DOI:
10.1039/D3RA01216F
(Paper)
RSC Adv., 2023,
13, 15126-15131
Red, green, and blue radio-luminescent polymer dots doped with heteroleptic tris-cyclometalated iridium complexes†
Received
23rd February 2023
, Accepted 11th May 2023
First published on 17th May 2023
Abstract
In this study, we synthesized radioexcitable luminescent polymer dots (P-dots) doped with heteroleptic tris-cyclometalated iridium complexes that emit red, green, and blue light. We investigated the luminescence properties of these P-dots under X-ray and electron beam irradiation, revealing their potential as new organic scintillators.
Introduction
Radiation is widely used in medicine, industry, and scientific research.1,2 For example, it is used in medical applications such as imaging and radiotherapy.3 Among these medical applications, linear accelerators are used to deliver radiation, including hard X-rays and electron beams, to visualize and induce cell death in targeted areas.4 The advantages of using X-rays and electron beams include their high biological permeability, which allows non-invasive imaging, high resolution and is already used in many clinical settings.5 There is substantial interest in using these technologies to develop novel imaging and therapeutic modalities by using photo-functional nanomaterials. In fact, recent studies have demonstrated the potential applications of radiation for photodynamic therapy6 and optogenetics7,8 by using photo-functional materials. Regarding photo-functional (e.g., radio-luminescent) nanomaterials for photo-induced therapeutic applications, scintillators are commonly used as the light-emitting materials.9 We and other research groups have developed radio-luminescent nanomaterials from inorganic and organic building blocks.10–14 For instance, we previously reported ∼500 nm yellow emission from polymer dots (P-dots) by using hard X-ray irradiation at a 50-kVp tube voltage with a homoleptic tris-cyclometalated iridium complex.15 However, the emission color and excitation radiation sources remain limited; thus, it is crucial to further develop these materials in terms of the emission color and radiation source variation.16 On the other hand, iridium complexes are characterised by changing their luminescent colour and other properties by converting their molecular structure like heteroleptic iridium complexes in various ways, and it was thought that multicolourisation might be possible by changing their structure, compared to organic scintillators such as anthracene. Here, we have developed iridium complex doped P-dots that emit over a range of wavelengths and investigated their radioluminescence by using hard X-rays and an electron beam as excitation sources.
