
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stable Fe/CeO2 catalyst for the reverse water gas shift reaction in the presence of H2S†
Ryo Watanabe *a,
Fumiya Karasawaa,
Chikamasa Yokoyamaa,
Kazumasa Oshimab,
Masahiro Kishidab,
Masahiro Horic,
Yukinori Onoc,
Shigeo Satokawad,
Priyanka Vermaa and
Choji Fukuhara*a
*a,
Fumiya Karasawaa,
Chikamasa Yokoyamaa,
Kazumasa Oshimab,
Masahiro Kishidab,
Masahiro Horic,
Yukinori Onoc,
Shigeo Satokawad,
Priyanka Vermaa and
Choji Fukuhara*a
aDepartment of Applied Chemistry and Biochemical Engineering, Graduate School of Engineering, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu, Shizuoka 432-8561, Japan. E-mail: watanabe.ryo@shizuoka.ac.jp; fukuhara.choji@shizuoka.ac.jp
bDepartment of Chemical Engineering, Faculty of Engineering, Kyushu University, 744 Motooka Nishi-ku, Fukuoka-shi, Fukuoka 819-0395, Japan
cResearch Institute of Electronics, Shizuoka University, 3-5-1 Johoku, Naka-ku, Hamamatsu, Shizuoka 432-8561, Japan
dDepartment of Materials and Life Science, Faculty of Science and Technology, Seikei University, 3-3-1 Kichijoji Kitamachi, Musashino-shi, 180-8633, Tokyo, Japan
First published on 12th April 2023
Abstract
This study focused on evaluating the catalytic properties for the reverse water gas shift reaction (RWGS: CO2 + H2 → CO + H2O ΔH0 = 42.1 kJ mol−1) in the presence of hydrogen sulfide (H2S) over a Fe/CeO2 catalyst, commercial Cu–Zn catalyst for the WGS reaction (MDC-7), and Co–Mo catalyst for hydrocarbon desulfurization. The Fe/CeO2 catalyst exhibited a relatively high catalytic activity to RWGS, compared to the commercial MDC-7 and Co–Mo catalysts. In addition, the Fe/CeO2 catalyst showed stable performance in the RWGS environment that contained high concentrations of H2S. The role of co-feeding H2S was investigated over the Fe/CeO2 catalyst by the temperature programmed reaction (TPR) of CO2 and H2 in the presence of H2S. The result of TPR indicated that the co-feeding H2S might enhance RWGS performance due to H2S acting as the hydrogen source to reduce CO2.
1. Introduction
Most of the world's natural gas resources are in the form of sour gas, which contains high concentrations of hydrogen sulfide (H2S) and carbon dioxide (CO2), with typical compositions of 0–30 vol% H2S and 0–80 vol% CO2.1–3 Sour gas is widely distributed worldwide, including in the Middle East, Canada, Northern Europe, and China. High concentrations of H2S (over 30 vol%) is contained in large gas fields in Saudi Arabia, Abu Dhabi, and offshore areas in Iran.4 Moreover, biogas, a renewable energy source that has attracted much attention in recent years, contains a relatively high concentration of H2S.5–7 In order to use the hydrocarbons in natural gas or methane in biogas as fuel, pollutants (H2S and CO2) that exist beyond allowable limits must be removed. For example, in natural gas, the heavy hydrocarbons are condensed and then passed through a scrubbing unit typically composed of liquid amine-based adsorbents to remove the sour gas components (H2S and CO2), in a process called “sweetening”.8 However, the liquid amine sorbents cannot control the H2S/CO2 selectivity because the concentration of these contaminants varies depending on the source of the natural gas.9,10 In this regard, acid gas remediation is costly, and it is often uneconomical to extract and use natural gas resources that contain high concentrations of acid gases. In many cases, most of the recovered CO2 and H2S is buried in landfills.11,12 This has necessitated the exploration of new strategies for the active use of C and S-containing substances (CO2 and H2S) to address the critical issues of global climate change and sustainability. H2S, one of the sour gas components, is known to cause catalytic deactivation; hence, its academic value and economic benefits are expected to be significant if S-resistant catalysts can be used to directly convert the CO2 in sour natural gas into value-added products.There has been extensive research on high-performance catalysts for CO2 conversion in the presence of H2S. Sharma et al. reported that MoS2 was effective for CO2 hydrogenation under relatively