
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBODIPY-based near-infrared semiconducting polymer dot for selective yellow laser-excited cell imaging†
Lei Chen ab,
Yifei Jiangb,
Shihan Xub,
Jicheng Zhangb,
Seung-Ryoung Jungb,
Jiangbo Yub,
Xuanjun Zhangc and
Daniel T. Chiu*b
ab,
Yifei Jiangb,
Shihan Xub,
Jicheng Zhangb,
Seung-Ryoung Jungb,
Jiangbo Yub,
Xuanjun Zhangc and
Daniel T. Chiu*b
aDepartment of Biomedical Engineering, Sun Yat-Sen University, Shenzhen, 518107, China
bDepartment of Chemistry and Bioengineering, University of Washington, Seattle, Washington 98195, USA. E-mail: chiu@uw.edu
cDepartment of Health Sciences, University of Macau, Taipa, Macau SAR 999078, China
First published on 22nd May 2023
Abstract
Semiconducting polymer dots (Pdots) with both narrow-band absorption and emission are desirable for multiplexed bioassay applications, but such Pdots with absorption peaks beyond 400 nm are difficult to achieve. Here we describe a donor–energy transfer unit–acceptor (D–ETU–A) design strategy to produce a BODIPY-based Pdot that exhibits simultaneously narrow absorption and emission bands. A green BODIPY (GBDP) unit was employed as the main building block of the polymer backbone, conferring a strong, narrow-band absorption around 551 nm. An NIR720 acceptor provides narrow-band NIR emission. The small Stokes shift of the GBDP donor allows introduction of a benzofurazan-based ETU, resulting in a ternary Pdot with a fluorescence quantum yield of 23.2%, the most efficient yellow-laser excitable Pdot. Due to the strong absorbance band centered at 551 nm and weak absorbance at 405 nm and 488 nm, the Pdot showed high single-particle brightness when excited by a 561 nm (yellow) laser, and selective yellow laser excitation when used to label MCF cells, with much greater brightness when excited at 561 nm than at 405 nm or 488 nm.
1. Introduction
Semiconducting polymer dots (Pdots) are a class of small (usually < ∼30 nm) fluorescent probes for in vitro and in vivo imaging, biosensors, optical theranostics, and other biomedical applications.1–17 Pdots provide ultrahigh brightness,18,19 excellent photostability,20,21 low cellular toxicity,22 good water dispersibility, tunable photophysical properties, and easy bioconjugation.23 Pdots are also capable of multi-color emission at the single-particle level, with tunable relative fluorescence intensities,24 a capability that is useful for developing spectrally-barcoded probes for multiplexed assays. However, the design of such probes is limited by the availability of fluorophores with distinct excitation and emission. We previously developed a series of Pdots with narrow-band emission by incorporating green and red BODIPY dyes into a blue-emitting polyfluorene (PF) backbone.25 By adjusting the relative content of the narrow-emission BODIPY dyes in the backbone, a set of 20 spectral-intensity-encoded Pdots with 405 nm laser excitation were obtained for cell barcoding and tracking.26 However, it is difficult to obtain many more Pdot barcodes by simply increasing the fluorescent components due to the emissive spectra overlap problem.The number of Pdot barcodes available for multiplex assays could be further increased by integrating additional Pdots with narrow absorption near other commonly used laser sources (488, 561, and 633 nm). For example, by designing another set of 20 Pdot barcodes which can be excited by a 561 nm laser while exhibiting weak fluorescence when excited by a 405 nm laser, the number of fluorescence-encoded cells in a single detection could be doubled. One limitation of existing Pdots is their broad absorption bands,27,28 which result in comparable emission intensities when excited by different light sources. Another limitation is the lack of bright, long-wavelength excitable (LWE) Pdots (λabs > 520 nm). Blue laser (405 or 488 nm)-excitable PF- or PFBT-based Pdots with high fluorescence quantum yield (Φf > 30%) have been achieved,29–31 but most LWE Pdots have shown low Φf (<10%),28,32–36 mainly due to aggregation-induced self-quenching in the solid-like Pdot state. A few LWE conjugated polymer nanoparticles with higher Φf have been reported,37,38 but these approaches have often involved dilution of the conjugated polymer content in the nanoparticles.
Therefore, there remains a need for bright LWE Pdots with narrow-band absorption and emission, with λabs near the commonly-used laser excitation wavelengths of 561 nm or 633 nm. Here we employ a donor–energy transfer unit–acceptor (D–ETU–A) design strategy with the novel use of a BODIPY unit as the predominant building block of a Pdot polymer backbone to produce LWE (561 nm) Pdot with simultaneously narrow absorption and emission bands, a large Stokes shift (165 nm), and a high fluorescence quantum yield (Φf = 23.2%).