Results and discussion
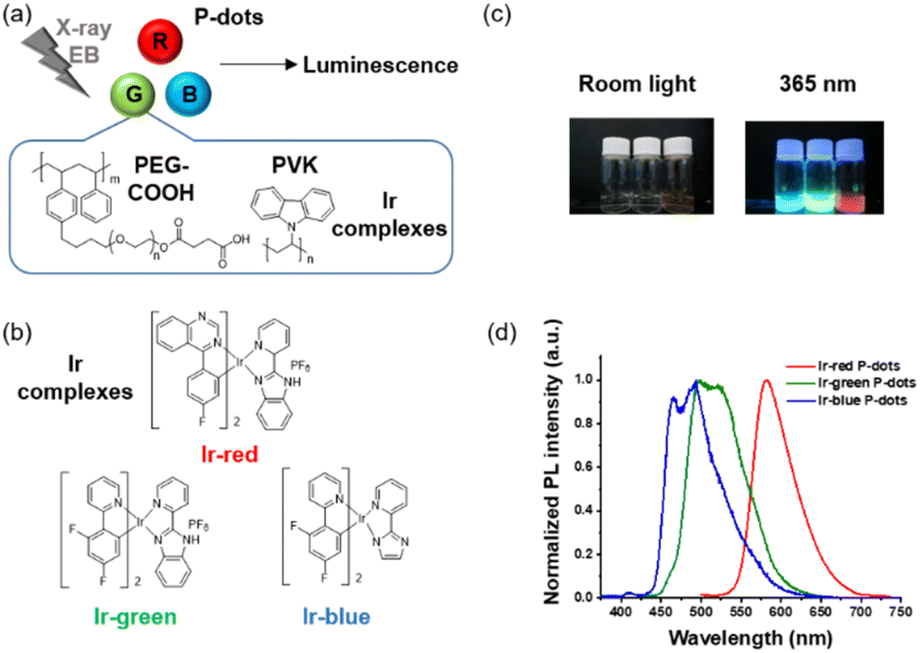
We synthesized three derivatives of heteroleptic tris-cyclometalated iridium complexes to develop three color variations (red, green, and blue) of iridium complex doped P-dots (Fig. 1a and b). P-dots are organic nanoparticles composed of polymers and no clear precipitation was observed after synthesis,17 confirming that their stability is equivalent to that of carbon dots, which have been synthesised frequently recently.18 Basically, we used three heteroleptic iridium complexes (Ir-red, Ir-green, and Ir-blue) with two cyclometalating bidentate ligands (C^N) and one bidentate ancillary ligand (N^N), to change the emission color to three primary colors, red, green, and blue. Specifically, Ir-red has fluorophenyl quinazoline on the cyclometalating ligands with 2-(2-pyridyl)benzimidazole (pybi) for the ancillary ligand. Ir-green and Ir-blue consist of two phenylpyridines with two fluorine functional groups as electron-withdrawing groups on the cyclometalating ligands to stabilize the HOMO level with pybi and 2-(1H-imidazol-2-yl)pyridine (pyim) as the ancillary ligand of Ir-green and Ir-blue, respectively, to change their colors. Compared with pyim, pybi is a π-extended ancillary ligand for stabilizing the LUMO level. Ir-blue is neutral, and that Ir-green and Ir-red are cationic iridium complexes, depending on the synthetic conditions (see ESI†). In addition to the emitter design, heteroleptic tris-cyclometalated iridium complexes also meet the following solubility requirement for synthesizing iridium complex doped P-dots. The iridium complexes must be soluble in organic solvents such as tetrahydrofuran (THF), but not in water. Subsequently, we selected three appropriate heteroleptic tris-cyclometalated iridium complexes. Detailed synthetic schemes and structural characterizations of the iridium complexes are provided in Fig. S1–S8.†
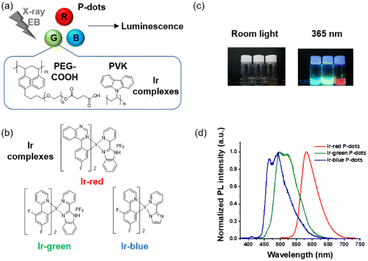 |
