DOI:
10.1039/D3RA01701J
(Paper)
RSC Adv., 2023,
13, 18229-18237
Preparation of a TiO2/PEDOT nanorod film with enhanced electrochromic properties†
Received
15th March 2023
, Accepted 12th June 2023
First published on 16th June 2023
Abstract
The designed growth of titanium dioxide (TiO2)/poly(3,4-ethylenedioxythiophene) (PEDOT) nanorod arrays has been achieved by the combination of hydrothermal and electrodeposition methods. Due to the use of one-dimensional (1D) TiO2 nanorod arrays as the template of the nanocomposites (TiO2/PEDOT), the surface area of the active materials is enlarged and the diffusion distance of the ions is shortened. The nanorod structure also contributes to increasing the length of PEDOT conjugated chains and facilitates the transfer of electrons in the conjugated chains. Consequently, the TiO2/PEDOT film delivers a shorter response time (∼0.5 s), higher transmittance contrast (∼55.5%) and long-cycle stability compared to the pure PEDOT film. In addition, the TiO2/PEDOT electrode is further developed to be a smart bi-functional electrochromic device exhibiting energy storage performance. We expect that this work may lead to new designs for powerful intelligent electrochromic energy storage devices.
1. Introduction
Electrochromism refers to reversibly changing the optical properties of a material under the conditions of applying a certain voltage.1,2 Electrochromic materials have low driving voltage, abundant colour change, high coloration efficiency and have been widely used in smart windows, military camouflage and other energy-efficient devices.3,4 The major electrochromic materials could be classified as transition metal oxides, organic small molecules and conjugated conductive polymers (CPs).5,6 Recently, conjugated CPs have attracted great attention due to their good conductive ability, multicolor change, high flexibility and ease of processing.7–11 Poly(3,4-ethylenedioxythiophene) (PEDOT) has been extensively studied as one of the most promising conjugated CPs for electrochromic applications.12 It is well known that PEDOT has high conductivity, environmental stability and electrochemical stability.13,14 However, pure PEDOT prepared by traditional chemical oxidative polymerization or electropolymerization methods exhibits low optical and electrochemical properties.15–19 Hence, the electrochromic properties of pure PEDOT need to be improved for daily applications. According to the studies, enhancing the length of the conjugated chains of CPs can improve the electrical conductivity and reactive sites, which in turn improves the electrochromic properties.20–22 However, it is still a challenge to regulate and quantification the growth of conjugate systems for constructing long conjugated chains.
The one-dimensional (1D) nanostructured materials such as nanowires, nanorods and nanotubes can provide wide surface areas and short diffusion distances for ion transport, which leads to more active sites and short charge transfers time.23–26 Fu et al.27 prepared the polyaniline (PANI)/titanium dioxide (TiO2) nanorods through electrochemical polymerization of aniline onto the TiO2 nanorods array film. The PANI/TiO2 nanorods hybrid film showed short response time at different states (0.7 s/2.6 s) and had a long-term stability after cyclic voltammetry scans for 200 circles. Chen et al.28 designed one-dimensional (1D) growth of ZnO/PEDOT core/shell hybrid nanotube arrays by electropolymerization technique. The composite materials had high transmittance contrast and color efficiency improved to 122.2 cm2 C−1.
However, little attention has been paid to use 1D TiO2 nanorods as the template to construct nanorods coated with the PEDOT through electrochemical polymerization in current reports, which can achieve the effect of extending the PEDOT conjugated chain. Herein, a facile and effective approach is developed for improving the electrochromic performance of the pure PEDOT by coating onto the 1D TiO2 nanorods. Due to the 1D TiO2 nanorod arrays as the template of the nanocomposite films (TiO2/PEDOT), the surface area of active materials is enlarged and the diffusion distance of ions is shortened. The conjugated chain and charge capacity of PEDOT also are enhanced. A longer conjugated chain of PEDOT obtained on TiO2 nanorod array was confirmed by the calculation based on quantum chemistry theory. The nanorod arrays provide to inflated space for PEDOT to work in a long-term cycle. Consequently, the TiO2/PEDOT film electrode demonstrates a superior bi-functional electrochromic and electrochemical properties when compared with the pure PEDOT film. Finally, a smart bi-function electrochromic device is developed based on the TiO2/PEDOT electrode exhibiting visual energy storage level.
2. Experimental
2.1 Preparation of TiO2 nanorod arrays film electrodes
The TiO2 nanorods array film electrodes were prepared by hydrothermal method. Approximately 300 μL of C16H36O4Ti was slowly dropped into 12 mL of HCl. Deionized water (DIW) (12 mL) was added gradually while the mixture was being continuously stirred for 10 min in turn to obtain the reaction precursor solution. The fluorine tin oxide (FTO) conducting glass was used as the substrate for the preparation of TiO2 nanorods array film electrodes. The FTO glasses were followed by ultrasonic cleaning in acetone, ethanol and DIW for 20 min in turn. The reaction precursor solution and treated FTO substrates were transferred to a Teflon-lined stainless steel autoclave. The FTO substrates were submerged in the solution and placed against the wall of the Teflon-liner at an angle with the FTO side facing down. The autoclave was maintained at the temperature of 150 °C for 4 h, then naturally cooled down to room temperature. Finally, the samples were washed and dried at room temperature.
2.2 Preparation of TiO2/PEDOT nanocomposite films
The PEDOT electrodes were deposited on the FTO glass covered with TiO2 nanorod arrays via an electrochemical polymerization in the electrolyte. The electrolyte was composed of 0.015 mol L−1 EDOT, 0.1 mol L−1 LiClO4 in a solvent of acetonitrile. Three-electrode system containing the FTO glass covered with TiO2 nanorod arrays as the working electrode, a Pt sheet as the counter electrode, and an Ag/AgCl (sat. KCl) electrode as the reference electrode were used during the electropolymerization. The electropolymerization was performed with a Princeton Versa STAT 4 electrochemical workstation at room temperature. After applying a constant current of 2.0 mA for a while (90 s (180 mC), 120 s (240 mC) and 150 s (300 mC)), where the mC represents the charge quantity, a blue thin film was formed on the substrate. The electrodes were rinsed with ethanol and then dried in air at room temperature. As a contrast, the pure PEDOT was prepared by the same method without TiO2. The whole procedure was showed in Scheme 1.
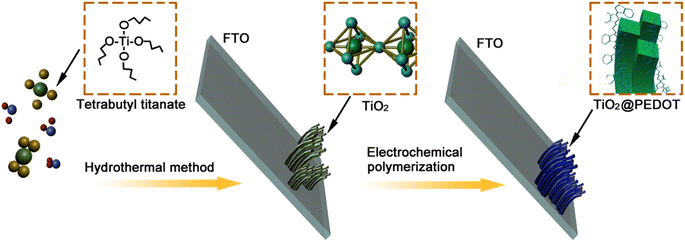 |
| | Scheme 1 Schematic diagram of the preparation of TiO2/PEDOT nanorod arrays film. | |
2.3 Characterizations
The X-ray diffraction (XRD) patterns was measured by a Bruker D8 XRD diffractometer with Cu Kα radiation (λ = 1.5418 Å). Fourier transform infrared (FTIR) spectra were recorded using a Shimadzu IR Prestige-21 FT-IR spectrophotometer. The morphologies and element compositions of TiO2/PEDOT were studied using field-emission scanning electron microscopy (FESEM) (JEOL FEG JSM 7001F), and transmission electron microscopy (TEM) attached with an energy dispersive spectrometer (EDS mapping) (JEOLJEM-2100F).
2.4 Electrochemical and electrochromic measurements
The electrochemical properties of TiO2/PEDOT were studied in a three-electrode system using a Princeton Versa STAT 4 electrochemical workstation at room temperature. A three-electrode system was used, consisting of the TiO2/PEDOT electrode as the working electrode, a platinum foil as the counter electrode and an Ag/AgCl (sat. KCl) as the reference electrode. 1 mol L−1 LiClO4 in the propylene carbonate (PC) solution was used as electrolyte. The electrochromic performance of PEDOT was studied by measuring in situ optical transmittance spectra through an ultraviolet-visible spectrophotometer (Beijing Purkinje General Instrument Co. Ltd, TU-1810 PC) coupled with electrochemical workstation.