2. Results and discussion
As shown in Fig. 1, the designed ternary D–ETU–A copolymer, PFBDP-NIR720 (P1), is composed of a green emissive BODIPY (GBDP) unit as predominant light absorber (D), 4,7-bis(4-octyloxy-2-thienyl)-2,1,3-benzooxadiazole (DOTBO) as the ETU, and a previously reported NIR720 emitter18 as the acceptor (A).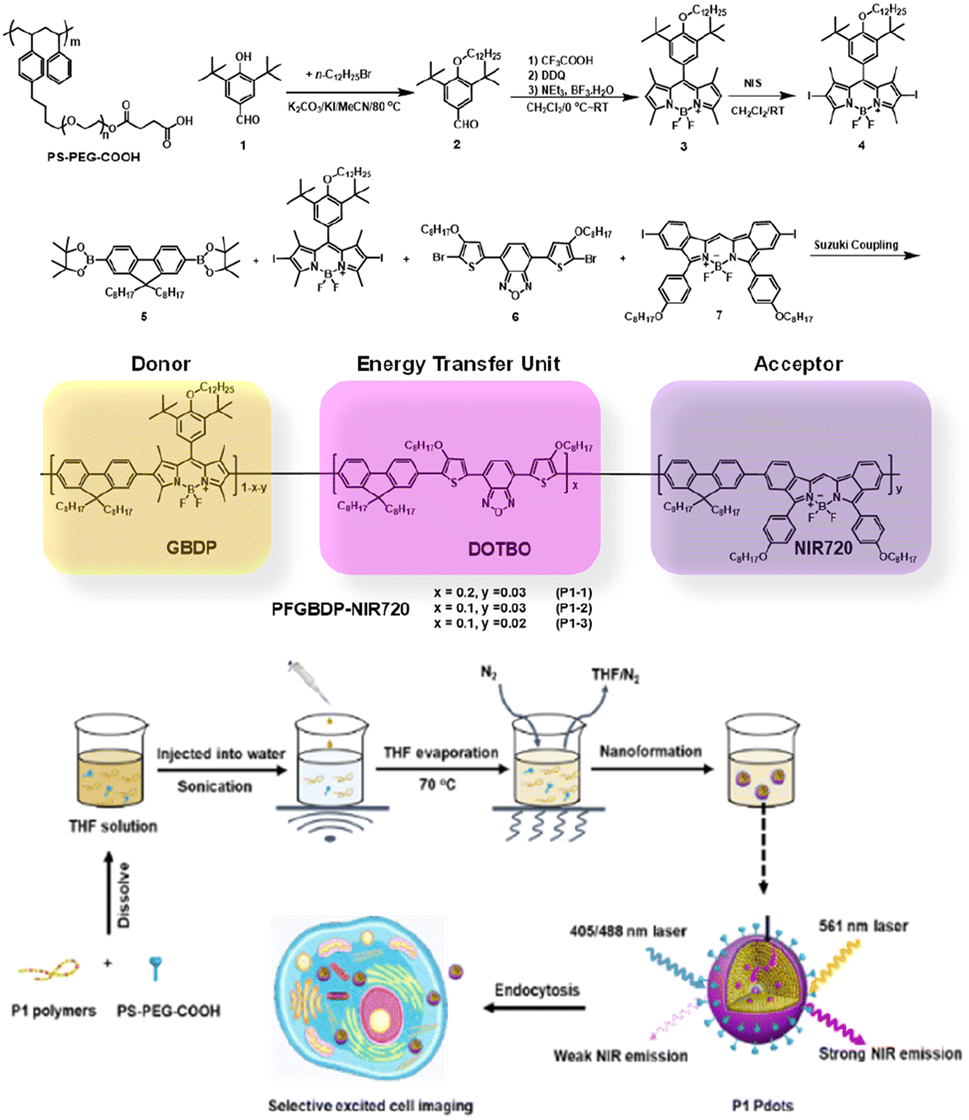 | ||
| Fig. 1 The chemical structure of PS-PEG-COOH and synthesis routes of P1 polymers. And the schematic illustration of P1 Pdot formation and its application for selective excited cell imaging. | ||
The corresponding P1 Pdots were obtained by using a nanoprecipitation method developed previously in our lab.31 Briefly, as shown in Fig. 1, conjugated polymer was co-dissolved with an optimized amount (20 wt%) of the amphiphilic polymer carboxyl-functionalized polystyrene-g-poly(ethylene oxide) (PS-PEG-COOH) in tetrahydrofuran (THF), and the mixture was quickly injected into Di-water under sonication. Organic THF solvent was then removed by blowing nitrogen gas into the mixture at ∼70 °C. The resulting solution was filtered with a 0.22 μm cellulose membrane filter to remove aggregates to obtain a Pdot solution at 0.01 mg mL−1.