| | Fig. 1 (a) Schematic of radiation-excited luminescence from P-dots doped with iridium complexes. (b) Chemical structures of iridium complexes. (c) Photograph of photoluminescence from P-dot solution under room light and 365 nm UV light irradiation. (d) Normalized photoluminescence spectra of P-dot solution excited at 365 nm. | |
First, we characterized the photochemical properties of these monomeric iridium complexes in organic solvents. The absorption spectra at room temperature are broad in the UV and visible regions (Fig. S9 and Table S1†). The photoluminescence spectra excited at 365 nm at room temperature are shown in Fig. S10.† The emission maxima and absolute quantum yields of Ir-red, Ir-green, and Ir-red are also listed in Tables S1 and S2.† In the presence of dissolved oxygen, the photoluminescence was quenched and the absolute photoluminescence quantum yields dramatically decreased to one-fifth, one-ninth, and one-thirtieth, respectively, for Ir-red, Ir-green, and Ir-blue (Fig. S10†). We measured the photoluminescence lifetimes of the iridium complexes in the absence and presence of molecular oxygen (Fig. S11 and Table S3†). The quenching rate constants were close to the diffusion-controlled limits of the solvents, suggesting that the triplet excited state is quenched within 1 μs. To demonstrate the quenching of the triplet excited state in Ir monomer complexes, we elucidated the triplet excited state of Ir monomer complexes in toluene by using nanosecond pulse radiolysis, that is a radiation chemistry technique for observing the triplet excited state of organic molecules in aromatic solvents.
The transient absorption spectra of the iridium complexes during pulse radiolysis in toluene are shown in Fig. S12.† The absorption maxima at the triplet excited state were at 510, 470, and 440 nm for Ir-red, Ir-green, and Ir-blue, respectively. We also investigated the molecular-oxygen-quenching characteristics of the triplet state by laser flash photolysis (Fig. S13†). We observed quenching of the triplet excited state in Ir monomer complexes in organic solvents in accordance with the photoluminescence lifetime measurements (Table S4†).
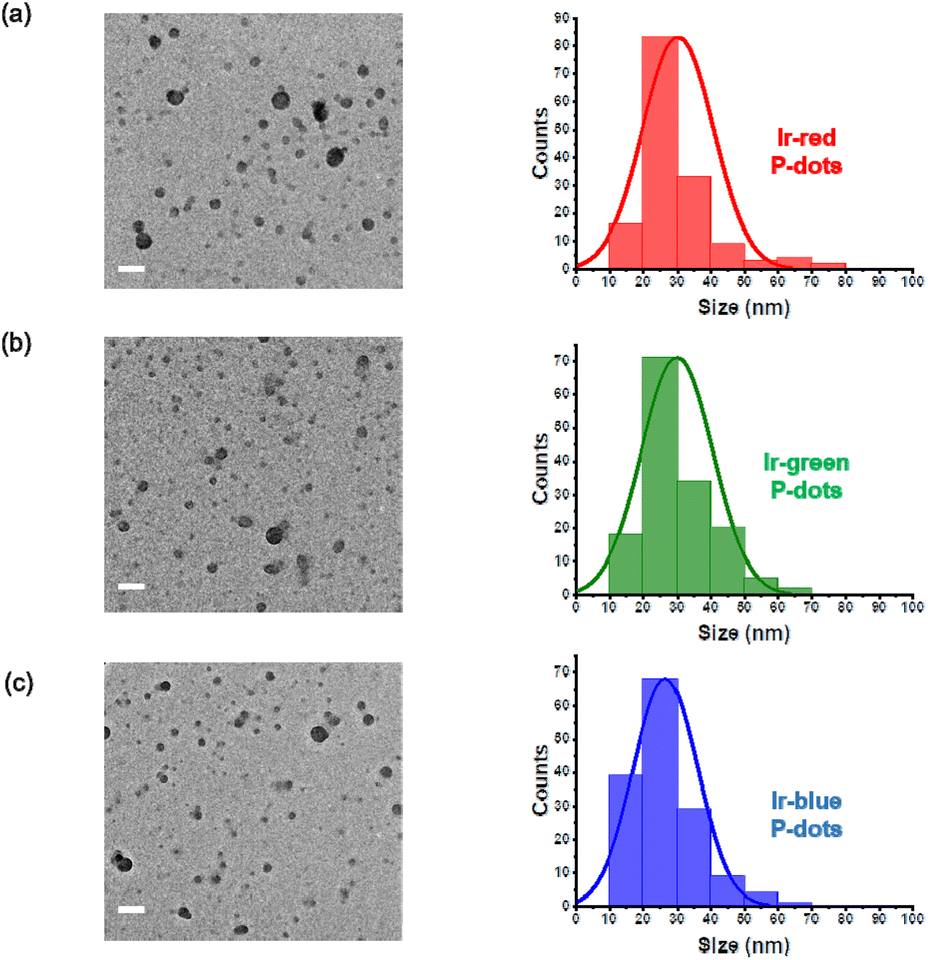
