
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled mechano-luminescence properties of SrGa2O4:Tb3+ co-doping with Dy3+ and Eu3+ ions†
Chaochao Wanga,
Zhichao Liu*a,
Alexey Nikolaevich Yakovlevb,
Tingting Hub,
Tatiana Grigorievna Cherkasovab,
Xiaodie Zhua,
Ya Liua,
Jian Zhanga,
Daiyuan Liua and
Xue Yu *c
*c
aCollege of Materials Science and Engineering, Key Laboratory of Advanced Materials of Yunnan Province, Kunming University of Science and Technology, Kunming 650093, P.R. China. E-mail: yuyu6593@126.com; liuzhichzo1028@126.com
bT.F. Gorbachev Kuzbass State Technical University, 28, Vesennyaya Street, Kemerovo, 650000, Russia
cSchool of Mechanical Engineering, Institute for Advanced Materials Deformation and Damage from Multi-Scale, Chengdu University, Chengdu, 610106, P.R. China
First published on 31st May 2023
Abstract
Trap-controlled mechano-luminescence (ML) featuring photon emission under mechanical stimuli provides promising applications such as dynamic imaging of force, integrated optical sensing, information storage, and anti-counterfeiting encryption. However, the corresponding emission with a single color still limits the application of ML materials. Here, a trap-controlled ML phosphor of SrGa2O4:Tb3+ (SGO:Tb3+) with a green-emission is investigated with an adjustable ML color. The relationship between ML and thermoluminescence (TL) is verified by co-doping with Dy3+ and Eu3+ ions for the manipulation of the constructed traps. Accordingly, the as-explored ML phosphor with multicolor output is employed to create encrypted anticounterfeiting patterns, which produces bright and spatially resolvable optical codes under the single-point dynamic pressure of a ballpoint pen. Hence, it provides a new approach to achieve ML with multicolor and gives us an insight into understanding the mechanism of the ML procedure.
1. Introduction
Mechano-luminescence (ML) is a light-emitting phenomenon when the corresponding ML materials are stimulated by various mechanics, such as compressing, stretching, bending and rubbing, as well as in response to tides and wind.1–4 The explored ML composites are generally composed of organic polymers and inorganic phosphors, which are potentially employed for constructing optoelectrical devices that are free from optical or electrical sources for operation.5,6 Herein, structural damage diagnosis platforms, E-signature systems, artificial skins, and smart wearable devices based on the fascinating ML behavior have been developed.7–11Notably, ML material with green emission color has drawn particularly attention because green is one of the most easily captured colors for human eyes.12 At present, three kinds of systems, i.e., ZnS:Cu, CaZnOS:Tb3+, and SrAl2O4:Eu2+, Dy3+, has been studied as green ML materials with high performance.13–15 However, ZnS:Cu and CaZnOS:Tb3+ contain the sulfur source, resulting in environmental pollution during synthesis.16 For the system of SrAl2O4:Eu2+, Dy3+, the poor stability in humidity causes inevitable interference on the measurement and analysis of ML signal due to a hydrolyze reaction with hydroxide ion proceeds and an equilibrium reaction between Sr3Al2(OH)12 and [Sr3Al2(OH)11]+ occurred.17 In recent years, significant progress has been made by the Xu group to render SrAl2O4:Eu2+ nanoparticles water-resistant, for example by modifying the nanoparticle surface with pyrophosphoric acid.18 It is confirmed that depositing a layer of aluminum phosphate layer on SrAl2O4:Eu2+ nanoparticles could protect against the humidity for at least one month. In addition, finding materials with relatively better waterproof properties could simplify the synthesis process and facilitates large-scale application.19,20 Thus, developing novel ML materials with green emission color towards environmental protection, steady and high ML efficiency is meaningful.21 Furthermore, the manipulation of the emission color of ML with stable output and high brightness indicates a more fascinating characteristic for practical application, which is an urgent task as well.15,22,23
Herein, we preliminarily aimed at SrGa2O4 (SGO), which possesses a low-dimensional structure, as the ideal host for the low synthesis temperature, high stability, and reasonable conductivity.24 In addition, SGO possessed an open structure with tunnel-like cavities running throughout the crystal lattice. The tunnel was generated through the link of Sr2+ sites, which was located in the one-dimensional space structure surrounded by a GaO4 tetrahedral.25 It was reasonable that the conduction along the tunnel (a crystal direction) is much larger than that along other directions (b and c directions), which could be beneficial to the formation of the trap level and excited state level and especially could provide an effective transmission path in one direction between luminescent centers and trapping centers.26 For the doped ions, Tb3+ is a well-known activation ion with green emission color.27 The dopant of Tb3+ ions has the advantages of large absorption coefficient, high luminescence intensity, and stable luminescence.28 Therefore, it is feasible to achieve green ML by doping Tb3+ ions in the SGO host.
In this work, an efficient ML material SGO:Tb3+ with green emission has been successfully synthesized by a high-temperature solid phase method. Moreover, the ML properties of SGO:Tb3+ can be enhanced by co-doping Dy3+ ions, while the corresponding ML color could be regulated accordingly. Additionally, the relationship between the ML and traps is further investigated by co-doping with Eu3+ ions. Hereby, the multicolor ML phosphors employed to create optically encoded graphics for anticounterfeiting applications is confirmed.
