
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of diarylmethyl thioethers via a DABCO-catalyzed 1,6-conjugate addition reaction of p-quinone methides with organosulfur reagents†
Ming-Shan Shuai‡
,
Xiang Guan‡,
Xing-Hai Fei,
Mao Zhang,
Xiao-Zhong Fu,
Bin He and
Yong-Long Zhao*
and
Yong-Long Zhao*
State Key Laboratory of Functions and Applications of Medicinal Plants, School of Pharmacy, and Engineering Research Center for the Development and Application of Ethnic Medicine and TCM (Ministry of Education), Guizhou Medical University, Guiyang 550004, People's Republic of China. E-mail: zhaoyl05@126.com
First published on 26th April 2023
Abstract
A rapid and simple method was developed for the synthesis of diarylmethyl thioethers via a DABCO-catalyzed 1,6-conjugate addition reaction of para-quinone methides (p-QMs) with organosulfur reagents. A series of diarylmethyl thioethers were synthesized at 13–85% yields by this method. After that, the antibacterial activities of synthesized diarylmethyl thioethers and their derivatives were evaluated. The MIC range (μg mL−1) against Staphylococcus aureus ATCC 25923 and clinically isolated methicillin-resistant S. aureus was 8–128 and 64–128, respectively.
Introduction
Diarylmethyl thioether is an important building block for drugs and bioactive molecules.1–4 For example, bioactive molecules such as antitumor agent (1),1 inhibitor of the pathogenic yeast Candida albicans (2),2 inhibitor of human mitotic kinesin Eg5 (3)3 and antimalarial agent (4)4 all contain diarylmethyl thioether structures (Fig. 1). Various C–S bond-forming methods, mainly involving catalytic strategies, have been developed for the synthesis of thioethers,5 but these reported methods have defects due to the use of thiols with strong smells, air-sensitivity and strong coordinating ability. Among them, reported examples involving the synthesis of diarylmethyl thioethers are rare. In 2005, a simple reaction of diarylmethyl bromide and thiols catalyzed by an inexpensive ionic liquid, 1-pentyl-3-methylimidazolium bromide ([pmIm]Br), without a solvent at room temperature was developed by Ranu and colleagues to realize the synthesis of diarylmethyl thioethers.6 More importantly, the method stated above was also extended for the direct conversion of diarylmethyl alcohols and ethers to diarylmethyl thioethers in the presence of tert-butyl bromide (1.5 equiv) at 60 °C (Scheme 1a). After that, various reaction systems (e.g., BF3·OEt2 (5 mol%), CH2Cl2, r.t.;7 ZnCl2 (10 mol%), DCM, r.t.;8 TeaMs, 80 °C (microwave irradiation);9 and HClO4–SiO2 (5 mol%), r.t.10) were developed to realize the direct conversion of diarylmethyl alcohols to diarylmethyl thioethers. However, the problem of using strong-smelling thiols must be solved, but some reaction systems are complex and have limited applications (Scheme 1a). In 2014 and 2018, Walsh and colleagues11 and Peng and coworkers12 reported the direct arylation of benzyl thioethers with aryl bromides or Knochel-type arylzinc reagents, respectively, to realize the synthesis of diarylmethyl thioethers. However, a complex reaction system (such as a catalytic system of NiXantPhos/[PdCl(allyl)]2 in the presence of LiN(SiMe3)2 or NaN(SiMe3)2 (2.0 equiv), using Knochel-type arylzinc reagents and a stoichiometric oxidant) is needed (Scheme 1b). Therefore, a more efficient and “green” approach for the synthesis of structurally diverse diarylmethyl thioethers is highly desired.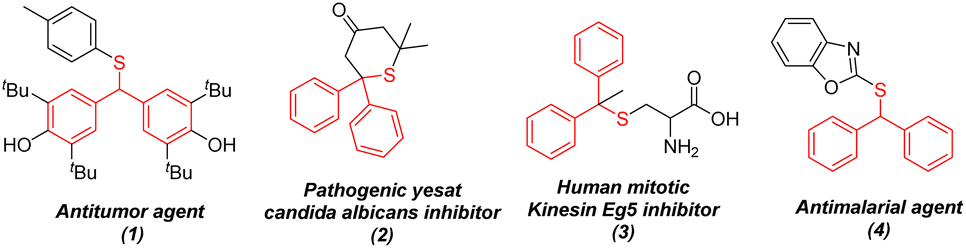 | ||
| Fig. 1 Representative bioactive molecules containing a diarylmethyl-thioether fragment. | ||
 | ||
| Scheme 1 Methods for the synthesis of diarylmethyl thioethers. | ||
para-Quinone methides (p-QMs) are composed of a cyclohexadiene in a para-conjugation with a carbonyl group and an exo-methylene component. p-QMs are found widely in natural products and drug molecules.13 Owing to their unique structure, p-QMs undergo resonance between neutral and zwitterionic structures, exhibit remarkable chemical reactivity as a Michael receptor, and have been subjected successfully to 1,6-addition or annulation reactions with carbon-, phosphorus-, or nitrogen-centered nucleophiles under various catalytic systems to form new C–C bonds and C–N bonds.14 Thiols (as the nucleophilic reagent) have undergone a 1,6-conjugate addition reaction to p-QMs by a catalytic method, which provides an effective way to synthesize new diarylmethyl thioethers. Recent studies have reported that this type of reaction can be employed to synthesize diarylmethyl thioethers under a series of catalytic systems (such as (C4H12N2)2[BiCl6]Cl·H2O,13 TfOH15 or UO2(NO3)2·6H2O),16 but some defects are unavoidable due to the use of thiols (Scheme 1c). In 2021,17 a convenient and highly efficient 1,6-conjugate addition of sodium aryl/alkyl sulfinates with p-QMs was developed by the group of Xiong, Tang and Wong to synthesize various diarylmethyl thioethers. This strategy avoids the use of thiols and transition metals, but the reaction system of stoichiometric triphenylphosphine and sulfuric acid at 80 °C under an N2 atmosphere are needed (Scheme 1d). Notably, in 2022,18 the teams of Xie, Wu and Nie team reported a catalyst-free 1,6-conjugate addition reaction between p-QMs and various sulfur-based nucleophiles. They provided diarylmethane derivatives in high-to-excellent yields. However, this method could be optimized further based on whether the reaction time could be shorter or if several specific product categories could be obtained by substrate expansion (Scheme 1c). Therefore, although significant advances have been made in the synthesis of diarylmethyl thioethers via a 1,6-conjugate addition reaction of p-QMs with thiols or sodium aryl/alkyl sulfinates, a more efficient and greener approach involving the use of other ideal organic sulfur reagents and simple reaction conditions needs to be developed further. 1,4-Dithiane-2,5-diol, as a mercaptoacetaldehyde dimer, is more convenient and efficient than common organic thiols and used widely in organic synthesis,19 especially in the diverse synthesis of thiophenes structures.20 With our continuing interest in organic sulfur chemistry,21,22 we herein describe a DABCO-catalyzed 1,6-conjugate addition reaction of p-QMs with 1,4-dithiane-2,5-diol to realize the synthesis of diarylmethyl thioethers (Scheme 1e).