3. Results and discussion
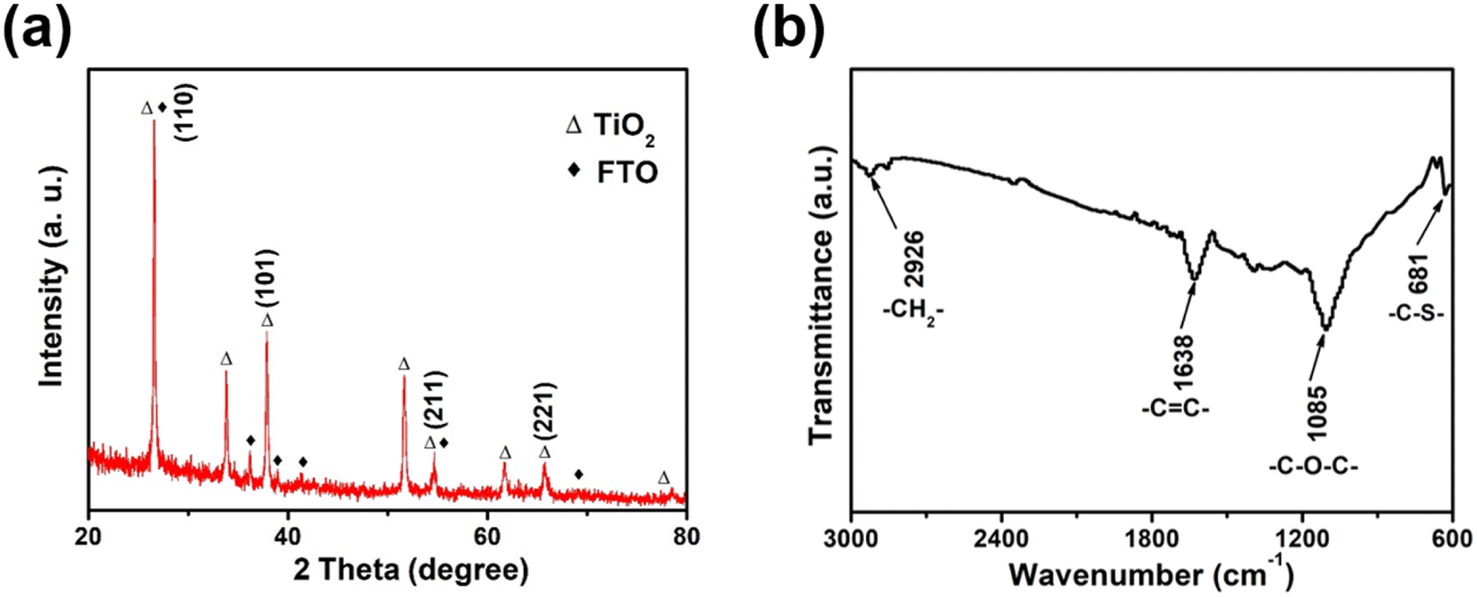
The XRD pattern of the synthesized TiO2 nanorods demonstrated distinct characteristic peaks showed in Fig. 1a, except for the characteristic peak of the FTO substrate (JCPDS 77-0451), the remaining diffraction peaks corresponded well to the rutile phase of TiO2 (JCPDS card no. 75-1748).29,30 The major XRD peaks located at about 26.62°, 36.44° and 54.62° were assigned to the TiO2 (110), (101) and (211) crystal plane.29,30 The molecule structure of TiO2/PEDOT was examined using the FTIR spectra (Fig. 1b). The peaks at 1085 and 681 cm−1 were ascribed to the stretching vibrations of C–O–C and C–S groups, respectively. The peak ascribing to C–H stretching vibration in ethylenedioxy group of EDOT was observed at 2926 cm−1. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of in thiophene ring located at 1638 cm−1. Meanwhile, the main functional groups of TiO2/PEDOT film are the same as EDOT monomer and bare PEDOT (Fig. S1†). The above results confirmed the formation of electrochromic PEDOT on the TiO2 nanorod arrays.31,32 The morphological structure of the 1D TiO2 nanorod arrays were monitored by using SEM. As shown in Fig. 2a, the prepared TiO2 revealed a nanorod arrays morphology, which were nearly perpendicular to the FTO substrate. The TiO2/PEDOT nanocomposites exhibited a uniform PEDOT coating on the outer surface of the TiO2 nanorod by electropolymerization (Fig. 2b), which were benefit for achieving a barrier-free electron conduction and increasing the contact area with the electrolyte. Meanwhile, it is beneficial to promote the formation of a longer system of conjugated chains of PEDOT molecules. The pare PEDOT on FTO prepared by electropolymerization exhibits dense morphology (Fig. S2†). The diameter of the TiO2/PEDOT nanorod ranged from 43.91–53.88 nm, and its cross section had a square shape (inset of the Fig. 2b). However, there are still differences in the diameter and morphological structure of the composite nanorods for the 180 and 300 mC films compared to the 240 mC film (Fig. S3†). This is due to the difference in electrodeposition time, resulting in difference of PEDOT content present on the nanorods and in their gap. This ultimately resulted in a thinner nanorod for the 180 mC film and a thicker diameter for the 300 mC film (Fig. S3a and c†). In addition, the PEDOT of the 300 mC film had been deposited onto the surface, making it too dense between the nanorods. A sparse active layer is not conducive to the increase of active sites, while a dense active layer is an obstacle to the deeper transport of ions. The 240 mC film morphology is optimized in terms of the structure of the active layer (Fig. S3b†), resulting in relatively good electrochromic properties. The magnified TEM images were employed for further study of element type and distribution. As shown in Fig. 2c, the elemental mapping images confirmed distribution of the major element C, O, S and Ti from the TiO2/PEDOT. It was demonstrated that the PEDOT was successfully and uniform coated on the surface of TiO2 nanorods to form TiO2/PEDOT nanocomposites.
C stretching vibrations of in thiophene ring located at 1638 cm−1. Meanwhile, the main functional groups of TiO2/PEDOT film are the same as EDOT monomer and bare PEDOT (Fig. S1†). The above results confirmed the formation of electrochromic PEDOT on the TiO2 nanorod arrays.31,32 The morphological structure of the 1D TiO2 nanorod arrays were monitored by using SEM. As shown in Fig. 2a, the prepared TiO2 revealed a nanorod arrays morphology, which were nearly perpendicular to the FTO substrate. The TiO2/PEDOT nanocomposites exhibited a uniform PEDOT coating on the outer surface of the TiO2 nanorod by electropolymerization (Fig. 2b), which were benefit for achieving a barrier-free electron conduction and increasing the contact area with the electrolyte. Meanwhile, it is beneficial to promote the formation of a longer system of conjugated chains of PEDOT molecules. The pare PEDOT on FTO prepared by electropolymerization exhibits dense morphology (Fig. S2†). The diameter of the TiO2/PEDOT nanorod ranged from 43.91–53.88 nm, and its cross section had a square shape (inset of the Fig. 2b). However, there are still differences in the diameter and morphological structure of the composite nanorods for the 180 and 300 mC films compared to the 240 mC film (Fig. S3†). This is due to the difference in electrodeposition time, resulting in difference of PEDOT content present on the nanorods and in their gap. This ultimately resulted in a thinner nanorod for the 180 mC film and a thicker diameter for the 300 mC film (Fig. S3a and c†). In addition, the PEDOT of the 300 mC film had been deposited onto the surface, making it too dense between the nanorods. A sparse active layer is not conducive to the increase of active sites, while a dense active layer is an obstacle to the deeper transport of ions. The 240 mC film morphology is optimized in terms of the structure of the active layer (Fig. S3b†), resulting in relatively good electrochromic properties. The magnified TEM images were employed for further study of element type and distribution. As shown in Fig. 2c, the elemental mapping images confirmed distribution of the major element C, O, S and Ti from the TiO2/PEDOT. It was demonstrated that the PEDOT was successfully and uniform coated on the surface of TiO2 nanorods to form TiO2/PEDOT nanocomposites.