The GBDP unit was chosen as the main building block because in its core structure, the coordination of the boron atom holds the dipyrromethene ligand in a highly rigid and planar conformation, giving GBDP a sharp and intense absorption band with a peak at around 500 nm, which is attributed to the S0–S1 (π–π*) transition. The full width at half maximum of this main absorption band (FWHMabs) is as small as 19 nm (Fig. 2a). Relatively weaker broad absorption bands in the shorter wavelength region can be assigned to the S0–Sn (n ≥ 2) (π–π*) transition.39 Due to the strong and narrow absorption of the BODIPY core, its alternative copolymer derivatives usually exhibit a narrow and S0–S1 transition-dominated absorption band as well.40–43
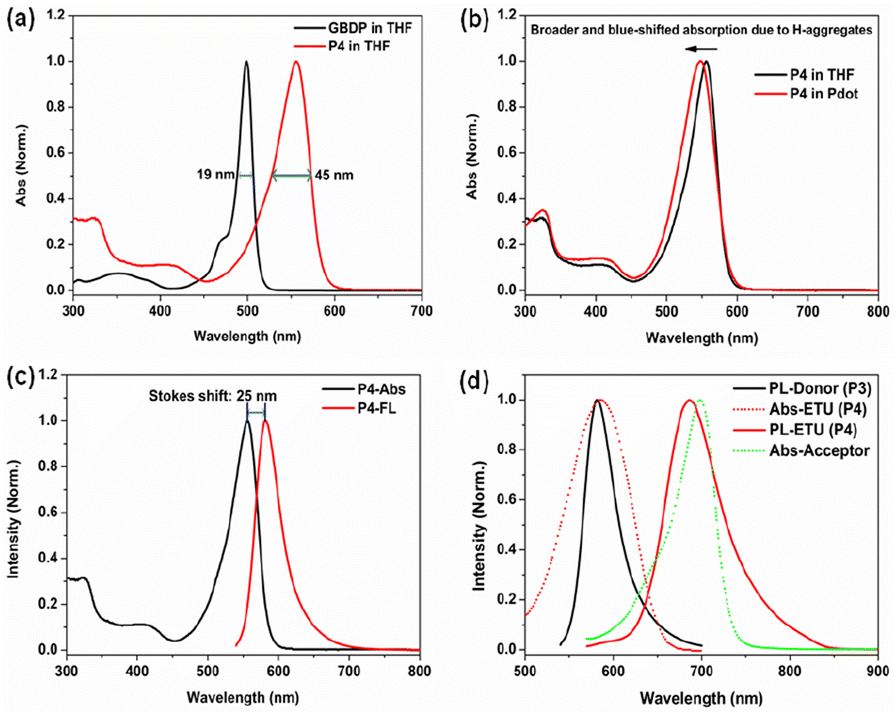 | ||
| Fig. 2 (a) Absorption spectra of GBDP dye and PFBDP polymer (P4) in diluted THF solution; (b) absorption spectra of P4 in THF and in the Pdot state; (c) Stokes shift of P4 in THF solution; (d) spectral overlap of donor emission and ETU absorption, and of ETU emission and acceptor absorption. | ||
The GBDP segment was alternatively copolymerized with 9,9-dioxylfluorene to form PFBDP (P4, Scheme S1†), acting as the predominant absorber in the P1 Pdot. Pure P4 polymer in diluted THF solution exhibits a pronounced bathochromic shift of the absorption maximum (57 nm) relative to the initial GBDP dye because of its significant extension of π-conjugation (Fig. 2a). In addition, the main absorption band of the P4 polymer becomes broader than that of GBDP dye (45 nm vs. 19 nm) as a result of the extended π-conjugation and reduced molecular rigidity.43 Compared to P4 polymer in THF, P4 Pdot in aqueous solution shows a broader S0–S1 absorption band (FWHMabs: 57 nm vs. 45 nm) and a much lower Φf (1.2% vs. 65.6%) due to formation of non-emissive H-aggregates (Fig. 2b).
