Azizi, M. Yousefi and F. Jadidi-Niaragh, The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer, Biomed. Pharmacother., 2018, 108, 1415–1424 CrossRef CAS PubMed.
- D. T. Fisher, M. M. Appenheimer and S. S. Evans, The two faces of IL-6 in the tumor microenvironment, Semin. Immunol., 2014, 26(1), 38–47 CrossRef CAS PubMed.
- J. Coward, H. Kulbe, P. Chakravarty, D. Leader, V. Vassileva, D. A. Leinster, R. Thompson, T. Schioppa, J. Nemeth and J. Vermeulen, et al., Interleukin-6 as a therapeutic target in human ovarian cancer, Clin. Cancer Res., 2011, 17(18), 6083–6096 CrossRef CAS PubMed.
- J. Coward, H. Kulbe, P. Chakravarty, D. Leader, V. Vassileva, D. A. Leinster, R. Thompson, T. Schioppa, J. Nemeth and J. Vermeulen, et al., Interleukin-6 as a Therapeutic Target in Human Ovarian Cancer, Clin. Cancer Res., 2011, 17(18), 6083–6096 CrossRef CAS PubMed.
- I. Szulc-Kielbik, M. Kielbik, M. Nowak and M. Klink, The implication of IL-6 in the invasiveness and chemoresistance of ovarian cancer cells. Systematic review of its potential role as a biomarker in ovarian cancer patients, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1876(2), 188639 CrossRef CAS PubMed.
- A. Fernández-Medarde and E. Santos, Ras in cancer and developmental diseases, Genes Cancer, 2011, 2(3), 344–358, DOI:10.1177/1947601911411084.
- A. D. Cox and C. J. Der, Ras history: The saga continues, Small GTPases, 2010, 1(1), 2–27 CrossRef PubMed.
- L. Huang, Z. Guo, F. Wang and L. Fu, KRAS mutation: from undruggable to druggable in cancer, Signal Transduction Targeted Ther., 2021, 6(1), 386 CrossRef CAS PubMed.
- B. Ancrile, K. H. Lim and C. M. Counter, Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis, Genes Dev., 2007, 21(14), 1714–1719 CrossRef CAS PubMed.
- A. E. R. Kartikasari, C. S. Huertas, A. Mitchell and M. Plebanski, Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis, Front. Oncol., 2021, 11, 692142 CrossRef PubMed.
- A. M. Waters, I. Ozkan-Dagliyan, A. V. Vaseva, N. Fer, L. A. Strathern, G. A. Hobbs, B. Tessier-Cloutier, W. K. Gillette, R. Bagni and G. R. Whiteley, et al., Evaluation of the selectivity and sensitivity of isoform- and mutation-specific RAS antibodies, Sci. Signaling, 2017, 10(498), eaao3332 CrossRef PubMed.
- A. M. Waters and C. J. Der, Validation of Isoform- and Mutation-Specific RAS Antibodies, Methods Mol. Biol., 2021, 2262, 91–103 CrossRef CAS PubMed.
- P. Flecknell, Replacement, reduction and refinement, Altex, 2002, 19(2), 73–78 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data were included: synthesis of ultrasound-responsive PEGylated mesoporous silica nanoparticles (MSN-PEG) (Scheme S1); size distribution of mesoporous silica nanoparticle (MSN) calculated from TEM images (Fig. S1); TEM image of MSN-PEG after ultrasound treatment and heating at 50 °C for 30 min (Fig. S2); fluorescence microscope images of OVCAR-3 cells after 4 h treatment of 75 μg mL−1 TOP-MSN-PEG with and without an additional 20 h of incubation (Fig. S3); cytotoxicity of the cells incubated with the supernatant obtained from ultrasound-stimulated TOP-loaded MSN-PEG solution (Fig. S4); N2 adsorption/desorption characterization (Table S1); summary of temperature increase of the cell supernatant and the cell viability post ultrasound treatments with various ultrasound parameters (Table S2). See DOI: https://doi.org/10.1039/d3ra02092d |
| This journal is © The Royal Society of Chemistry 2023 |