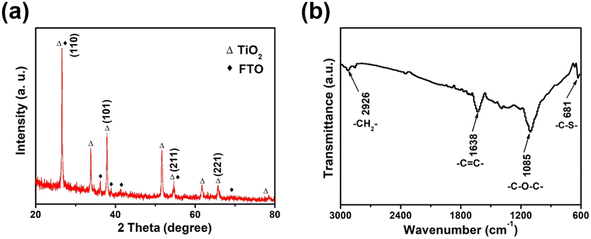 |
| | Fig. 1 (a) XRD pattern and (b) FTIR spectra of TiO2/PEDOT film. | |
 |
| | Fig. 2 Top-view scanning electron microscope (SEM) images of (a) TiO2 and (b) TiO2/PEDOT nanorod arrays. (Inset) The TiO2/PEDOT nanorod arrays details of partial enlarged. (c) TEM images and EDS elemental maps of the TiO2/PEDOT nanorod. | |
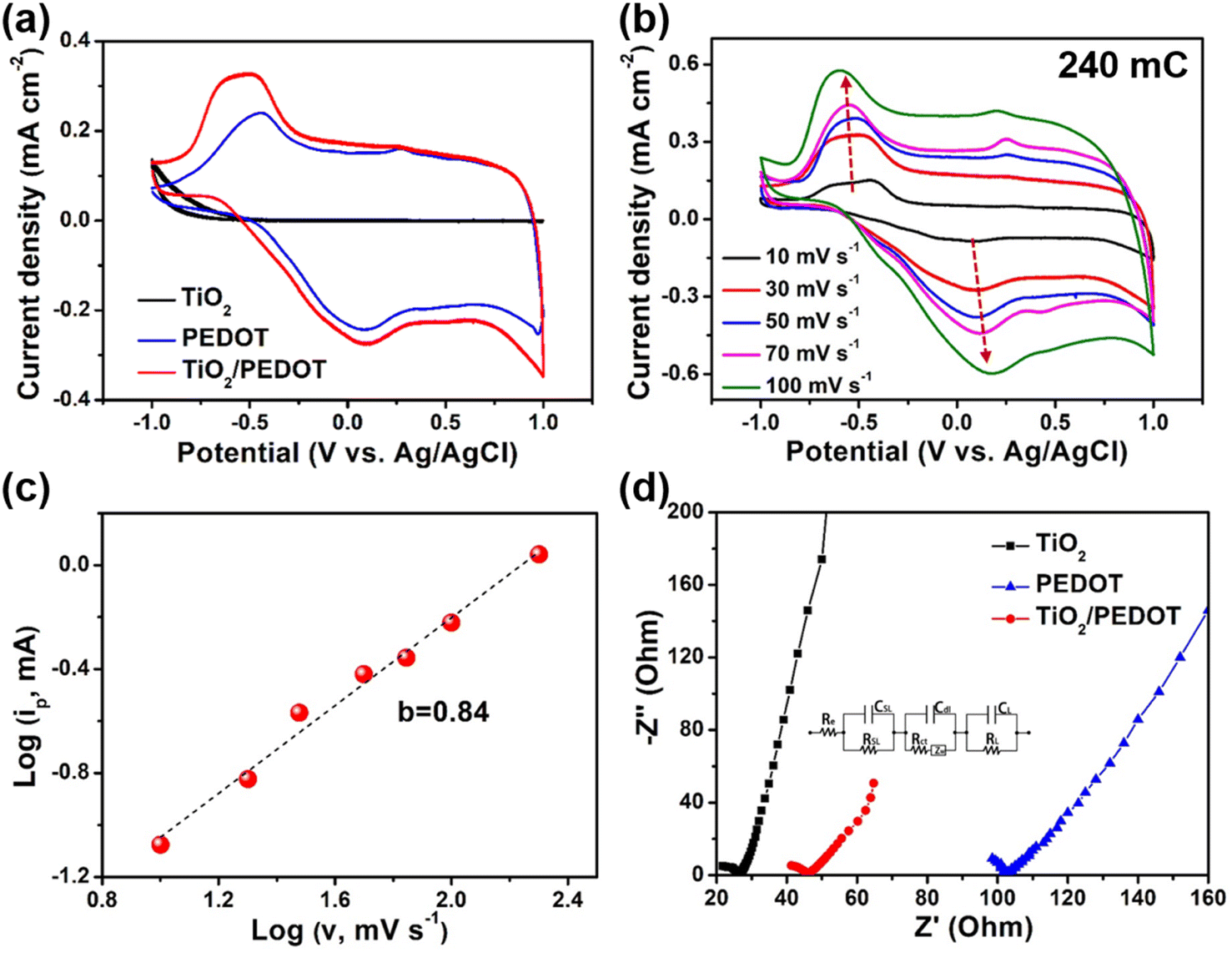
To understand the electrochemical behavior of composite films in 1 M LiClO4/PC, the cyclic voltammetry (CV) curves were measured in the potential of −1 V to 1 V (Fig. 3a). The CV curves recorded at a scan rate of 30 mV s−1 demonstrated two pairs of redox peaks, which were ascribed to the redox reactions of PEDOT. By comparison, the redox peaks of TiO2/PEDOT (prepared at 240 mC) film were much stronger that those of the pure PEDOT film, indicating a rapid charge injection/extraction process (Fig. S4†).33 Compared with the sharp and narrow maximum oxidation peak of PEDOT, the maximum oxidation peak of TiO2/PEDOT showed obvious hysteresis. It showed that ions have hysteresis phenomenon in the deep doping process, the ions in the active layer will hinder the further doping anions, and the voltage needed to be increased to maintain the further doping process, so a wide oxidation peak phenomenon was generated.34–36 Meanwhile, the TiO2/PEDOT had a larger enclosed area of the CV curve compared with PEDOT and TiO2, suggesting a higher charge capacity. It was worth mentioned that TiO2 contributed little to the charge capacity of the whole electrode. This may be related to the higher onset oxidation potential. The CV curves of TiO2/PEDOT (240 mC) performed at different potential scan rates range from 10 to 100 mV s−1 were showed in Fig. 3b. It can be concluded from the figure that the CV curves of the TiO2/PEDOT remained largely unchanged at different scanning rates, illustrating that this material had good reversibility and stability. Moreover, the electrochemical reaction dynamics could be analyzed through the CV behaviors at various scan rates according to the power law:
Here
i was the peak current (mA),
v was the scan rate (mV s
−1),
a and
b were the adjustable parameters. Convert
(1) to
(2):
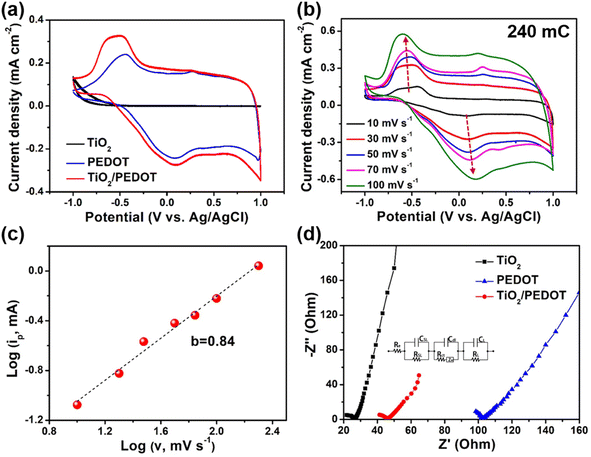 |
| | Fig. 3 Electrochemical performance of TiO2, PEDOT and TiO2/PEDOT (prepared at 240 mC) films. (a) Cyclic voltammetry (CV) curves of TiO2, PEDOT and TiO2/PEDOT films at 30 mV s−1. (b) CV curves of the TiO2/PEDOT (240 mC) at different scan rates. (c) The fitting curve of the relationship between log(ip) and log(v) of TiO2/PEDOT. (d) Nyquist curves for fitting electrochemical impedance spectra (EIS) plots of TiO2, PEDOT and TiO2/PEDOT. (Inset) The equivalent circuit for fitting EIS plots. | |
The b value was a key parameter, which was determined that the charge storage was dominated by the diffusion processes (b ≈ 0.5) or the surface capacitance (b ≈ 1.0). The obtained b value of TiO2/PEDOT was ∼0.84 (Fig. 3c), indicating a capacitance-controlled charge storage of the TiO2/PEDOT.37 Fig. 3d demonstrated Nyquist plots of TiO2, PEDOT and TiO2/PEDOT. The charge transfer resistance (Rct) of TiO2, PEDOT and TiO2/PEDOT were 8.55, 24.2 and 11.1 Ω, respectively. These observations suggested that the incorporation of 1D TiO2 nanorod into PEDOT effectively increased electronic conduction, provided a more direct path for electrons.38,39 Meanwhile, the TiO2 nanorod also could act as a transport path for ions to provide smooth and convenient ion transfer, thereby enhancing the accessibility of the electrolyte to the surface of PEDOT, resulting in fast kinetics of electrochemical reaction.