high concentrations (<2500 ppm) of H2S.13 They suggested that H2S acts as a “conduit” for the active hydrogen required for the hydrogenation of CO2, contributing to the enhancement of activity. Guilera et al. investigated the S resistance of Ni/Al2O3 doped with rare-earth oxides of CeO2 and La2O3 for the low-temperature hydrogenation of CO2.14,15 They reported that even in an atmosphere containing low concentrations (0.4 ppm) of H2S, the Ni-based catalysts were not deactivated by sintering or S poisoning. To the best of our knowledge, there have only been a few reports on S-resistant CO2 conversion catalysts. Our group studied the dehydrogenation of alkanes in the presence of high concentrations of H2S (several tens of vol%), and found that Fe-based catalysts showed high activity and high selectivity with a stable performance.16,17
Therefore, we focused on Fe as a component as it is effective in water gas shift (WGS) reactions, and CeO2 as the support because of its excellent functionality for CO2 conversion.18–22 By employing an Fe-supported CeO2 (Fe/CeO2) catalyst in the reverse water gas shift (RWGS) reaction, a highly stable performance is expected even when the reaction atmosphere contains a high concentration of H2S. In this study, the characteristics of the RWGS reaction using the Fe/CeO2 catalyst with/without H2S co-feeding were compared with those of the reaction using a commercial Cu–ZnO catalyst (MDC-7) and Co–Mo-based desulfurization catalyst, which are known to exhibit catalytic performance even in a high concentration of H2S in the atmosphere. In addition, the stability of the Fe/CeO2 catalyst was investigated over a reaction time of 12 h while co-feeding H2S. The structure of the catalyst after the reaction was qualitatively investigated by X-ray diffraction (XRD) and STEM-EDX mapping to estimate the structure of the Fe and Ce component phases in the RWGS atmosphere in the presence of H2S. Furthermore, to identify the effect of H2S on the RWGS, the reaction mechanism was estimated by the temperature programmed reaction (TPR) with CO2 and H2 in the presence of H2S.
2. Experimental
2.1. Preparation and characterization of the catalyst
The Fe/CeO2 catalyst was prepared by an impregnation method. The CeO2 (JRC-CEO-2) catalyst support was provided by the Catalysis Society of Japan. First, 2.0 g of CeO2 was immersed in 30 mL of distilled water for 6 h, following which 1.624 g of Fe(NO3)3·9H2O was added and stirred for an additional 2 h. The Fe-based catalyst was then calcined at 500 °C for 1 h. The loading amount of Fe was 10 wt%. MDC-7 (Clariant Catalysts K.K.), with 34 wt% Cu, was used for comparison with the Fe/CeO2 catalyst. The reference Co–Mo-based catalyst was prepared by a previously reported method.23 Some promoters of Zn and P components were included in the reference Co–Mo-based catalyst. The crystal structures of the Fe species and Ce species in the Fe/CeO2 catalyst were determined via XRD (Ultima IV, Rigaku) using a Cu Kα radiation source. The elemental mapping of the Fe/CeO2 catalyst after the RWGS with H2S co-feeding was performed by STEM-EDX (JEM-2100F, JEOL, Japan). The element maps of the Fe, Ce, S, and O components of the Fe/CeO2 catalyst were collected for investigating the dispersion of the Fe components and coverage of the S atoms on the catalyst.2.2. Evaluation of catalytic performance
Catalytic reaction tests were conducted at ambient pressure in a fixed bed reactor. After placing the catalyst in the center of the reaction tube, H2 reduction (100 vol%) was carried out at 500 °C for 1 h. After purging the reduction gas, the reaction temperature was set at 400 to 600 °C, the catalyst amount was 75 mg, and the reaction gas with the composition CO2/H2/H2S/He was supplied at 40/40/x/20 − x mL min−1 (x = 0–10). The gas was quantified by an FID gas chromatograph (GC-2014, Shimadzu, Japan) and FPD gas chromatograph (GC-2030, Shimadzu, Japan).2.3. Temperature programmed reaction method