Result and discussion
Chemistry
To test our initial hypothesis, we selected 1,4-dithiane-2,5-diol 2 as a nucleophilic reagent to attack the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of p-QMs 1a. Hence, a 1,6-conjugated addition reaction was undertaken to construct a diarylmethyl sulfide, which was used as the model reaction to screen the condition (Table 1). At the first stage of our study, we envisaged screening a base with the best catalytic effect to facilitate the reaction. Initially, the organic base was used as the cut-in point, and 1a and 2 were dissolved in the same solvent: CHCl3. The catalytic effects of organic bases (DABCO, t-BuOK, DEA, TEA, DBU) and alkaloids (quinine, quinidine and cinchonine) on the reaction were tested at room temperature. The yield of the target reaction could reach 68% if 10 mol% DABCO was used as a base catalyst (Table 1, entry 1). When t-BuOK and DEA were used as the catalytic base, respectively, the yields were <10% (Table 1, entries 2–3). When TEA or DBU was used as the catalytic base, the yield of the reaction was only 33% and 37%, respectively (Table 1, entries 4–5). Using the catalysts quinine, quinidine, and cinchonine, the yields were 35%, 49%, and 60%, respectively. The yields were low-to-moderate, and the reaction took a long time (Table 1, entries 6–8). Next, we investigated the catalytic effect of a few inorganic bases: K2CO3, Cs2CO3, NaOH, and LiOH. Except for the reaction catalyzed by K2CO3, the yield reached 51% (Table 1, entry 9), and the yields of the other inorganic bases were <10% (Table 1, entries 10–12). Therefore, we identified DABCO as the best-performing base. In addition to CHCl3, we continued to investigate the effects of other solvents (acetone, toluene, CH2Cl2, and dimethyl sulfoxide (DMSO)) on the reaction (Table 1, entries 13–16). The yields of toluene, CH2Cl2, and DMSO as solvents were 26%, 57%, and 22%, respectively, which were not as good as those of CHCl3 (Table 1, entries 14–16). However, when acetone was used as the solvent for the reaction, although the yield was only slightly higher than that when CHCl3 was used as the solvent (∼69%), the reaction rate was improved greatly: the reaction was almost complete in only 8 min (Table 1, entry 13). Simultaneously, with the concept of green chemistry, we also tried to use water as a solvent for the reaction but, unfortunately, our reactants barely dissolved in water. Therefore, we chose acetone as the best solvent for the reaction. Subsequently, we also tried to explore the necessity of DABCO. That is, 1a and 2 were dissolved directly in acetone to react without DABCO at room temperature. As the reaction time was extended, the yield was reduced to 57% (Table 1, entry 17). This finding revealed that a small amount of DABCO was necessary for the reaction, which could improve the reaction rate and yield greatly, and have a positive role in promoting the reaction. We also studied the effect of the reaction temperature on the reaction. When the reaction temperature was 50 °C, the reaction was faster, but the yield was less than ideal (42%) (Table 1, entry 18). When the reaction temperature was 0 °C, the reaction rate was reduced greatly and, after 2 h of reaction, the reaction of the raw materials was incomplete, and the yield was reduced to 58% (Table 1, entry 19). These data indicated that the reaction can be carried out sufficiently well at room temperature. The reaction conditions were simple and mild, and there was no strict requirement for the temperature. Because 2 was a sulfur dimer, we used 0.6 equiv of 2 to participate in the reaction according to our previous conditional screening. Then, we added 2 to 1 equiv to ensure that 1a had reacted completely, and discovered that the yield of the reaction was increased to 78% and the reaction rate was fast under the identical other conditions. After only 7 min of reaction, almost all of 1a had reacted completely (Table 1, entry 20). To check the effect of the reaction upon different loading of DABCO, we examined the effects of 5 mol% and 15 mol% DABCO on the reaction yield, respectively. The yield was 65% when using 5 mol% base (Table 1, entry 21), and 69% when using 15 mol% base (Table 1, entry 22), both of which were lower than the yield (78%) when using 10 mol% base. Therefore, we considered that 10 mol% of the base was the best condition. In short, after simple screening of conditions, DABCO was the best base (entry 1), acetone was the optimal reaction solvent (entry 13), and the reaction was rapid at room temperature. Next, we tried to increase the equivalent of 1,4-dithiane-2,5-diol 2 to 1 equiv (entry 20): the yield was increased to 78%, and the reaction was completed in only 7 min.
C bond of p-QMs 1a. Hence, a 1,6-conjugated addition reaction was undertaken to construct a diarylmethyl sulfide, which was used as the model reaction to screen the condition (Table 1). At the first stage of our study, we envisaged screening a base with the best catalytic effect to facilitate the reaction. Initially, the organic base was used as the cut-in point, and 1a and 2 were dissolved in the same solvent: CHCl3. The catalytic effects of organic bases (DABCO, t-BuOK, DEA, TEA, DBU) and alkaloids (quinine, quinidine and cinchonine) on the reaction were tested at room temperature. The yield of the target reaction could reach 68% if 10 mol% DABCO was used as a base catalyst (Table 1, entry 1). When t-BuOK and DEA were used as the catalytic base, respectively, the yields were <10% (Table 1, entries 2–3). When TEA or DBU was used as the catalytic base, the yield of the reaction was only 33% and 37%, respectively (Table 1, entries 4–5). Using the catalysts quinine, quinidine, and cinchonine, the yields were 35%, 49%, and 60%, respectively. The yields were low-to-moderate, and the reaction took a long time (Table 1, entries 6–8). Next, we investigated the catalytic effect of a few inorganic bases: K2CO3, Cs2CO3, NaOH, and LiOH. Except for the reaction catalyzed by K2CO3, the yield reached 51% (Table 1, entry 9), and the yields of the other inorganic bases were <10% (Table 1, entries 10–12). Therefore, we identified DABCO as the best-performing base. In addition to CHCl3, we continued to investigate the effects of other solvents (acetone, toluene, CH2Cl2, and dimethyl sulfoxide (DMSO)) on the reaction (Table 1, entries 13–16). The yields of toluene, CH2Cl2, and DMSO as solvents were 26%, 57%, and 22%, respectively, which were not as good as those of CHCl3 (Table 1, entries 14–16). However, when acetone was used as the solvent for the reaction, although the yield was only slightly higher than that when CHCl3 was used as the solvent (∼69%), the reaction rate was improved greatly: the reaction was almost complete in only 8 min (Table 1, entry 13). Simultaneously, with the concept of green chemistry, we also tried to use water as a solvent for the reaction but, unfortunately, our reactants barely dissolved in water. Therefore, we chose acetone as the best solvent for the reaction. Subsequently, we also tried to explore the necessity of DABCO. That is, 1a and 2 were dissolved directly in acetone to react without DABCO at room temperature. As the reaction time was extended, the yield was reduced to 57% (Table 1, entry 17). This finding revealed that a small amount of DABCO was necessary for the reaction, which could improve the reaction rate and yield greatly, and have a positive role in promoting the reaction. We also studied the effect of the reaction temperature on the reaction. When the reaction temperature was 50 °C, the reaction was faster, but the yield was less than ideal (42%) (Table 1, entry 18). When the reaction temperature was 0 °C, the reaction rate was reduced greatly and, after 2 h of reaction, the reaction of the raw materials was incomplete, and the yield was reduced to 58% (Table 1, entry 19). These data indicated that the reaction can be carried out sufficiently well at room temperature. The reaction conditions were simple and mild, and there was no strict requirement for the temperature. Because 2 was a sulfur dimer, we used 0.6 equiv of 2 to participate in the reaction according to our previous conditional screening. Then, we added 2 to 1 equiv to ensure that 1a had reacted completely, and discovered that the yield of the reaction was increased to 78% and the reaction rate was fast under the identical other conditions. After only 7 min of reaction, almost all of 1a had reacted completely (Table 1, entry 20). To check the effect of the reaction upon different loading of DABCO, we examined the effects of 5 mol% and 15 mol% DABCO on the reaction yield, respectively. The yield was 65% when using 5 mol% base (Table 1, entry 21), and 69% when using 15 mol% base (Table 1, entry 22), both of which were lower than the yield (78%) when using 10 mol% base. Therefore, we considered that 10 mol% of the base was the best condition. In short, after simple screening of conditions, DABCO was the best base (entry 1), acetone was the optimal reaction solvent (entry 13), and the reaction was rapid at room temperature. Next, we tried to increase the equivalent of 1,4-dithiane-2,5-diol 2 to 1 equiv (entry 20): the yield was increased to 78%, and the reaction was completed in only 7 min.
