DOI:
10.1039/D3RA01893H
(Paper)
RSC Adv., 2023,
13, 19119-19129
Design, synthesis, and biological evaluation of morpholinopyrimidine derivatives as anti-inflammatory agents†
Received
23rd March 2023
, Accepted 1st June 2023
First published on 27th June 2023
Abstract
Here, we outline the synthesis of a few 2-methoxy-6-((4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)(phenyl)methyl)phenol derivatives and assess their anti-inflammatory activity in macrophage cells that have been stimulated by LPS. Among these newly synthesized morpholinopyrimidine derivatives, 2-methoxy-6-((4-methoxyphenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)phenol (V4) and 2-((4-fluorophenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)-6-methoxyphenol (V8) are two of the most active compounds which can inhibit the production of NO at non-cytotoxic concentrations. Our findings also showed that compounds V4 and V8 dramatically reduced iNOS and cyclooxygenase mRNA expression (COX-2) in LPS-stimulated RAW 264.7 macrophage cells; western blot analysis showed that the test compounds decreased the amount of iNOS and COX-2 protein expression, hence inhibiting the inflammatory response. We find through molecular docking studies that the chemicals had a strong affinity for the iNOS and COX-2 active sites and formed hydrophobic interactions with them. Therefore, use of these compounds could be suggested as a novel therapeutic strategy for inflammation-associated disorders.
1. Introduction
The body naturally responds to damage or infection by causing inflammation to protect itself. Redness, swelling, discomfort, heat, and loss of function may all serve as indicators. Through the removal of toxic substances and the restoration of damaged tissue, inflammation aids the body's ability to heal. However, if it is not under control, inflammation can also cause heart disease, stroke, cancer, and arthritis.1 Inflammation can be short-term or long-term. Short-term inflammation acts as an automatic defense response but its persistence can lead to chronic inflammation which can be a cause of diabetes, cancer, cardiovascular diseases, and many other illnesses.2,3 Prostaglandins (PGs), nitric oxide (NO), interleukin 6 (IL-6), interleukin 1β (IL-1β), and tumour necrosis factor-alpha (TNF-α) are a few examples of pro-inflammatory mediators that increase in concentration with rising inflammation. Pro-inflammatory mediators like PGs and NO are produced by cyclooxygenases (COXs) and nitric oxide synthase (NOS) respectively.4 Nitric oxide (NO) is a radical effector of the innate immune system that plays a significant role in both acute and chronic inflammation.5,6 It also reacts with various molecules/groups like superoxide or thiol to produce compounds like nitrosothiols, dinitrogen trioxide, peroxynitrite anion, and nitrogen dioxide. An isoform of nitric oxide synthase (iNOS) catalyzes the reaction that yields most of the nitric oxide (NO) from the amino acid L-arginine. It is a step-by-step reaction in which NO is released in the form of a by-product.7 iNOS is usually silenced in most of the tissue, preventing it from expressing itself.8 It can be triggered by several signaling pathways which get activated in response to external stimuli. Lipopolysaccharide (LPS) is one of the stimuli that play a key role in activating the toll-like receptor 4 (TLR4) and the following signaling cascade.9 Significant amounts of NO and other free radicals are produced when iNOS is activated in macrophages during the inflammatory phase.10
Likewise, the significant role of prostaglandins in an inflammation process is also underlined. The main enzyme system responsible for the formation of prostaglandins from arachidonic acid is cycloxygenase (COX) which found in two isoforms i.e., COX-1 and COX-2. COX-1 is active in majority of the cells whereas COX-2 get expressed only during an inflammatory response. The studies highlighted that the COX-2 isoform is primarily accountable for the production of pro inflammatory regulator prostaglandin (PG).11 Inhibiting these pro inflammatory molecules will open up new ways to treat inflammation. The first NSAIDs (non-steroidal anti-inflammatory drugs) to be adopted in therapeutic contexts were aspirin, diclofenac, and indomethacin. These competitive inhibitor drugs act on COX enzyme and block the synthesis of prostaglandins. This property of NSAIDs was first identified by Vane in 1971.12 In the late 1990's, COXIB (also known as COX-2 inhibitor) was alternatively introduced to overcome the gastro intestinal side effects caused by the NSAIDs, while treating inflammation. However the risk of serious cardiovascular diseases associated with it limited its use13 and the search for the new anti-inflammatory chemical entities continues.
Inflammation is a complex process involving multiple cells and molecules, and the inhibition of COX-1 or COX-2 alone may not be sufficient to control inflammation in all cases. Nitric oxide (NO) is one such molecule that plays a crucial role in inflammation, and its levels need to be tightly regulated to ensure a balanced immune response. The enzyme iNOS, which is activated during inflammation, is on account of the excessive release of NO by macrophages.14 By targeting iNOS, it is possible to reduce the levels of NO and thus control inflammation. Several drugs have been developed that target iNOS, but their efficacy and safety need to be further evaluated in clinical trials.
The development of new, more potent anti-inflammatory drugs has been a major focus of study. Usually, it can be done in two ways, (a) by substituting, altering, or modifying the pre-existing drugs or (b) by developing a completely new drug. To develop new drugs that operate well as anti-inflammatory agents, attempts have been made to hybridize chemical elements with biological processes. Such a logical approach can alleviate the negative effects of an existing medication. One of the newest approaches of drug development, known as molecular hybridization (MH), relies on combining various pharmacophores for the development of novel hybrids. Typically, the resultant hybrid molecules may show enhanced activity and affinity in comparison to the parent scaffold due to the resulting dual action mode.15 Having reduced adverse side effects. This has so far resulted in the development of several new anti-inflammatory scaffolds.
Organic compounds, especially heterocyclic compounds have primarily contributed for the development of library of bioactive compounds. The library of bioactive compounds shown a wide range of applications in the field of medicinal chemistry.16–19 According to the FDA (Food and Drug Administration) database, nitrogen-based heterocyclic compounds are present in over 60% of small molecule drugs, supporting the significance of nitrogen-based heterocycles.20 The studies signify that benzhydrylpiperazine scaffolds are important scaffolds found in various bioactive compounds with antihistaminic,21 anti-leishmanial,22 antibacterial,23 and anticancer,24 antimicrobial25 and anti-inflammatory activity.26,27 The benzhydryl piperazine has received increased attention in the design of new anti-inflammatory agents. Based on the literature found it as an anti-inflammatory agent.28,29 Similarly, six-membered aromatic heterocyclic motif pyrimidine underlined in several natural and synthetic bioactive small molecules. There are several studies which utilizes pyrimidine for the development of library of pharmacologically active agents.30 From the literature survey, numerous pharmacological properties of pyrimidine derivatives have been reported, including antifungal,31 antibacterial,32 analgesic,33 antileishmanial,34 antihypertensive,35 antiviral,36 antipyretic,37 antidiabetic,38 antioxidant,38 anticonvulsant,39 antihistaminics,40 and anti-inflammatory.41,42 The anti-inflammatory action of pyrimidines has received increased attention in literature publications. Researchers are focusing on the synthesis of substituted pyrimidines as anti-inflammatory drugs due to the enormous diversity in their chemistry. Many pyrimidine analogs have previously received anti-inflammatory medication approval and are being used in clinical settings, including afloqualone, proquazone, epirizole, and tofacitinib.43
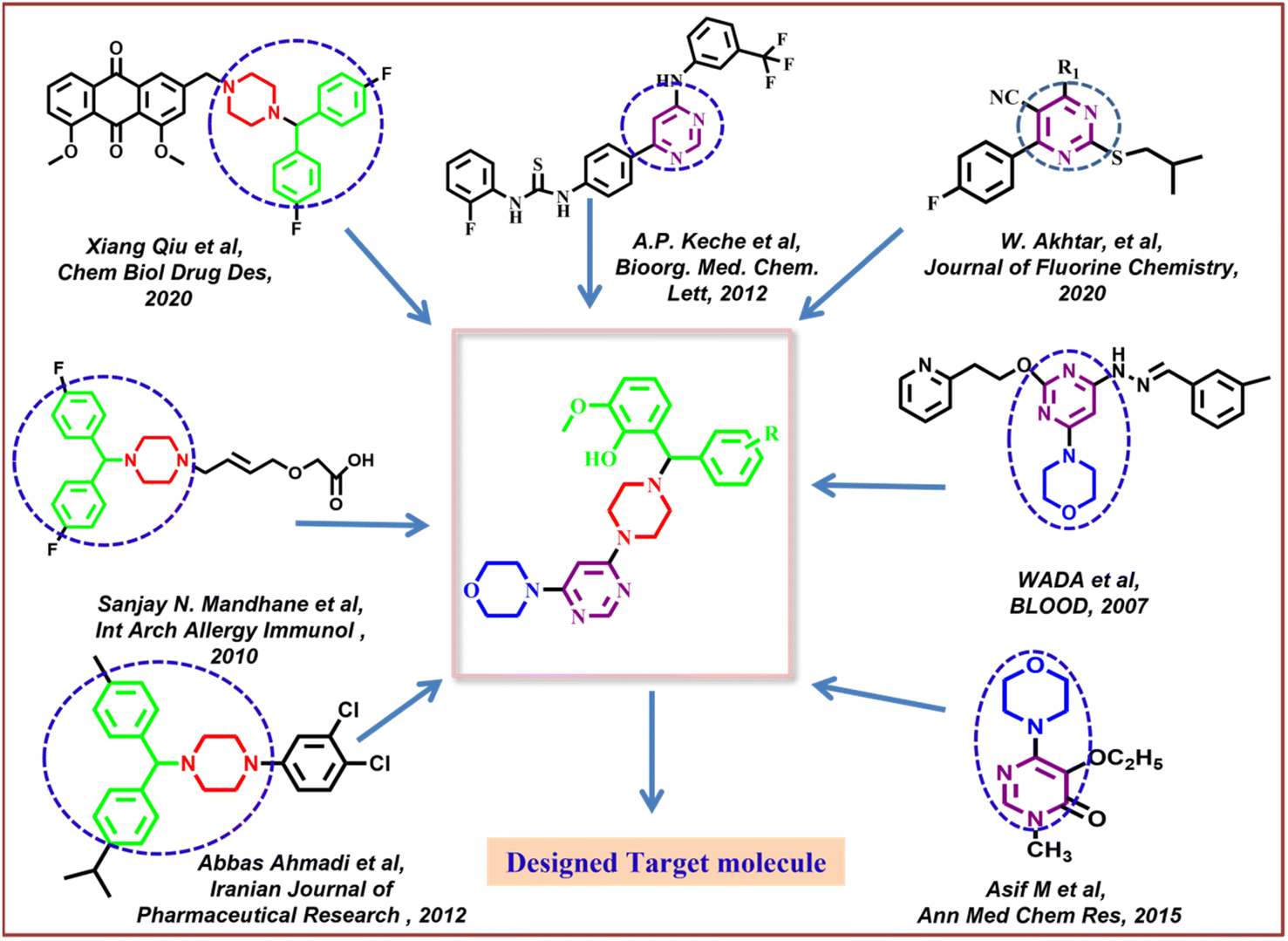
Along with pyrimidine, morpholine-clubbed pyrimidines were mostly studied as PI3K/Akt/mTOR pathway inhibitors.44,45 Liture revealed that, a morphilinopyramidine-based candidate drug called STA5326 is now being tested in phase 2 clinical studies on people with rheumatoid arthritis and Crohn's disease.46 In Fig. 1, a few compounds containing morpholine, pyrimidine, and benzhydryl piperazine units were displayed as good leads, encouraging us to apply these moieties in molecular hybridization for constructing our targeted molecules.