To obtain an efficient PFBDP-based Pdot, it was necessary to introduce an energy-transfer unit to compete with H-aggregates and to maximally capture the excitation energy of PFBDP. The small Stokes shift of P4 (25 nm, in THF solution) (Fig. 2c) provided sufficient spectral width to add the DOTBO ETU, which was selected due to its ideal spectral overlap with the PFBDP energy donor and NIR720 acceptor (Fig. 2d). A small amount of NIR720 was incorporated into the polymer backbone, resulting in the ternary D–ETU–A P1 polymer. Three P1 variants with different ETU and NIR720 composition (recording based on the feeding ratios) were characterized: P1-1 (20 mol% ETU, 3 mol% NIR720), P1-2 (10 mol% ETU, 3 mol% NIR720), and P1-3 (10 mol% ETU, 2 mol% NIR720) (Fig. 1).
The properties of the three P1 variant Pdots are listed in Table 1. The three Pdots showed an average diameter of 21–25 nm based on dynamic light scattering (DLS). Of the three P1 variants, P1-2 exhibited the best performance, with the highest Φf (23.2%), lowest relative absorption intensity in the UV/blue region, and most complete energy transfer from the ETU to the NIR720 emitter (Fig. S1†). The Φf of P1-x series Pdots is determined by the trade-off between energy transfer efficiency and aggregation-induced fluorescence quenching. The light absorbed by PFGBDP host cannot be fully utilized with low ETU and NIR720 emitter content. On the other hand, high ETU and NIR720 emitter content will lead to aggregation-induced fluorescence quenching. The P1-2 Pdot with a feeding ratio of x = 0.1 and y = 0.03 optimized this trade-off. Based on these results, P1-2 Pdot was selected for further characterization.
| Pdot | λabs [nm] | λPL [nm] | Φf [%] | Size [nm] |
|---|---|---|---|---|
| P1-1 | 550 | 715 | 14.5 | 22.5 |
| P1-2 | 551 | 715 | 23.2 | 21.8 |
| P1-3 | 551 | 715 | 19.8 | 24.6 |
As shown in Fig. 3a, the number-average diameter of hydrated P1-2 Pdot was ∼22 nm evaluated by DLS. Fig. 3b shows the profile of dried P1-2 Pdot on cupper mesh substrate by high-resolution transmission electron microscopy. The P1-2 Pdot has a dominant narrow absorption band with λabs at 551 nm, close to the 561 nm laser source (Fig. 3c). The FWHMabs of P1-2 is only 56 nm, much narrower than that of PFBT-NIR720 (P2, 104 nm) and PFDHTBT-NIR720 (P3, 112 nm), which are commonly used in NIR Pdots (Fig. S2†).18 Compared to the pure P4 Pdot, the relative absorbance of the P1-2 Pdot in the short wavelength region (300–450 nm) is slightly stronger, mainly because of the introduction of the DOTBO unit, but is still much lower than that of existing non-BODIPY-based Pdots. The building blocks used in previously reported Pots include extended π-conjugated repeated units (such as PPV derivatives), and strong electron donating and electron withdrawing repeated units (such as PFDHTBT). These types of polymers have non-rigid and twisted molecular structures, and usually exhibit a broader and relatively weaker S0–S1 (π–π* transition or intramolecular charge transfer) absorption band in the longer wavelength region than our P1 polymers. Because of the excellent spectral overlap of the D/ETU and ETU/A in P1-2, sufficient cascade energy transfer occurred to produce a narrow emission band with a FWHMemi of 46 nm.
