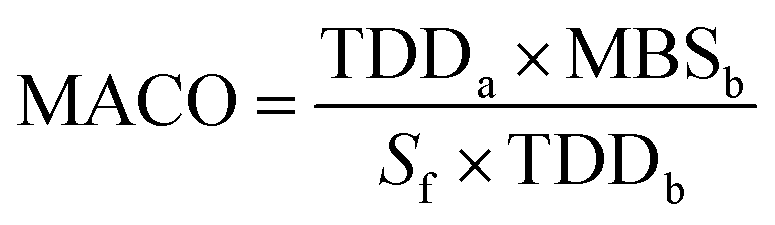DOI:
10.1039/D3RA01211E
(Paper)
RSC Adv., 2023,
13, 11642-11651
A novel smartphone HPTLC assaying platform versus traditional densitometric method for simultaneous quantification of alfuzosin and solifenacin in their dosage forms as well as monitoring content uniformity and drug residues on the manufacturing equipment†
Received
23rd February 2023
, Accepted 31st March 2023
First published on 14th April 2023
Abstract
The large popularity and rapid technology of smartphones have opened new avenues for their integration into different analytical methodologies and drug quality monitoring as a portable, easily accessible, and user-friendly detector. Herein, a novel and portable smartphone-based high-performance thin layer chromatographic (HPTLC) approach is proposed for the simultaneous analysis of two urological drugs, alfuzosin and solifenacin, which treat benign prostatic hyperplasia accompanied by overactive bladder syndrome. First, chromatographic separation was accomplished using an ecofriendly mobile phase, then the developed plates were visualized using Dragendorff's reagent and photographed via a smartphone's rear-facing camera fixed on a fabricated two-illumination-source chamber. The intensities of the drug spots were quantified using open-source image analysis software ImageJ over the concentration ranges of 2.0 to 30.0 μg per band for both drugs with acceptable results in ICH validation parameters. To improve the method's accuracy and reproducibility, various construction and shooting key parameters were investigated and optimized. Moreover, the study was extended to compare the obtained results with those of a benchtop densitometric method using a Camag TLC Scanner 3 at 215.0 nm; the densitometric method provided an additional assessment tool for peak purity and was capable of assaying lower drug concentrations over a linearity range of 0.2–8.0 μg per band for alfuzosin and 0.1–6.0 μg per band for solifenacin. The fast, simple, reliable, green merits of the proposed HPTLC/smartphone method suggest that it is an excellent platform for assaying marketed combined capsules and assuring their content uniformity. Moreover, the high sensitivity of the densitometric method was used, for the first time, to determine the residual content of the cited drugs on manufacturing equipment surfaces for cleaning validation. Finally, the environmental impact of the developed methods was evaluated based on green analytical chemistry principles.
Introduction
Adopting smartphone-based analytical methods is considered a pioneering strategy to face the current global economic crisis and can hamper the availability of expensive detectors or adherence to their maintenance schedule. One successful application of smartphones is their integration with high-performance thin-layer chromatography (HPTLC). Researchers' efforts have been directed to ameliorate the bulky size, high price and massive operating power consumption of the traditional HPTLC benchtop densitometric scanner by employing the astounding capabilities of smartphones as inexpensive, portable and easily accessible detectors.1 Smartphone-based HPTLC involves the rapid capture of developed HPTLC plates using a smartphone camera with the possibility of illuminating the plates with various light sources. Digital image analysis is then employed to gauge the spot colour intensity of calibration standards so that the concentration of unknown samples can be found via the obtained regression equation. Such an approach has been broadly applied and has proven its validity for the determination of pharmaceutical drugs2,3 and medicinal plants4,5 as well as monitoring counterfeit drugs.6,7
The digital image analysis is executed through either various chemometric manipulations or commercially available mobile applications and computer programs.5,6,8 One of the most common and modern software packages is ImageJ, an open-source image analysis software developed by the National Institutes of Health (NIH) organization.9,10 ImageJ is well-established software for the quantification of electrophoretic gels and has been recently integrated with TLC imaging analysis.2,4,11 It is characterized by a user-friendly interface that allows straightforward application without requiring sophisticated manipulation steps or highly trained personnel. In comparison to other mobile applications for image analysis, ImageJ is superior in dealing with different image formats regardless of the smartphone operating system. It is also compatible with most computer operating systems, including Windows, Mac OS and Linux and is devoid of any copyright restrictions. Moreover, the software's validity in generating reliable and reproducible results is assured by NIH as an authorized scientific organization rather than by uncertified application developers.10