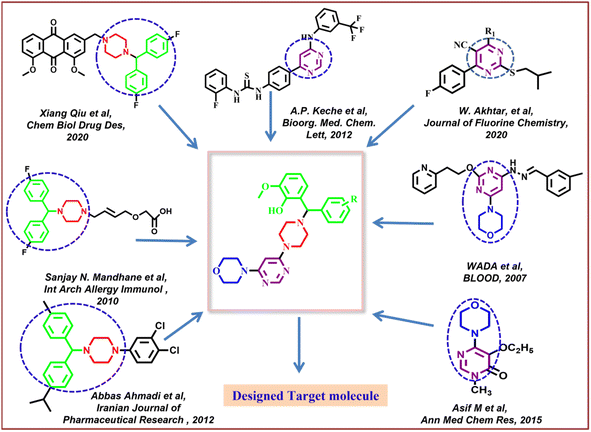 |
| | Fig. 1 Design strategy for target compounds. | |
After taking into account all of these facts, the present study disclosed the synthesis of new 2-methoxy-6-((substituted phenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)phenol derivatives by employing the synthetic protocol of multicomponent Petasis reaction. The synthetic compounds underwent in vitro anti-inflammation and cytotoxicity evaluation. Hence, our study significantly contributed into the domain of development of new anti-inflammation agents for potential future therapeutic advancement.
2. Results and discussion
2.1. Chemistry
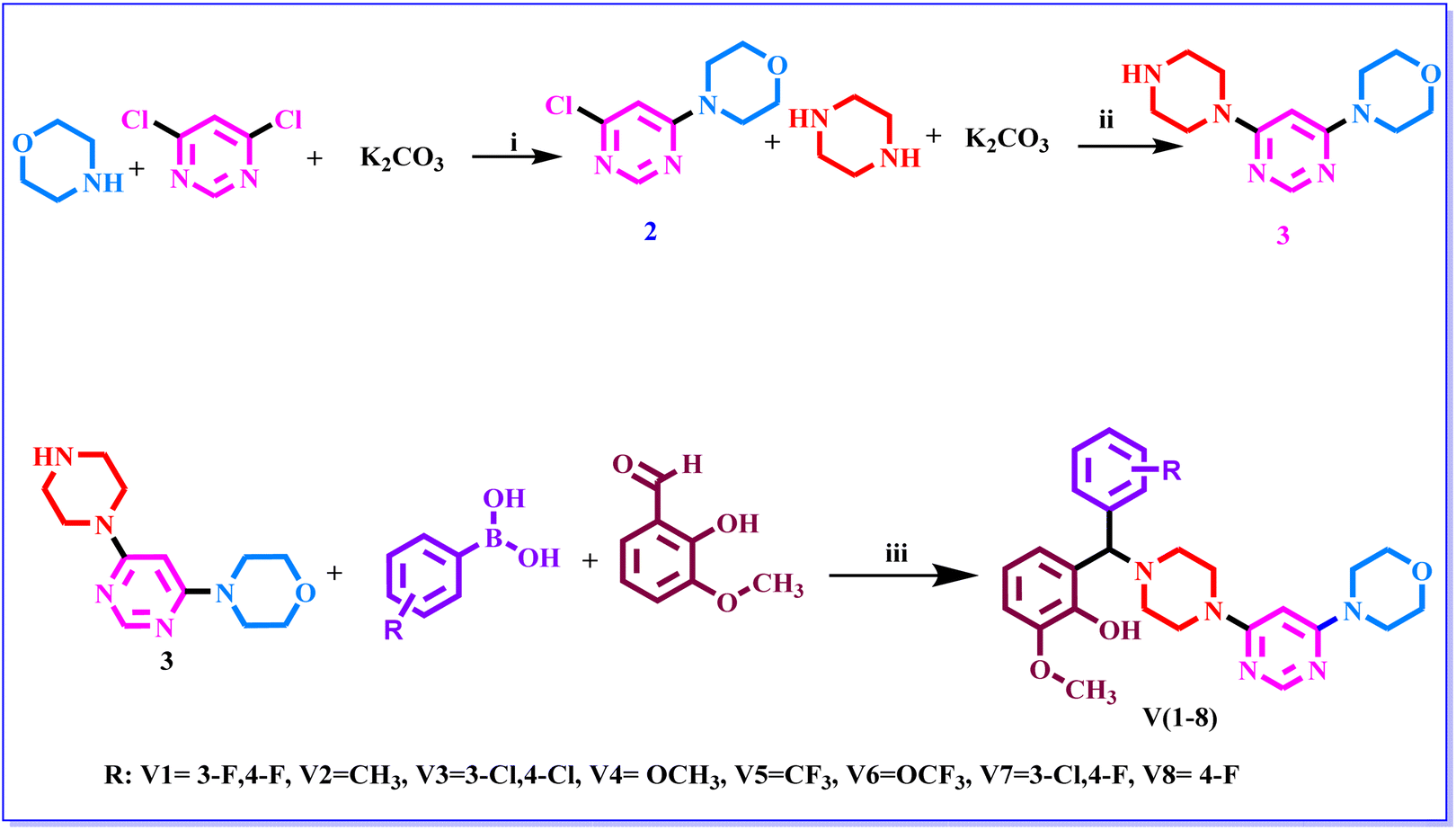
The three-step synthetic protocol as indicated in Scheme 1 was followed to synthesise a new series of 2-methoxy-6-((substituted phenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)phenol (V1–V8). Thin layer chromatography (TLC) was used to determine the purity of the compounds using the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethylacetate–n-hexane solvent system. Sophisticated instrumentation techniques such as HRMS, NMR, and elemental analysis were used investigate the molecular weight, structure, and purity of the synthesized compounds (ESI†). Further, the synthesized compounds were subjected to investigate the melting point. The findings of the elemental analysis and spectrum data have been found to be entirely consistent with the synthesised compounds. The solvent for the NMR study was CDCl3-d. Given that such solvents can't be completely deuterated. As a result, the peaks of these solvents' non-deuterated types were additionally identified in the spectra. In the CDCl3 analysis, these peaks were seen in the range of 7.21 to 7.33 ppm. The presence of trace amount of moisture was also identified by an additional peak in the range of 1–2 ppm. Proton NMR study of compounds V1–V8 revealed that the singlet signal with δ value at 4.38–4.47 ppm was observed for CH protons. Furthermore, the 2nd and 5th aromatic protons of the pyrimidine ring were identified at around 8.1 and 5.44–5.47 ppm respectively. The proton of phenolic hydroxy group was observed ∼11 ppm while singlet at around 3.79–3.89 ppm for three protons confirmed the presence of methoxy group at phenyl ring. Few spectra revealed that the protons of morpholine and piperazine group were overlapped each other, therefore, they were not able to precisely distinguished (Fig. 2).
:1 ethylacetate–n-hexane solvent system. Sophisticated instrumentation techniques such as HRMS, NMR, and elemental analysis were used investigate the molecular weight, structure, and purity of the synthesized compounds (ESI†). Further, the synthesized compounds were subjected to investigate the melting point. The findings of the elemental analysis and spectrum data have been found to be entirely consistent with the synthesised compounds. The solvent for the NMR study was CDCl3-d. Given that such solvents can't be completely deuterated. As a result, the peaks of these solvents' non-deuterated types were additionally identified in the spectra. In the CDCl3 analysis, these peaks were seen in the range of 7.21 to 7.33 ppm. The presence of trace amount of moisture was also identified by an additional peak in the range of 1–2 ppm. Proton NMR study of compounds V1–V8 revealed that the singlet signal with δ value at 4.38–4.47 ppm was observed for CH protons. Furthermore, the 2nd and 5th aromatic protons of the pyrimidine ring were identified at around 8.1 and 5.44–5.47 ppm respectively. The proton of phenolic hydroxy group was observed ∼11 ppm while singlet at around 3.79–3.89 ppm for three protons confirmed the presence of methoxy group at phenyl ring. Few spectra revealed that the protons of morpholine and piperazine group were overlapped each other, therefore, they were not able to precisely distinguished (Fig. 2).