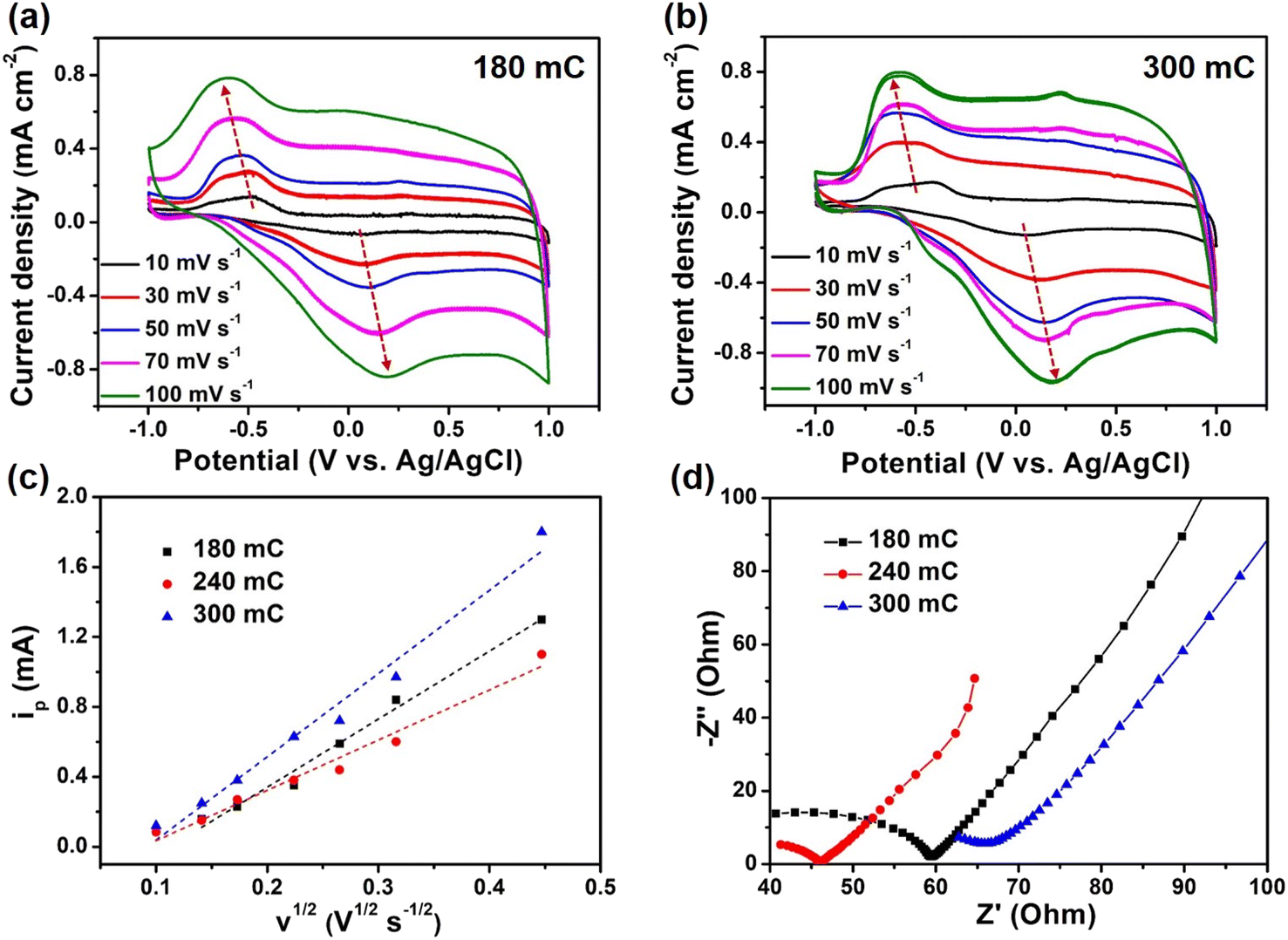
Moreover, we also explored the effects of charge quantity by electrodeposition of PEDOT on the electrochemical performance of composite films. Compared with the 240 mC film, the redox peaks of 180 mC and 300 mC films had a larger deviation, which indicated that the electrochemical reversibility was poor (Fig. 4a and b). More importantly, with the gradual increase of scan rate, the redox peak of 240 mC film still existed, while 180 mC and 300 mC films gradually weaken. It was deduced that the 240 mC film has more electrochemical active sites and stable structure. The relationships between the oxidation peak current versus the square root of potential scan rate for the three samples were showed in Fig. 4c. We observed a linear relationship between the peak current density and the square root of the scan rate, indicating the electrochemical reversibility and revealing a nondiffusion-controlled behavior.40,41 Based on the Randles Sevcik equation,42 the diffusion coefficients of composite films prepared at 180 mC, 240 mC and 300 mC could be calculated. The diffusion coefficient D of 180 mC, 240 mC and 300 mC films were determined to be 2.1 × 10−10, 1.1 × 10−10 and 3.3 × 10−10 cm2 s−1, respectively, much higher than that of pure PEDOT (6.5 × 10−11 cm2 s−1). This indicated that the diffusion behavior of ions in the active layer of the composite films were better than that of PEDOT film. Furthermore, the Rct value of 240 mC film (11.1 Ω) was lower than that of 180 mC and 300 mC films (17.1 and 16.6 Ω) (Fig. 4d). Since the electropolymeric time was the key to influence the growth of PEDOT, the amount of PEDOT on the surface of TiO2 nanorods was thin or dense in 180 mC and 300 mC films, thereby affecting the transport of ions and electrons. Therefore, the fine structure system of composite films would facilitate a fast electrochemical reaction kinetics and thus contribute to an enhanced electrochromic performance.