2. Experimental
2.1. Synthesis of powders
A series of rare earths doped SrGa2O4 powders (SGO:xTb3+ (x = 0, 0.001, 0.003, 0.005, 0.007, 0.009), SGO:Tb3+, yDy3+(y = 0.001, 0.002, 0.003, 0.004, 0.005) and SGO:0.005Tb3+, 0.001Eu3+, SGO:0.001Eu3+) were synthesized by a high-temperature solid-state method. The raw materials of SrCO3 (99.95%), Ga2O3 (99.99%), Tb4O7 (99.99%), Dy2O3 (99.99%), and Eu2O3 (99.99%) were accurately weighed by stoichiometric ratios. Then, the mixture was dispersed in a certain amount of alcohol and fully ground in the agate mortar. After that, the mixture was transferred into a corundum crucible and placed in a muffle furnace. The mixture was kept at 1200 °C for 6 h in a reducing atmosphere composed of nitrogen and hydrogen (95% N2 and 5% H2 in volume ratio). After cooling to room temperature, the sample was taken out and SGO:Tb3+ was finally obtained.2.2. Preparation of composites films
Polydimethylsiloxane (PDMS) was chosen as the flexible matrix for the as-obtained powders to facilitate the ML measurement and analysis. First, 2.0 g of PDMS precursor, 0.2 g of curing agent, and 1.0 g of the as-obtained powders were mixed. The phosphor slurry was poured onto a thin glass plate and flattened to a smooth film using a scraper. The phosphor@PDMS composite films were firmed in an oven at 60 °C for 3 h. The thickness of the composite films was set to be 1–2 mm.2.3. Characterizations
X-ray diffractometer (XRD) of Rigaku Model D/max-2200 was used to analyze the crystalline structure of the phosphor. The working voltage is maintained at 220 V, and the excitation source is Cu Kα (1.15418 Å) with a scanning step of 0.02° and a scanning speed of 10° min−1 in a scanning range of 10°–65°. Rietveld refinement of XRD data by using the General Structure Analysis System (GSAS). Scanning Electron Microscope (SEM) image, Energy Dispersive Spectroscopy (EDS), and the corresponding element mapping of SGO:0.005Tb3+, 0.004Dy3+ powders were characterized by a field emission scanning electron microscopy (Tescan China, Ltd). Photoluminescence (PL), photoluminescence excitation (PLE), and long persistent luminescence (LPL) spectra were recorded using a time-resolved fluorescence spectrometer (Hitachi F-7000) with a 150 W Xe 900 lamp as the excitation source. The LPL curves were measured using PR 305 phosphorophotometer (Zhejiang University Sensing Instruments Co., Ltd, Hangzhou, China). The samples were UV (254 nm) irradiated for 20 min before measurement, and the LPL duration after stopping excitation 15 s was recorded. The thermoluminescence (TL) curve was detected by a FJ-427A TL measuring instrument from 300 to 523 K with a heating rate of 1 K s−1 after the samples were exposed to UV for 20 min, the weight of the measured samples was constant (0.0020 g). The rubbing experiments of the rare earths doped SrGa2O4/PDMS elastomer were operated on a rotary friction testing machine (MS-T3001) using the stainless-steel rod (radius: 0.2 mm) as the friction pair measured in the 1st hour after UV irradiation for 20 min. The applied load and rotational speed were set to 5 N and 200 rpm, respectively. The produced ML signals during mechanic stimuli were collected through a spectrometer (Ocean Optics USB2000 + XR1-ES). All digital photographs were taken by a digital camera (Nikon D7100).3. Results and discussion