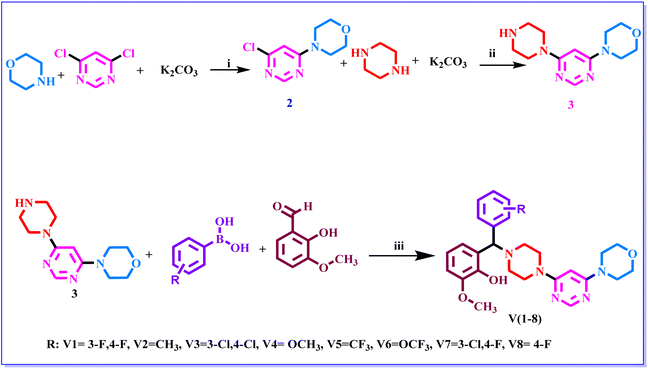 |
| | Scheme 1 The synthetic route of compounds. Reagent and conditions: (i) DMF, RT, K2CO3, 4 h. (ii) 1,4-dioxane, 100 °C, K2CO3 20 h (iii) 1,4-dioxane, 110 °C, 48 h. | |
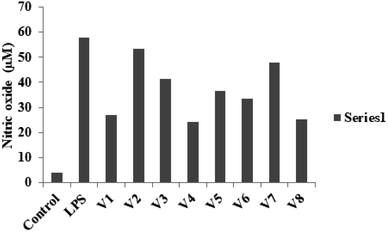 |
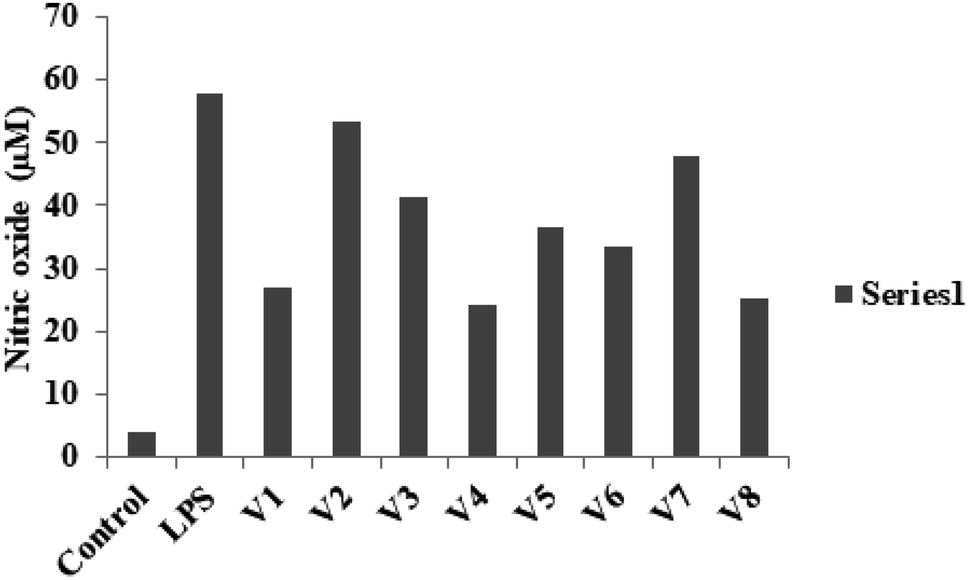
| | Fig. 2 NO production was determined at 12.5 μM concentration of different derivatives on LPS-stimulated RAW 264.7 cells. | |
2.2. Biological evaluation
2.2.1 Evaluation of the anti-inflammatory properties. Inflammatory diseases or injuries can be caused by an overproduction of the inflammatory intermediate NO. The ability of various substances to prevent NO generation is widely used to determine their anti-inflammatory efficacy.47 In the current study, we used a Griess assay to determine if our compounds might reduce NO production in RAW 264.7 macrophages that had been stimulated with LPS. None of the test compounds show toxicity at the 12.5 μM concentration when they were treated RAW 264.7 cells (Fig. 3). The results revealed that the cells treated with 100 ng mL−1 LPS showed a substantial upsurge in NO release as compared with untreated cells. When these cells were treated with V1–V8, the NO generation was dramatically reduced. Among the synthetic compounds, compounds V4 and V8 had the most NO-inhibiting effects. It was demonstrated that compounds containing an aromatic ring with an electron-withdrawing group (V4 and V8) have the ability to inhibit the production of NO. The inhibitory potency was raised by adding a methoxy and a fluoro group to the R position. The inhibitory activity of compounds having an aromatic ring substituted with electron-donating groups was, however, less effective.
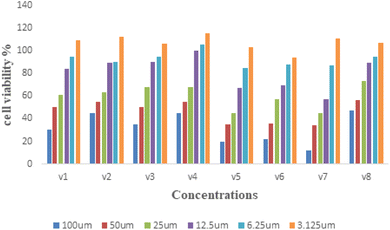 |
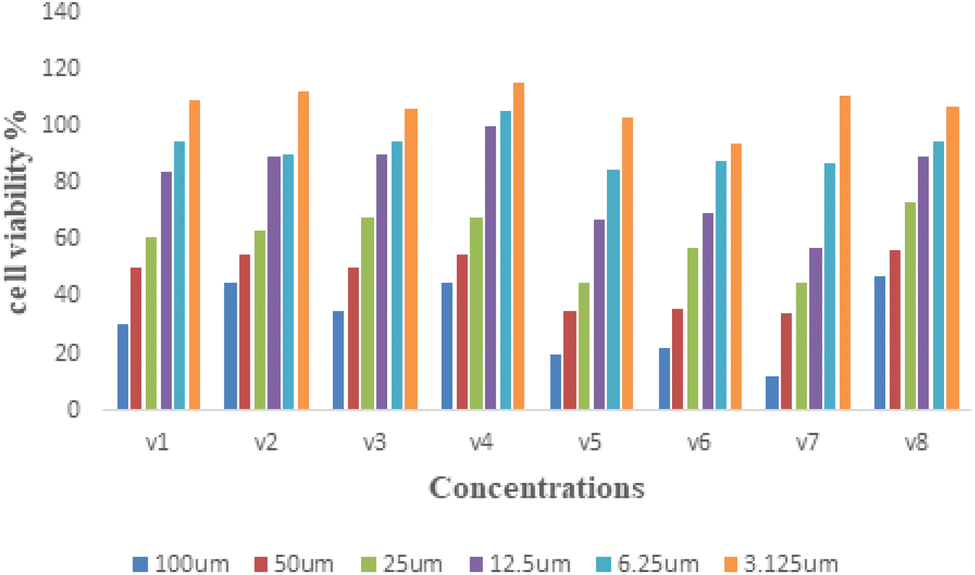
| | Fig. 3 Cytotoxicity assessment of treatment with V1–V8 compounds. | |
2.2.2 Assessment of the cytotoxicity. The MTT assay was used to determine the toxicity of the test compounds on cell growth. Fig. 3 depicts the results of a 24 hours exposure experiment using RAW 264.7 cells with test compounds at concentration of 3.125–100 μM. There was no noticeable cytotoxicity in the cells at a lower concentration below 50 μM. As a result, additional evaluations of different bioactivities were conducted on RAW 264.7 cells for 24 h.
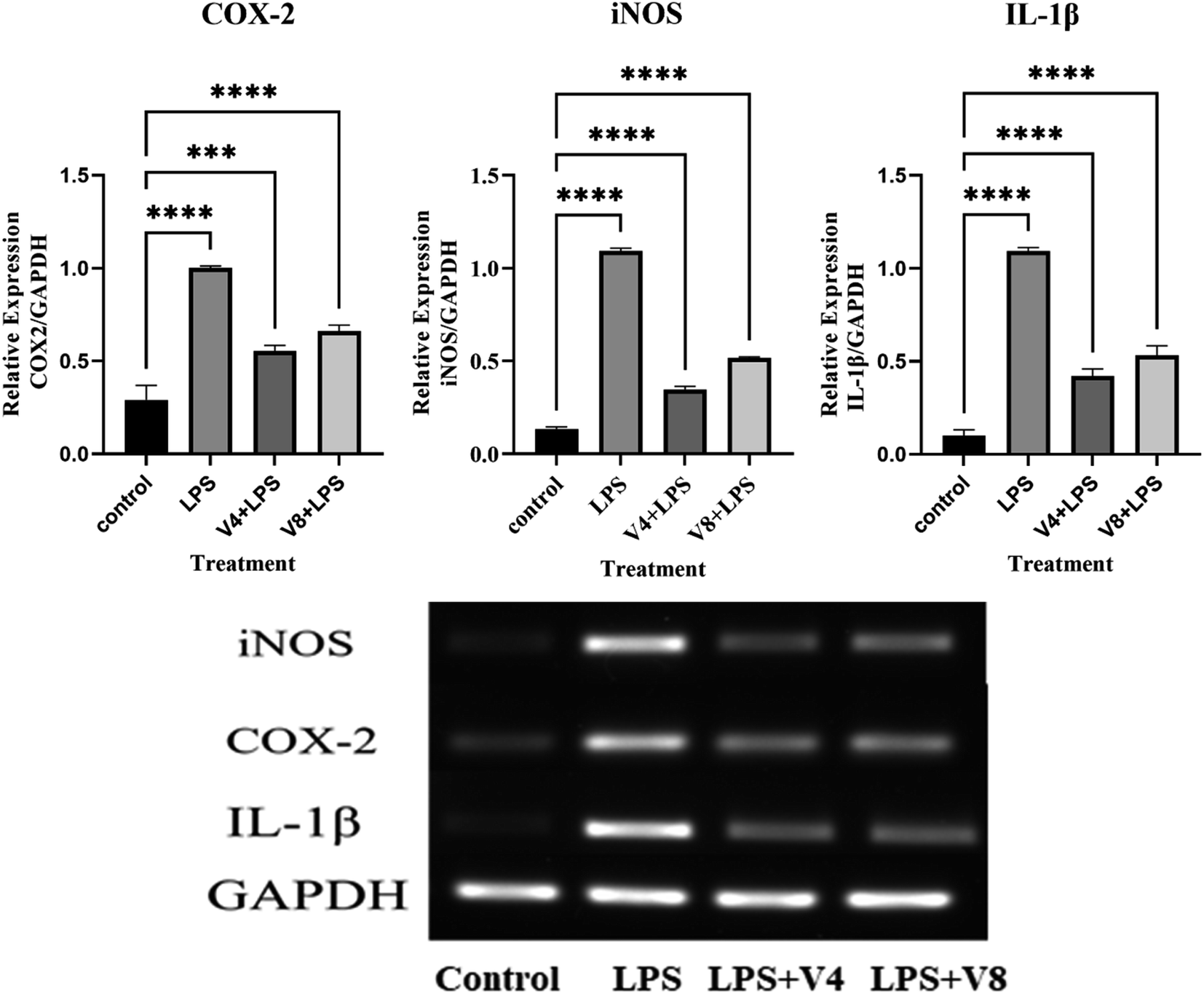
2.2.3 Effects on mRNA expression. Inflammatory diseases are also linked to inflammatory cytokines such iNOS, IL-1β, and COX-2. In order to ascertain if the substances V4 and V8 inhibited the transcriptional expression of these cytokines, we performed a quantitative PCR study. The findings demonstrated that the expression of the iNOS, IL-1β, and COX-2 genes was dramatically decreased by compounds V4 and V8 (Fig. 4). To further examine the anti-inflammatory activity, we measured their protein levels. It was shown that treating the LPS-induced RAW 264.7 cells with 12.5 μM concentrations of the compounds significantly reduced the most noticeable elevations of iNOS and COX-2 protein levels. Thus our findings revealed that, compounds V4 and V8 substantially reduced the expression of iNOS and COX-2 in LPS-induced RAW 264.7 cells at the mRNA and protein levels.