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Temp (°C) | Timeb | Yieldc (%) |
| a Unless specified otherwise, all reactions were carried out with 0.10 mmol (1 equiv) of 1a, 0.60 mmol (0.6 equiv) of 2 and 10 mol% base in 1 mL of solvent.b The reaction time was judged by TLC.c Isolated yield after silica-gel column chromatography.d 0.10 mmol (1 equiv) of 2 was used.e 5 mol% of DABCO was used.f 15 mol% of DABCO was used. | |||||
| 1 | DABCO | CHCl3 | r.t. | 19 h | 68 |
| 2 | t-BuOK | CHCl3 | r.t. | 5 h | <10 |
| 3 | DEA | CHCl3 | r.t. | 25 h | <10 |
| 4 | TEA | CHCl3 | r.t. | 25 h | 33 |
| 5 | DBU | CHCl3 | r.t. | 3 min | 37 |
| 6 | Quinine | CHCl3 | r.t. | 19 h | 35 |
| 7 | Quinidine | CHCl3 | r.t. | 46 h | 49 |
| 8 | Cinchonine | CHCl3 | r.t. | 46 h | 60 |
| 9 | K2CO3 | CHCl3 | r.t. | 4 h | 51 |
| 10 | Cs2CO3 | CHCl3 | r.t. | 5 h | <10 |
| 11 | NaOH | CHCl3 | r.t. | 23 h | <10 |
| 12 | LiOH | CHCl3 | r.t. | 23 h | <10 |
| 13 | DABCO | Acetone | r.t. | 8 min | 69 |
| 14 | DABCO | Toluene | r.t. | 25 h | 26 |
| 15 | DABCO | CH2Cl2 | r.t. | 1 h | 57 |
| 16 | DABCO | DMSO | r.t. | 5 h | 22 |
| 17 | — | Acetone | r.t. | 1 h | 57 |
| 18 | DABCO | Acetone | 50 | 6 min | 42 |
| 19 | DABCO | Acetone | 0 | 2 h | 58 |
| 20d | DABCO | Acetone | r.t. | 7 min | 78 |
| 21d,e | DABCO | Acetone | r.t. | 15 min | 65 |
| 22d,f | DABCO | Acetone | r.t. | 8 min | 69 |

With the established optimal reaction conditions, we further expanded the application range of the reaction by changing the R group of p-QMs 1, thereby checking the generality of the reaction (Table 2). First, we took p-QMs 1 with phenyl as the basic substrate structure, and connected –F, –Cl and –Br (3b–3d) in the vicinal position of the phenyl group, respectively, to explore the influence of the electron-withdrawing induction effect on the yield of the target compounds. The strength of the induction of the electron-withdrawing group on the benzene ring of p-QMs had little effect on the yield of the target compounds, and all of them could elicit yields ranging from 55% to 61%. Then, the influence of the steric effects of the bromine substituent on the yield of the target compounds was investigated when it was substituted in ortho-, meta- and para-positions, respectively. Irrespective of where the bromine atom was substituted in the benzene ring, the yield changed only slightly, from 53% to 55% (3d–3f), which was lower than the yield of unsubstituted 3a (78%). However, substitution of strong electron-withdrawing groups on the benzene ring and the different substitution positions influenced the yield of the target products obviously. The yield range of nitro groups substituted at different positions was 13–85% (3o–3q), that of ortho-nitro groups was 13% (3o), and that of meta-nitro groups was 85% (3p). This vast difference may have been influenced by induction effects and steric effects. Conversely, we investigated the influence on the yield (3g and 3j) when the methyl group and methoxy group were substituted on the benzene ring. The target compound produced by attaching the electron-donating group had a higher yield compared with that obtained by attachment of the electron-withdrawing group. Combined with the changes in yield of other weak electron-donating substituents (3l and 3t), we found that the stronger the conjugated effect of the electron-donating substituent, the higher the yield of the target compound. We also investigated the effect of different positions of electron-donating substituents on the benzene ring on the yield of the target compound (3g–3i and 3j–3k). A substituent in the para position could reduce the yield slightly, and both were lower than the yield (78%) obtained with unsubstituted phenyl p-QMs (3a). Also, two methyl groups were added to the benzene ring of p-QMs (3r), the yield was comparable with that of 3a. Finally, we also tried to synthesize other target thioether compounds (3m, 3n and 3s) substituted by other heteroaryl groups, and obtained moderate yields (62–66%) and at a slower rate, which took 30 min to complete. Twenty diaryl thioether derivatives (3a–3t) were obtained by reacting p-QMs 1 substituted with different aryl or substituted aryl groups with 2, and the yield was 13–85%. Our method could readily tolerate various substitutions on the aromatic ring. Comparison of the influences of electron-withdrawing groups, electron-donating groups and substitution sites in substituted aryl groups revealed the yield to be mostly not greatly changed, which indicated that our method was only slightly affected by the induction effect and steric effect.
| a All reactions were carried out with 0.10 mmol of 1, 0.10 mmol of 2 and 10 mol% DABCO in 1 mL of acetone at room temperature. Isolated yield after silica-gel column chromatography. |
|---|
 |
In addition, according to the ideas provided by some literature,17,18 we proposed a possible reaction mechanism for this 1,6-conjugate addition reaction of p-QM (1) with an organic sulfur reagent (Scheme 2). First, zwitterionic intermediate A was formed via a 1,6-conjugate addition process of p-QM (1) with catalyst DABCO. Then, intermediate A could react readily with thiol groups in organic sulfur reagents (e.g., mercaptoacetaldehyde 2a generated by 1,4-dithiane-2,5-diol 2) to generate the corresponding zwitterionic intermediate B. Finally, the desired diarylmethyl sulfides 3 were generated via a proton-transfer process of B.
 | ||
| Scheme 2 Proposed mechanism of the reaction. | ||
In addition, inspired by the literature,23 we briefly attempted the reaction of p-QMs 1a with another organic sulfur reagent: methyl thioglycolate 4. We obtained another diarylmethyl thioether 5a containing an ester group, and the yield was 96% (Scheme 3). Also, we used the sulfide product 5b prepared in this way as a starting material to conduct the cyclization reaction. First, 5b was hydrolyzed in a large dose of sodium hydroxide, and then allowed to react directly with sodium acetate and acetic anhydride. Finally, the cyclization product 6 was obtained at a yield of 57% (Scheme 4). To illustrate the practicality and scalability of our method, we continued to attempt the 1,6-conjugate addition reaction between the other two organosulfur reagents and p-QM under standard conditions (Scheme 5). The reaction of p-QM 1a with benzyl mercaptan 7 was completed in 8 min and up to 97% yield of desired diarylmethyl thioether 8 was obtained. The reaction with ethanethiol 9 took only 30 s to complete, with the yield of desired product 10 reaching 98%.