Second, we performed structural characterizations by dynamic light scattering (DLS), zeta potential measurements, and transmission electron microscopy (TEM) (Fig. 2, Tables S5 and S6†). DLS measurements indicate hydrodynamic diameters of approximately 100 nm for the three P-dots prepared in this study (Table S5†). The zeta potential of the P-dots was approximately −20 mV, indicating the negatively charged outer surface of the P-dots because of the carboxyl groups of the amphiphilic PEG polymers (Table S5†). We also observed TEM images to examine their core structures (Fig. 2). The average sizes of the P-dots are listed in Table S6.† A rough breakdown of the compositional breakdown from the structure results estimated that approximately 5000 Ir complexes and 100 polymers (PVG and PEG together) are doped per particle. The aforementioned results indicate that we successfully synthesized the P-dots by co-precipitation.
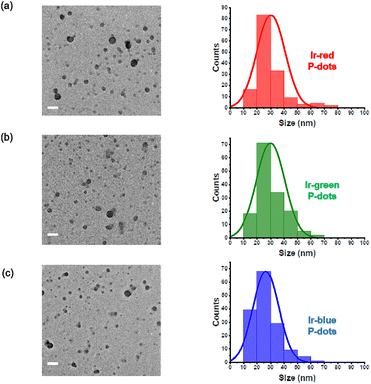 |
| | Fig. 2 TEM images and histogram of P-dots sizes. (a) Ir-red P-dots, (b) Ir-green P-dots, and (c) Ir-blue P-dots. Scale bar is 100 nm. | |
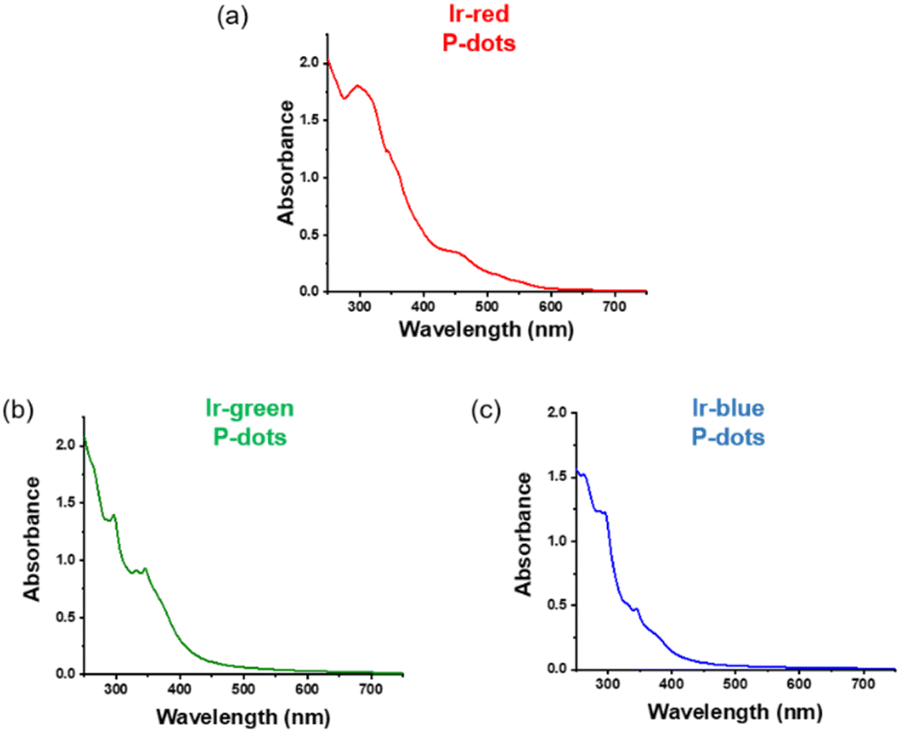
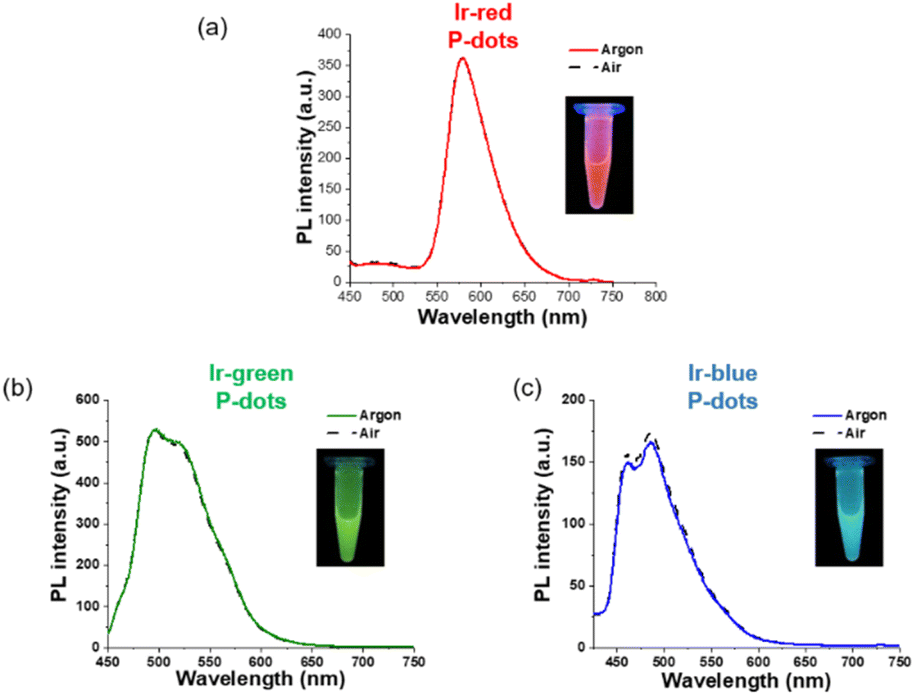
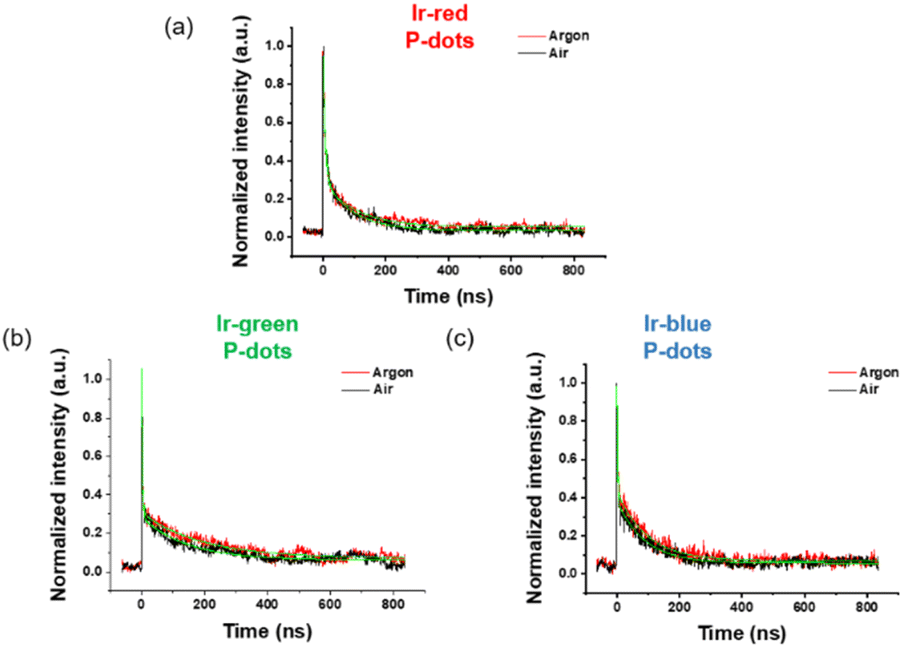
Third, we investigated photochemical properties of P-dots. The photoluminescence and absorption spectra in P-dots are shown in Fig. 1c, d, 3 and 4. The photoluminescence colors of P-dots were, respectively, red, green, and blue for Ir-red P-dots, Ir-green P-dots, and Ir-blue P-dots, respectively, upon 365 nm light irradiation (Fig. 1c). The photoluminescence quantum yields upon UV light irradiation are summarized in Table S2.† The values were smaller than those of corresponding monomeric iridium complexes, but independent of dissolved molecular oxygen. This finding is important for subsequent imaging. We measured the photoluminescence lifetimes of the P-dots in the absence and presence of molecular oxygen (Fig. 5 and Table S7†). P-dots exhibited photoluminescence lifetime shorter than the corresponding iridium monomeric complexes, probably because of aggregation induced quenching.19 We also conducted laser flash photolysis by the randomly interleaved pulse train (RIPT) method with a picosecond laser to further investigate the triplet excited state lifetime (Fig. S14†).20 The lifetime of the triplet excited state was also much shorter than that of the Ir monomer complexes, whereas we were unable to measure Ir-red P-dots because of an insufficient detection limit (Table S8†). Regardless, we will conduct a detailed study of this quenching in the future. These results reconfirm that oxygen has little effect on luminescence in P-dots, which is favorable for imaging.