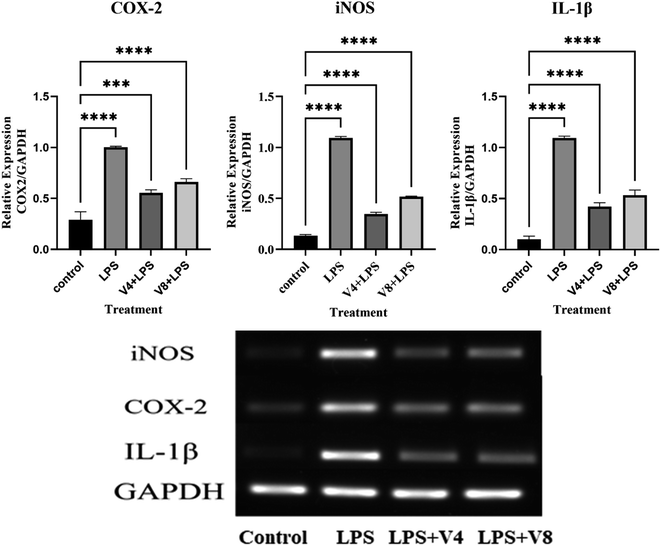 |
| | Fig. 4 Effects of target compounds on mRNA levels of iNOS, IL-1β and COX-2 in LPS-induced RAW 264.7 cells. | |
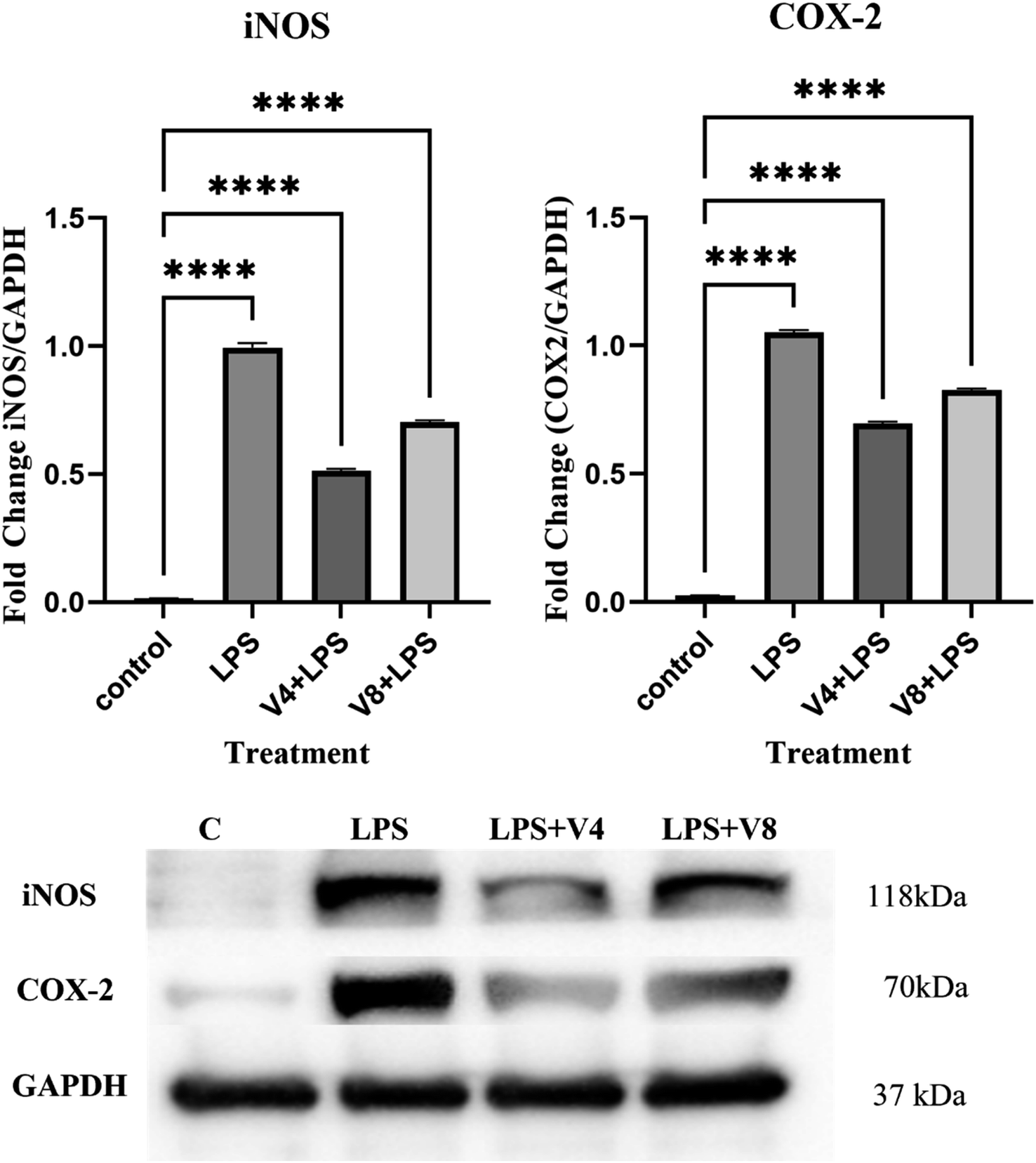
2.2.4 Effects on protein expression. The production of inflammatory mediators in the course of LPS and other triggers is regulated by two key enzymes, nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2). Hence, using compounds V4 and V8 (12.5 μM) and LPS (100 ng mL−1), we treated RAW 264.7 cells for 24 hours to determine how well they could modify the expression of iNOS and COX-2 that was caused by LPS. By using western blotting, the expression of these two proteins was evaluated. It was observed that the level of iNOS and COX-2 proteins was significantly increased after LPS treatment, however, this LPS-induced upregulation was inhibited by compounds V4 and V8, as demonstrated in Fig. 5. iNOS is the main catalyst for NO generation in conditions of high immunological reactivity, whereas COX-2 controls prostaglandin synthesis. As a result, persistent inflammation may be aided by the overexpression of COX-2 and/or iNOS. Our findings imply that compounds V4 and V8 can be promising anti-inflammatory drugs that can inhibit LPS-induced signaling activity since they can limit the overexpression of both of these proteins in an inflammatory situation.
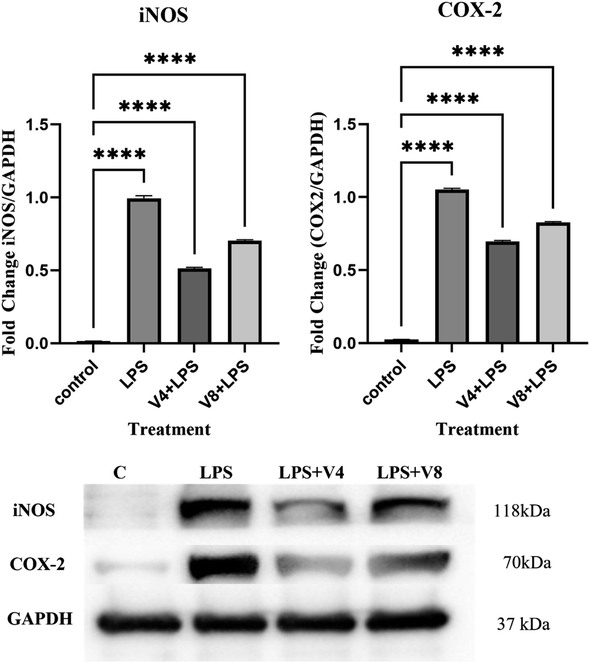 |
| | Fig. 5 The impact of compounds V4 and V8 on LPS-induced iNOS and COX-2 protein expressions in RAW 264.7 cells following a 24 h treatment with a combination of LPS. | |
3. Molecular docking studies
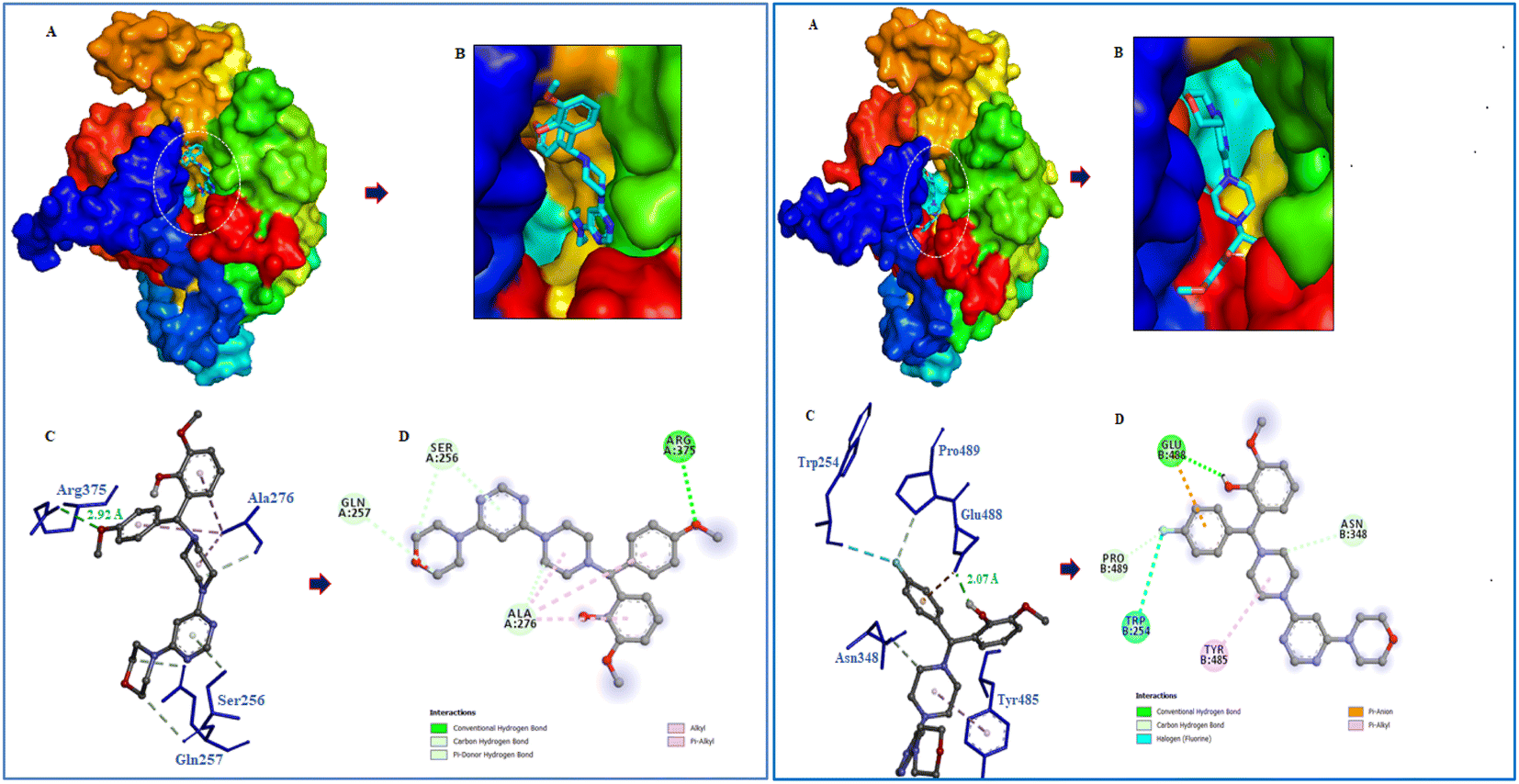
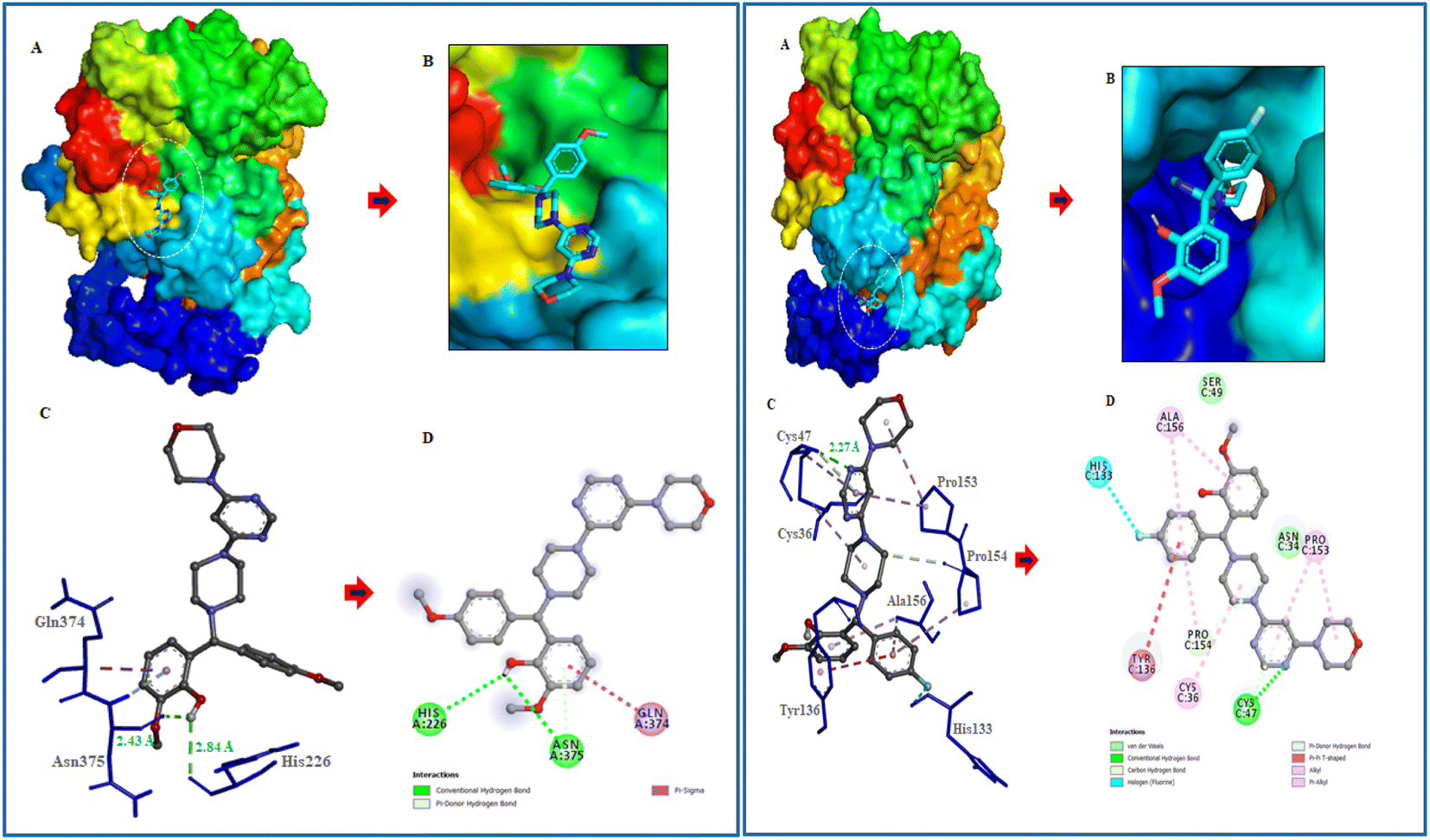
Following the development of morpholinopyrimidine derivatives, a molecular docking study using the AutoDock vina program was conducted in order to comprehend the biological outcomes of our active compounds. The COX-2 and iNOS active sites have been investigated for possible modes of binding for the two most potent compounds (V4 and V8). The calculated binding free energies of the test compound were shown in Table 1. The docking results demonstrated that the compounds V4 and V8 binds with the iNOS with binding free energy below −8.0 kcal mol−1 and COX-2 with binding free energy below −10.0 kcal mol−1. The investigation of their binding poses disclosed that both compounds were well-fitted at the binding site of the target proteins. Protein-ligand interactions investigation deduced that the compound V4 showed hydrogen bonding interactions with Arg375 and hydrophobic interactions with Ala276, Ser256, and Gln257 residues of iNOS, as shown in Fig. 6. Further, it was involved in hydrogen bonding interactions with His226, Asp375 and hydrophobic interactions with Gln374 residues of COX-2 as shown in Fig. 7.
Table 1 Binding energies of NO and COX-2 inhibitors to the active cavities of iNOS (PDB code: 3E6T) and COX-2 (PDB code: 1CX2) and targeting residues of the binding site
| Compound |
Binding free energy (kcal mol−1) |
Residues |
| iNOS |
COX-2 |
iNOS |
COX-2 |
| V4 |
−8.8 |
−10.0 |
Arg 375, Ala276, Ser256, Gln257 |
His226, Asp375, Gln374 |
| V8 |
−8.7 |
−10.3 |
Glu488, Trp254, Asn348, Pro489, Tyr485 |
Cys47, His133, Pro154, Tyr136, Ala156, Pro153 |
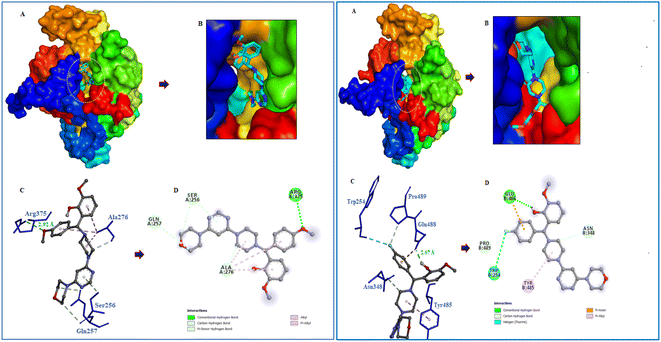 |
| | Fig. 6 Molecular interactions of iNOS (PDB ID: 3E6T) with V4 and V8. (A) The potential surface binding cavity of protein in complex with the ligand. (B) Zoomed view of the surface complex. (C) Interacting residues of protein with the ligand. (D) 2D representation of protein residues interacted with the ligand. | |
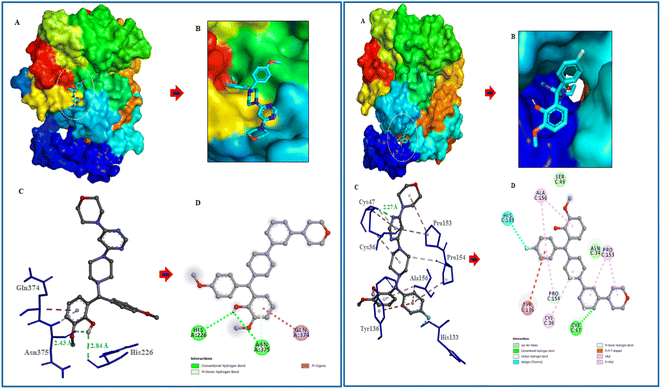 |