Fig. 1a shows the representative X-ray diffraction (XRD) patterns of SGO, SGO:0.005Tb3+, and SGO:0.005Tb3+, 0.004Dy3+, respectively. It is observed that all diffraction peaks match well with the standard PDF card (PDF#72-0222) of β-SrGa2O4, indicating that the as-synthesized samples are single phase and the doped of Tb3+ and Dy3+ ions into SGO hardly bring any impurity peaks. Besides, the Rietveld refinement results of SGO:0.005Tb3+, 0.004Dy3+, as illustrated in Fig. S1,† have been calculated and the reliability parameters of refinement are Rwp = 7.86%, Rp = 5.63%, and χ2 = 2.941%. Fig. 1b presents the crystal structure of SGO, which is monoclinic, space group P21/C (14) and Z = 8.29 In addition, SGO has three crystallographically independent cation sites, namely, two different sites of Sr2+ (marked as Sr1 and Sr2) and one of Ga3+ with Sr2+ sites surrounded by GaO4 tetrahedrons.30 The structure of the SGO phase possesses a distinct open structure with tunnel-like cavities (a crystal direction) running throughout the host lattice.31 Compared the ionic radius of Sr2+, Tb3+ and Dy3+ and combining the Hume-Rothery principle,28,32 it is determined that Tb3+ and Dy3+ ions are inclined to occupy the lattice position of Sr2+ ions. SEM and EDS mappings are further employed to investigate the morphology and chemical composition of the SGO:Tb3+, Dy3+. The EDS mappings in Fig. 1c and d confirm that the sample is composed of Sr, Ga, O, Tb, and Dy, and the corresponding elements are well distributed within the particle.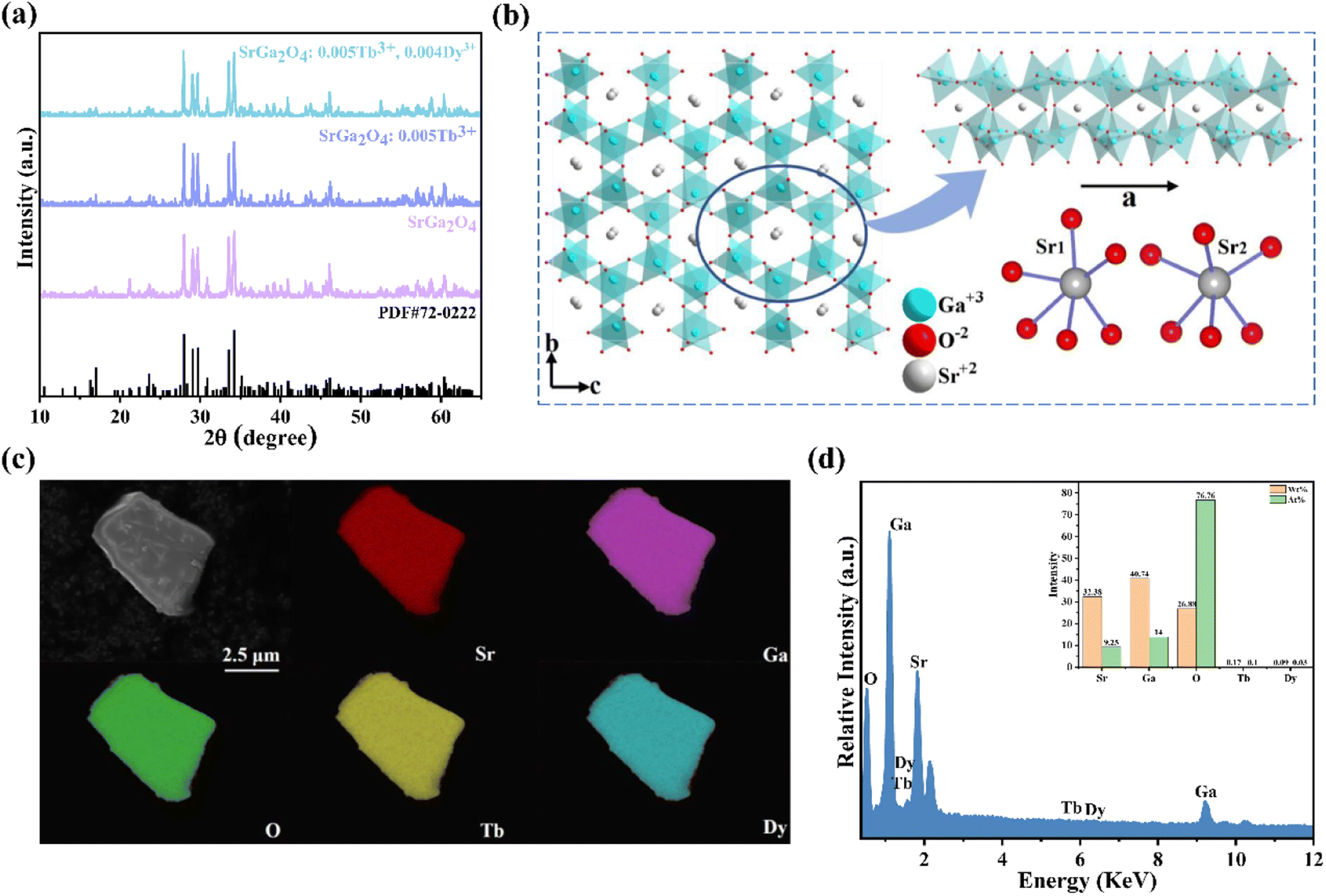 | ||
| Fig. 1 (a) XRD patterns of SGO, SGO:Tb3+ and SGO:Tb3+, Dy3+, respectively; (b) the crystal structure of SGO; (c) the elemental mappings and (d) the EDS of SGO:0.005Tb3+, 0.004Dy3+ (the inset in (d) is the corresponding the elemental composition ratio). | ||
Fig. 2a shows the PLE and PL spectra of the SGO sample. SGO shows an asymmetry band of the PLE spectrum, which is attributed to the contribution of both host absorption and the O2−–Ga3+ charge transfer band. A blue broad band centered at 398 nm under the excitation at 238 nm is termed from the 4T1–4A2 transition of electrons in d orbits of Ga3+ ions.33,34 Similar to Zn2+ ions, Ge4+ ions and In3+ ions, Ga3+ ions have an electronic configuration of 3d10. 3d orbital is full. However, it has been reported that one electron in 3d orbital will be excited to 4s orbital when Ga3+ ions are excited.35 Moreover, the XRD patterns of Sr1−xGa2O4:xTb3+ are presented in Fig. S2a,† indicating the Sr1−xGa2O4:xTb3+ are single phase. Fig. 2b illustrates the PLE and PL spectra of SGO:0.005Tb3+ phosphors. Under 248 nm excitation, the emission spectrum has two emission band from blue to green emissions in the region of 380–460 and 490–630 nm, due to the 5D3 → 7FJ (J = 6, 5, 4, 3) and 5D4 → 7FJ (J = 6, 5, 4, 3) transitions of Tb3+ ions. While monitoring the emission of 549 nm originated from the characteristic transition of 5D4 → 7F5 of Tb3+ ions, it can be observed that the excitation spectrum is made up of a broad peak in the region of 200–300 nm and a series of sharp peaks from 320 to 380 nm, which belongs to the charge transfer band (CTB) of O2−–Tb3+ and the 4f–4f transitions of Tb3+ ions, respectively.36,37 Besides, the emission band of SGO spectral overlaps with the excitation band of Tb3+ ions, which indicates the energy transfer from SGO host to Tb3+ ions according to Dexter's theory.38 The inserted decay curves of Sr1−xGa2O4:xTb3+ (λex = 238 nm; λem = 398 nm) in Fig. 2c provide a clear indication of energy transfer from the host to the Tb3+ ions. The decay curves of Sr1−xGa2O4:xTb3+ samples can be fitted well by the single-exponential decay mode as the following equation (eqn (1)):39
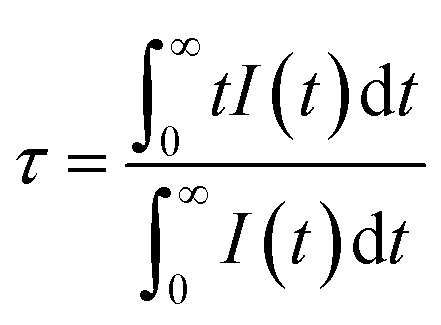 | (1) |
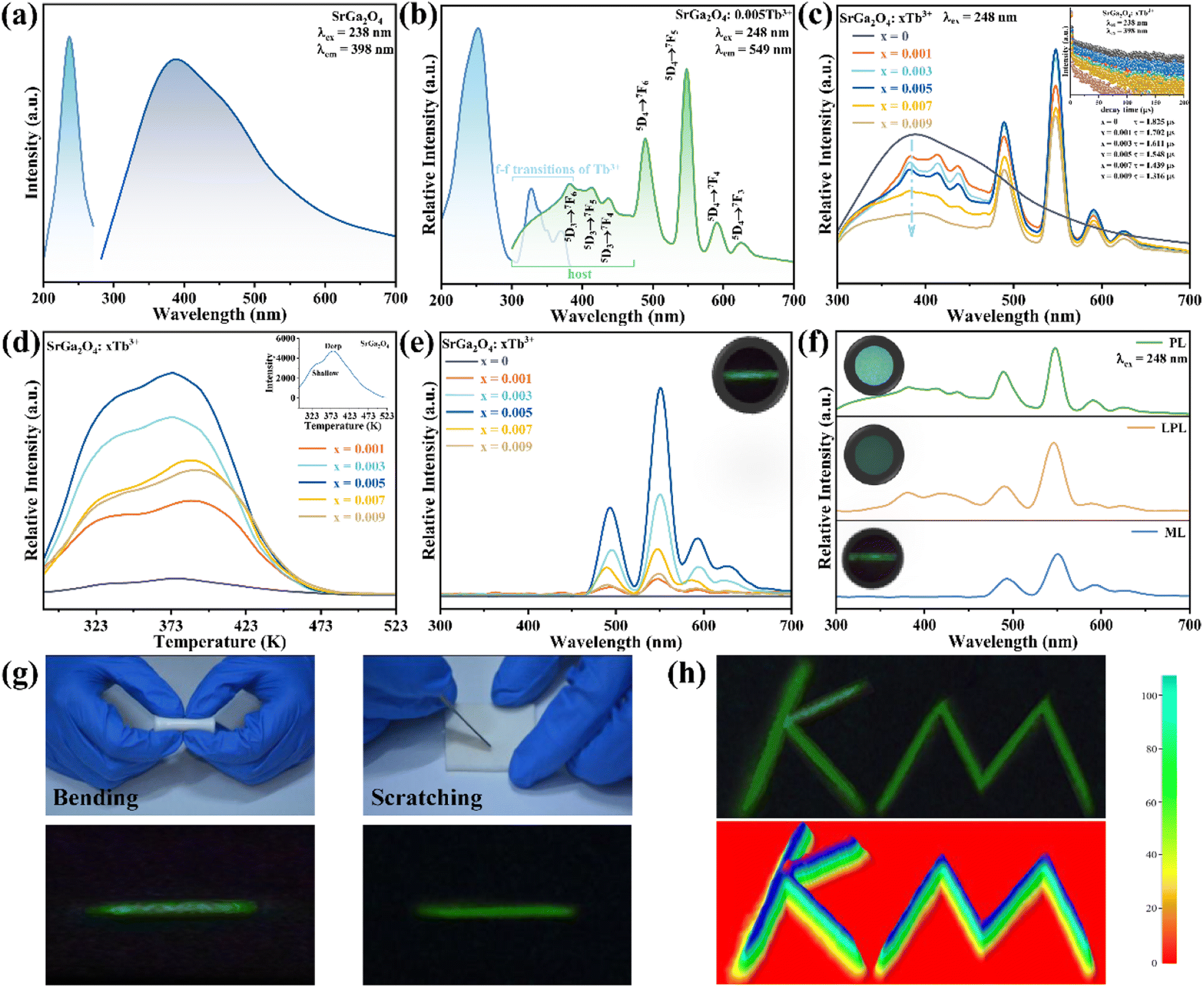 | ||