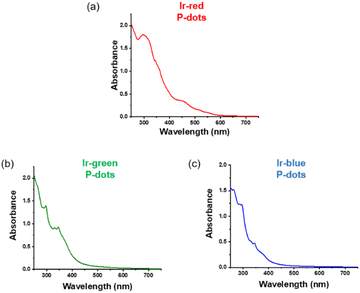 |
| | Fig. 3 Absorption spectra of iridium complexes doped P-dots. (a) Ir-red P-dots, (b) Ir-green P-dots, and (c) Ir-blue P-dots. | |
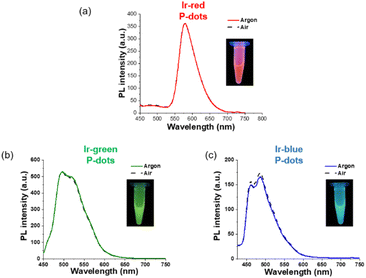 |
| | Fig. 4 PL spectra of iridium complex doped P-dots. (a) Ir-red P-dots, (b) Ir-green P-dots, and (c) Ir-blue P-dots, under argon (solid) and air (dashed). The inset shows a photograph of the luminescence of each sample under UV irradiation. | |
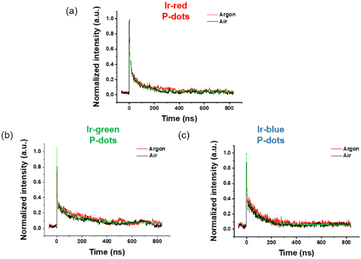 |
| | Fig. 5 Lifetime of iridium complex doped P-dots. (a) Ir-red P-dots, (b) Ir-green P-dots, and (c) Ir-blue P-dots under air (black) and argon (red). | |
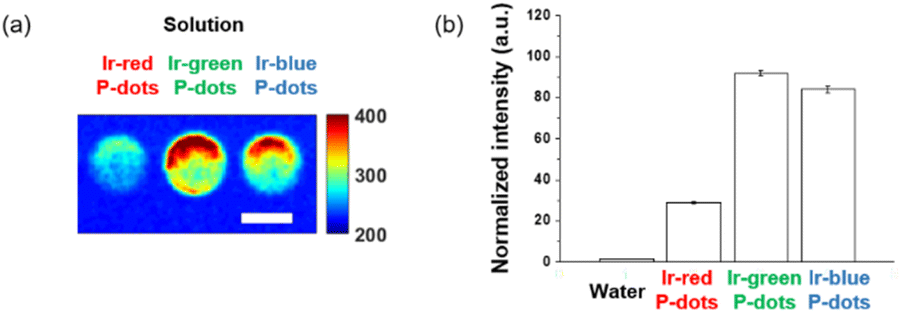
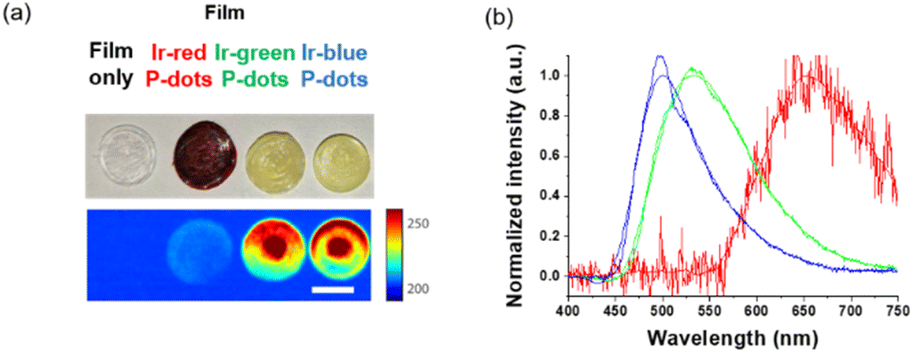
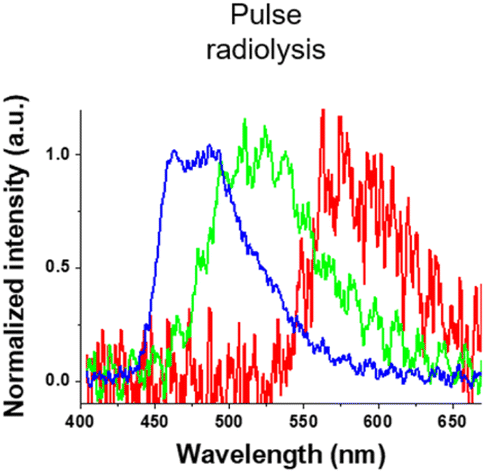
Finally, we investigated luminescence under radiation excitation (Fig. 6a). We placed the samples into a 96-well plate and irradiated them with hard X-rays (60 kVp tube voltage), whereas we observed the luminescence with an electron-multiplying charge-coupled-device (EM-CCD) camera. Hard X-ray beam was directed perpendicularly to the viewing axis, resulting in evident nonuniformity in the emission intensity. We counted the average emission from each well (Fig. 6b). Consistent with the quantum yields of the samples under UV excitation, the Ir-green and Ir-blue P-dots exhibited more intense luminescence than Ir-red P-dots under hard X-ray excitation. We observed the same tendency for the film samples (Fig. 7a). The hard X-ray luminescence spectra of the film samples indicate their emission in the red, green, and blue range (Fig. 7b). In the set-up we used in this study, the spectrum is noisy in Ir-red P-dots, but this is probably because the emission itself is so low and close to the detection limit that it was very difficult to obtain a signal itself. We also conducted pulse radiolysis studies to investigate their emission under electron beam irradiation (Fig. 8). We used an electron beam from a linear accelerator (28 MeV) as an excitation source and the resulting luminescence was detected with a multichannel spectroscopic detector. We clearly observed the luminescence spectra during pulse radiolysis (Fig. 8). Under radiation excitation, hard X-ray and electron beams ionize H2O in a manner that forms radicals and ionizing electrons. These ionizing electrons deposit energy into surrounding molecules, leading to further ionization or excitation, the latter of which can result in luminescence. These results clearly indicate that radiation-induced luminescence in visible light wavelengths from P-dots doped with heteroleptic tris-cyclometalated iridium complexes. Further mechanistic studies are in progress to understand the origin of the luminescence.