 | ||
| Scheme 3 Synthesis of an ester-containing thioether. | ||
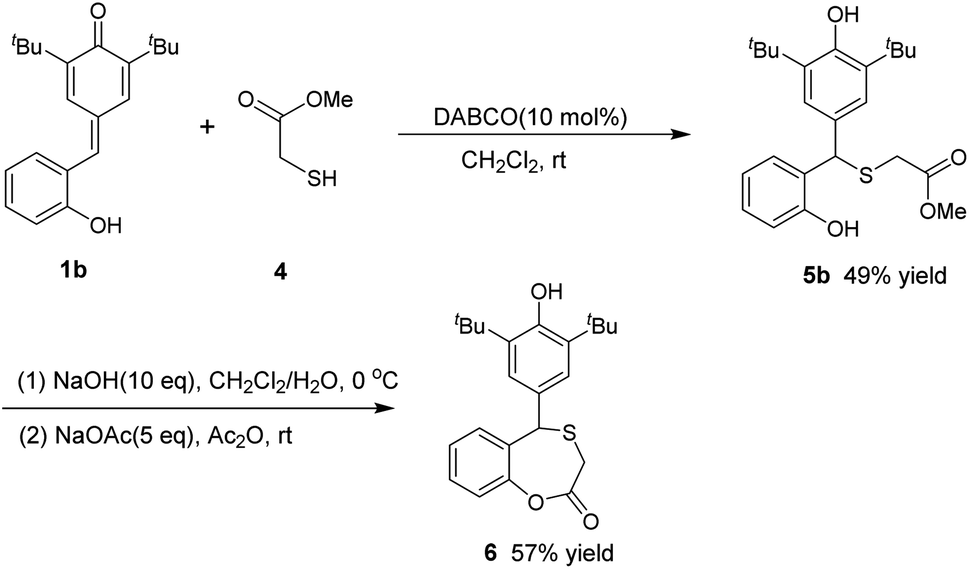 | ||
| Scheme 4 Synthesis of an ester-containing thioether and cyclic thioether. | ||
 | ||
| Scheme 5 1,6-Conjugate addition reaction of p-QM with other organic sulfur reagents. | ||
Antibacterial activity
Due to the extensive biological activity of p-QMs and thioether structural fragments, including antibacterial activity, this could indicate the potential antibacterial activity of diarylmethyl thioethers. Therefore, we preliminarily explored the minimum inhibitory concentration (MIC) of our newly synthesized diarylmethyl thioethers. We assayed the in vitro activity of compounds 3a–3t, 5a, 5b and 6 against Gram-positive bacteria (Staphylococcus aureus ATCC 25923 and clinically isolated methicillin-resistant S. aureus (MRSA)), Gram-negative bacteria (Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 9027) and fungi (Candida albicans BNCC 259769 and C. albicans BNCC 333935). The MIC value was employed as an indicator of antibacterial efficacy to detect synthesized diarylmethyl thioethers. A 96-well plate micro-double dilution method was used, with levofloxacin or fluconazole as the positive control under identical assay conditions for assessment of antimicrobial susceptibility (see ESI†).The compounds had no obvious inhibitory effect on tested Gram-negative bacteria (E. coli, P. aeruginosa) and C. albicans. However, most of them had certain inhibitory effects on tested Gram-positive bacteria. For the antibacterial activities against S. aureus ATCC 25923, the MIC of 3a was up to 16 μg mL−1, and 6 was up to 8 μg mL−1. Besides, some of the compounds also showed inhibition of MRSA, with a MIC of 64–128 μg mL−1. Compound 6 had the best antibacterial effect against S. aureus ATCC 25923, but showed no significant activity against MRSA. Furthermore, we compared the activity of the synthesized thioether compounds containing an ester group with those containing an aldehyde group. Under identical substitution conditions, compounds containing an aldehyde group had certain antibacterial activity, and compounds containing an ester group also had activity, but the activity effect was weaker than that of compounds containing an aldehyde group (3a vs. 5a). Meanwhile, 5b was inactive against all bacteria, demonstrating that an ester group might have a negative effect on antibacterial action.
Conclusions
We developed a rapid and simple method for the synthesis of diarylmethyl thioethers via a DABCO-catalyzed 1,6-conjugate addition reaction of p-QMs with an organosulfur reagent. We synthesized a series of diarylmethyl thioethers with 13–98% yield. Then, we synthesized two types of derivative thioether products by p-QMs (thioether-containing ester groups and cyclized thioether) and the 1,6-conjugate addition reaction with other organic sulfur reagents was attempted using this method. After preliminary evaluation of the antibacterial activity of these compounds, most of the synthesized diarylmethyl thioethers showed some antibacterial activity against S. aureus. Although these synthesized compounds showed only slight antibacterial activity against S. aureus (including MRSA), they also had the potential of further modification and development for antibacterial applications.Experimental section
Chemistry
The chemical reagents and solvents used in synthesis and separation were purchased from commercial suppliers (e.g., Energy-Chemical, Macklin, Xilong SCIENTIFIC) and used without further purification. Solvents were of analytical grade. p-QMs 1, as the raw material, was synthesized according to the literature.24 All reactions were monitored by analytical thin-layer chromatography (TLC). TLC was carried out on silica-gel plates with F-254 indicator. Products were visualized by irradiation with ultraviolet light and/or by iodine fumigation. Flash column chromatography was carried out over silica gel (200–300 mesh).Proton nuclear magnetic resonance (1H NMR) and 13C NMR spectroscopy were carried out on a JNM-ECS400 or Avance III HD spectrometer (400 or 600 MHz for 1H; 100 or 150 MHz for 13C). Spectra were recorded in CDCl3 as the solvent at room temperature. Chemical shifts (δ) are reported in ppm with respect to residual solvent signals or TMS as an internal standard (CDCl3, 7.26 ppm, singlet for 1H NMR; CDCl3, 77.16 ppm, triplet for 13C NMR). The coupling constant J is expressed in Hz. The following abbreviations are used to indicate the multiplicity: s, singlet; d, doublet; t, triplet; m, multiplet; dd, doublet of doublet.
High-resolution mass spectroscopy (HRMS) was done on a WaterXevo G2S QTOF mass spectrometer (ESI) or Thermo Fisher (Vanquish (UPLC)-Q-Exactive Plus) mass instrument (ESI).