To clarify the reaction mechanism of the RWGS, TPR analysis was carried out with the following steps. First, the Fe/CeO2 catalyst of 100 mg was pretreated with the gas composition of H2/Ar = 25/5 mL min−1 at 500 °C. After 1 h of the pretreatment, the reduced gas was purged by He, following the catalyst temperature was decreased to 40 °C. Then, CO2 with co-feeding H2S (gas composition: CO2/H2S/Ar/He = 1/1/5/93 mL min−1) was supplied to the catalyst with increasing temperature from 40 °C to 800 °C. After that, the catalyst temperature was decreased to 40 °C, again. Subsequently, H2 with co-feeding H2S (gas composition: H2/H2S/Ar/He = 1/1/5/93 mL min−1) was supplied with increasing temperature from 40 °C to 800 °C. The produced gases in these operations were monitored by a quadrupole mass spectrometer (QMS, HIDEN ANALYTICAL, UK).3. Results and discussion
3.1. Effect of H2S on RWGS performance
The performance of the Fe/CeO2 catalyst was evaluated and compared with those of the reference MDC-7 and Co–Mo catalysts. The Co–Mo catalyst has been reported to be highly active in WGS reactions involving H2S. Hence, this catalyst was used as a desulfurization catalyst, and its RWGS performance was evaluated. Fig. 1 shows the effect of the H2S concentration on the CO2 conversion of the Fe/CeO2, and commercial MDC-7 and Co–Mo catalysts at various reaction temperatures. The RWGS reaction selectively proceeded since the selectivity to CO was ca. 100% under all conditions. For the Fe/CeO2 catalyst, the CO2 conversion increased with increasing reaction temperature, and reached 29.6% at 600 °C. Compared to its RWGS performance with no H2S, there was no significant decrease in the catalytic performance at H2S concentrations of 1, 5 and 10%. In other words, the performance of the Fe/CeO2 catalyst was not adversely affected by H2S. There has been no report on the catalyst for RWGS almost not degrading under a high concentration of H2S. The performance of the MDC-7 catalyst was superior to that of the Fe/CeO2 catalyst. However, there was no significant increase in activity with increasing temperature, which is possibly attributed to the sintering and oxidation of the active Cu component by the water vapor produced during the progress of the RWGS reaction.24 In addition, the co-feeding of H2S greatly decreased the RWGS activity on the MDC-7 catalyst, where the CO2 conversion was approximately 1.7% at 600 °C under 1 vol% H2S. Interestingly, the degradation of the Cu-based catalyst progressed with the coexistence of H2S, while that of the Fe/CeO2 catalyst was almost negligible. For the Co–Mo catalyst, in a H2S-free atmosphere, the WGS performance was low, but with increasing H2S concentration, improved. In particular, the highest WGS performance was observed under 10 vol% H2S. Although the Co–Mo catalyst was effective under a high concentration of H2S, the Fe/CeO2 catalyst showed better performance under a low concentration of H2S, which indicated that the Fe/CeO2 catalyst could be applicable to the biogas. | ||
| Fig. 1 Effect of H2S co-feeding on the RWGS performance of (a) Fe/CeO2, (b) MDC-7, and (c) Co–Mo catalysts. The reaction conditions were as follows: the catalyst amount was 75 mg, gas composition was CO2/H2/H2S/He = 40/40/x/20 − x mL min−1 (x = 0–10), and reaction temp. ranged from 400 to 600 °C. Note that sel. of CO was 100%. | ||
To evaluate the durability of the Fe/CeO2 and Co–Mo catalysts, long-term RWGS reaction was tested under 1 vol% of H2S. Moreover, to determine the role of the Fe component and CeO2 support, RWGS tests were carried out over the Fe/SiO2 and bare CeO2 catalysts. Fig. 2 shows the CO2 conversion over the Fe/CeO2 and Co–Mo catalysts with the reaction time. When H2S was supplied, the CO2 conversion was high for 12 h over the Fe/CeO2 catalyst. Since the products mostly contained CO, the RWGS selectively proceeded over the Fe/CeO2 catalyst. Compared with the RWGS performance of the Co–Mo catalyst, that of the Fe/CeO2 catalyst was superior, with high stability. The Fe/SiO2 and bare CeO2 catalysts were less active than the Fe/CeO2 catalysts, indicating that both, Fe and CeO2, are required for the RWGS when co-feeding H2S. Since CeO2 had a low activity and Fe/SiO2 exhibited almost no activity, it was assumed that there might be an interaction between the Fe and CeO2 components.