| | Fig. 7 Molecular interactions of COX-2 (PDB ID: 1CX2) with V4 and V8. (A) The potential surface binding cavity of protein in complex with the ligand. (B) Zoomed view of the surface complex. (C) Interacting residues of protein with the ligand. (D) 2D representation of protein residues interacted with the ligand. | |
Similarly, compound V8 showed hydrogen bonding interactions with Glu488, halogen bonding with Trp254, and hydrophobic interactions with Asn348, Pro489, and Tyr485 residues of iNOS as shown in Fig. 6. This compound also showed hydrogen bonding interactions with Cys47, halogen bonding with His133, hydrophobic interactions with Pro154, and hydrophobic interactions with Ala156 and Pro153 residues of COX-2. The pi–pi stacking between Tyr136 and the phenyl ring of the compound also noticed as shown in Fig. 7.
4. Physicochemical parameters
The most important factor in medication design after potency and selectivity is optimizing the administration route. Verifying that a molecule complies with the Lipinski and Veber guidelines is a reliable method for predicting a drug's oral bioavailability. According to Lipinski's guidelines, a compound had to satisfy a minimum of three of the following requirements to be taken orally: have a molecular weight (MW) of no more than 500, an octanol–water partition coefficient (logP) of no more than 5, a maximum of 5 H-bond donor atoms (HBD), and a maximum of 10 H-bond acceptor atoms (HBA). Veber then proposes further guidelines for drug bioavailability, including (a) the number of rotatable bonds (nRB) <10, and (b) the maximum polar surface area (PSA) of 140 Å2. For the compounds with striking biological activity, the Lipinski and Veber rules were determined using the free access website: https://www.molinspiration.com/cgi-bin/properties. Table 2 displays the calculated Lipinski's and Veber's parameters for V4 and V8. Since all substances follow the Lipinski and Veber principles, they may likely have a high oral bioavailability.
Table 2 Calculated Lipinski and Veber parameters for compounds V4 and V8
| Comp |
MW |
logP |
HBD |
HBA |
nVs |
nRB |
PSA |
| *Limpinski |
<500 |
<5 |
<5 |
<10 |
— |
— |
— |
| **Veber |
— |
— |
— |
— |
— |
<10 |
<140 Å2 |
| V4 |
491.59 |
3.20 |
1 |
9 |
0 |
7 |
83.43 |
| V8 |
479.56 |
3.30 |
1 |
8 |
0 |
6 |
74.19 |
5. Conclusion
The present study reported the synthesis and anti-inflammatory screening of eight new (V1–V8) phenyl morpholinopyrimidine derivatives. The ability of each synthesized compound to knock down LPS-induced NO generation in RAW 264.7 macrophages at a concentration of 12.5 μM was tested. Among the test compounds, V4 and V8 were able to reduce the formation of the inflammatory mediator NO. In macrophage cells, both of these compounds were non-cytotoxic at the tested concentrations (maximum 50 μM). The docking study revealed that the molecules were bind to the target protein iNOS and COX-2 with strong binding free energy up to −10 kcal mol−1. Although certain aspects of the mechanisms of these two lead compounds' NO and COX-2 inhibitory activity have been established by molecular docking studies, western blot, and qPCR study, further investigation is needed to fully understand their mechanisms of action as therapeutic agents. Our findings are consistent with the hypothesis that while compounds V4 and V8 predominantly restrict NO generation, compound V4 also has significant anti-inflammatory properties as evidenced by decreased iNOS, COX-2 expression, and NO production. With aim to develop potential anti-inflammatory drug, the current research also illustrates a novel technique for developing and synthesizing a more effective scaffold.