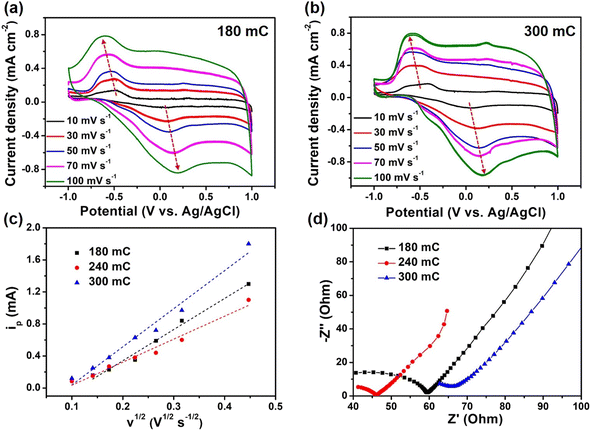 |
| | Fig. 4 Electrochemical performance of composite films. CV curves of (a) 180 mC and (b) 300 mC films at different scan rates. (c) The fitting curve of the relationship between ip and v1/2 and (d) Nyquist plots of 180 mC, 240 mC and 300 mC films. | |
It has been shown that the increasement of the conjugated chains in conjugated CPs molecules could boost their electrochromic performance.20,21 Because long and stable conjugated chains can promote the speed and quantity of charge transport in conjugated CPs.20,21 Therefore, the length of PEDOT conjugate chains grown from TiO2 nanorod arrays was quantified by quantum chemistry theory, and the promotion effect of longer conjugate chains on electrochromism was further verified by electrochromic tests. The UV-vis absorption spectra showed that the maximum absorption peaks (λmax) of TiO2/PEDOT (240 mC) and PEDOT were located at 585 and 505 nm (Fig. 5a). The λmax of the 180 mC and 300 mC films (547 and 550 nm) were also significantly larger than PEDOT. The lengthen of PEDOT conjugated chain assisted by TiO2 nanorod was studied in detail via a calculation based on quantum chemistry theory.22,43–45 According to the eqn (3) as follow:
| |
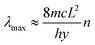 | (3) |
where
c was the speed of light (3.0 × 10
8 m s
−1),
m was the electron mass (9.1 × 10
−31 kg),
h was the Planck constant (6.63 × 10
−34 kg m
2 s
−1) and
n was the number of conjugate chain links. Each link of PEDOT had
y = 6 (four electrons from the p orbital of α carbon and two from the p orbital of sulfur). The bond lengths of C–C and C
C were ∼1.54 Å and ∼1.37 Å, respectively, and
L was estimated to be 2.0–3.0 Å. All the above values were substituted into
eqn (3), and it can be calculated as follows (the result was an integer):
n(TiO
2/PEDOT) = 27 (or 12),
n(PEDOT) = 23 (or 10). Based on the calculation, it could be estimated that each PEDOT conjugated long chain on TiO
2/PEDOT was about 6.0–8.0 Å longer than that of the pure PEDOT. However, 6.0–8.0 Å was only the length difference of this single conjugate chain. Considering the correlation between the overall conjugate chain and electrode performance of PEDOT, this difference will be enlarged by increasing the amount of active materials on the electrode. The above calculations and experimental results both verify the shift of
λmax to higher wavelengths, which indicate an increase in the number of conjugated chain links during polymerization of PEDOT.
46,47 It confirms that the length of conjugated chain was assisted by TiO
2 nanorod. The longer structure of the conjugated chain facilitates the transfer of electrons in the conjugated chain and improves the conductivity.
Fig. 5b showed the response time characteristics of the films at 900 nm. In this work, the response time for coloring and bleaching of 180, 240 and 300 mC films were 0.86/0.85, 0.5/0.5, 2.0/2.0 s, respectively, which were shorter than that of PEDOT (1.4/2.4 s) and TiO
2 (1.5/2.5 s). Prolonging the conjugated chains of PEDOT can be used as a promising strategy to improve the electrochromic performance.
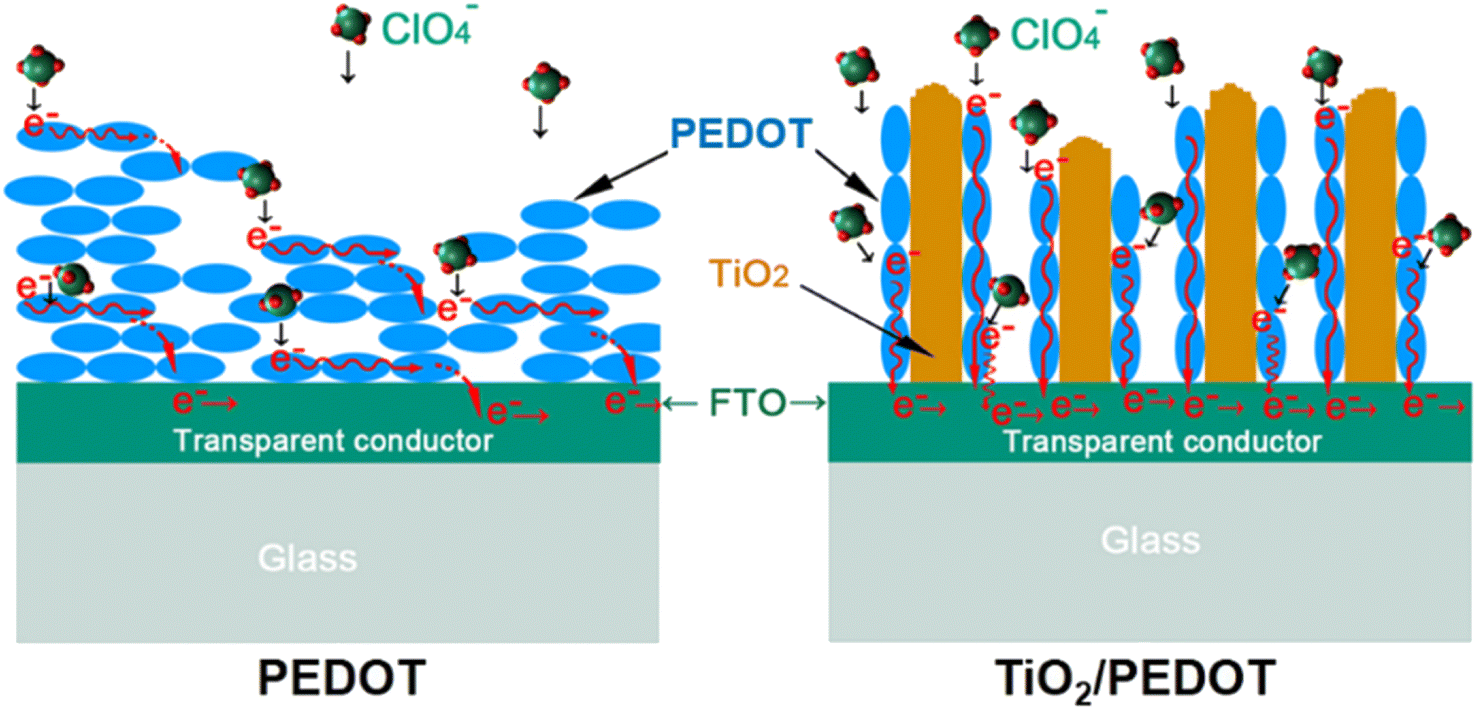
48 Scheme 2 reflected the growth and electron transport of conjugated chains of PEDOT on different substrates (FTO-glass or TiO
2-FTO-glass). The conjugated chains of PEDOT were stacked along the surface of FTO to form the dense layer (
Scheme 2 left), while the conjugated chains of PEDOT of the TiO
2/PEDOT were polymerized and grown along the nanorods (
Scheme 2 right).