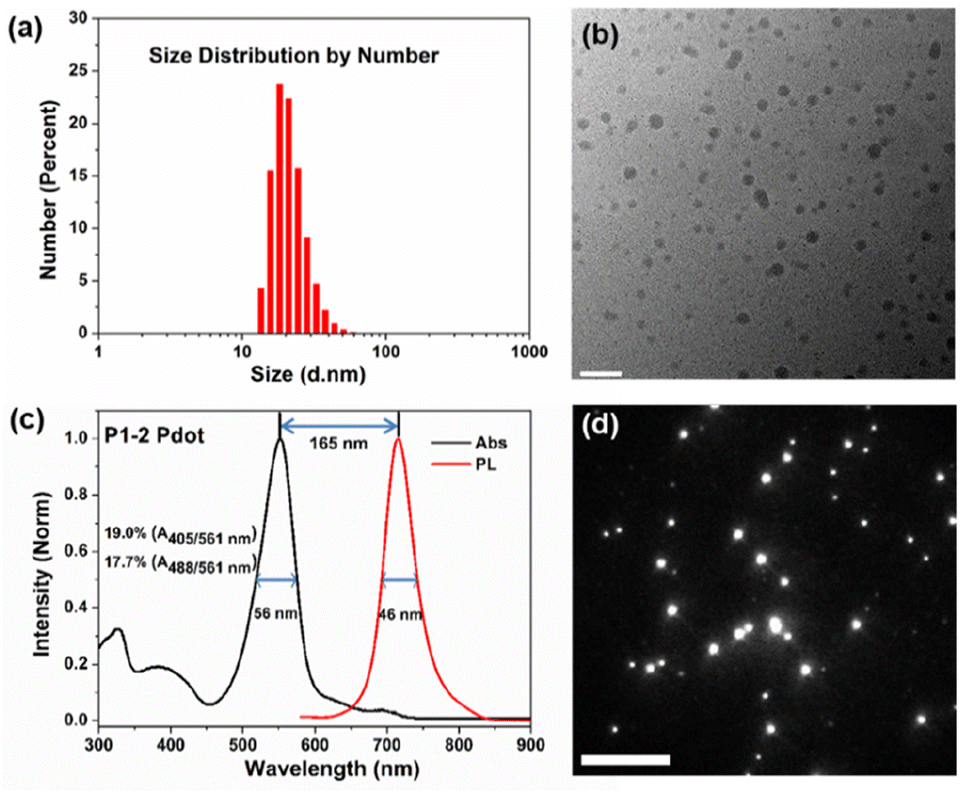 | ||
| Fig. 3 Characterization of P1-2 Pdot, the best performing of the three P1 Pdot variants. (a) and (b) Size distribution characterized by DLS (a) and TEM (b, scale bar = 100 nm); (c) absorption and emission spectra; (d) single-particle fluorescence imaging using a wide-field total internal reflection fluorescence microscope (scale bar = 100 μm). | ||
The GBDP unit also gives the P1-2 Pdot a larger molar extinction coefficient (ε) than that of existing NIR Pdots. The maximum absorbance (Amax) of the P1-2 Pdot at 0.005 mg mL−1 is 0.3, ∼1.9 times that of the PFBT-based Pdot and 2.7 times that of the PFDHTBT-based Pdot (Table S1†). Because single-particle brightness is proportional to the product of absorption cross-section and Φf, higher ε leads to a brighter Pdot. Single-particle fluorescence imaging of P1-2 Pdots was acquired by using a wide-field total internal reflection (TIRF) microscope (Fig. 3d), with a mean-averaged single-particle brightness of 3.0 × 103 photons per frame. The P1-2 Pdot also exhibits a large Stokes shift of 165 nm, a favourable feature for minimizing fluorescence noise from the excitation source.
To further evaluate the brightness of the P1-2 Pdot, we compared it with that of a currently used fluorescence probe PE-Cy5.5. The absorption and fluorescence spectra of PE-Cy5.5 are shown in Fig. S3.† The single-particle brightness of the P1-2 Pdot and PE-Cy5.5 were assessed by using a custom single-particle flow analyzer.44 Using this system and 561 nm laser excitation, the mean single-particle brightness of the P1-2 Pdot is ∼6.8 times that of the PE-Cy5.5 probe (Fig. 4a). The main purpose in designing the P1-2 polymer was to obtain a bright Pdot which can be selectively excited by a 561 nm laser. Therefore, we also acquired the single-particle brightness of the P1-2 Pdot when excited by 488 nm and 405 nm lasers at the same laser power density. The mean brightness ratios were 13.4 for I561 nm/I488 nm and 18.2 for I561 nm/I405 nm, indicating minimal cross excitation at 405 nm and 488 nm (Fig. 4b).