6. Experimental
6.1 Chemistry
The reagents and chemicals in AR grade were procured from commercial suppliers and used without purification. The silica gel coated aluminium TLC plates of Merck Millipore (TLC Silica gel 60 F254) were used to observed the chemical reactions and the synthesized compounds were purified by column chromatography using silica gel (200–400 mesh, SRL, India) as stationary phase. Bruker's maXis-II ETD mass-spectrometer was used to record molecular weight at high-resolution. NMR spectra of the compounds were recorded in CDCl3 or d6-DMSO using Bruker 400/600 MHz spectrometers.
6.1.1 General procedure for the synthesis of 4-(6-chloropyrimidin-4-yl) morpholine (2). To a solution of morpholine (0.034 mol) and DMF in 50 mL round bottom flask, K2CO3 (3 mmol) was added and stirred for 20 to 30 minutes followed by addition of 4,6 dichloropyrimidine (0.033 mol) and stirred at room temperature for 4–6 h. After the completion of the reaction, the mixture was brought to room temperature. The obtained reaction mixture was diluted with ethylacetate and washed with water (100 mL × 3) and the organic layer was dried over anhydrous Na2SO4. The organic solvent was removed under vacuum and the crude was then purified by column chromatography using 40:60 ethyl acetate/hexane as mobile phase to obtain intermediate 2.
6.1.2 General procedure for the synthesis of 4-(6-(piperazin-1-yl) pyrimidin-4-yl) morpholine (3). To a stirring solution of piperazine (0.075 mol) in 30 mL, dioxane was added with K2CO3 (3 mmol) and stirred for 20 to 30 minutes followed by addition of 4-(6-chloropyrimidin-4-yl) morpholine (2) (0.015 mol) and refluxed for 18–20 h at 80–110 °C. After the completion of the reaction, the mixture was brought to room temperature and dioxane was removed under vacuum. The crude obtained was diluted with chloroform and washed with water (100 mL × 3). The organic layer was then separated and dried over anhydrous Na2SO4. The crude mixture was then purified by column chromatography using 10:90 MeOH/chloroform solvent as mobile phase. The yield of the product was found to be 70%.
6.1.3 General procedure for the preparation of 2-((substituted phenyl) (4-(6-morpholinopyrimidin-4-yl) piperazin-1-yl) methyl)-6-methoxyphenol (V1–V8). Compound 3 (0.002 mol) was added to o-vanilline (0.0019 mol) in anhydrous 1,4 dioxane (20 mL). After 5 minutes add different substituted boronic acid (0.003 mol) and the reaction mixture was refluxed at 80–90 °C for 45–48 h, Scheme 1. The reaction was monitored via TLC. The mixture was cooled to room temperature following the process. Under vacuum, the reaction mixture was concentrated, and the resulting crude was diluted with ethyl acetate and washed three times with water (100 mL). After being separated, the organic layer was dried over anhydrous Na2SO4. The target compounds were subsequently purified from the crude mixture using column chromatography with a mobile phase made up of a 40:60 ethyl acetate:hexane solvent.
6.1.3.1 2-((3,4-Difluorophenyl) (4-(6-morpholinopyrimidin-4-yl) piperazin-1-yl) methyl)-6-methoxyphenol (V1). Yield 70%; mp 110–120 °C. 1H NMR (500 MHz, chloroform-d) δ 10.81 (s, 1H), 8.25 (s, 1H), 7.32 (s, 1H), 7.18 (s, 1H), 7.13–7.06 (m, 1H), 6.81–6.73 (m, 2H), 6.63 (s, 1H), 5.51 (s, 1H), 4.47 (s, 1H), 3.89 (s, 3H), 3.79–3.75 (m, 4H), 3.67 (d, J = 33.8 Hz, 4H), 3.55 (t, J = 5.0 Hz, 4H), 2.60 (d, J = 67.2 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 185.09, 180.04, 155.08, 147.44, 144.50, 136.86, 124.57, 120.66, 119.79, 117.77, 117.63, 117.22, 117.21, 110.70, 81.15, 76.01, 66.45, 55.93, 51.27, 44.71, 44.40. HRMS: (ESI, m/z): [M + H]+ calcd for C26H29F2N5O3 497.224; found 498.231; anal. calcd for C26H29F2N5O3; C, 62.77; H, 5.88; F, 7.64; N, 14.08; O, 9.65%; found C, 62.67; H, 5.86; F, 7.62; N, 14.05; O, 9.63%.
6.1.3.2 2-Methoxy-6-((4-(6-morpholinopyrimidin-4-yl) piperazin-1-yl) (p-tolyl)methyl)phenol (V2). Yield 65%; mp 80–100 °C. 1H NMR (500 MHz, chloroform-d) δ 8.16 (d, J = 13.2 Hz, 1H), 7.03 (d, J = 7.8 Hz, 2H), 6.81 (dd, J = 31.9, 7.5 Hz, 2H), 6.69 (t, J = 7.8 Hz, 1H), 6.61 (d, J = 7.9 Hz, 1H), 6.49 (dd, J = 13.8, 8.8 Hz, 1H), 5.48 (s, 1H), 4.37 (s, 1H), 3.79 (d, J = 16.5 Hz, 3H), 3.72–3.65 (m, 4H), 3.45 (dt, J = 17.7, 4.9 Hz, 8H), 2.51 (d, J = 59.2 Hz, 4H), 2.23 (s, 3H).13C NMR (126 MHz, CDCl3) δ 162.48, 161.98, 155.12, 149.05, 147.27, 144.49, 142.84, 138.46, 136.93, 135.14, 128.59, 128.30, 127.42, 126.34, 124.34, 120.06, 119.11, 118.62, 117.31, 113.34, 109.40, 80.48, 74.66, 65.51, 53.91, 53.51, 49.39, 43.45, 43.18, 20.06. HRMS: (ESI, m/z): [M + H]+ calcd for: C27H33N5O3 475.59; found 475.241; anal. calcd for C27H33N5O C, 68.19; H, 6.99; N, 14.73; O, 10.09%; found C, 68.23; H, 6.99; N, 14.73; O, 10.09%.
6.1.3.3 2-((3,4-Dichlorophenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)-6-methoxyphenol (V3). Yield 60%; mp 100–125 °C. 1H NMR (500 MHz, chloroform-d) δ 10.59 (s, 1H), 8.24 (s, 1H), 7.52 (s, 1H), 7.43–7.31 (m, 2H), 6.82–6.70 (m, 2H), 6.64 (s, 1H), 5.51 (s, 1H), 4.47 (s, 1H), 3.89 (s, 3H), 3.80–3.74 (m, 4H), 3.65 (s, 4H), 3.54 (t, J = 4.9 Hz, 4H), 2.60 (d, J = 64.3 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 155.06, 147.62, 145.32, 132.93, 132.21, 131.05, 130.24, 127.57, 123.79, 120.63, 119.82, 117.64, 117.34, 110.73, 81.23, 73.43, 66.47, 55.94, 51.31, 44.67, 44.35, 38.63. HRMS: (ESI, m/z): [M + H]+ calcd for C26H29Cl2N5O3 529.16; found 530.1720; anal. calcd for C26H29Cl2N5O3; C, 58.87; H, 5.51; Cl, 13.37; N, 13.20; O, 9.05% found C, 58.90; H, 5.51; Cl, 13.38; N, 13.21; O, 9.05%.
6.1.3.4 2-Methoxy-6-((4-methoxyphenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)phenol (V4). Yield 70%; mp 90–100 °C. 1H NMR (500 MHz, chloroform-d) δ 11.76 (s, 1H), 8.15 (s, 1H), 7.28 (d, J = 8.2 Hz, 2H), 6.76 (d, J = 8.8 Hz, 2H), 6.70 (d, J = 8.1 Hz, 1H), 6.64 (t, J = 7.9 Hz, 1H), 6.53 (s, 1H), 5.44 (s, 1H), 4.38 (s, 1H), 3.82 (s, 4H), 3.69 (d, J = 9.0 Hz, 6H), 3.58 (d, J = 32.3 Hz, 4H), 3.45 (t, J = 4.8 Hz, 4H), 2.53 (d, J = 62.8 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 162.29, 161.76, 158.39, 156.15, 147.25, 144.39, 130.06, 128.76, 124.31, 120.03, 118.29, 113.23, 112.62, 109.39, 80.39, 74.10, 65.50, 54.86, 54.23, 50.15, 43.50, 43.15. HRMS: (ESI, m/z): [M + H]+ calcd for C27H33N5O4 491.25; found 492.2605; anal. calcd for C27H33N5O4; C, 65.97; H, 6.77; N, 14.25; O, 13.02%; found C, 65.87; H, 6.75; N, 14.22; O, 13.00%.
6.1.3.5 2-Methoxy-6-((4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)(4-(trifluoromethyl)phenyl)methyl)phenol (V5). Yield 70%; mp 100–120 °C. 1H NMR (500 MHz, chloroform-d) δ 10.92 (s, 1H), 8.16 (s, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.48 (d, J = 8.1 Hz, 2H), 6.70 (d, J = 7.9 Hz, 1H), 6.65 (t, J = 7.9 Hz, 1H), 6.56 (d, J = 7.7 Hz, 1H), 5.45 (s, 1H), 4.47 (s, 1H), 3.80 (s, 3H), 3.67 (t, J = 4.7 Hz, 4H), 3.57 (d, J = 24.4 Hz, 4H), 3.44 (t, J = 4.8 Hz, 4H), 2.52 (d, J = 79.2 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 162.40, 161.90, 156.29, 147.28, 144.08, 142.74, 129.33, 129.07, 127.66, 124.95, 123.98, 123.59, 121.82, 119.73, 118.66, 109.69, 80.48, 73.71, 65.49, 54.87, 50.39, 43.46, 43.15. HRMS: (ESI, m/z): [M + H]+ calcd for C27H30F3N5O3 529.23; found 530.2374; anal. calcd for; C27H30F3N5O3 C, 61.24; H, 5.71; F, 10.76; N, 13.23; O, 9.06%; found C, 61.10; H, 5.70; F, 10.77; N, 13.20; O, 9.05%.