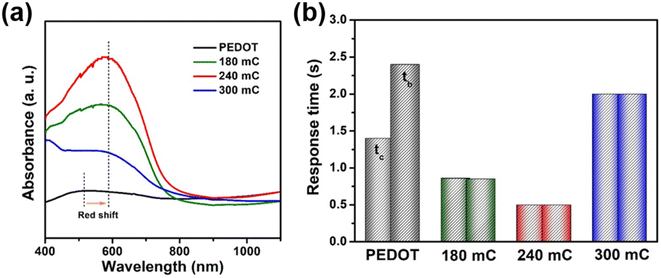 |
| | Fig. 5 (a) Adsorbance spectra and (b) in situ transmittance response of the PEDOT and composite films. | |
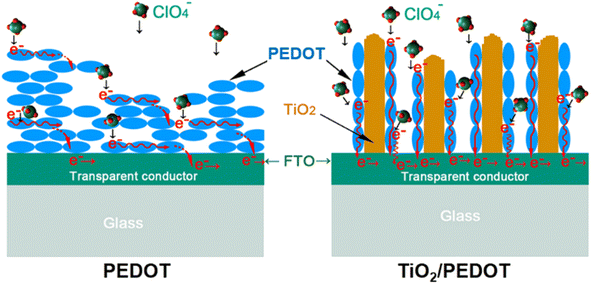 |
| | Scheme 2 Schematic diagram of the electron conduction along the conjugated chain of PEDOT and TiO2/PEDOT electrodes. | |
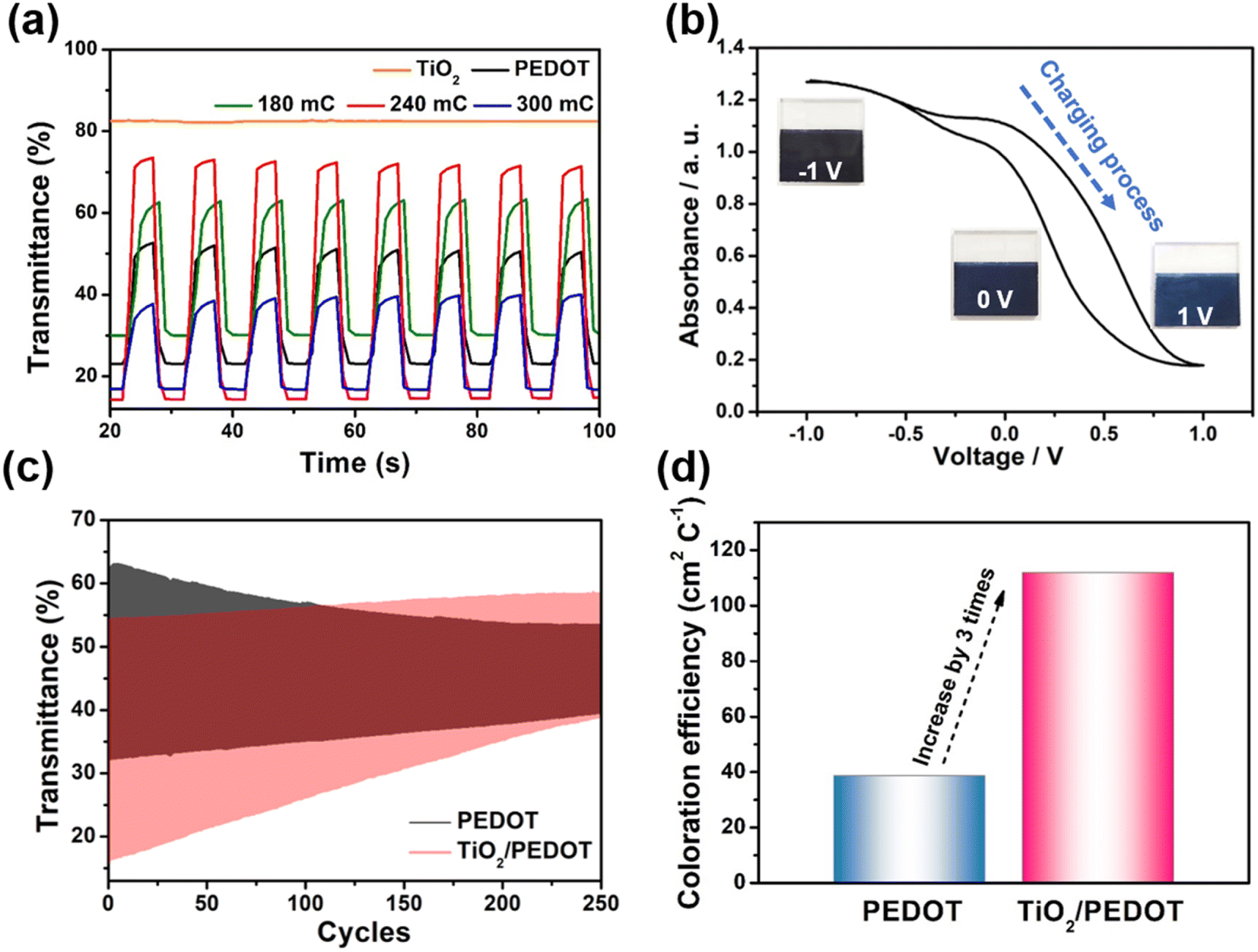
Therefore, the TiO2/PEDOT was more conductive to PEDOT forming longer molecular conjugated chains, thus the corresponding λmax was red shifted. More importantly, the nanorod structure inherited from TiO2 could act as a transport path for electric charge to provide smooth and convenient electric charge transfer, thereby enhancing the accessibility of the electrolyte to the above PEDOT active regions, resulting in fast kinetics of electrochemical reactions and more active sites.49 In the dense pure PEDOT film, the redox reaction area is confined to a thin surface layer due to the limited diffusion of electric charge in the dense PEDOT film, thus reducing the response time and utilization of active sites.50 Fig. 6a showed the optical modulation of the TiO2, PEDOT and composite films applying the potential of −1 and 1 V in the wavelength of 900 nm. The transmittance modulation of 240 mC (TiO2/PEDOT) reached 55.5% which was higher than that of pure PEDOT (28.1%). Compared with PEDOT, the colored 240 mC had a lower transmittance while the bleached had a higher value. We also explored the effects of different charge quantity of electrodeposition PEDOT on the optical modulation. The 180 mC and 300 mC films were poor than the 240 mC film. The possible reason was that the thickness of the PEDOT layer can seriously affect charge transport and corresponding optical modulation. Note that there was a negligible optical modulation of the TiO2 nanosheet arrays substrate. Based on different doping states of PEDOT, the TiO2/PEDOT demonstrated multiple colors in different energy storage status. To further evaluate the relationship between absorbance and energy storage/release during the electrochemical test process, the corresponding in situ absorbance of the TiO2/PEDOT electrode is measured and is shown in Fig. 6b. Obviously, the absorbance of the TiO2/PEDOT electrode decreased from ∼1.2 to ∼0.2 when it was charged to 1 V, corresponding to the color change from dark blue to light blue, and then gradually colored during the inverse discharging process (inset of Fig. 6b). Inset of the Fig. 6b showed photographs of the TiO2/PEDOT (240 mC) in the bleached (right) and colored state (left) by applying a voltage of 1, 0 and −1 V, respectively. It could reflect different energy levels at different voltages. The electrode appeared light blue in fully charged state (1 V). The color of TiO2/PEDOT gradually deepened with the discharging. A deep blue was achieved when the electrode reached the fully discharged state (−1 V). This electrochromic phenomenon provided a platform to realize visual status of PEDOT-based electrochemical devices. The above results clearly confirmed the possibility of quantitatively monitoring the energy storage level via the optical absorbance/transmittance, this can be regarded as a convenient indicator for the energy status of PEDOT-based electrochemical devices. The chronoamperometry (CA) curves at the potential step from −1 to 1 V for 10 s were showed in Fig. S5.† It could be seen from the CA curves that the TiO2/PEDOT (240 mC) exhibited a higher transient current density than that of the pure TiO2 and PEDOT films, also demonstrating that there were more active sites in TiO2/PEDOT film for ion insertion/extraction.51,52 The TiO2/PEDOT demonstrated a better cycling stability of alternating coloration and bleaching. The ΔT could still maintain of its initial value after 250 cycles, which was much higher than that of PEDOT (Fig. 6c). Note that the bleached state of TiO2/PEDOT (240 mC) remained almost unchanged while the transmittance of colored state increased dramatically during the cycling, which was mainly because there were ion traps inside the materials during doping and undoping process.53 Ion trapping were generated because the chemical bonds of conjugated chains were destroyed in the process of doping and undoping. When the anions in the active layer were gradually captured by the ion traps in the inner layer of the PEDOT, it was difficult to carry out the undoping process, which leaded to the increase of Tc. With the gradual increase of anions in the inner layer, the doping process was hindered. The doping process could occur in the surface layer of the PEDOT, the Tb was increased slightly.53 Coloration efficiency (CE) was defined as the change in optical density (OD) per unit of charge (Q) inserted into (extracted out) the electrochromic materials at a certain wavelength, according to the following formula:54
| | |
CE = ΔOD/ΔQ = log(Tb/Tc)/ΔQ
| (4) |
where ΔOD was the change in optical density that could obtained by the transmittance of colored (
Tc) and bleached (
Tb) state, and Δ
Q was the varied charge density during the redox process. The calculated CE of the TiO
2/PEDOT (240 mC) was 111.98 cm
2 C
−1, which was approximately three times higher than that of PEDOT film (
Fig. 6d).