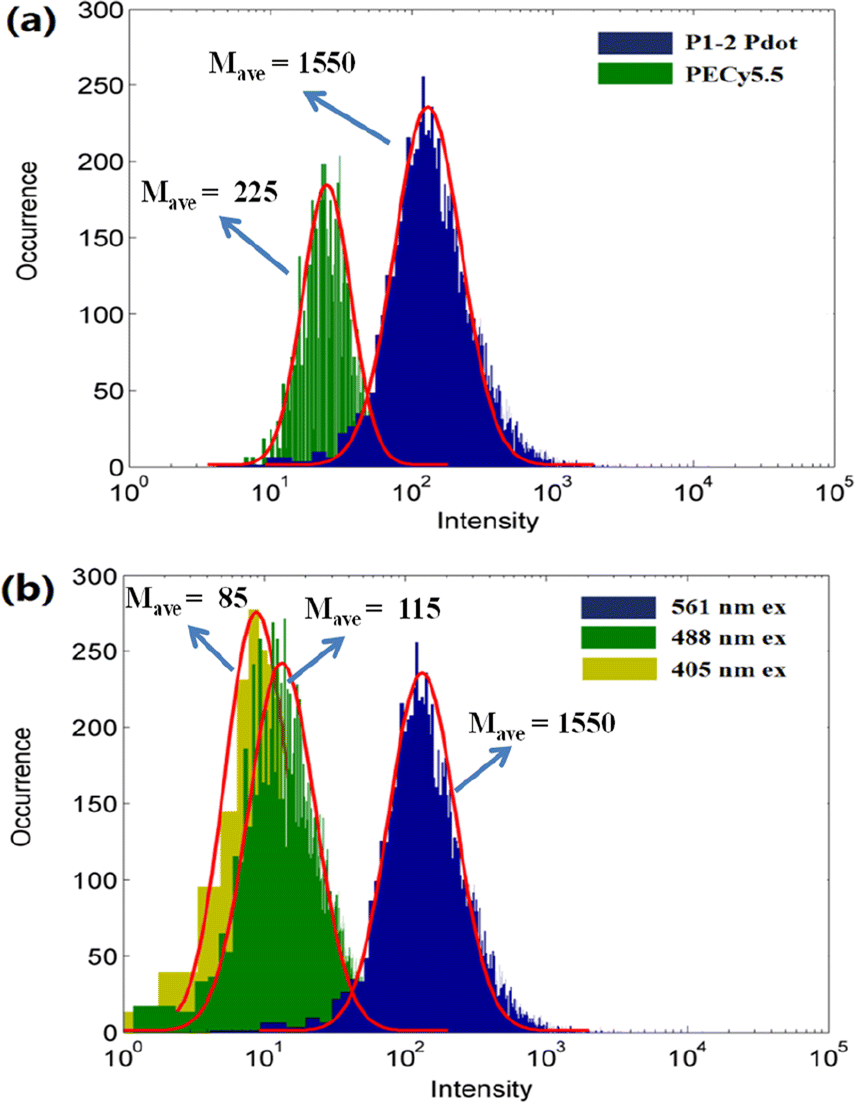 | ||
| Fig. 4 Single-particle brightness of (a) the P1-2 Pdot and PE-Cy5.5 excited using a 561 nm laser, and (b) the P1-2 Pdot excited using 405, 488, and 561 nm lasers. | ||
To test the capability of the P1-2 Pdot for use in multi-excitation encoding applications, we allowed MCF-7 breast cancer cells to take up the P1-2 Pdot via endocytosis. Confocal fluorescence microscopy images of Pdot-labeled MCF-7 cells were acquired at different excitation wavelengths (Fig. 5). The cells were much brighter when excited with a 561 nm laser than with a 488 nm or 405 nm laser: the cell brightness ratio was 9.4 for I561 nm/I488 nm and 12.5 for I561 nm/I405 nm (Fig. S4†). These results are consistent with the single-particle flow results and indicate that the P1-2 Pdot possesses excitation selectivity and is suitable for use in multi-excitation-based fluorescence encoding.
 | ||
| Fig. 5 Confocal fluorescence microscopy images of MCF-7 cells labeled with the P1-2 Pdot (scale bar = 50 μm): (a) bright-field image, and corresponding fluorescence images with laser excitation at (b) 561 nm, (c) 488 nm, and (d) 405 nm, showing selective excitation at 561 nm. | ||
3. Conclusions
Here we report an efficient 561 nm laser-excitable NIR Pdot, achieved via the novel use of a BODIPY unit as the primary absorber in a Pdot. This ternary D–ETU–A type Pdot exhibits simultaneously narrow absorption and emission bands, together with high single-particle brightness and a large Stokes shift. Due to the strong, narrow absorption around 551 nm, the Pdot is much brighter when excited by a 561 nm laser than when excited by a 488 nm or 405 nm laser. This property makes the Pdot can be utilized for selective yellow laser excited living cell imaging. Furthermore, we are trying to adopt this BODIPY-based D–ETU–A design strategy to develop 561 nm laser excited Pdot system with distinct emission spectra for multiplexed bioanalysis such as cell barcoding we previously reported22 or mRNA-FISH application.Conflicts of interest
The authors declare the following competing financial interests: D. T. Chiu and J. Yu have financial interest in Lamprogen Inc., which has licensed the technology from the University of Washington.Acknowledgements
We gratefully acknowledge support of this work by the National Institutes of Health (R01MH115767) and the University of Washington.References
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086 CrossRef CAS PubMed.