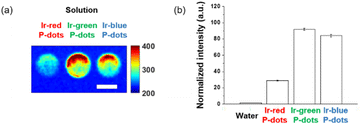 |
| | Fig. 6 Hard X-ray excited luminescence of P-dot solutions. (a) X-ray luminescence image. Scale bar is 5 mm. (b) Image intensity quantification of water background, lr-red P-dots, lr-green P-dots, and lr-blue P-dots. | |
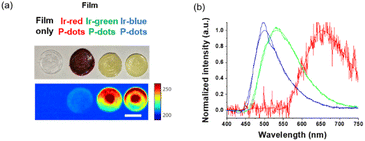 |
| | Fig. 7 Hard X-ray excited luminescence of P-dot films. (a) Brightfield photograph (top) and hard X-ray luminescence image (bottom). Scale bar is 7 mm. (b) Hard X-ray luminescence spectra of lr-red P-dots, lr-green P-dots, and lr-blue P-dots embedded in films. | |
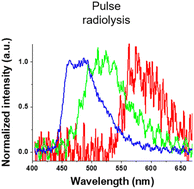 |
| | Fig. 8 Luminescence spectra at 0 ns after an electron pulse with a gate time of 500 ns for Ir-red P-dots (red), Ir-green P-dots (green), and Ir-blue P-dots (blue). | |
Conclusions
In this study, we demonstrated color variation of the radio-luminescence from P-dots doped with heteroleptic tris-cyclometalated iridium complexes. Consistent with previous studies, the relatively long photoluminescence under UV-visible irradiation is an important consideration for nano-sized scintillators. Here, we confirmed luminescence of three colors upon hard X-ray and electron beam irradiation over a wide range of visible light wavelengths, which is pertinent to further research in imaging and sensing of radiation.
Experimental section
Synthesis of P-dots
Iridium complex doped P-dots were prepared by co-precipitation.21 Typically, the iridium complexes and polymers were dissolved in THF at the concentration of 1 mg mL−1. Then, the iridium complexes and polymers were diluted in THF to form a mixture consisting of iridium complexes (500 μg mL−1; 5.1 × 10−4 mol dm−3 (Ir-red), 5.5 × 10−4 mol dm−3 (Ir-green), 7.0 × 10−4 mol dm−3 (Ir-blue)), PEG (5.5 × 10−6 mol dm−3) and PVK (4.0 × 10−6 mol dm−3). The mixture was sonicated to homogeneity. THF solution (0.3 mL) was added into 1 mL of Milli-Q water under sonication. The THF was evaporated by centrifugal evaporation (speed vac.). Finally, water was added to make a final volume of 1 mL of to prepare a P-dot solution. The prepared P-dot solution was stored at 4 °C for immediate use or lyophilized with an FDU-1200 lyophilizer (EYELA, Japan) for storage.
Preparation of P-dots films
Films were prepared by embedding P-dots into polymer matrix. To make polymer matrix, 0.5 g of PVA (n = 1700, TCI, Japan) was dissolved in 5 mL of Milli-Q water at 90 °C under vigorous stirring. After that, 250 μL of PEG (Mw = 200, TCI, Japan) was added into PVA solution to form uniform polymer matrix under continuous stirring at 90 °C. The P-dots solutions were concentrated 18 times and 1 mL of concentrated P-dots solution was mixed with 1 mL of polymer matrix by vigorous stirring to form a uniform mixture. To prepare the films, 200 μL of the polymer mixture was placed on to a polystyrene dish and dried at 50 °C. Then 5 pieces of films were attached together to make thick (1.5 mm) films with a diameter of about 12 mm. The films embedded with P-dots were irradiated under hard X-ray for imaging. Film only with PEG and PVA was used for comparison.
Absolute quantum yield measurements
The absolute quantum yield of Ir-blue P-dots and Ir-green P-dots were measured with a Quantaurus-QY absolute quantum yield spectrometer (Hamamatsu Photonics K.K., Japan). The sample was measured at the excitation wavelength of 350 nm.