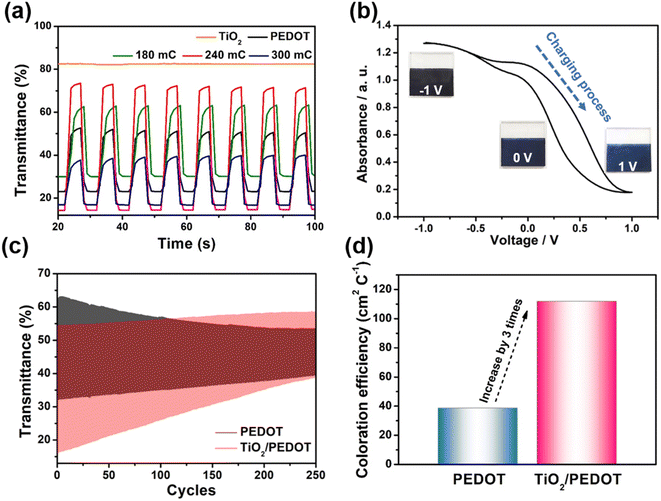 |
| | Fig. 6 (a) Optical modulation of the TiO2, PEDOT and composite films. (b) In situ absorbance with potential of the TiO2/PEDOT (240 mC) in the range of −1–1 V. (Inset) Photographs of color transition during a charging/discharging process for the TiO2/PEDOT (240 mC). (c) Cycling stability and (d) the calculated coloration efficiencies of PEDOT and TiO2/PEDOT (240 mC). | |
Furthermore, the TiO2/PEDOT (240 mC) electrodes were assembled to the symmetrical electrochromic device. An open circuit voltage (OCV) of 1.45 V enabled the device to power a light bulb (1 V/0.4 W) (Fig. 7a and b). It was indicated that the TiO2/PEDOT electrode exhibited excellent electrochemical energy storage behavior for real-world applications. And then, we have calculated the specific capacitance values of electrode and device by referring to electrochemical characterization methods in other literature.55,56 Although both the electrode and the device have some electrochemical energy storage capacity (Fig. S6 and S7†) and stable redox reversibility (Fig. S8†), the electrochemical energy storage performance needs to be further improved compared to other reported literature.
 |
| | Fig. 7 (a) The digital photo shows the OCV of the symmetrical electrochromic device. (b) The digital photo of a bulb lit up by the symmetrical electrochromic device. | |
4. Conclusions
In this work, we had successfully demonstrated an effective strategy to obtain both electrochromic and electrochemistry of PEDOT-based electrochromic materials. The TiO2/PEDOT nanorod arrays had been achieved by the combination of hydrothermal and electrodeposition methods. The one-dimensional (1D) TiO2 nanorod arrays promoted the extension of the conjugated chain of PEDOT and this was further confirmed by detailed calculations in quantum chemical theory. This conjugated chain extension could effectively shorten the ion diffusion length and provided more active regions for charge transfer reactions. The nanorod arrays provided the inflated space for PEDOT to work in a long-term cycle. As a result, the TiO2/PEDOT film exhibited short response time (∼0.5 s), high transmittance contrast (55.5%@900 nm) and long-cycle stability compared to pure PEDOT film. Moreover, the TiO2/PEDOT electrode was further developed to be a smart bi-function electrochromic device exhibiting energy storage level. It was optimistic that the 1D TiO2/PEDOT nanorod array electrode could be applied in e-books, smart windows and other energy-efficient displays.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Natural Science Foundation of Beijing Municipality (2222045).
Notes and references
- V. K. Thakur, G. Ding, J. Ma, P. S. Lee and X. Lu, Adv. Mater., 2012, 24, 4071–4096 CrossRef CAS PubMed.
- D. Louloudakis, D. Vernardou, G. Papadimitropoulos, D. Davazoglou and E. Koudoumas, Adv. Mater. Lett., 2018, 9, 578–584 CrossRef CAS.
- S. Mishra, S. Lambora, P. Yogi, P. R. Sagdeo and R. Kumar, ACS Appl. Nano Mater., 2018, 1, 3715–3723 CrossRef CAS.
- Q. Guo, X. Zhao, Z. Li, B. Wang, D. Wang and G. Nie, ACS Appl. Energy Mater., 2020, 3, 2727–2736 CrossRef CAS.
- P. Yang, P. Sun and W. Mai, Mater. Today, 2016, 19, 394–402 CrossRef CAS.
- Q. Guo, J. Li, B. Zhang, G. Nie and D. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6491–6501 CrossRef CAS PubMed.
- Q. Guo, X. Zhao, Z. Li, D. Wang and G. Nie, Chem. Eng. J., 2020, 384, 123370 CrossRef CAS.
- Q. Li, B. Wang, H. Zou, Q. Guo and G. Nie, J. Alloys Compd., 2022, 921, 166140 CrossRef CAS.
- B. Carbas, Polymer, 2022, 254, 125040 CrossRef.
- B. Carbas and E. Ergun, Eur. Polym. J., 2022, 175, 111363 CrossRef.
- P. Beaujuge and J. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- B. Zhuang, X. Wang, Q. Zhang, J. Liu, Y. Jin and H. Wang, Sol. Energy Mater. Sol. Cells, 2021, 232, 111357 CrossRef CAS.
- F. Wang, X. Zhang, Y. Ma and W. Yang, Appl. Surf. Sci., 2018, 427, 1038–1045 CrossRef CAS.
- F. Hu, B. Yan, G. Sun, J. Xu, Y. Gu, S. Lin, S. Zhang, B. Liu and S. Chen, ACS Appl. Nano Mater., 2019, 2, 3154–3160 CrossRef CAS.
- J. Zheng, L. Chen, S. Liu, C. Sun, X. Hu and S. Zhou, Mater. Res. Express, 2019, 6, 1150h8 CrossRef.
- S. Zhang, S. Chen, F. Yang, F. Hu, Y. Zhao, B. Yan, H. Jiang and Y. Cao, J. Mater. Sci. Mater. Electron., 2019, 30, 3994–4005 CrossRef CAS.
- J. P. Lock, J. L. Lutkenhaus, N. S. Zacharia, S. Im, P. T. Hammond and K. Karen, Synth. Met., 2007, 157, 894–898 CrossRef CAS.
- E. Ergun and B. Carbas, Mater. Today Commun., 2022, 32, 103888 CrossRef.
- E. Poverenov, M. Li, A. Bitler and M. Bendikov, Chem. Mater., 2010, 22, 4019–4025 CrossRef CAS.
- J. Duchet, R. Legras and S. Demoustier-Champagne, Synth. Met., 1998, 98, 113–122 CrossRef CAS.
- P. M. Carrasco, H. J. Grande, M. Cortazar, J. M. Alberdi, J. Areizaga and J. A. Pomposo, Synth. Met., 2006, 156, 420–425 CrossRef CAS.
- B. Y. Zhuang, X. Q. Wang, F. S. Li, Q. Q. Zhang, J. B. Liu, Y. Y. Sun, H. W. Zhao and H. Wang, Sol. Energy Mater. Sol. Cells, 2022, 238, 111621 CrossRef CAS.
- H. Yu, Y. Li, L. Zhao, G. Li, J. Li, H. Rong and Z. Liu, Mater. Lett., 2016, 169, 65–68 CrossRef CAS.
- X. Wang, B. Liu, J. Tang, G. Dai, B. Dong, L. Cao, R. Gao and G. Su, Sol. Energy Mater. Sol. Cells, 2019, 191, 108–116 CrossRef CAS.
- K. Tang, Y. Zhang, Y. Shi, J. Cui, X. Shu, Y. Wang, Y. Qin, J. Liu, H. Tan and Y. Wu, Electrochim. Acta, 2020, 330, 135189 CrossRef CAS.
- Z. Wei, J. Xu, J. Hou, W. Zhou and S. Pu, J. Mater. Sci., 2006, 41, 3923–3930 CrossRef CAS.