6.1.3.6 2-Methoxy-6-((4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)(4-(trifluoromethoxy)phenyl)methyl)phenol (V6). Yield 80%; mp 90–100 °C. 1H NMR (500 MHz, chloroform-d) δ 11.12 (s, 1H), 8.16 (s, 1H), 7.41 (d, J = 8.2 Hz, 2H), 7.08 (d, J = 8.2 Hz, 2H), 6.71 (d, J = 8.9 Hz, 1H), 6.66 (t, J = 7.9 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 5.45 (s, 1H), 4.42 (s, 1H), 3.82 (s, 3H), 3.68 (t, J = 4.9 Hz, 4H), 3.57 (d, J = 27.8 Hz, 4H), 3.45 (d, J = 5.0 Hz, 4H), 2.60 (s, 4H). 13C NMR (126 MHz, CDCl3) δ 163.33, 162.80, 157.20, 148.90, 148.34, 145.15, 138.20, 129.88, 124.84, 123.53, 121.34, 120.84, 119.60, 110.66, 81.46, 74.65, 66.51, 55.89, 51.36, 44.52, 44.20. HRMS: (ESI, m/z): [M + H]+ calcd for C27H30F3N5O4 545.22; found 546.2323; anal. calcd for C27H30F3N5O4 C, 59.44; H, 5.54; F, 10.45; N, 12.84; O, 11.73%; found C, 59.36; H, 5.53; F, 10.43; N, 12.82; O, 11.71%.
6.1.3.7 2-((3-Chloro-4-fluorophenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)-6-methoxyphenol (V7). Yield 70%; mp 80–90 °C. 1H NMR (500 MHz, chloroform-d) δ 10.83 (s, 1H), 8.16 (s, 1H), 7.41 (d, J = 7.0 Hz, 1H), 7.24 (d, J = 44.8 Hz, 1H), 7.01 (d, J = 8.5 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.67 (t, J = 7.8 Hz, 1H), 6.55 (d, J = 7.3 Hz, 1H), 5.45 (s, 1H), 4.39 (s, 1H), 3.82 (s, 3H), 3.68 (s, 4H), 3.58 (d, J = 26.3 Hz, 4H), 3.46 (t, J = 4.9 Hz, 4H), 2.52 (d, J = 70.3 Hz, 4H). 13C NMR V7 (126 MHz, CDCl3) δ 162.19, 161.64, 157.69, 155.70, 147.27, 143.98, 129.48, 127.12, 123.96, 123.57, 120.41, 119.66, 118.68, 116.17, 116.00, 109.67, 80.39, 72.91, 65.48, 54.89, 50.31, 43.52, 43.19. HRMS: (ESI, m/z): [M + H]+ calcd for C26H29ClFN5O3 513.19; found 514.2016; anal. calcd for C26H29ClFN5O3; C, 60.76; H, 5.69; Cl, 6.90; F, 3.70; N, 13.63; O, 9.34%; found C, 60.73; H, 5.68; Cl, 6.89; F, 3.69; N, 13.62; O, 9.33%.
6.1.3.8 2-((4-Fluorophenyl)(4-(6-morpholinopyrimidin-4-yl)piperazin-1-yl)methyl)-6-methoxyphenol (V8). Yield 70%; mp 90–110 °C. 1H NMR (500 MHz, chloroform-d) δ 11.42 (s, 1H), 8.13 (s, 1H), 7.34 (d, J = 5.1 Hz, 2H), 6.92 (s, 2H), 6.65 (s, 1H), 6.51 (s, 1H), 5.45 (s, 1H), 4.41 (d, J = 12.2 Hz, 1H), 3.81 (d, J = 11.4 Hz, 3H), 3.68 (d, J = 4.9 Hz, 4H), 3.57 (d, J = 36.4 Hz, 4H), 3.45 (t, J = 4.9 Hz, 4H), 2.51 (d, J = 72.7 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 163.41, 162.91, 161.46, 157.32, 148.34, 145.24, 135.22, 130.14, 125.10, 120.90, 119.47, 116.41, 110.57, 81.46, 74.84, 66.52, 56.94, 51.29, 44.17. HRMS: (ESI, m/z): [M + H]+ calcd for C26H30FN5O3 479.56; found 479.23223; anal. calcd for C26H30FN5O3; C, 65.12; H, 6.31; F, 3.96; N, 14.60; O, 10.01%; found C, 65.16; H, 6.31; F, 3.96; N, 14.61; O, 10.01%.
6.2 Biological evaluation
6.2.1 Cell culture. The National Centre for Cell Sciences (NCCS, Pune, India) supplied the murine macrophage RAW 264.7 cells, which were cultured in Dulbecco's Modified Eagles' medium (Gibco, Carlsbad, CA, USA) with 10% Foetal Bovine Serum and 1% antibiotic-antimycotic supplements at the temperature of 37 °C.
6.2.2 MTT assay. The MTT assay was used to determine the in vitro cytotoxicity of test substances on RAW cells. 96-well plates were seeded with 3000–4000 cells per well, depending on their doubling rates, and the cells were left to adhere for 24 hours at 37 °C and 5% CO2. After the 24 hours incubation period, cells were treated with a test substance, (DMSO) alone serving as a control. After 24 hours, the medium was removed, and 100 μL of culture media containing 5 mg mL−1 MTT (thiazol blue tetrazolium bromide) was added to each well. The cells were then left to incubate for an additional 4 hours at 37 °C. Later, the MTT-containing media was withdrawn, and 100 μL of DMSO was added to each well to dissolve the formazan product that had been crystallized. Using a microplate reader (iMARKBioRad, USA), the plates were read at 570 nm. Using the formula 100 − [(mean OD of treated cell × 100)/mean OD of vehicle-treated cells (DMSO)], the percent growth inhibition was computed.
6.2.3 Nitric oxide assay. In a 24-well culture plate, 70–80% of RAW 264.7 cells were plated, and the cells were then incubated for 24 hours at 37 °C with 5% CO2. After, the supernatants were removed and replaced with fresh DMEM bearing 100 ng mL−1 LPS (100 ng mL−1, Escherichia coli 055: B5, Sigma, MO, USA) in the presence or absence of the test compounds (12.5 μM) for 24 h. The supernatant was then harvested and the NO levels were determined immediately by using the Griess method according to the test kit's instructions. Following incubation with Griess reagents 1 and 2, the sample's absorbance was measured at 540 nm using a microplate reader, and the NO levels were estimated using a standard curve with a known quantity of sodium nitrite.
6.2.4 RNA extraction. RAW 264.7 cells were seeded in 6-well plates. Further, LPS (100 ng mL−1) and treatment of substances V4 and V8 were given to cells for 24 h incubation. Total RNA was extracted using the TRIZOL reagent (Invitrogen) following the manufacturer's instructions after the 24 hours incubation period. Cells were briefly rinsed twice with 2 mL of ice-cold 1 × PBS, then 1 mL of Trizol was added. Cells were treated with Trizol for 10 minutes at room temperature, after which samples were transferred to 1.5 mL Eppendorf centrifuge tubes, and 120 μL of chloroform was then added. The tubes were then vigorously inverted for 15 seconds, incubated for 10 minutes, then centrifuged for 15 minutes at 4 °C at 12000 rpm. New 1.5 mL microcentrifuge tubes were used to transfer the aqueous phase from the old ones. RNA precipitated by adding 300 μL of ice-chilled isopropanol. At 4 °C samples were centrifuged for 5 minutes at 8000 rpm. After removing the supernatant, the pellet was washed thrice with 600 μL of 75% ethanol. After centrifuging the samples at 8000 rpm for 5 min at 4 °C, the pellet was air-dried and then resuspended in 40 μL of DEPC-treated water. The RNA was kept at −80 °C. A Nanodrop was used to measure the concentration of RNA (Thermo Scientific, USA).
6.2.5 cDNA synthesis. According to the manufacturer's instructions, cDNA was created from RNA. By using the iScript cDNA synthesis kit, a cDNA library of the mRNAs from the transcriptome was produced (Bio-Rad, Hercules, CA, USA). The produced cDNA was kept at −20 °C until needed.
6.2.6 Semiquantitative PCR. According to the manufacturer's instructions, cDNA was created from RNA. By using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA), a cDNA library of the mRNAs from the transcriptome was produced. The produced cDNA was kept at −20 °C until needed. The target genes were amplified using specific gene primers
| iNOS (forward): 5′-GAGCGAGTTGTGGATTGTC-3′ |
| iNOS (reverse): 5′-CTCCTTTGAGCCCTTTGT-3′ |
| COX 2 (forward): 5′-GAAGGACTGAGATCAAATTCTC-3′ |
| COX 2 (reverse): 5′-ATGACAGAGGAGTCATTGAG-3′ |
| IL1β (forward): 5′-GGTGTGTGACGTTCCCATTA-3′ |
| IL1β (reverse): 5′-CCCAAGGCCACAGGTATTT-3′ |
| GAPDH (forward): 5′-CTGAACGGGAAGCTCAC-3′ |
| GAPDH (reverse): 5′-ATACTTGGCAGGTTTCTCC-3′ |
Each reaction was run in triplicate, and specific gene expression levels were normalized to GAPDH levels. One cycle of PCR at 95 °C for three minutes was followed by 35 cycles of denaturation at 95 °C for 25 seconds and annealing at 54 °C for 20 seconds and extension at 72 °C for 30 s.
6.2.7 Western blot analysis. The RIPA lysis buffer was used to extract the total protein, which was then measured using the Bradford assay. After being allowed to separate on 10% SDS-PAGE, the recovered protein was then transferred to a polyvinylidene (PVDF) membrane (Bio-Rad). Intermittent washing in Tris-buffered saline with 1% Tween-20 was used while sequentially probing with a specific primary antibody, followed by an HRP-conjugated secondary antibody. The PVDF membrane was then treated with enhanced chemiluminescence (ECL) (Bio-Rad) substrate to see chemiluminescence. GAPDH (1:10000 dilutions; ABclonal Germany), inducible nitric oxide synthase (iNOS) (1:2000; Biolegend) Cyclooxygenase (COX-2)(1:2000; Affinity Biosciences) were used as the primary antibodies. Depending on the host specificity, HRP-conjugated secondary antibodies (1:10000 dilutions; Santa Cruz Biotechnology) were used in their place.
6.2.8 Statistical analyses. Software called GraphPad Prism version 6 (San Diego, CA, USA) was used for the statistical analysis. Data are shown as the mean ± SEM. The data were analyzed using an unpaired t-test, and real-time gene expression analysis was analyzed using a student's t-test. 95% confidence intervals were used to determine statistical significance (p-values <0.05).