Photoluminescence lifetime measurements
The time profiles of photoluminescence decay were measured for monomers (CH2Cl2) and P-dots (H2O) with an EasyLife fluorescence lifetime fluorometer (Optical Building Blocks, NJ, USA). The sample was excited with a 403 nm LED light source equipped with long pass filters; 550 nm for Ir-red, 455 nm for Ir-green, and 495 nm for Ir-blue. The O2 effect was studied by degassing under argon flow for 10 min. The rate constant kq for O2 quenching of the triplet states in the iridium complexes (CH2Cl2) was calculated by:
where kobs is the decay constant in air-saturated solution and kTD is the decay constant of the triplet state in the absence of O2. The O2 concentration in air-equilibrated solution was 2.2 × 10−3 mol dm−3 in CH2Cl2 and 1.9 × 10−3 mol dm−3 in acetonitrile.22
Nanosecond laser flash photolysis for transient absorption measurements of Ir monomer complexes
Nanosecond transient absorption measurement was conducted as previously described.23 In brief, the sample was irradiated with a 355 nm Nd3+:YAG nanosecond laser (5 ns full width at half maximum) and the transient absorption was probed with multi-channel spectrometer equipped with a pulsed xenon source lamp.
RIPT method for transient absorption measurements of P-dots20
Transient absorption was measured with a picoTAS system at UNISOKU (Hirakata, Osaka).24 Briefly, a 355 nm picosecond YAG laser (EKSPLA, PL2210A, fwhm 25 ps, 1 kHz, 6 μJ per pulse) was used for excitation.
Hard X-ray excited luminescence measurements
X-ray luminescence was produced by irradiating the samples with a small-animal X-ray irradiator (X-RAD SmART system, PXI, North Branford, CT, USA) and measured with an EMCCD camera mounted perpendicular to the radiation beam axis. The X-ray beam (60 kVp and 20 mA, unfiltered) was collimated with a 2.5 cm diameter round aperture and directed toward the side of the 96-well plate, each containing 200 μL of sample in solution. We acquired the X-ray luminescence images as series of 20 frames, each taken with a 1 s exposure, an electron gain of 50, and no pixel binning. We used MATLAB (MathWorks, Natick, MA, USA) to remove radiation noise from the raw camera images and quantify the image intensity.
X-ray luminescence spectra were acquired with another X-ray irradiator (PXI XRAD 320, North Branford, CT, USA) and measured with an optical spectrometer (Sensline, Avantes, Lafayette, CO, USA). The X-ray beam (60 kVp and 20 mA, unfiltered) was aimed at the film samples immediately beneath the X-ray aperture to maximize the absorbed dose rate and emitting light intensity. The film samples were placed on top of the distal end of a 1 mm-core-diameter optical fiber whereas the proximal end was coupled to the spectrometer. Each film sample was measured 3× with an exposure time of 5 s each for averaging. The spectra signals were then corrected by subtracting a background signal (non-doped control film) followed by spectral intensity correction to rectify the non-uniform sensitivity of the spectrometer at various wavelengths. The spectra were then normalized and smoothed with a Savitzky–Golay filter in MATLAB.
Pulse radiolysis for transient absorption and luminescence measurements
Pulse radiolysis was performed with an electron pulse (28 MeV, 8 ns, 0.7 kGy) from the L-band LINAC at SANKEN, Osaka University.25 An electron beam was used for as the excitation source; transient absorption or luminescence was measured with a multi-channel spectrometer or photomultiplier tube equipped with a monochromator.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the members of the Radiation Laboratory of SANKEN, Osaka University, for running the linear accelerator and Dr Tatsuo Nakagawa at UNISOKU for the RIPT method on transient absorption measurements of P-dots. This work was partly supported by JSPS KAKENHI Grant Number JP17K19103 and the Initiative for realizing diversity in the research environment project at Osaka University to YO. MY acknowledges a financial support from the Cooperative Research Program “Network Joint Research Center for Materials and Devices”. The authors gratefully acknowledge funding through Stanford Molecular Imaging Scholars (SMIS) Program (NIH T32 CA118681) to HTMN. We thank Michael Scott Long, PhD, from Edanz for editing a draft of this manuscript (https://jp.edanz.com/ac).
References
- C. Dujardin, E. Auffray, E. Bourret-Courchesne, P. Dorenbos, P. Lecoq, M. Nikl, A. N. Vasil’ev, A. Yoshikawa and R. Y. Zhu, IEEE Trans. Nucl. Sci., 2018, 65, 1977–1997 CAS.
- C. Bilynsky, N. Millot and A.-L. Papa, Bioeng. Transl. Med., 2022, 7, e10256 Search PubMed.
- J. Xie, M. Zhao, C. Wang, Y. Yong, Z. Gu and Y. Zhao, Adv. Healthcare Mater., 2021, 10, 2001615 CrossRef CAS PubMed.
- O. Piccolo, J. D. Lincoln, N. Melong, B. C. Orr, N. R. Fernandez, J. Borsavage, J. N. Berman, J. Robar and M. N. Ha, Sci. Rep., 2022, 12, 1559 CrossRef CAS PubMed.