 | ||
| Fig. 2 Performances of Fe/CeO2, Co–Mo, Fe/SiO2, and CeO2 catalysts for the RWGS with 1 vol% H2S at 600 °C. Note that sel. of CO was 100%. | ||
3.2. Structure characterization
To investigate the structure of the Fe/CeO2 catalyst, XRD and STEM-EDX analyses were performed on the catalysts after the RWGS with H2S co-feeding (Fig. 3(a) and (b), respectively). From Fig. 3(a), regardless of the change in the H2S concentration, no diffraction peaks of the Fe component are observed and only the Ce component is detected. Further, only CeO2 is detected when the H2S concentrations are 0 vol% and 1 vol%, and the peak intensity of CeO2 decreases with increasing H2S supply. When the H2S concentration is further increased from 1 vol% to 10 vol%, the CeO2 phase transforms into oxysulfates of Ce2O2S and Ce2OxSy. From the atomic mapping of Fig. 3(b), the Fe species are dispersed on the CeO2 support; however, there are some areas where the Fe species are heavily segregated. The Fe mapping is visually similar to the S mapping, which suggests the absorption of S species on the Fe species in the catalyst. ESI 1† shows the thermodynamically stable phases of the Fe component from 25 °C to 700 °C in the reaction simulated gas atmosphere of CO and H2, and H2S. Since Fe0.877S (major phase) and FeS (minor phase) are the stable phases of Fe species at the reaction temperature from 25 °C to 700 °C, the S identified from the STEM-EDX mapping is assumed to be incorporated in a sulfide phase. | ||
| Fig. 3 (a) XRD pattern and (b)–(g) STEM-EDX mapping of the Fe/CeO2 catalyst after RWGS with H2S co-feeding. | ||
Fig. 4 shows the Raman spectra of the Fe/CeO2 catalyst after RWGS with different hydrogen sulfide concentrations (1 vol% and 10 vol%). Some peaks attributed to pyrrhotite (FeSx) were observed at 213 cm−1, 277 cm−1, and 391 cm−1 for the Fe/CeO2 catalyst after RWGS containing 1 vol% and 10 vol% of H2S.26 The results are roughly in agreement with those inferred from thermodynamic calculations, as shown in ESI 1.†
 | ||
| Fig. 4 Raman spectra of the Fe/CeO2 after RWGS with H2S co-feeding. | ||
3.3. Role of H2S