General procedure for the synthesis of diarylmethyl thioethers
In a reaction tube with a Teflon™-coated stir bar, p-QMs 1 (0.10 mmol), 1,4-dithiane-2,5-diol 2 (0.10 mmol) and DABCO (0.01 mmol, 10 mol%) were dissolved in 1 mL of acetone. The reaction mixtures were stirred for 5–30 min at room temperature (as judged by TLC). Crude mixtures were purified by flash chromatography to obtain the product 3.General procedure for the synthesis of sulfide compounds containing ester groups (5a and 5b)
In a reaction tube with a Teflon-coated stir bar, p-QMs 1 (0.10 mmol), methyl mercaptoacetate 4 (0.15 mmol) and DABCO (0.01 mmol, 10 mol%) were dissolved in 1 mL of CH2Cl2. The reaction mixtures were stirred for 20 min at room temperature (as judged by TLC). Crude mixtures were purified by flash chromatography to obtain the product 5.General procedure for the synthesis of cyclized derivative 6
In a reaction tube with a Teflon-coated stir bar, compound 5b (0.10 mmol) was dissolved in 0.5 mL of CH2Cl2. A small amount of aqueous solution in which 10 equiv of NaOH (1 mmol) were dissolved was dropped to react at 0 °C for 3 h (as judged by TLC). The reaction mixture was diluted with DCM and acidified with 10% hydrochloric acid solution. Then, the mixture was extracted thrice with DCM, the extracted organic layer was dried with anhydrous sodium sulfate and concentrated under reduced pressure. We added 5 equiv of sodium acetate to the concentrated mixture, and dissolved it in 1 mL of acetic anhydride to react at room temperature for 22 h (as judged by TLC). The mixture was extracted thrice with DCM, the extracted organic layer was dried with anhydrous sodium sulfate and concentrated under reduced pressure. Finally, the mixture was purified by flash column chromatography to afford the desired products 6.General procedure for the synthesis of other thioether products (8)
In a reaction tube with a Teflon-coated stir bar, p-QMs 1 (0.10 mmol), benzyl mercaptan 7a (or ethanethiol 7b) (0.10 mmol) and DABCO (0.01 mmol, 10 mol%) were dissolved in 1 mL of acetone. The reaction mixtures were stirred at room temperature (as judged by TLC). Crude mixtures were purified by flash chromatography to obtain the product 8a (or 8b).2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(phenyl)methyl)thio)acetaldehyde (3a)
This compound was prepared via the general procedure as a pale-yellow oily liquid (28.7 mg, yield 78%). 1H NMR (600 MHz, CDCl3) δ 9.42 (t, J = 3.2 Hz, 1H), 7.45 (d, J = 7.2 Hz, 2H), 7.34 (t, J = 7.7 Hz, 2H), 7.24 (s, 1H), 7.19 (s, 2H), 5.18 (s, 1H), 4.98 (s, 1H), 3.12–3.03 (m, 2H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 194.9, 153.3, 140.8, 136.1, 130.5, 128.8, 128.6, 127.6, 125.2, 54.4, 42.0, 34.5, 30.4; HRMS (ESI) m/z calcd. for C23H30NaO2S [M + Na]+ 393.18587, found 393.18568.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(2-fluorophenyl)methyl)thio)acetaldehyde (3b)
This compound was prepared via the general procedure as a pale-yellow oily liquid (23.5 mg, yield 61%). 1H NMR (400 MHz, CDCl3) δ 9.43 (s, 1H), 7.65 (td, J = 7.6, 1.7 Hz, 1H), 7.26 (s, 1H), 7.21 (s, 2H), 7.16 (td, J = 7.5, 1.2 Hz, 1H), 7.03 (ddd, J = 9.6, 8.1, 1.2 Hz, 1H), 5.38 (s, 1H), 5.19 (s, 1H), 3.12 (dd, J = 4.4, 3.3 Hz, 2H), 1.41 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 195.0, 160.3 (d, J = 245.8 Hz), 153.4, 136.2, 129.1 (d, J = 8.5 Hz), 128.3, 128.2, 125.2, 124.5 (d, J = 3.8 Hz), 115.9, 115.7, 46.4, 42.2, 34.5, 30.4; 19F NMR (565 MHz, CDCl3) δ = −117.22 ppm; HRMS (ESI) m/z calcd. for C23H29FNaO2S [M + Na]+ 411.17645, found 411.17601.2-(((2-Chlorophenyl)(3,5-di-tert-butyl-4-hydroxyphenyl)methyl)thio)acetaldehyde (3c)
This compound was prepared via the general procedure as a pale-yellow oily liquid (22.9 mg, yield 57%). 1H NMR (600 MHz, CDCl3) δ 9.45 (t, J = 3.2 Hz, 1H), 7.74 (dd, J = 7.9, 1.4 Hz, 1H), 7.35 (dd, J = 8.0, 1.1 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.22 (s, 2H), 7.20–7.17 (m, 1H), 5.57 (s, 1H), 5.19 (s, 1H), 3.12 (dd, J = 5.4, 3.2 Hz, 2H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 195.1, 153.4, 138.6, 136.1, 134.0, 129.9, 129.7, 129.4, 128.7, 127.3, 125.3, 50.1, 42.3, 34.5, 30.4; HRMS (ESI) m/z calcd. for C23H29ClNaO2S [M + Na]+ 427.14690, found 427.14609.2-(((2-Bromophenyl)(3,5-di-tert-butyl-4-hydroxyphenyl)methyl)thio)acetaldehyde (3d)
This compound was prepared via the general procedure as a pale-yellow oily liquid (24.8 mg, yield 55%). 1H NMR (400 MHz, CDCl3) δ 9.46 (dt, J = 6.4, 3.2 Hz, 1H), 7.74 (dd, J = 7.8, 1.7 Hz, 1H), 7.55 (dd, J = 8.0, 1.2 Hz, 1H), 7.36–7.31 (m, 1H), 7.24 (s, 2H), 7.13–7.08 (m, 1H), 5.57 (s, 1H), 5.19 (s, 1H), 3.14–3.12 (m, 2H), 1.41 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 195.1, 153.4, 140.2, 136.1, 133.3, 130.0, 129.4, 129.0, 128.0, 125.3, 124.8, 52.8, 42.3, 34.5, 30.4; HRMS (ESI) m/z calcd. for C23H29BrNaO2S [M + Na]+ 471.09638, found 471.09540.2-(((3-Bromophenyl)(3,5-di-tert-butyl-4-hydroxyphenyl)methyl)thio)acetaldehyde (3e)
This compound was prepared via the general procedure as a pale-yellow oily liquid (23.7 mg, yield 53%). 1H NMR (400 MHz, CDCl3) δ 9.44 (ddd, J = 4.5, 2.9, 1.0 Hz, 1H), 7.61 (t, J = 1.8 Hz, 1H), 7.41–7.36 (m, 2H), 7.23–7.18 (m, 1H), 7.15 (s, 2H), 5.21 (d, J = 0.5 Hz, 1H), 4.92 (s, 1H), 3.09 (ddd, J = 15.9, 2.9, 0.9 Hz, 2H), 1.41 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 194.4, 153.5, 143.2, 136.3, 131.7, 130.7, 130.3, 129.8, 127.2, 125.1, 122.8, 53.7, 41.9, 34.6, 30.4; HRMS (ESI) m/z calcd. for C23H29BrNaO2S [M + Na]+ 471.09638, found 471.09591.2-(((4-Bromophenyl)(3,5-di-tert-butyl-4-hydroxyphenyl)methyl)thio)acetaldehyde (3f)