| Fig. 2 PLE and PL spectra of (a) SGO, and (b) SGO:0.005Tb3+, respectively; (c) PL spectra recorded under 248 nm excitation, the inset shows the fluorescence decay curves (λem = 395 nm, and λex = 238 nm); (d) TL curves recorded immediately after UV pre-radiation 20 min; (e) ML spectra of Sr1−xGa2O4:xTb3+ (x = 0, 0.001, 0.003, 0.005, 0.007, and 0.009) samples, respectively; (f) PL, LPL and ML spectra of SGO:0.005Tb3+; (g) ML images of SGO:0.005Tb3+/PDMS composites under the stimulation of bending and scratching; (h) visualization and pseudo-colour figure of pressure distribution of handwritten signature. | ||
The peak position of the TL curve is roughly proportional to the trap depth and the following function (eqn (2)) is used to calculate the trap depth (E):42–44
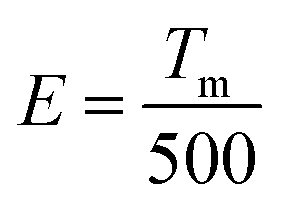 | (2) |
In the SGO host, some intrinsic defects, such as 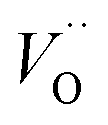 and
and 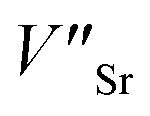 exist. Considering that Tb3+ ions replace the Sr2+ site in the SGO host matrix, three Sr2+ ions are replaced by two Tb3+ ions to balance the charge of the host matrix, creating two positive defects and one negative defect
exist. Considering that Tb3+ ions replace the Sr2+ site in the SGO host matrix, three Sr2+ ions are replaced by two Tb3+ ions to balance the charge of the host matrix, creating two positive defects and one negative defect 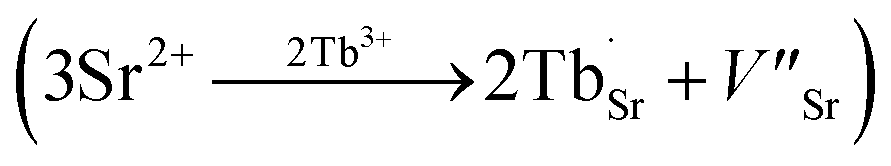 . Therefore, two extra electron traps
. Therefore, two extra electron traps  are formed. The low-dimensional structure provides one-dimensional tunnel-like cavities running throughout the crystal lattice, which could be propitious to the interaction among these defect states and further yield bright ML.
are formed. The low-dimensional structure provides one-dimensional tunnel-like cavities running throughout the crystal lattice, which could be propitious to the interaction among these defect states and further yield bright ML.
To enhance the ML output, Dy3+ is co-doped into the SGO:Tb3+ host matrix. XRD patterns of the SGO:0.005Tb3+, yDy3+ (x = 0, 0.001, 0.003, 0.005, 0.007, and 0.009) are shown in Fig. S2b,† illustrating the synthesized phase without impurities. Compared with the PL spectrum of SGO:Tb3+, the PL spectrum of SGO:0.005Tb3+, 0.004Dy3+ in Fig. 3a shows significant difference in the range of 380–460 nm under the excitation of 248 nm for the observation of the emission originated from the host. Besides, the typical 5D3 → 7FJ (J = 6, 5, 4) transitions of Tb3+ ions are clearly observed.46 Fig. 3b shows the PL spectra of SGO:0.005Tb3+, yDy3+ as the function of the concentration of Dy3+ ions, where SGO:0.005Tb3+, 0.004Dy3+ displays the strongest emission intensity. Fig. S3a† exhibits the TL curves of SGO:0.005Tb3+, yDy3+. It's noted that shallow trap and deep trap have different quenching concentration after UV irradiation for 20 min, indicating that outstanding LPL and ML performance is observed with different co-doping concentration. The normalized TL curves, plotted in Fig. 3c, present that the intensity of deep traps is drastically increased relative to that of shallow traps. Moreover, the TL range is gradually broadened with the increase of Dy3+ ions content, which suggests that the influence of co-doped Dy3+ on deep trap and shallow trap is distinct. In addition, as shown in Fig. S3b,† the optimized LPL is observed in the SGO:0.005Tb3+, 0.001Dy3+ sample, while the strongest ML is obtained from SGO:0.005Tb3+, 0.004Dy3+, as presented in Fig. 3d, indicating that deep trap and shallow trap contribute to ML and LPL, respectively. Furthermore, it is fascinating to notice that the ML color of SGO:0.005Tb3+, yDy3+ changes from green (y = 0) to cyan (y = 0.004) owing to the appearance of 5D3 → 7FJ (J = 6, 5, 4) transition of Tb3+ ions after the introduction of Dy3+ ions.