To clarify the role of H2S in the Fe/CeO2 catalyst on the RWGS reaction, a TPR analyses were performed. Fig. 5(a) and (b) respectively show the profile of product during the CO2-TPR and H2-TPR analyses. The products were detected by a quadrupole mass spectrometer, where the temperature was increased by 10 °C min−1, along with supplying 1 vol%-CO2 or 1 vol%-H2 in the presence of 1 vol%-H2S to the Fe/CeO2 catalyst. The shaded area indicated the amount change of each gas; positive area represents generation, and negative area represents consumption. By flowing CO2 to the Fe/CeO2 catalyst, CO is observed to form at from 180 °C to 350 °C. At the same temperature of the formation of CO, the production of H2 and H2O and the consumption of H2S were observed. The above phenomena can be understood as follows; H2 generated by sulfurization of the Fe/CeO2 catalyst is thought to have reduced CO2 to form CO. Similarly, at temperatures above 350 °C, H2S was consumed to generate H2, and CO2 was reduced to CO. The reason for the consumption of H2S and CO2 in both low (180–350 °C) and high temperature (T > 350 °C) regions is thought to be due to the formation of the lattice sulfide ion (S2−) by feeding H2S on both the surface and the bulk of the catalyst. As confirmed by Fig. 5(b), H2 was consumed at around 250 °C by the reaction with lattice S2− (H2 + S2− → H2S + VS). In this experiment, H2-TPR is performed after CO2-TPR; the regeneration of lattice S2− from H2S during CO2-TPR was considered to contribute the formation of H2S at low temperature from 250 to 400 °C. Furthermore, the formation of H2O can be observed at temperatures above 500 °C. The result meant that a lattice oxygen was generated by catalyst oxidation (CO2 + Vox → CO + O2−) in CO2-TPR, and followed by the consumption of generated lattice oxygen by H2 to produce H2O.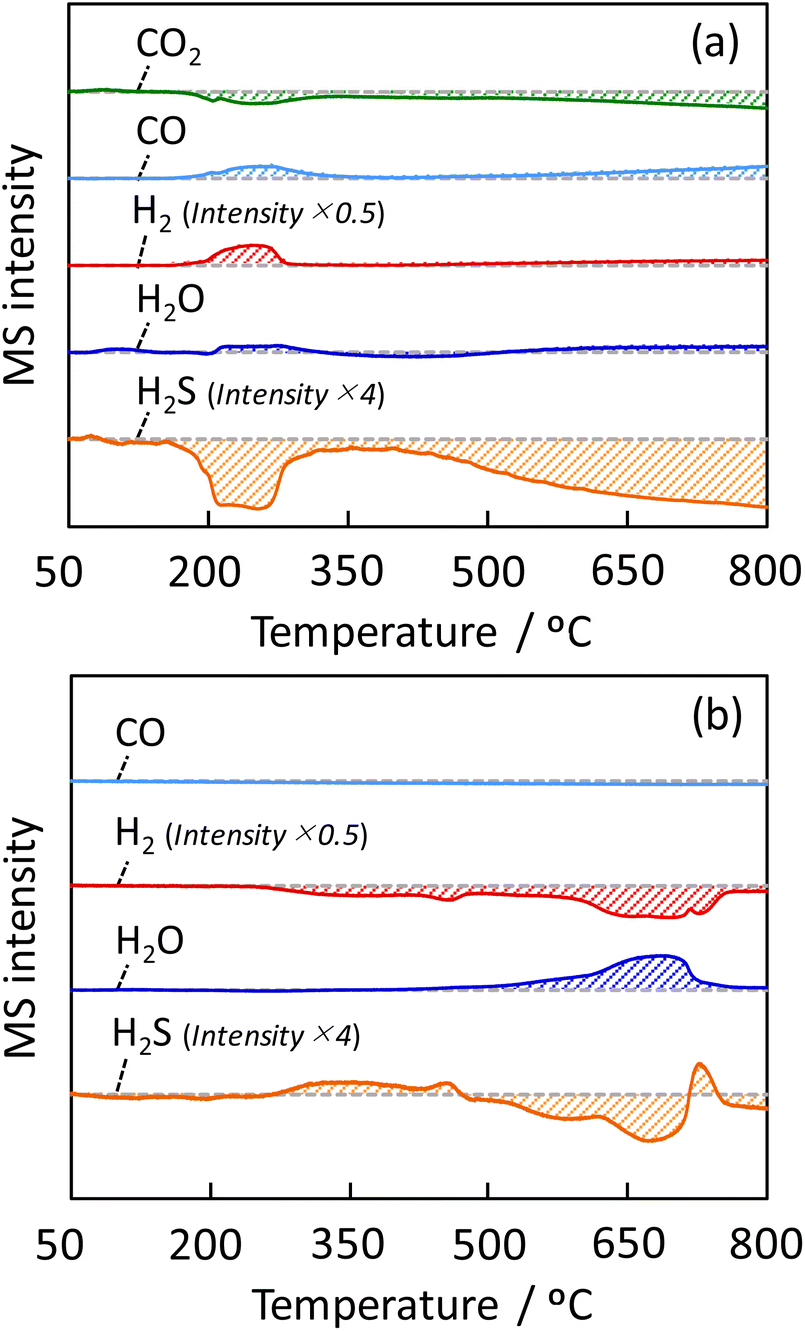 | ||
| Fig. 5 Products behavior in TPR with (a) CO2 and (b) H2 in the presence of H2S over the Fe/CeO2 catalyst. The H2 and H2S intensities were adjusted because the sensitivity was quite different from the other products. | ||
Fig. 6 exhibits the ESR signal of oxygen and/or sulfur vacancy after TPR with CO2 and H2. The vacancy was confirmed at g = 2.03 before the reaction, due formation of lattice vacancy in CeO2.25 After TPR with CO2 and H2 with H2S, the peak at g = 2.03 was not also confirmed, indicating that the vacancy was replenished as S2−/O2− by H2S/CO2. Based on these data, H2S might act as a sulfidizing-agent in the Fe/CeO2 catalyst, meaning that generated lattice vacancy was replenished rapidly by S2−. To confirm this assumption, H2S was supplied to the Fe/CeO2 catalyst before the reaction. It is clear from this figure that the peak at g = 2.03 originating from defects has almost disappeared. The result suggests that the following reaction (H2S + V → S2− + H2) proceeds and restores lattice S2− to the vacancy.