This compound was prepared via the general procedure as a pale-yellow sticky solid (24.7 mg, 55%). 1H NMR (600 MHz, CDCl3) δ 9.43 (t, J = 2.9 Hz, 1H), 7.46 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 7.15 (s, 2H), 5.20 (s, 1H), 4.93 (s, 1H), 3.11 (dd, J = 14.7, 2.8 Hz, 1H), 3.03 (dd, J = 14.7, 3.7 Hz, 1H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 194.4, 153.5, 140.0, 136.3, 131.9, 130.3, 130.0, 125.1, 121.5, 53.6, 41.9, 34.6, 30.4.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(o-tolyl)methyl)thio)acetaldehyde (3g)
This compound was prepared via the general procedure as a pale-yellow sticky solid (27.6 mg, 72%). 1H NMR (600 MHz, CDCl3) δ 9.45 (t, J = 3.4 Hz, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.25–7.22 (m, 1H), 7.17 (s, 2H), 7.15 (dd, J = 6.6, 1.1 Hz, 2H), 5.21 (s, 1H), 5.17 (s, 1H), 3.10 (dd, J = 14.6, 3.2 Hz, 1H), 3.06 (dd, J = 14.6, 3.5 Hz, 1H), 2.36 (s, 3H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 194.7, 153.2, 138.6, 136.4, 136.0, 130.8, 129.8, 128.1, 127.4, 126.4, 125.5, 50.3, 42.1, 34.5, 30.4, 19.8; HRMS (ESI) m/z calcd. for C24H32NaO2S [M + Na]+ 407.20152, found 407.20062.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(m-tolyl)methyl)thio)acetaldehyde (3h)
This compound was prepared via the general procedure as a pale-yellow oily liquid (28.9 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 9.40 (td, J = 3.5, 0.9 Hz, 1H), 7.25–7.24 (m, 2H), 7.23–7.21 (m, 1H), 7.19 (s, 2H), 7.05 (d, J = 7.3 Hz, 1H), 5.17 (s, 1H), 4.93 (s, 1H), 3.06 (ddd, J = 6.8, 3.1, 0.8 Hz, 2H), 2.33 (s, 3H), 1.40 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 195.0, 153.3, 140.7, 138.4, 136.0, 130.6, 129.3, 128.7, 128.4, 125.5, 125.2, 54.3, 42.1, 34.5, 30.4, 21.6; HRMS (ESI) m/z calcd. for C24H32NaO2S [M + Na]+ 407.20152, found 407.20067.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(p-tolyl)methyl)thio)acetaldehyde (3i)
This compound was prepared via the general procedure as a pale-yellow oily liquid (21.9 mg, 57%). 1H NMR (600 MHz, CDCl3) δ 9.41 (t, J = 3.3 Hz, 1H), 7.33 (d, J = 8.0 Hz, 2H), 7.19 (s, 2H), 7.14 (d, J = 8.0 Hz, 2H), 5.17 (s, 1H), 4.95 (s, 1H), 3.11–3.02 (m, 2H), 2.32 (s, 3H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 195.0, 153.3, 137.8, 137.3, 136.0, 130.7, 129.5, 128.4, 125.1, 54.1, 42.1, 34.5, 30.4, 21.2; HRMS (ESI) m/z calcd. for C24H32NaO2S [M + Na]+ 407.20152, found 407.20042.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(2-methoxyphenyl)methyl)thio)acetaldehyde (3j)
This compound was prepared via the general procedure as a pale-yellow oily liquid (30.7 mg, 77%). 1H NMR (400 MHz, CDCl3) δ 9.41 (t, J = 3.2 Hz, 1H), 7.59 (dd, J = 7.7, 1.6 Hz, 1H), 7.24 (s, 3H), 6.96 (td, J = 7.6, 1.0 Hz, 1H), 6.86–6.83 (m, 1H), 5.56 (s, 1H), 5.15 (s, 1H), 3.82 (s, 3H), 3.08 (t, J = 3.1 Hz, 2H), 1.41 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 195.9, 156.7, 153.1, 135.8, 130.6, 129.3, 128.9, 128.5, 125.3, 120.8, 110.9, 55.7, 46.4, 42.3, 34.5, 30.4; HRMS (ESI) m/z calcd. for C24H32NaO3S [M + Na]+ 423.19644, found 423.19562.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(4-methoxyphenyl)methyl)thio)acetaldehyde (3k)
This compound was prepared via the general procedure as a pale-yellow oily liquid (28.7 mg, 72%). 1H NMR (400 MHz, CDCl3) δ 9.42 (dd, J = 3.6, 3.0 Hz, 1H), 7.39–7.36 (m, 2H), 7.20 (s, 2H), 6.88 (d, J = 8.8 Hz, 2H), 5.19 (s, 1H), 4.96 (s, 1H), 3.80 (s, 3H), 3.07 (dd, J = 11.3, 3.0 Hz, 2H), 1.42 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 194.9, 158.9, 153.2, 136.0, 132.8, 130.8, 129.7, 125.1, 114.0, 55.4, 53.7, 42.0, 34.5, 30.3; HRMS (ESI) m/z calcd. for C24H32NaO3S [M + Na]+ 423.19644, found 423.19603.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(naphthalen-2-yl)methyl)thio)acetaldehyde (3l)
This compound was prepared via the general procedure as a pale-yellow oily liquid (27.7 mg, 66%). 1H NMR (400 MHz, CDCl3) δ 9.45–9.41 (m, 1H), 7.84–7.80 (m, 3H), 7.62 (dd, J = 8.6, 1.8 Hz, 1H), 7.48–7.45 (m, 2H), 7.25 (s, 3H), 5.19 (s, 1H), 5.14 (s, 1H), 3.13 (dd, J = 14.8, 2.7 Hz, 1H), 3.04 (dd, J = 14.8, 3.8 Hz, 1H), 1.40 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 194.8, 153.4, 138.1, 136.1, 133.3, 132.8, 130.4, 128.7, 128.1, 127.8, 127.5, 126.4 (d, J = 2 Hz), 126.2, 125.2, 54.4, 41.9, 34.5, 30.3; HRMS (ESI) m/z calcd. for C27H32NaO2S [M + Na]+ 443.20152, found 443.20093.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(furan-2-yl)methyl)thio)acetaldehyde (3m)
This compound was prepared via the general procedure as a yellow sticky solid (22.2 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 9.37 (td, J = 3.1, 0.6 Hz, 1H), 7.41–7.40 (m, 1H), 7.24 (s, 2H), 6.34–6.30 (m, 1H), 6.26 (td, J = 3.2, 1.9 Hz, 1H), 5.23 (s, 1H), 4.98 (s, 1H), 3.15 (dd, J = 3.2, 0.4 Hz, 2H), 1.42 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 195.2, 153.7, 153.1, 142.7, 136.2, 128.4, 125.3, 110.5, 108.8, 47.3, 41.9, 34.5, 30.4; HRMS (ESI) m/z calcd. for C21H28NaO3S [M + Na]+ 383.16514, found 383.16368.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(thiophen-2-yl)methyl)thio)acetaldehyde (3n)
This compound was prepared via the general procedure as a yellow oily liquid (24.2 mg, 64%). 1H NMR (400 MHz, CDCl3) δ 9.40 (d, J = 3.2 Hz, 1H), 7.25–7.22 (m, 3H), 7.00–6.99 (m, 1H), 6.92–6.90 (m, 1H), 5.23 (s, 1H), 5.20 (s, 1H), 3.12 (s, 2H), 1.42 (s, 18H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.6, 145.3, 136.2, 130.4, 126.7, 126.6, 125.7, 125.1, 49.6, 42.3, 34.5, 30.3; HRMS (ESI) m/z calcd. for C21H28NaO2S2 [M + Na]+ 399.14229, found 399.14079.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(2-nitrophenyl)methyl)thio)acetaldehyde (3o)