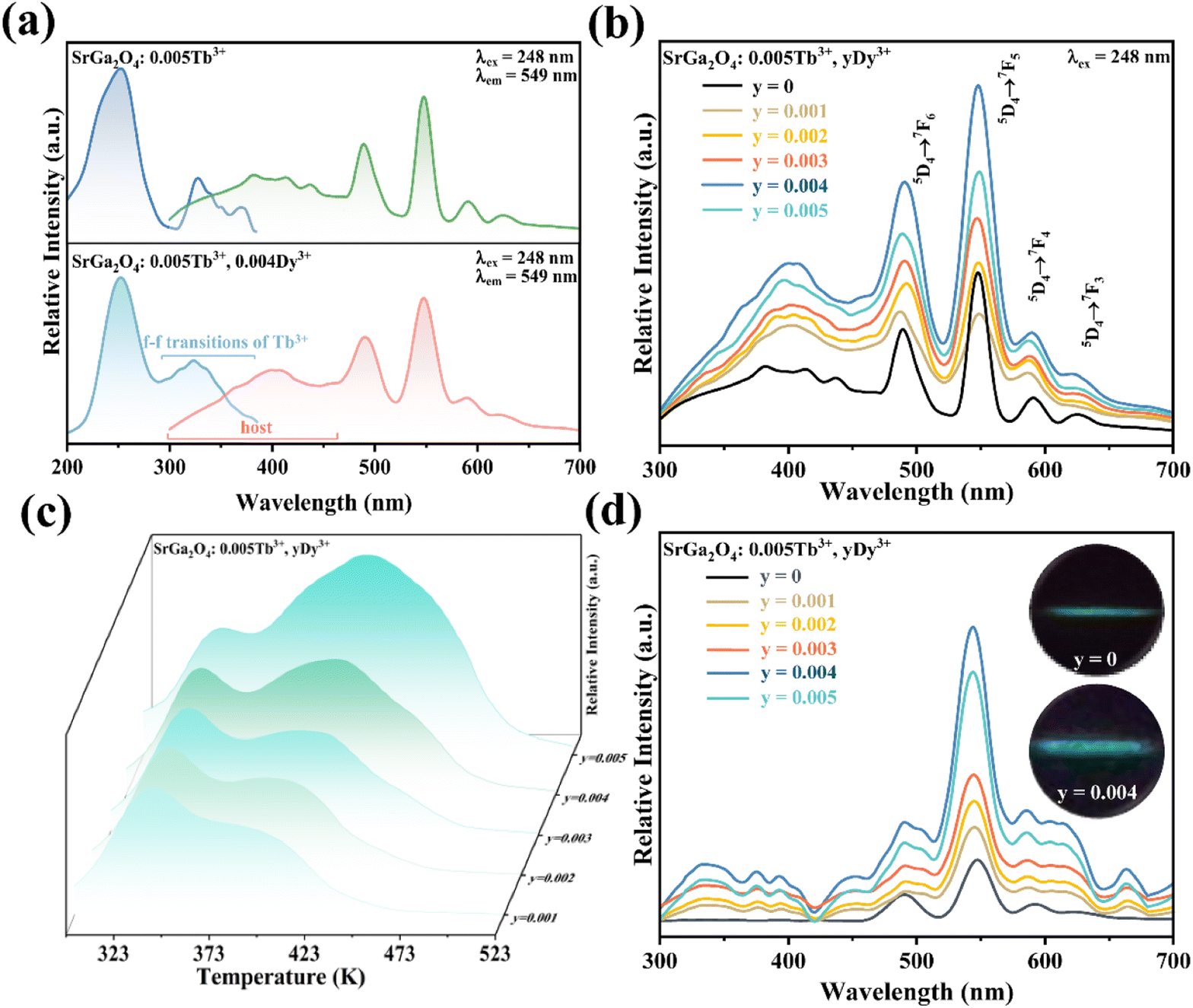 | ||
| Fig. 3 (a) PLE and PL spectra of SGO:0.005Tb3+, and SGO:0.005Tb3+, 0.004Dy3+, respectively; (b) PL spectra, (c) normalized TL curves, (d) ML spectra of Sr0.995−yGa2O4:0.005Tb3+, yDy3+ (y = 0.001, 0.002, 0.003, 0.004, and 0.005); inset in (d) shows the ML images of SGO:0.005Tb3+ and SGO:0.005Tb3+, 0.004Dy3+, respectively. | ||
To further investigate the role of traps in the ML procedure, Eu3+ ions are introduced into the SGO:Tb3+ sample. XRD patterns in Fig. S2c† confirm the phase purity of the as-obtained corresponding sample. PL spectra of SGO:Tb3+, Eu3+ in Fig. 4a display the characteristic emission peaks of Tb3+ and Eu3+ ions, proving the successful incorporation of Tb3+ and Eu3+ ions in the SGO host matrix. Compared with the TL curves of SGO:Tb3+ recorded in Fig. 4b, the incorporation of Eu3+ ions obviously quenches the TL intensity of SGO:Tb3+, which deteriorates LPL performance, as depicted in Fig. 4c. It is obviously that SGO:Tb3+, Eu3+ presents a weak LPL, which is closely related to the decreased concentration of traps. ML spectra of SGO:Tb3+ before and after co-doping Eu3+ ions are recorded in Fig. 4d, which clearly presented a disappear in ML. Eu3+ ions in SGO efficiently quenches the trap anomaly for using of Tb3+ ions, leading to the disappearance of ML.
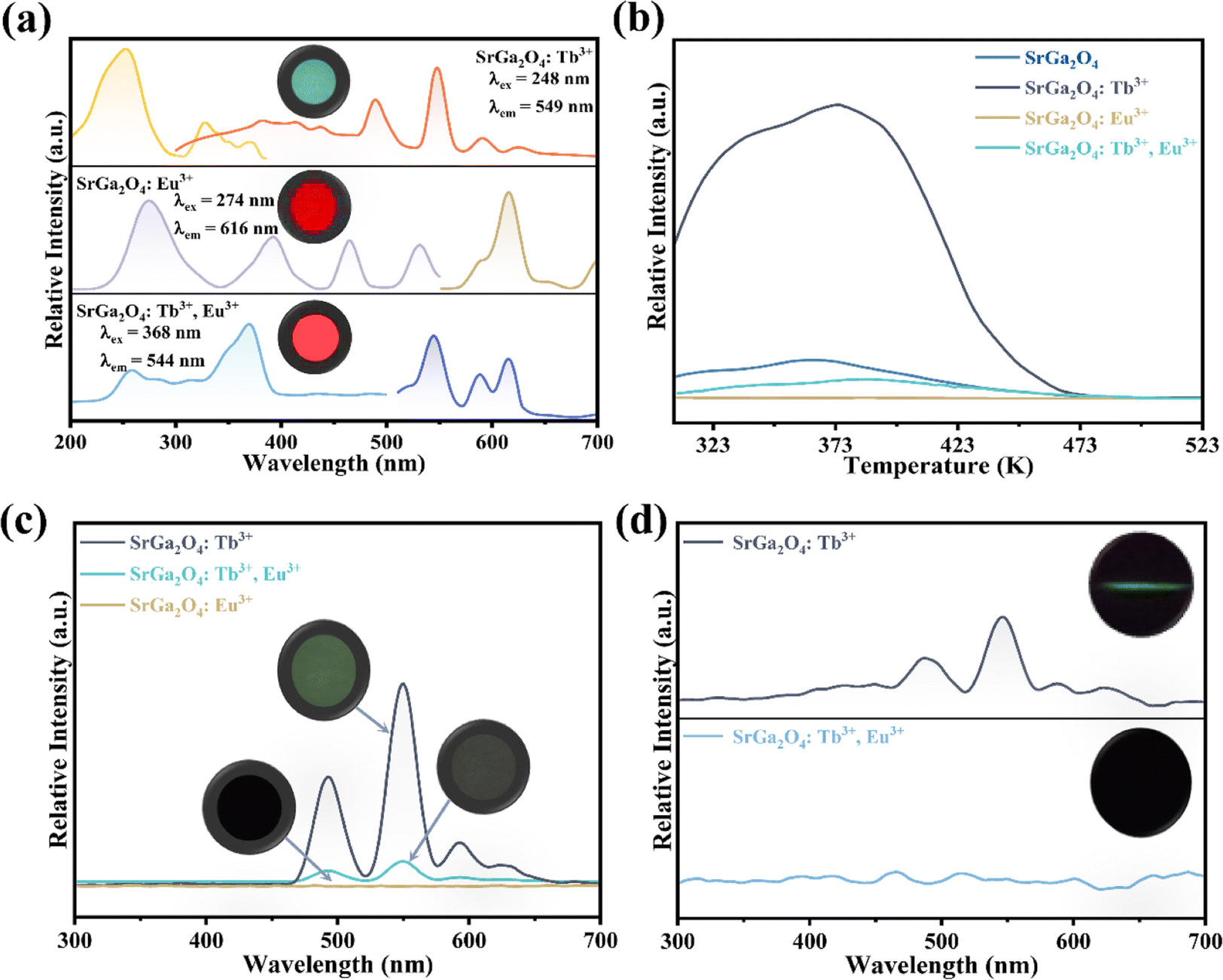 | ||
| Fig. 4 (a) PLE and PL spectra, (b) TL curves and (c) LPL spectra after UV lamp pre-radiation for 20 min of SGO:0.005Tb3+, SGO:0.002Eu3+, and SGO:0.005Tb3+, 0.002Eu3+, respectively; (d) ML spectra of SGO:0.005Tb3+ and SGO:0.005Tb3+, 0.002Eu3+ samples, inset shows the corresponding ML image. | ||
Fig. 5a records the change of the ML intensity over time after pre-radiation 20 min under UV excitation. It can be found that the ML decreases gradually with the extension of time, which is related to the gradual release of trap, as shown in Fig. S4.† After five days, the ML is still observed, indicating that the trap has a strong storage capacity. Fig. 5b presents TL curves of SGO:0.005Tb3+, 0.004Dy3+ recorded at different friction times after UV irradiation 20 min. It can be found that carriers captured by traps is gradually released with the extension of friction time. All the evidence suggests that the release of trapped carriers under the mechanical stimuli should be the origination of ML in SGO:0.005Tb3+, 0.004Dy3+. Based on the PL, ML, and TL analysis as well as the force-dependent behaviors, the luminescent mechanisms of SGO:0.005Tb3+, 0.004Dy3+ are supposed in