- X. Fu, C. Jia, Z. Wan, X. Weng, J. Xie and L. Deng, Org. Electron., 2014, 15, 2702–2709 CrossRef CAS.
- L. Chen, H. Zhang, S. Liu, C. Sun, X. Hu and S. Zhou, Surf. Interface Anal., 2020, 52, 389–395 CrossRef CAS.
- C. Wang, X. Zhang, C. Shao, Y. Zhang, J. Yang, P. Sun, X. Liu, H. Liu, Y. Liu, T. Xie and D. Wang, J. Colloid Interf. Sci., 2011, 363, 157–164 CrossRef CAS PubMed.
- Z. Chen, J. Yang, X. Yang, Y. Zhao, J. Kang, F. Yang, Y. Zhang, M. Cheng, G. Wang and Q. Duanmu, Appl. Organomet. Chem., 2018, 32, e4356 Search PubMed.
- H. Zhou, W. Yao, G. Li, J. Wang and Y. Lu, Carbon, 2013, 59, 495–502 CrossRef CAS.
- J. Mathiyarasu, S. Senthilkumar, K. L. N. Phani and V. Yegnaraman, J. Nanosci. Nanotechnol., 2007, 7, 2206–2210 CrossRef CAS PubMed.
- J. M. Skowroński and T. Rozmanowski, J. Solid State Electrochem., 2013, 17, 949–960 CrossRef.
- C. C. Bof Bufon, J. Vollmer, T. Heinzel, P. Espindola, H. John and J. Heinze, J. Phys. Chem. B, 2005, 109, 19191–19199 CrossRef CAS PubMed.
- R. Hillman, S. J. Daisley and S. Bruckenstein, Phys. Chem. Chem. Phys., 2007, 9, 2379–2388 RSC.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- K. Xu, Q. Zhang, Z. Hao, Y. Tang, H. Wang, J. Liu and H. Yan, Sol. Energy Mater. Sol. Cells, 2020, 206, 110330 CrossRef CAS.
- B. Liu and E. S. Aydil, J. Am. Chem. Soc., 2009, 131, 3985–3990 CrossRef CAS PubMed.
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. C. Fitzmorris, C. Wang, J. Zhang and Y. Li, Nano Lett., 2011, 11, 3026–3033 CrossRef CAS PubMed.
- C. Zhang, Y. Xu, N. Wang, Y. Xu, W. Xiang, M. Ouyang and C. Ma, Electrochim. Acta, 2009, 55, 13–18 CrossRef CAS.
- B. Wang, J. Zhao, C. Cui, J. Liu and Q. He, Sol. Energy Mater. Sol. Cells, 2012, 98, 161–167 CrossRef CAS.
- J. Y. Go and S. I. Pyun, J. Solid State Electrochem., 2007, 11, 323–334 CrossRef CAS.
- N. Laskin, Physics, 2002, 66, 249–264 Search PubMed.
- K. Beauchard, J. Math. Pure Appl., 2005, 84, 851–956 CrossRef.
- P. Marquet, Prog. Phys., 2016, 12, 318–322 CAS.
- S. Oztemiz, G. Beaucage, O. Ceylan and H. B. Mark Jr., J. Solid State Electrochem., 2004, 8, 928–931 CrossRef CAS.
- C. Pozo-Gonzalo, J. A. Pomposo, J. A. Alduncin, M. Salsamendi, A. I. Mikhaleva, L. B. Krivdin and B. A. Trofimov, Electrochim. Acta, 2007, 52, 4784–4791 CrossRef.
- K. Fu, R. Lv, B. Na, S. Zou, R. Zeng, B. Wang and H. Liu, Polymer, 2019, 184, 121900 CrossRef CAS.
- T. Zhu, J. Zhou, Z. Li, S. Li, W. Si and S. Zhuo, J. Mater. Chem. A, 2014, 2, 12545–12551 RSC.
- G.-F. Cai, J.-P. Tu, D. Zhou, L. Li, J.-H. Zhang, X.-L. Wang and C.-D. Gu, CrystEngComm, 2014, 16, 6866–6872 RSC.
- Y. Chen, Z. Bi, X. Li, X. Xu, S. Zhang and X. Hu, Electrochim. Acta, 2017, 224, 534–540 CrossRef CAS.
- S. R. Bathe, M. S. Illa, R. Narayan and P. Basak, ChemNanoMat, 2018, 4, 203–212 CrossRef CAS.
- R.-T. Wen, M. A. Arvizu, M. Morales-Luna, C. G. Granqvista and G. A. Niklasson, Chem. Mater., 2016, 28, 4670–4676 CrossRef CAS.
- Z. Xie, Q. Zhang, Q. Liu, J. Zhai and X. Diao, Thin Solid Films, 2018, 653, 188–193 CrossRef CAS.
- A. Banerjee, S. Bhatnagar, K. K. Upadhyay, P. Yadav and S. Ogale, ACS Appl. Mater. Interfaces, 2014, 6, 18844–18852 CrossRef CAS PubMed.
- W. An, L. Liu, Y. Gao, Y. Liu and J. Liu, RSC Adv., 2016, 6, 75251–75257 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Kailing Zhou and
Hao Wang
,
Kailing Zhou and
Hao Wang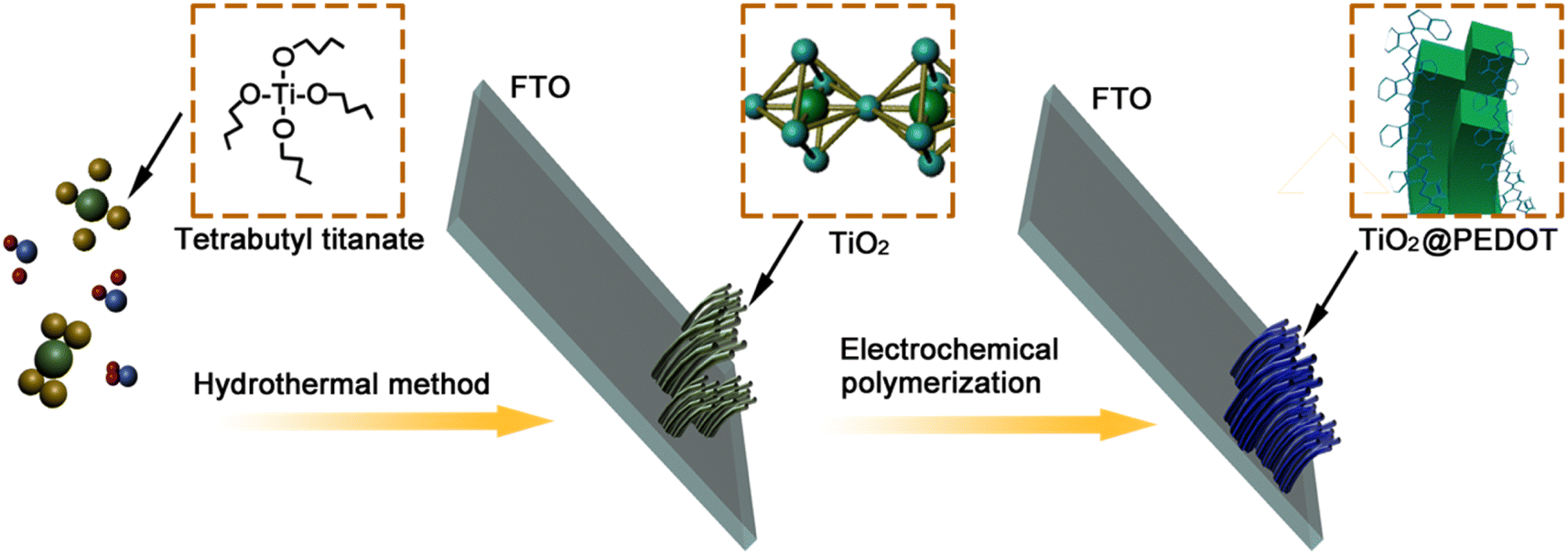

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i = log
i = log