- H. Chen, M. M. Rogalski and J. N. Anker, Phys. Chem. Chem. Phys., 2012, 14, 13469–13486 RSC.
- L. He, X. Yu and W. Li, ACS Nano, 2022, 16, 19691–19721 CrossRef CAS PubMed.
- A. F. Bartley, M. Fischer, M. E. Bagley, J. A. Barnes, M. K. Burdette, K. E. Cannon, M. S. Bolding, S. H. Foulger, L. L. McMahon and J. P. Weick, J. Neural Eng., 2021, 18, 046036 CrossRef PubMed.
- Z. Chen, V. Tsytsarev, Y. Z. Finfrock, O. A. Antipova, Z. Cai, H. Arakawa, F. W. Lischka, B. M. Hooks, R. Wilton and D. Wang, ACS Nano, 2021, 15, 5201–5208 CrossRef CAS PubMed.
- J. Perego, I. Villa, A. Pedrini, E. C. Padovani, R. Crapanzano, A. Vedda, C. Dujardin, C. X. Bezuidenhout, S. Bracco, P. E. Sozzani, A. Comotti, L. Gironi, M. Beretta, M. Salomoni, N. Kratochwil, S. Gundacker, E. Auffray, F. Meinardi and A. Monguzzi, Nat. Photonics, 2021, 15, 393–400 CrossRef CAS.
- Q. Chen, J. Wu, X. Ou, B. Huang, J. Almutlaq, A. A. Zhumekenov, X. Guan, S. Han, L. Liang, Z. Yi, J. Li, X. Xie, Y. Wang, Y. Li, D. Fan, D. B. L. Teh, A. H. All, O. F. Mohammed, O. M. Bakr, T. Wu, M. Bettinelli, H. Yang, W. Huang and X. Liu, Nature, 2018, 561, 88–93 CrossRef CAS PubMed.
- Z. Liu, K. O. Jung, R. Takahata, M. Sakamoto, T. Teranishi, M. Fujitsuka, G. Pratx and Y. Osakada, RSC Adv., 2020, 10, 13824–13829 RSC.
- D. J. Naczynski, C. Sun, S. Turkcan, C. Jenkins, A. L. Koh, D. Ikeda, G. Pratx and L. Xing, Nano Lett., 2015, 15, 96–102 CrossRef CAS PubMed.
- Y. Osakada, G. Pratx, C. Sun, M. Sakamoto, M. Ahmad, O. Volotskova, Q. Ong, T. Teranishi, Y. Harada, L. Xing and B. Cui, Chem. Commun., 2014, 50, 3549–3551 RSC.
- C. Sun, G. Pratx, M. Carpenter Colin, H. Liu, Z. Cheng, S. Gambhir Sanjiv and L. Xing, Adv. Mater., 2011, 23, H195–H199 CrossRef CAS PubMed.
- Y. Osakada, G. Pratx, L. Hanson, P. E. Solomon, L. Xing and B. Cui, Chem. Commun., 2013, 49, 4319–4321 RSC.
- D.-L. Ma, C. Wu, W. Tang, A.-R. Gupta, F.-W. Lee, G. Li and C.-H. Leung, J. Mater. Chem. B, 2018, 6, 537–544 RSC.
- Y. Osakada, L. Hanson and B. Cui, Chem. Commun., 2012, 48, 3285–3287 RSC.
- X. Wang, Y. Zhang, J. Li, G. Liu, M. Gao, S. Ren, B. Liu, L. Zhang, G. Han, J. Yu, H. Zhao and F. Rosei, Small Methods, 2022, 6, 2101470 CrossRef CAS PubMed.
- S. Takayasu, T. Suzuki and K. Shinozaki, J. Phys. Chem. B, 2013, 117, 9449–9456 CrossRef CAS PubMed.
- T. Nakagawa, K. Okamoto, H. Hanada and R. Katoh, Opt. Lett., 2016, 41, 1498–1501 CrossRef PubMed.
- Y. Osakada, T. Fukaminato, Y. Ichinose, M. Fujitsuka, Y. Harada and T. Majima, Chem.–Asian J., 2017, 12, 2660–2665 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi, M. T. Gandolfi and Editors, Handbook of photochemistry, 3rd edn, 2006 Search PubMed.
- Y. Osakada, K. Kawai, M. Fujitsuka and T. Majima, Chem. Commun., 2008, 2656–2658 RSC.
- M. Fujitsuka, C. Lu, B. Zhuang, E. Kayahara, S. Yamago and T. Majima, J. Phys. Chem. A, 2019, 123, 4737–4742 CrossRef CAS PubMed.
- Y. Wei, S. Samori, S. Tojo, M. Fujitsuka, J.-S. Lin, C.-T. Chen and T. Majima, J. Am. Chem. Soc., 2009, 131, 6698–6707 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and general methods. Synthesis of starting compounds, all NMR, and characterization of iridium complex monomers. See DOI: https://doi.org/10.1039/d3ra01216f |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Daiki Asanumaa,
Sachiko Tojoa,
Minoru Yamaji
b,
Daiki Asanumaa,
Sachiko Tojoa,
Minoru Yamaji