Fig. 5c. Electrons are excited to the 5L10 level of Tb3+ ions and then relaxed to the 5D3 and 5D4 levels to generate PL (① and ②). During UV irradiation, the electrons excited from the ground level are captured by trapping centers (shallow and deep traps).47 After the UV is switched off, carriers captured in the shallow traps are easily released under thermal stimulation at room temperature to generate LPL.48 Under stress stimuli, the electrons in deep traps could be released to the conduction band (CB) first and then to the 5F7, 5D3 and 5D4 level of Tb3+ ions (③ and ④). Finally, the electrons recombine with holes in 7FJ (J = 3, 4, 5, 6) levels, producing the as-observed cyan ML.
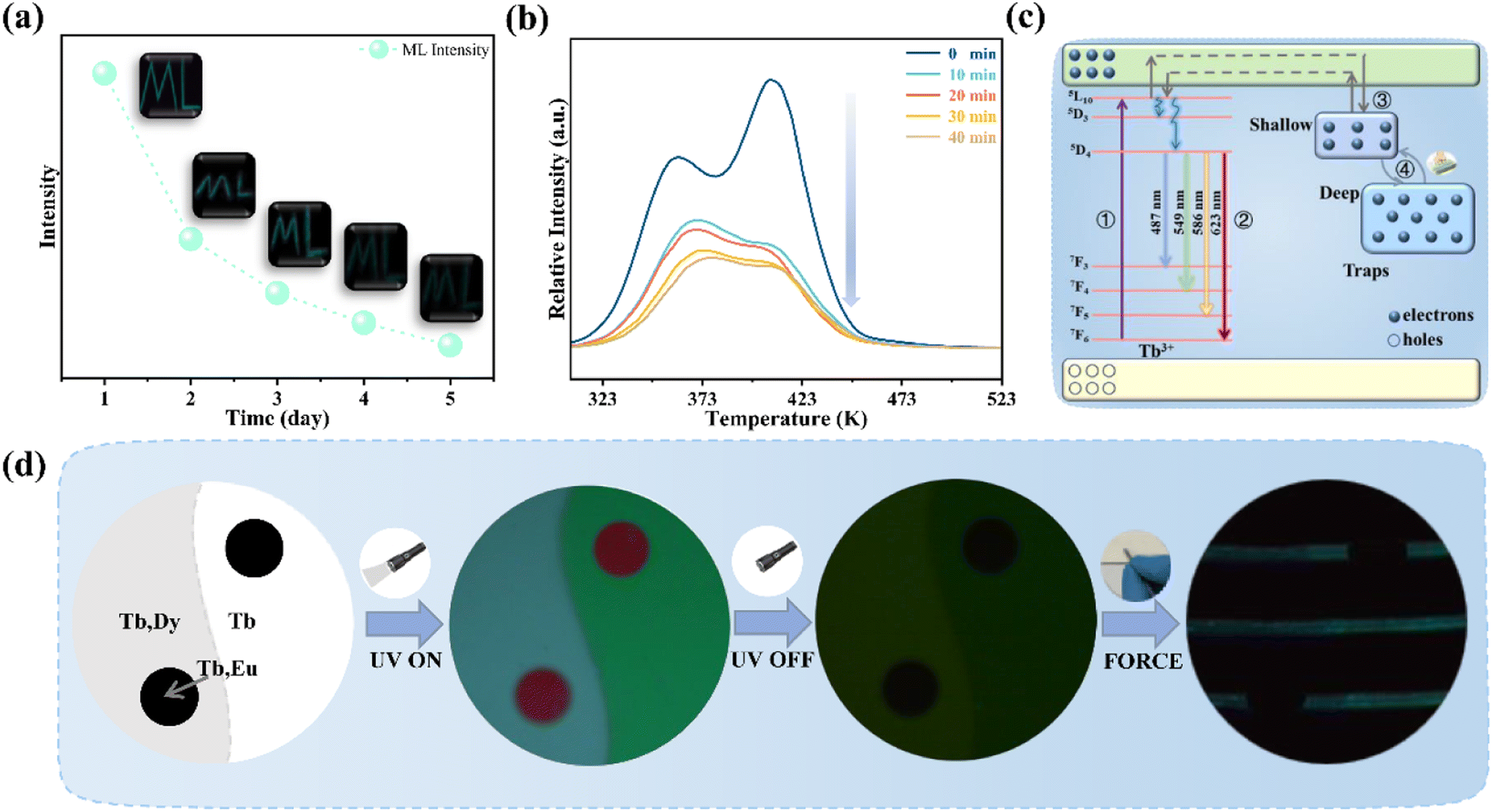 | ||
| Fig. 5 (a) ML intensity of SGO:0.005Tb3+, 0.004Dy3+ recorded after deposited in air for 1, 2, 3, 4, and 5 days; (b) time-dependent TL curves of SGO:0.005Tb3+, 0.004Dy3+ under 5 N load after UV irradiation 20 min; (c) luminescence diagram of SGO:Tb3+, Dy3+; (d) the photo of the anti-counterfeiting based on the as-obtained SGO:Tb3+, SGO:Tb3+, Dy3+ and SGO:Tb3+, Eu3+. | ||
Herein, we prepared an anti-counterfeiting mold of a pattern of “Yin-Yang”, where SGO:Tb3+, Dy3+/PDMS is embedded at the Yang half (left side) and SGO:Tb3+/PDMS at the Yin half (right side), while the circle in both Yin and Yang part are embedded with SGO:Tb3+, Eu3+/PDMS. The Yin-Yang pattern is not observable in either dark or nature light (Fig. 5d) before UV irradiation. Due to the difference of the luminous center and carrier concentration of the powder in the three area, the Yin-Yang pattern present distinguishable PL, LPL, and ML over time due to the clear contrast in brightness and color among the pattern. This distinguishable optical conversion model provides great convenience for anti-counterfeiting applications.
4. Conclusion
In summary, SGO:Tb3+ phosphors with ML properties are successfully synthesized by the solid-state method. SGO:Tb3+ exhibits 5D4 → 7FJ (J = 6, 5, 4, 3) green emissions of Tb3+ ions under force stimulation. The co-doped Dy3+ and Eu3+ ions have a significant influence on the manipulation of the concentration of traps, which confirms the ML of SGO:Tb3+ is critically related to the constructed traps. In addition, the ML color changes from green to cyan by co-doping Dy3+ ions. Herein, an anti-counterfeiting application has been proposed, which suggests that the as-explored material has a good application prospect in advanced multiple anti-counterfeiting.Author contributions