7. Molecular docking
Molecular docking studies were performed using AutoDock vina 1.5.7.48 The RCSB Protein Data Bank (https://www.rcsb.org/) were used to obtain the crystal structure of target proteins iNOS (PDB ID: 3E6T) and COX-2 (PDB ID: 1CX2). The associated water molecules were deleated from the downloaded protein and prepared by adding polar hydrogen atoms and the Kollman charges and saved as a .pdbqt file. A grid box was generated over the docking region and the information was saved. Further, the ligands were prepared and saved as the .pdbqt file. Finally, Discovery Studio Visualizer was used to visualize the protein–ligand interactions and their binding orientations.
Conflicts of interest
The authors declare that they have no known competing financial interests.
Acknowledgements
The author Sadaf Fatima extremely thankful to the University Grants Commission, Government of India for financial assistance through Central University PhD Students Fellowship. The Indian Council of Medical Research supported Almaz Zaki by providing JRF [grant no. 3/1/2/JRF-2019/HRD-061(23934)]. Hari Madhav thank to the Indian Council of Medical Research, Government of India for providing financial assistance as Senior Research Fellowship (award no. 45/02/2020-Nan/BMS) during the study.
References
- P. M. Ridker, Can. Med. Assoc. J., 2017, 189, E382–E383 CrossRef PubMed.
- L. Chen, H. Deng, H. Cui, J. Fang, Z. Zuo, J. Deng, Y. Li, X. Wang and L. Zhao, Oncotarget, 2018, 9, 7204–7218 CrossRef PubMed.
- S. Qian, O. Golubnitschaja and X. Zhan, EPMA J., 2019, 10, 365–381 CrossRef PubMed.
- K. Muniandy, S. Gothai, K. M. H. Badran, S. Suresh Kumar, N. M. Esa and P. Arulselvan, J. Immunol. Res., 2018, 2018, 1–12 CrossRef PubMed.
- C. Nathan and M. U. Shiloh, Proc. Natl. Acad. Sci., 2000, 97, 8841–8848 CrossRef CAS PubMed.
- P. Tripathi, P. Tripathi, L. Kashyap and V. Singh, FEMS Immunol. Med. Microbiol., 2007, 51, 443–452 CrossRef CAS PubMed.
- S. F. Kim, Nitric Oxide, 2011, 25, 255–264 CrossRef CAS PubMed.
- F. Z. Mónica, K. Bian and F. Murad, 2016, pp. 1–27.
- E. M. Palsson-McDermott and L. A. J. O'Neill, Immunology, 2004, 113, 153–162 CrossRef CAS PubMed.
- U. Forstermann and W. C. Sessa, Eur. Heart J., 2012, 33, 829–837 CrossRef PubMed.
- O. Morteau, Arch. Immunol. Ther. Exp., 2000, 48, 473–480 CAS.
- J. R. Vane, Nat. Cell Biol., 1971, 231, 232–235 CAS.
- J. A. Cairns, Can. J. Cardiol., 2007, 23, 125–131 CrossRef CAS PubMed.
- Y. Zhang, L. Luo, C. Han, H. Lv, D. Chen, G. Shen, K. Wu, S. Pan and F. Ye, Molecules, 2017, 22, 1960 CrossRef PubMed.
- V. S. Murthy, Y. Tamboli, V. S. Krishna, D. Sriram, F. X. Zhang, G. W. Zamponi and V. Vijayakumar, ACS Omega, 2021, 6, 9731–9740 CrossRef CAS PubMed.
- E. Jameel, H. Madhav, P. Agrawal, M. K. Raza, S. Ahmedi, A. Rahman, N. Shahid, K. Shaheen, C. H. Gajra, A. Khan, M. Z. Malik, M. A. Imam, M. Kalamuddin, J. Kumar, D. Gupta, S. M. Nayeem, N. Manzoor, A. Mohammad, P. Malhotra and N. Hoda, J. Biomol. Struct. Dyn., 2023, 1–22 CrossRef PubMed.
- H. Madhav, S. A. Abdel-Rahman, M. A. Hashmi, M. A. Rahman, M. Rehan, K. Pal, S. M. Nayeem, M. T. Gabr and N. Hoda, Eur. J. Med. Chem., 2023, 254, 115354 CrossRef CAS PubMed.
- H. Madhav, E. Jameel, M. Rehan and N. Hoda, RSC Med. Chem., 2022, 13, 258–279 RSC.
- H. Madhav and N. Hoda, Eur. J. Med. Chem., 2021, 210, 112955 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- R. Baltzly, S. DuBreuil, W. S. Ide and E. Lorz, J. Org. Chem., 1949, 14, 775–782 CrossRef CAS.
- V. de Melo Mendes, A. G. Tempone and S. E. Treiger Borborema, Acta Trop., 2019, 195, 6–14 CrossRef CAS PubMed.
- S. Govindaiah, S. Sreenivasa, R. A. Ramakrishna, T. M. C. Rao and H. Nagabhushana, ChemistrySelect, 2018, 3, 8111–8117 CrossRef CAS.
- D. Roy and G. Panda, ACS Omega, 2020, 5, 19–30 CrossRef CAS PubMed.
- C. S. Ananda Kumar, K. Vinaya, J. Narendra Sharath Chandra, N. R. Thimmegowda, S. B. Benaka Prasad, C. T. Sadashiva and K. S. Rangappa, J. Enzyme Inhib. Med. Chem., 2008, 23, 462–469 CrossRef CAS PubMed.
- C. S. Ananda Kumar, V. Bantal, K. Ramesha, C. Raj, K. Mahadevaiah, S. B. Benaka Prasad, S. Naveen and M. Malavalli, J. Appl. Chem., 2017, 2017, 282–290 Search PubMed.
- A. Ahmadi, M. Khalili, S. Chavrogh and B. Nahri-Niknafs, Iran. J. Pharm. Res., 2012, 11, 1027–1037 CAS.
- S. N. Mandhane, J. H. Shah, P. C. Bahekar, S. V. Mehetre, C. A. Pawar, A. S. Bagad, G. U. Chidrewar, C. T. Rao and T. Rajamannar, Int. Arch. Allergy Immunol., 2010, 151, 56–69 CrossRef CAS PubMed.
- X. Qiu, H. Pei, H. Ni, Z. Su, Y. Li, Z. Yang, C. Dou, L. Chen and L. Wan, Chem. Biol. Drug Des., 2021, 97, 358–371 CrossRef CAS PubMed.
- S. Mohana Roopan and R. Sompalle, Synth. Commun., 2016, 46, 645–672 CrossRef.
- N. Agarwal, S. K. Raghuwanshi, D. N. Upadhyay, P. K. Shukla and V. J. Ram, Bioorg. Med. Chem. Lett., 2000, 10, 703–706 CrossRef CAS PubMed.
- P. Sharma, N. Rane and V. Gurram, Bioorg. Med. Chem. Lett., 2004, 14, 4185–4190 CrossRef CAS PubMed.
- S. Vega, J. Alonso, J. A. Diaz and F. Junquera, J. Heterocycl. Chem., 1990, 27, 269–273 CrossRef CAS.
- V. Ram, N. Haque and P. Guru, Eur. J. Med. Chem., 1992, 27, 851–855 CrossRef CAS.
- A. M. Farghaly, O. M. AboulWafa, Y. A. M. Elshaier, W. A. Badawi, H. H. Haridy and H. A. E. Mubarak, Med. Chem. Res., 2019, 28, 360–379 CrossRef CAS.
- M. M. M. Ramiz, W. A. El-Sayed, E. Hagag and A. A.-H. Abdel-Rahman, J. Heterocycl. Chem., 2011, 48, 1028–1038 CrossRef CAS.
- O. Bruno, A. Ranise, F. Bondavalli, P. Schenone, M. D'Amico, A. Filippelli, W. Filippelli and F. Rossi, Farmaco, 1993, 48, 949–966 CAS.
- V. Ramya, S. Vembu, G. Ariharasivakumar and M. Gopalakrishnan, Drug Res., 2017, 67, 515–526 CrossRef CAS PubMed.
- O. S. Hanna Severina, A. Khairulin, N. Voloshchuk and V. Georgiyants, J. Appl. Pharm. Sci., 2019, 9, 12–19 Search PubMed.
- K. Sirisha, G. Achaiah and A. R. Ram Rao, Indian J. Pharm. Sci., 2014, 76, 519–528 CAS.
- A. P. Keche, G. D. Hatnapure, R. H. Tale, A. H. Rodge, S. S. Birajdar and V. M. Kamble, Bioorg. Med. Chem. Lett., 2012, 22, 3445–3448 CrossRef CAS PubMed.
- W. Akhtar, L. M. Nainwal, M. F. Khan, G. Verma, G. Chashoo, A. Bakht, M. Iqbal, M. Akhtar, M. Shaquiquzzaman and M. M. Alam, J. Fluorine Chem., 2020, 236, 109579 CrossRef CAS.
- M. J. Alam, O. Alam, N. Shrivastava, M. J. Naim and P. Alam, Int. J. Pharmacol., 2015, 2, 55–69 CAS.
- C. M. Dehnhardt, A. M. Venkatesan, E. Delos Santos, Z. Chen, O. Santos, S. Ayral-Kaloustian, N. Brooijmans, R. Mallon, I. Hollander, L. Feldberg, J. Lucas, I. Chaudhary, K. Yu, J. Gibbons, R. Abraham and T. S. Mansour, J. Med. Chem., 2010, 53, 798–810 CrossRef CAS PubMed.
- K. M. Foote, J. W. M. Nissink, T. McGuire, P. Turner, S. Guichard, J. W. T. Yates, A. Lau, K. Blades, D. Heathcote, R. Odedra, G. Wilkinson, Z. Wilson, C. M. Wood and P. J. Jewsbury, J. Med. Chem., 2018, 61, 9889–9907 CrossRef CAS PubMed.
- Y. Wada, R. Lu, D. Zhou, J. Chu, T. Przewloka, S. Zhang, L. Li, Y. Wu, J. Qin, V. Balasubramanyam, J. Barsoum and M. Ono, Blood, 2007, 109, 1156–1164 CrossRef CAS PubMed.
- L. Sautebin, Fitoterapia, 2000, 71, S48–S57 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2009, 31, 455–461 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Bibi Shaguftah Khatoonad,
Abdur Rahmana,
Mohd Wasif Manhasb,
Nasimul Hoda
a,
Bibi Shaguftah Khatoonad,
Abdur Rahmana,
Mohd Wasif Manhasb,
Nasimul Hoda