 | ||
| Fig. 6 ESR signal of the Fe/CeO2 catalyst before TPR (after H2 reduction), after H2S feeding, after CO2-TPR and H2-RPR. | ||
The role of H2S was discussed from the viewpoint of reaction mechanism. The reaction pathway envisioned is that CO2 is activated on the surface of the CeO2 support27 and reduced to CO by reacting with hydrogen activated by FeSx. The coexisting H2S is also expected to regenerate lattice S2− to produce activated H atoms, and this generated hydrogen species further activates CO2. In other words, the generated H2 from H2S on the catalyst could work as hydrogen source to CO2 reduction. In the case of Fe/CeO2, the activity does not change much when the gas-phase H2S concentration is changed because the bulk diffusion of lattice S2− is the rate-limiting factor. In the Co–Mo system, on the other hand, the performance is greatly enhanced by the H2S concentration, so we expect that H2S → H2 (active H species) +S2− at the catalyst surface is the rate-limiting factor.
4. Conclusions
The catalytic properties of Fe/CeO2 and MDC-7 catalysts for the RWGS reaction with H2S co-feeding were evaluated. MDC-7 showed a relatively high activity in the low-temperature range, but underwent significant deactivation during the RWGS with H2S co-feeding. The Fe/CeO2 catalyst similarly showed a relatively high activity for the RWGS, and showed a stable activity for 12 h in the reaction atmosphere containing H2S. The result TPR with CO2 and H2 indicated that co-feeding H2S might act as the hydrogen source for RWGS over the Fe/CeO2 catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Uncharted Territory Challenge 2050, New Energy and Industrial Technology Development Organization (NEDO), Japan.References
- W. F. J. Burgers, P. S. Northrop, H. S. Kheshgi and J. A. Valencia, Energy Procedia, 2011, 4, 2178–2184 CrossRef.
- W. Taifan and J. Baltrusaitis, Catal. Sci. Technol., 2017, 7, 2919–2929 RSC.
- N. W. Chakroun and A. F. Ghoniem, Int. J. Greenhouse Gas Control, 2015, 41, 163–173 CrossRef CAS.
- A. Hofmann, W. van Strien, and R. Malekzadeh, in, Abu Dhabi International Petroleum Exhibition & Conference, OnePetro, 2017 Search PubMed.
- Y. Belmabkhout, G. De Weireld and A. Sayari, Langmuir, 2009, 25(23), 13275–13278 CrossRef CAS PubMed.
- J. I. Huertas, N. Giraldo and S. Izquierdo, Mass transfer in chemical engineering processes, 2011 Search PubMed.