C. Wang and Z. Liu wrote this paper. X. Yu provided the funding. X. Yu and Z. Liu designed the experiments. C. Wang synthesized the compounds and prepared film. A. Yakovlev, T. Hu, T. Cherkasova, C. Wang, Y. Liu and X. Zhu performed the experiments and analyzed the data. J. Zhang and D. Liu took photos for ML. All authors edited the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Nature Science Foundation of China (NSFC) (61965012), the Science and Technology Major Project of Yunnan Province (202202AG050004, 202002AB080001), Yunnan Ten Thousand Talents Plan Young & Elite Talents Project (YNWR-QNBJ-2018-295), Sichuan Natural Science Foundation (2022JDJQ0030).References
- D. K. Patel, C. Bat-El, L. Etgar and S. Magdassi, Mater. Horiz., 2018, 5, 708–714 RSC.
- X. P. Zhang, L. L. Du, W. J. Zhao, Z. Zhao, Y. Xiong, X. W. He, P. F. Gao, P. Alam, C. Wang, Z. Li, J. Leng, J. X. Liu, C. Y. Zhou, J. W. Y. Lam, D. L. Phillips, G. Q. Zhang and B. Z. Tang, Nat. Commun., 2019, 10, 5161 CrossRef PubMed.
- Y. J. Xie and Z. Li, Chem, 2018, 4, 943–971 CAS.
- H. Guo, T. Wang, B. Liu, W. Gao, L. Xiu, Z. Cui, H. Zhang, Q. Ma, S. Wang, Z. Li, L. Guo, G. Yan, S. Yu, X. Yu, X. Xu and J. Qiu, J. Rare Earths, 2022, 40, 1173–1180 CrossRef CAS.
- L. K. Bao, X. J. Xu, Y. Zuo, J. Zhang, F. Liu, Y. F. Yang, F. Xu, X. M. Sun and H. S. Peng, Sci. Bull., 2019, 64, 151–157 Search PubMed.
- Y. X. Zhuang and R. J. Xie, Adv. Mater., 2021, 33, 2005925 CrossRef CAS.
- B. R. Tian, Z. F. Wang, A. T. Smith, Y. Q. Bai, J. Z. Li, N. Zhang, Z. H. Xue and L. Y. Sun, Nano Energy, 2021, 83, 8 Search PubMed.
- Z. C. Liu, X. Yu, L. Zhao, H. T. Tang, Z. F. Wang, X. D. Zhu, X. X. Yang, F. Zhao, J. B. Qiu and X. H. Xu, J. Mater. Chem. C, 2022, 10, 11697–11702 Search PubMed.
- J. J. Ning, Y. T. Zheng, Y. T. Ren, L. P. Li, X. Q. Shi, D. F. Peng and Y. M. Yang, Sci. Bull., 2022, 67, 707–715 CrossRef CAS.
- X. Qian, Z. R. Cai, M. Su, F. Y. Li, W. Fang, Y. D. Li, X. Zhou, Q. Y. Li, X. Q. Feng, W. B. Li, X. T. Hu, X. D. Wang, C. F. Pan and Y. L. Song, Adv. Mater., 2018, 30, 6 Search PubMed.
- T. Z. Zhan, C. N. Xu, O. Fukuda, H. Yamada and C. S. Li, Ultrason. Sonochem., 2011, 18, 436–439 CrossRef CAS PubMed.
- S. Y. Qin, J. L. Bian, Y. Han, Z. D. Ma, B. Liu, J. C. Zhang, X. H. Xu and Z. F. Wang, Mater. Res. Bull., 2022, 145, 7 CrossRef.
- J. Y. Li, Z. W. Zhang, X. X. Luo, L. P. Zhu and Z. L. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 4775–4782 CrossRef CAS.
- Y. J. Jiang, F. Wang, H. Zhou, Z. J. Fan, C. Wu, J. Zhang, B. Liu and Z. F. Wang, Mater. Sci. Eng., C, 2018, 92, 374–380 CrossRef CAS PubMed.