- J. Yu, Y. Rong, C.-T. Kuo, X.-H. Zhou and D. T. Chiu, Anal. Chem., 2017, 89, 42 CrossRef CAS PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38 RSC.
- Q. Zhao, X. Zhou, T. Cao, K. Y. Zhang, L. Yang, S. Liu, H. Liang, H. Yang, F. Li and W. Huang, Chem. Sci., 2015, 6, 1825 RSC.
- H.-S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699 RSC.
- B. Bao, P. Su, Z. Yang, X. Zhai, J. Zhang, Y. Xu, Y. Liu, B. Gu and L. Wang, Adv. Healthcare Mater., 2019, 8, 1900255 CrossRef PubMed.
- W. Tsai, C. Wang, C. Liao, a C. Yao, T. Kuo, M. Liu, C. Hsu, S. Lin, C. Wu, J. R. Pyle, J. Chen and Y. Chan, Chem. Sci., 2019, 10, 198 RSC.
- T. Senthilkumar, L. Zhou, Q. Gu, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 13114 CrossRef CAS PubMed.
- K. Zhang, Y. J. Gao, P. P. Yang, G. Bin Qi, J. P. Zhang, L. Wang and H. Wang, Adv. Healthcare Mater., 2018, 7, 1800344 CrossRef CAS PubMed.
- Z. Zhang, X. Fang, Z. Liu, H. Liu, D. Chen, S. He, J. Zheng, B. Yang, W. Qin, X. Zhang and C. Wu, Angew. Chem., Int. Ed., 2020, 59, 3691 CrossRef CAS PubMed.
- Y. Jiang and J. D. McNeill, Nat. Commun., 2018, 9, 4314 CrossRef PubMed.
- Q. Zhang, Z. Zhang, X. Hu, J. Sun and F. Gao, ACS Appl. Mater. Interfaces, 2022, 14, 179 CrossRef CAS PubMed.
- T. Li, M. Wu, Q. Wei, D. Xu, X. He, J. Wang, J. Wu and L. Chen, Biomacromolecules, 2023, 24, 1943 CrossRef CAS PubMed.
- H. Gao, N. Zhang, J. Hu, J. Pan, Y. Cheng, H. Chen and J. Xu, ACS Appl. Nano Mater., 2021, 4, 7244 CrossRef CAS.
- X. Bai, K. Wang, L. Chen, J. Zhou and J. Wang, J. Mater. Chem. B, 2022, 10, 6248 RSC.
- M. Wu, Q. Wei, C. Xian, C. Dai, X. He, C. Wu, G. Sun and L. Chen, Chin. Chem. Lett., 2023, 34, 107867 CrossRef CAS.
- Q. Wei, D. Xu, T. Li, X. He, J. Wang, Y. Zhao and L. Chen, Biosensors, 2022, 12, 1126 CrossRef CAS PubMed.
- L. Chen, D. Chen, Y. Jiang, J. Zhang, J. Yu, C. C. DuFort, S. R. Hingorani, X. Zhang, C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2019, 58, 7008 CrossRef CAS PubMed.
- J. Zhang, J. Yu, Y. Jiang and D. T. Chiu, ACS Appl. Mater. Interfaces, 2022, 14, 13631 CrossRef CAS PubMed.
- L. Chen, L. Wu, J. Yu, C. Kuo, T. Jian, I. Wu, Y. Rong and D. T. Chiu, Chem. Sci., 2017, 8, 7236 RSC.
- L. Wu, I.-C. Wu, C. C. DuFort, M. A. Carlson, X. Wu, L. Chen, C.-T. Kuo, Y. Qin, J. Yu, S. R. Hingorani and D. T. Chiu, J. Am. Chem. Soc., 2017, 139, 6911 CrossRef CAS PubMed.
- F. Ye, C. C. White, Y. Jin, X. Hu, S. Hayden, X. Zhang, X. Gao, T. J. Kavanagh and D. T. Chiu, Nanoscale, 2015, 7, 10085 RSC.
- D. Wang, J. Liu, Z. Liu, Z. Zhang, Z. Sun, C. Wu and G. Wang, ACS Appl. Nano Mater., 2020, 3, 2214 CrossRef CAS.
- W. Xu, S. Lu, M. Xu, Y. Jiang, Y. Wang and X. Chen, J. Mater. Chem. B, 2016, 4, 292 RSC.