This compound was prepared via the general procedure as a pale-yellow oily liquid (5.3 mg, 13%). 1H NMR (600 MHz, CDCl3) δ 9.42 (t, J = 3.1 Hz, 1H), 7.92 (dd, J = 7.9, 1.3 Hz, 1H), 7.83 (dd, J = 8.1, 1.3 Hz, 1H), 7.61 (td, J = 7.7, 1.3 Hz, 1H), 7.42–7.39 (m, 1H), 7.22 (s, 2H), 5.79 (s, 1H), 5.22 (s, 1H), 3.17 (dd, J = 15.1, 3.1 Hz, 1H), 3.11 (dd, J = 15.1, 3.1 Hz, 1H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 194.7, 153.7, 149.5, 136.3, 135.8, 133.1, 130.7, 128.8, 128.4, 125.4, 124.8, 48.3, 42.4, 34.6, 30.3; HRMS (ESI) m/z calcd. for C23H29NNaO4S [M + Na]+ 438.17095, found 438.16982.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(3-nitrophenyl)methyl)thio)acetaldehyde (3p)
This compound was prepared via the general procedure as a pale-yellow oily liquid (35.3 mg, 85%). 1H NMR (600 MHz, CDCl3) δ 9.46 (dd, J = 3.5, 2.9 Hz, 1H), 8.35 (t, J = 2.0 Hz, 1H), 8.12 (ddd, J = 8.2, 2.2, 0.9 Hz, 1H), 7.78 (d, J = 7.7 Hz, 1H), 7.52 (t, J = 8.0 Hz, 1H), 7.16 (s, 2H), 5.25 (s, 1H), 5.05 (s, 1H), 3.15 (dd, J = 14.8, 2.9 Hz, 1H), 3.05 (dd, J = 14.8, 3.7 Hz, 1H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 194.0, 153.8, 148.6, 143.3, 136.6, 134.7, 129.7, 129.2, 125.1, 123.5, 122.7, 53.4, 41.8, 34.6, 30.3.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(4-nitrophenyl)methyl)thio)acetaldehyde (3q)
This compound was prepared via the general procedure as a yellow solid (23.9 mg, 72%). m.p. 117–118 °C. 1H NMR (600 MHz, CDCl3) δ 9.46 (t, J = 2.9 Hz, 1H), 8.20 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.14 (s, 2H), 5.25 (s, 1H), 5.04 (s, 1H), 3.16 (dd, J = 14.7, 2.7 Hz, 1H), 3.04 (dd, J = 14.8, 3.7 Hz, 1H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 193.9, 153.8, 148.4, 147.2, 136.6, 129.5, 129.0, 125.1, 124.1, 53.4, 41.7, 34.6, 30.3.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(3,4-dimethylphenyl)methyl)thio)acetaldehyde (3r)
This compound was prepared via the general procedure as a pale-yellow oily liquid (30.9 mg, 78%). 1H NMR (600 MHz, CDCl3) δ 9.41 (t, J = 3.2 Hz, 1H), 7.21 (s, 2H), 7.19 (d, J = 8.6 Hz, 2H), 7.09 (d, J = 7.6 Hz, 1H), 5.16 (s, 1H), 4.92 (s, 1H), 3.07 (d, J = 3.0 Hz, 1H), 3.06 (d, J = 3.5 Hz, 1H), 2.25 (s, 3H), 2.23 (s, 3H), 1.41 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 195.1, 153.3, 138.2, 136.9, 136.0, 135.9, 130.9, 130.0, 129.8, 125.8, 125.1, 54.2, 42.1, 34.5, 30.4, 20.0, 19.6; HRMS (ESI) m/z calcd. for C25H34NaO2S [M + Na]+ 421.21717, found 421.21533.2-(((3,5-Di-tert-butyl-4-hydroxyphenyl)(thiazol-2-yl)methyl)thio)acetaldehyde (3s)
This compound was prepared via the general procedure as a yellow sticky mass (24.8 mg, 66%). 1H NMR (600 MHz, CDCl3) δ 9.43 (t, J = 3.1 Hz, 1H), 7.73 (d, J = 3.3 Hz, 1H), 7.30 (d, J = 3.2 Hz, 1H), 7.28 (s, 2H), 5.35 (s, 1H), 5.27 (s, 1H), 3.31 (dd, J = 15.3, 3.1 Hz, 1H), 3.24 (dd, J = 15.3, 3.1 Hz, 1H), 1.42 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 195.0, 171.7, 154.1, 142.8, 136.5, 128.7, 125.4, 120.2, 51.5, 42.2, 34.6, 30.3; HRMS (ESI) m/z calcd. for C20H27NNaO2S2 [M + Na]+ 400.13754, found 400.13674.2-(([1,1′-Biphenyl]-2-yl(3,5-di-tert-butyl-4-hydroxyphenyl)methyl)thio)acetaldehyde (3t)
This compound was prepared via the general procedure as an off-white solid (28.8 mg, 65%). m.p. 113–114 °C. 1H NMR (600 MHz, CDCl3) δ 9.18 (t, J = 3.2 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.41 (dd, J = 7.7, 1.4 Hz, 1H), 7.39 (d, J = 1.3 Hz, 1H), 7.38 (d, J = 2.2 Hz, 2H), 7.27 (td, J = 7.5, 1.3 Hz, 1H), 7.25–7.14 (m, 3H), 7.01 (s, 2H), 5.13 (s, 1H), 5.11 (s, 1H), 2.98 (dd, J = 3.1, 1.3 Hz, 2H), 1.37 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 195.5, 153.1, 142.2, 140.9, 138.6, 135.9, 130.8, 130.2, 129.5, 128.3, 128.2, 128.0, 127.5, 127.1, 125.2, 50.6, 42.4, 34.5, 30.4; HRMS (ESI) m/z calcd. for C29H34NaO2S [M + Na]+ 469.21717, found 469.21580.Methyl 2-(((3,5-di-tert-butyl-4-hydroxyphenyl)(phenyl)methyl)thio)acetate (5a)
This compound was prepared via the general procedure as a colorless oily liquid (38.4 mg, 96%). 1H NMR (600 MHz, CDCl3) δ 7.47 (d, J = 7.3 Hz, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.24 (dd, J = 3.7, 2.4 Hz, 1H), 7.20 (s, 2H), 5.29 (s, 1H), 5.15 (s, 1H), 3.66 (s, 3H), 3.07 (d, J = 3.7 Hz, 2H), 1.40 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 171.0, 153.2, 141.1, 135.9, 130.8, 128.7, 128.6, 127.4, 125.2, 54.7, 52.4, 34.5, 33.8, 30.4; HRMS (ESI) m/z calcd. for C24H32NaO3S [M + Na]+ 423.19644, found 423.19591.5-(3,5-Di-tert-butyl-4-hydroxyphenyl)-5H-benzo[f][1,4]oxathiepin-2(3H)-one (6)
This compound was prepared via the general procedure as an off-white solid (22.0 mg, 57%). m.p. 180–181 °C. 1H NMR (600 MHz, CDCl3) δ 7.32 (td, J = 7.7, 1.3 Hz, 1H), 7.29 (s, 2H), 7.22 (t, J = 7.6 Hz, 1H), 7.15 (d, J = 7.9 Hz, 1H), 7.00 (d, J = 7.7 Hz, 1H), 5.70 (s, 1H), 5.32 (s, 1H), 3.36 (d, J = 12.0 Hz, 1H), 3.11 (d, J = 12.0 Hz, 1H), 1.45 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 167.7, 154.2, 150.3, 136.5, 130.3, 129.3, 128.3, 127.5, 126.5, 125.8, 120.1, 46.6, 34.7, 30.7, 30.5; HRMS (ESI) m/z calcd. for C23H28NaO3S [M + Na]+ 407.16514, found 407.16424.4-((Benzylthio)(phenyl)methyl)-2,6-di-tert-butylphenol (8a)
This compound was prepared via the general procedure as a colorless oily liquid (40.7 mg, 97%). 1H NMR (600 MHz, CDCl3) δ 7.41 (d, J = 6.8 Hz, 2H), 7.33–7.26 (m, 4H), 7.24–7.18 (m, 4H), 7.15 (s, 2H), 5.12 (s, 1H), 4.87 (s, 1H), 3.52 (d, J = 2.8 Hz, 2H), 1.40 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 152.9, 141.9, 138.4, 135.8, 131.4, 129.2, 128.6, 128.5, 127.1, 127.0, 125.2, 53.8, 36.8, 34.5, 30.4.2,6-Di-tert-butyl-4-((ethylthio)(phenyl)methyl)phenol (8b)