- Y. Y. Du, Y. Jiang, T. Y. Sun, J. X. Zhao, B. L. Huang, D. F. Peng and F. Wang, Adv. Mater., 2019, 31, 8 Search PubMed.
- W. X. Wang, Z. N. Wang, J. C. Zhang, J. Y. Zhou, W. B. Dong and Y. H. Wang, Nano Energy, 2022, 94, 12 Search PubMed.
- J. Lin, Y. Huang, J. Zhang, F. J. Shi, S. Y. Wei, J. M. Gao, Z. X. Huang, X. X. Ding and C. C. Tang, Mater. Chem. Phys., 2008, 108, 440–444 CrossRef CAS.
- Y. Fujio, C. N. Xu, N. Terasaki and N. Ueno, J. Lumin., 2014, 148, 89–93 CrossRef CAS.
- J. C. Zhang, X. S. Wang, G. Marriott and C. N. Xu, Prog. Mater. Sci., 2019, 103, 678–742 CrossRef CAS.
- P. Yang, X. Yu, H. Yu, T. Jiang, D. Zhou and J. Qiu, J. Rare Earths, 2012, 30, 1208–1212 CrossRef CAS.
- Z. Li, Y. Yan, T. Wang, S. Wang, L. Guo, W. Feng, L. Zhao, Z. Wang, F. Zhao, J. Chen, Z. Zhang, X. Xu and X. Yu, J. Rare Earths, 2022 DOI:10.1016/j.jre.2022.10.002.
- J. Y. Yuan, Y. L. Yang, X. C. Yang, Y. T. Fan, T. Li, M. Huang, F. Zhang, Q. L. Li, J. T. Zhao and Z. J. Zhang, J. Mater. Chem. C, 2021, 9, 7689–7696 RSC.
- W. G. Hu, C. Zhang and Z. L. Wang, Nanotechnology, 2019, 30, 21 Search PubMed.
- T. Wang, X. H. Xu, D. C. Zhou, Y. Yang, J. B. Qiu and X. Yu, Inorg. Chem., 2016, 55, 894–901 CrossRef CAS PubMed.
- V. Kahlenberg, R. X. Fischer and C. S. J. Shaw, J. Solid State Chem., 2000, 153, 294–300 CrossRef CAS.
- F. W. Jiang, P. F. Jiang, M. F. Yue, W. L. Gao, R. H. Cong and T. Yang, J. Solid State Chem., 2017, 254, 195–199 CrossRef CAS.
- T. Wang, X. Xu, D. Zhou, J. Qiu and X. Yu, J. Rare Earths, 2015, 33, 361–365 CrossRef CAS.
- X. Yu, S. B. Wang, Y. C. Zhu, J. J. Liang, J. B. Qiu, X. H. Xu and W. Lu, J. Alloys Compd., 2017, 701, 774–779 CrossRef CAS.
- A.-R. Schulze and H. Müller-Buschbaum, Z. Naturforsch., 1981, 36, 892–893 CrossRef.
- X. Y. Cai, Z. F. Mu, S. Zhang, D. Y. Zhu, Q. Wang, Y. B. Yang, D. X. Luo and F. G. Wu, J. Lumin., 2018, 200, 169–174 CrossRef CAS.
- S. X. Liu, S. W. Ma, S. X. Wang and Z. M. Ye, J. Lumin., 2019, 210, 155–163 CrossRef CAS.
- J. Zhou, X. Yu, T. Wang, D. Zhou and J. Qiu, J. Rare Earths, 2017, 35, 241–246 CrossRef CAS.
- L. Wang, C. L. Wang, Y. Chen, Y. Jiang, L. Chen, J. Z. Xu, B. Y. Qu and H. T. Hintzen, Chem. Mater., 2022, 34, 10068–10076 CrossRef CAS.
- B. Qiao, Z. L. Tang, Z. T. Zhang and L. Chen, Key Eng. Mater., 2007, 336, 629–632 Search PubMed.
- M. E. Foley, R. W. Meulenberg, J. R. McBride and G. F. Strouse, Chem. Mater., 2015, 27, 8362–8374 CrossRef CAS.
- A. Mendoud, L. Guerbous, A. Boukerika, B. Boudine and N. Benrekaa, Opt. Mater., 2018, 75, 802–808 CrossRef CAS.
- N. D. Hebbar, K. S. Choudhari, N. Pathak, S. A. Shivashankar and S. D. Kulkarni, New J. Chem., 2022, 46, 7032–7042 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- N. Guo, Y. J. Huang, H. P. You, M. Yang, Y. H. Song, K. Liu and Y. H. Zheng, Inorg. Chem., 2010, 49, 10907–10913 CrossRef CAS.
- C. G. Zuo, J. Z. Huang, S. Y. Liu, A. G. Xiao, Y. M. Shen, X. Y. Zhang, Z. H. Zhou and L. G. Zhu, Spectrochim. Acta, Part A, 2017, 187, 181–185 CrossRef CAS PubMed.
- M. Grüne and S. Schweizer, Materials, 2023, 16, 2596 CrossRef PubMed.
- C. Shalgaonkar and A. Narlikar, J. Mater. Sci., 1972, 7, 1465–1471 CrossRef CAS.
- X. D. Zhu, T. Wang, Z. C. Liu, Y. Y. Cai, C. Wang, H. Y. Lv, Y. Liu, C. C. Wang, J. B. Qiu, X. H. Xu, H. Q. Ma and X. Yu, Inorg. Chem., 2022, 61, 3223–3229 CrossRef CAS PubMed.
- Y. Liu, H. Zhang, Z. C. Liu, Y. Y. Cai, C. Wang, H. Y. Lv, X. D. Zhu, C. C. Wang, X. Yu, J. B. Qiu, H. Q. Ma, L. Zhao and X. H. Xu, J. Mater. Chem. C, 2022, 10, 3547–3552 Search PubMed.
- X. Zhou, L. Ning, J. Qiao, Y. Zhao, P. Xiong and Z. Xia, Nat. Commun., 2022, 13, 7589 CrossRef CAS.
- U. Fawad, H. J. Kim and M. Khan, Radiat. Meas., 2016, 90, 319–324 CrossRef CAS.
- X. Q. Zhou, K. Han, Y. X. Wang, J. C. Jin, S. D. Jiang, Q. Y. Zhang and Z. G. Xia, Adv. Mater., 2023, 35, 2212022 CrossRef CAS PubMed.
- X. Fan, Z. Liu, X. Yang, W. Chen, W. Zeng, S. Tian, X. Yu, J. Qiu and X. Xu, J. Rare Earths, 2019, 37, 679–690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01985c |
| This journal is © The Royal Society of Chemistry 2023 |