- Y. Rong, C. Wu, J. Yu, X. Zhang, F. Ye, M. Zeigler, M. E. Gallina, I. C. Wu, Y. Zhang, Y. H. Chan, W. Sun, K. Uvdal and D. T. Chiu, ACS Nano, 2013, 7, 376 CrossRef CAS PubMed.
- C. T. Kuo, H. S. Peng, Y. Rong, J. Yu, W. Sun, B. Fujimoto and D. T. Chiu, Anal. Chem., 2017, 89, 6232 CrossRef CAS PubMed.
- C. S. Ke, C. C. Fang, J. Y. Yan, P. J. Tseng, J. R. Pyle, C. P. Chen, S. Y. Lin, J. Chen, X. Zhang and Y. H. Chan, ACS Nano, 2017, 11, 3166 CrossRef CAS PubMed.
- S. Y. Liou, C. S. Ke, J. H. Chen, Y. W. Luo, S. Y. Kuo, Y. H. Chen, C. C. Fang, C. Y. Wu, C. M. Chiang and Y. H. Chan, ACS Macro Lett., 2016, 5, 154 CrossRef CAS PubMed.
- X. Zhang, J. Yu, Y. Rong, F. Ye, D. T. Chiu and K. Uvdal, Chem. Sci., 2013, 4, 2143 RSC.
- R. Du, S. Cui, Z. Sun, M. Liu, Y. Zhang, Q. Wu, C. Wu, F. Guo and L. Zhao, Chem. Commun., 2017, 53, 8612 RSC.
- I. C. Wu, J. Yu, F. Ye, Y. Rong, M. E. Gallina, B. S. Fujimoto, Y. Zhang, Y. H. Chan, W. Sun, X. H. Zhou, C. Wu and D. T. Chiu, J. Am. Chem. Soc., 2015, 137, 173 CrossRef CAS PubMed.
- C. P. Chen, Y. C. Huang, S. Y. Liou, P. J. Wu, S. Y. Kuo and Y. H. Chan, ACS Appl. Mater. Interfaces, 2014, 6, 21585 CrossRef CAS PubMed.
- G. Hong, Y. Zou, A. L. Antaris, S. Diao, D. Wu, K. Cheng, X. Zhang, C. Chen, B. Liu, Y. He, J. Z. Wu, J. Yuan, B. Zhang, Z. Tao, C. Fukunaga and H. Dai, Nat. Commun., 2014, 5, 4206 CrossRef CAS PubMed.
- M. Liu, Z. Zhang, Y. C. Yang and Y. Chan, Angew. Chem., Int. Ed., 2021, 60, 983 CrossRef CAS PubMed.
- H. Pan, C. Wu, C. Lin, C. Hsu, Y. Tsai, P. Chowdhury, C. Wang, K. Chang, C. Yang, M. Liu, Y. Chen, S. Su, Y. Lee, H. K. Chiang, Y. Chan and P. Chou, J. Am. Chem. Soc., 2023, 145, 516 CrossRef CAS PubMed.
- M. Liu, T. Chen, J. R. Vicente, C. Yao, Y. Yang, C. Chen, P. Lin, Y. Ho, J. Chen, S. Lin and Y. Chan, ACS Appl. Bio Mater., 2020, 3, 3846 CrossRef CAS PubMed.
- H. Zhu, Y. Fang, X. Zhen, N. Wei, Y. Gao, K. Q. Luo, C. Xu, H. Duan, D. Ding, P. Chen and K. Pu, Chem. Sci., 2016, 7, 5118 RSC.
- S. Wang, J. Liu, G. Feng, L. G. Ng and B. Liu, Adv. Funct. Mater., 2019, 29, 1808365 CrossRef.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778 RSC.
- C. Thivierge, A. Loudet and K. Burgess, Macromolecules, 2011, 44, 4012 CrossRef CAS PubMed.
- A. Nagai and Y. Chujo, Macromolecules, 2010, 43, 193 CrossRef CAS.
- G. Meng, S. Velayudham, A. Smith, R. Luck and H. Liu, Macromolecules, 2009, 42, 1995 CrossRef CAS.
- V. R. Donuru, G. K. Vegesna, S. Velayudham, S. Green and H. Liu, Chem. Mater., 2009, 21, 2130 CrossRef CAS.
- S. R. Jung, R. Han, W. Sun, Y. Jiang, B. S. Fujimoto, J. Yu, C. T. Kuo, Y. Rong, X. H. Zhou and D. T. Chiu, Anal. Chem., 2018, 90, 6089 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01083j |
| This journal is © The Royal Society of Chemistry 2023 |