This compound was prepared via the general procedure as a colorless oily liquid (34.9 mg, 98%). 1H NMR (600 MHz, CDCl3) δ 7.45 (d, J = 8.0 Hz, 2H), 7.32 (t, J = 7.7 Hz, 2H), 7.24 (t, J = 7.2 Hz, 1H), 7.19 (s, 2H), 5.16 (s, 1H), 5.07 (s, 1H), 3.10 (q, J = 14.5 Hz, 2H), 2.18 (s, 3H), 1.40 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 153.2, 141.1, 136.0, 130.9, 128.7, 128.6, 127.4, 125.2, 54.5, 42.4, 34.5, 30.4, 28.6.Assays to measure antibacterial susceptibility
The MIC of the test compounds was determined using the broth micro-dilution procedure described in the Clinical and Laboratory Standards Institute (CLSI) guidelines.25 Strains of S. aureus ATCC 25923, E. coli ATCC 25922, P. aeruginosa ATCC 9027 and a clinically isolated multi-drug resistance strain of S. aureus (20151027077) were incubated in MHB for 16–24 h at 37 °C and diluted to 1 × 106 CFU mL−1. C. albicans BNCC 259769 and C. albicans BNCC 333935 were incubated in MHB for 20–24 h at 30 °C and diluted. Both compounds to be tested and positive control drugs were prepared in 1 mg mL−1 mother liquor with MHB. Then, twofold dilution of samples was done in 96-well plates (final concentration was 0.5 μg mL; 100 μL in each well). Next, 100 μL of the test microorganism was added to wells and incubation undertaken. Bacteria were incubated for 18 h at 37 °C. Fungi were incubated for 20 h at 30 °C. Finally, the MIC was calculated. Equivalent amounts of DMSO (5%) and medium were used successively as negative controls and blank control, respectively. Levofloxacin was used as the positive drug to determine the MIC of bacteria. Fluconazole was used as the positive drug to determine the MIC of fungi. All experiments were carried out in three replicates.Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
We thank the following for financial support for this work: National Natural Science Foundation of China (22061011), Guizhou Science and Technology Department of China (QKHJC[2020]1Y031), and the Cultivation Project of National Natural Science Foundation of Guizhou Medical University (20NSP054).Notes and references
- R. Y. T. Kao, J. L. Jenkins, K. A. Olson, M. E. Key, J. W. Fett and R. Shapiro, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10066 CrossRef CAS PubMed.
- K. A. Toenjes, S. M. Munsee, A. S. Ibrahim, R. Jeffrey, J. E. Edwards and D. I. Johnson, Antimicrob. Agents Chemother., 2005, 49, 963 CrossRef CAS PubMed.
- S. DeBonis, D. A. Skoufias, R.-L. Indorato, F. Liger, B. Marquet, C. Laggner, B. Joseph and F. Kozielski, J. Med. Chem., 2008, 51, 1115 CrossRef CAS PubMed.
- M. Goyal, P. Singh, A. Alam, S. K. Das, M. S. Iqbal, S. Dey, S. Bindu, C. Pal, S. K. Das, G. Panda and U. Bandyopadhyay, Free Radical Biol. Med., 2012, 53, 129 CrossRef CAS PubMed.
- P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807 CrossRef CAS PubMed.
- B. C. Ranu and R. Jana, Adv. Synth. Catal., 2005, 347, 1811 CrossRef CAS.
- P. Singh and R. K. Peddint, Synthesis, 2017, 49, 3633 CrossRef CAS.
- A. Singh, S. Gupta and J. M. Khurana, Org. Prep. Proced. Int., 2020, 52, 110 CrossRef CAS.
- J. M. Altimari, J. P. Delaney, L. Servinis, J. S. Squire, M. T. Thornton, S. K. Khosa, B. M. Long, M. D. Johnstone, C. L. Fleming, F. M. Pfeffer, S. M. Hickey, M. P. Wride, T. D. Ashton, B. L. Fox, N. Byrne and L. C. Henderson, Tetrahedron Lett., 2012, 53, 2035 CrossRef CAS.
- B. P. Bandgar, S. S. Gawande and D. B. Muley, Green Chem. Lett. Rev., 2010, 3, 49 CrossRef CAS.
- G. Frensch, N. Hussain, F. A. Marques and P. J. Walsh, Adv. Synth. Catal., 2014, 356, 2517 CrossRef CAS PubMed.
- Z.-H. Peng, Y.-L. Wang, Z. Yu, D.-Z. Zhao, L.-H. Song and C.-Y. Jiang, J. Org. Chem., 2018, 83, 7900 CrossRef CAS PubMed.
- X.-X. Liang, H.-Y. Xu, H.-L. Li, L.-Z. Chen and H.-F. Lu, Eur. J. Org. Chem., 2020, 2020, 217 CrossRef CAS.
- J.-Y. Wang, W.-J. Hao, S.-J. Tu and B. Jiang, Org. Chem. Front., 2020, 7, 1743 RSC.
- A. S. Jadhav and R. V. Anand, Eur. J. Org. Chem., 2017, 25, 3716 CrossRef.
- J.-P. Yu, S.-Y. Chen, K. Liu, L.-Y. Yuan, Y.-B. Zhao, Z.-F. Chai and L. Mei, Tetrahedron Lett., 2020, 61, 152076 CrossRef CAS.
- B.-Q. Xiong, S.-P. Xu, Y. Liu, K.-W. Tang and W.-Y. Wong, J. Org. Chem., 2021, 86, 1516 CrossRef CAS PubMed.
- L. Xie, X.-Y. Zang, W.-X. Liu, P. Wu, Z.-X. Wang, C.-Y. Zhao, Z.-Z. Gao, X.-J. Li, S.-Z. Nie and L.-G. Wu, Asian J. Org. Chem., 2022, 11, e202200524 CAS.
- F. Zamberlan, A. Fantinati and C. Trapella, Eur. J. Org. Chem., 2018, 25, 3248 CrossRef.
- S. S. Sajadikhah and M. T. Maghsoodlou, Chem. Heterocycl. Compd., 2018, 54, 581 CrossRef CAS.
- Y.-L. Zhao, X.-H. Fei, Y.-Q. Tang, P.-F. Xu, F.-F. Yang, B. He, X.-Z. Fu, Y.-Y. Yang, M. Zhou, Y.-H. Mao, Y.-X. Dong and C. Li, J. Org. Chem., 2019, 84, 8168 CrossRef CAS PubMed.
- X.-H. Fei, Y.-L. Zhao, F.-F. Yang, X. Guan, Z.-Q. Li, D.-P. Wang, M. Zhou, Y.-Y. Yang and B. He, Adv. Synth. Catal., 2021, 363, 3018 CrossRef CAS.
- C. Gharui, S. Prakash, D. Chopra and S. C. Pan, Org. Biomol. Chem., 2020, 18, 2828 RSC.
- Z.-L. Liu, H.-F. Xu, T.-F. Yao, J.-L. Zhang and L. Liu, Org. Lett., 2019, 21, 7539 CrossRef CAS PubMed.
- J. B. Pate, F. R. Cockerill III, P. A. Bradford, G. M. Eliopoulos, J. A. Hindler, S. G. Jenkin, J. S. Lewis II, B. Limbago, L. A. Miller, D. P. Nicolau, M. Powell, J. M. Swenson, M. M. Traczewski and J. D. Turn, Clin. Lab. Stand. Inst., 2015, 35, M07 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01815f |
| ‡ These authors made equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2023 |