- P. Iovane, F. Nanna, Y. Ding, B. Bikson and A. Molino, Fuel, 2014, 135, 352–358 CrossRef CAS.
- W. Y. Lee, S. Y. Park, K. B. Lee and S. C. Nam, Energy Fuels, 2020, 34(2), 1992–2000 CrossRef CAS.
- K. Huang, X. M. Zhang, X. B. Hu and Y. T. Wu, AIChE J., 2016, 62, 4480–4490 CrossRef CAS.
- K. Huang, D. N. Cai, Y. L. Chen, Y. T. Wu, X. B. Hu and Z. B. Zhang, ChemPlusChem, 2014, 79(2), 241–249 CrossRef CAS PubMed.
- C. Khan, R. Amin and G. Madden, J. Pet. Explor. Prod. Technol., 2013, 3, 55–60 CrossRef CAS.
- A. Y. Kaita, O. Ogolo, X. Wu, I. Mohammed and E. A. Akpan, J. Pet. Explor. Prod. Technol., 2020, 10, 1575–1589 CrossRef CAS.
- L. Sharma, R. Upadhyay, S. Rangarajan and J. Baltrusaitis, ACS Catal., 2019, 9, 10044–10059 CrossRef CAS.
- J. Guilera, J. D. Valle, A. Alarcón, J. A. Díaz and T. Andreu, J. CO2 Util., 2019, 30, 11–17 CrossRef CAS.
- A. Alarcón, J. Guilera, R. Soto and T. Andreu, Appl. Catal., B, 2020, 263, 118346 CrossRef.
- R. Watanabe, N. Hirata, Y. Yoda, Y. Fushimi and C. Fukuhara, J. Jpn. Pet. Inst., 2020, 63(4), 228–237 CrossRef CAS.
- R. Watanabe, N. Hirata, K. Miura, Y. Yoda, Y. Fushimi and C. Fukuhara, Appl. Catal., A, 2019, 587, 117238 CrossRef CAS.
- Y. Sekine, T. Chihara, R. Watanabe, Y. Sakamoto, M. Matsukata and E. Kikuchi, Catal. Lett., 2010, 140(3), 184–188 CrossRef CAS.
- R. Watanabe, Y. Sakamoto, K. Yamamuro, S. Tamura, E. Kikuchi and Y. Sekine, Appl. Catal., A, 2013, 457, 1–11 CrossRef CAS.
- C. Fukuhara, K. Hayakawa, Y. Suzuki, W. Kawasaki and R. Watanabe, Appl. Catal., A, 2017, 532, 12–18 CrossRef CAS.
- S. Ratchahat, M. Sudoh, Y. Suzuki, W. Kawasaki, R. Watanabe and C. Fukuhara, J. CO2 Util., 2018, 24, 210–219 CrossRef CAS.
- C. Fukuhara, A. Kamiyama, M. Itoh, N. Hirata, S. Ratchahat, M. Sudoh and R. Watanabe, Chem. Eng. Sci., 2020, 219, 115589 CrossRef CAS.
- N. Nakajima, Y. Seriguchi, M. Kato, M. Yoshinari, T. Yoshida and K. Watanabe, J. Jpn. Pet. Inst., 2013, 56(6), 388–394 CrossRef CAS.
- C.-S. Chen, W.-H. Chen and S.-S. Lin, Appl. Catal., A, 2004, 257, 97–106 CrossRef CAS.
- X. Chen, S. Zhan, D. Chen, C. Hea, S. Tian and Y. Xiong, Appl. Catal., B, 2021, 286(5), 119928 CrossRef CAS.
- Y. El Mendili, A. Abdelouas, H. El Hajja and J.-F. Bardeau, RSC Adv., 2013, 3, 26343 RSC.
- F. Cao, Y. Xiao, Z. Zhang, J. Li, Z. Xia, X. Hu, Y. Ma and Y. Qu, J. Catal., 2022, 414, 25–32 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01323e |
| This journal is © The Royal Society of Chemistry 2023 |