In addition, the concept of “cleaning validation” has currently been introduced in the pharmaceutical industry as an integrated part of good manufacturing practice (GMP) guidelines.12,13 It aims to verify the efficacy of cleaning procedures in removing drug residues from industrial equipment to prevent cross-contamination of subsequently manufactured pharmaceutical products.14 Cleaning validation is performed not only to comply with international regulatory authorities but also to assure patient safety, especially upon manufacturing narrow therapeutic index drugs.15 Two challenging prerequisites have to be fulfilled for accurate residual estimation.16 The first one is the selection of an appropriate sampling technique, as the Food and Drug Administration (FDA) recommends two different sampling protocols: direct surface sampling via swabbing technique or indirect sampling through collecting the rinsing solution after the cleaning process.14 Although the rinse method provides a good tool for sampling large surfaces and inaccessible areas in the equipment, it fails to collect residues that are physically occluded in manufacturing equipment or practically insoluble in the rinsing solution. Such limitations are resolved upon adopting the swabbing method, with the capability of sampling dried-out residues.12,17 However, it is of great importance to take numerous swabbing samples across equipment and to carefully evaluate various sampling parameters such as applied protocol, utilized solvents and sampling sites.16,18 The second prerequisite for good cleaning validation practice is developing a reliable analytical method.12 Such an analytical method has to be specific and sensitive enough to quantify traces of active analytes remaining after the cleaning of manufacturing equipment.14,16 HPTLC is considered a promising eco-friendly approach for cleaning validation in the realm of liquid chromatography. It allows the monitoring of multiple swab samples simultaneously with minimal energy, mobile phase consumption, and waste generation. Furthermore, HPTLC complies with the principles of green analytical chemistry through swapping hazardous solvents with more sustainable ones.19,20 A literature survey revealed the adoption of the high-performance liquid chromatographic (HPLC) technique for detecting residual content of drugs,21–24 but the applicability of the HPTLC technique as a greener analytical candidate for cleaning validation has not yet been reported in any published work.
Therefore, this work is dedicated to developing and optimizing an eco-friendly HPTLC method with two different detection approaches: the conventional densitometric scanner and the new smartphone-based one. This comparative study was performed for simultaneous analysis of two urological drugs, alfuzosin hydrochloride (ALF, Fig. S1A†) and solifenacin succinate (SOL, Fig. S1B†), prescribed for alleviating benign prostatic hyperplasia in concurrence with overactive bladder syndrome.25 A literature review revealed the determination of the two cited drugs by spectrophotometric,26 LC-MS/MS,27 HPLC,28 chiral HPLC,29 two stability-indicating chromatographic30,31 and potentiometric32 techniques. None of those methods explored the applicability of a smartphone camera for rapid, onsite quantification of the cited drugs or adopted eco-friendly solvents for their HPTLC separation. Consequently, two eco-friendly HPTLC methods are proposed to spotlight the feasibility of smartphones for drug monitoring and to highlight the merits and demerits of each method. ICH validation and statistical analysis with pharmacopeial methods for each drug were also conducted to evaluate the methods' performances. The proposed methods were used to assay ALF and SOL in their marketed combined tablets and to assure the content uniformity of dosage units. Finally, this work discusses, for the first time, the suitability of the HPTLC technique in monitoring drug residues in cleaning validation swab samples from manufacturing equipment surfaces.
Experimental
Chemicals and reagents
The ethyl acetate, ethanol, methanol and ammonia solution (25% w/w) were HPLC grade and purchased from Sigma-Aldrich, Darmstadt-Germany. Dragendorff's reagent was prepared by mixing 5 mL of solution A (1.7 g basic bismuth nitrate in 100 mL water/acetic acid (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)) and 5 mL of solution B (40 g potassium iodide in 100 mL water) with 20 mL acetic acid and 70 mL distilled water.33 Pure ALF (BN: AH018045196), manufactured by Hetero Labs, Telangana-India, was kindly donated by Eva Pharma, Cairo-Egypt. SOL (BN: 801802553), a product of Glenmark Pharmaceuticals Ltd., Mumbai-India, was generously supplied by Marcyrl Pharmaceuticals Ind., Cairo-Egypt. Applying their corresponding official methods34 revealed their potencies to be 100.45 ± 0.408% for ALF and 99.35 ± 1.706% for SOL. Commercially available Solitral capsules (BN: MPF18001SK), with labelled contents of 10 mg of ALF and 5 mg of SOL per capsule and manufactured by Sun Pharmaceutical Ind, Ltd., Mumbai-India, were purchased from a local Indian market.
:1)) and 5 mL of solution B (40 g potassium iodide in 100 mL water) with 20 mL acetic acid and 70 mL distilled water.33 Pure ALF (BN: AH018045196), manufactured by Hetero Labs, Telangana-India, was kindly donated by Eva Pharma, Cairo-Egypt. SOL (BN: 801802553), a product of Glenmark Pharmaceuticals Ltd., Mumbai-India, was generously supplied by Marcyrl Pharmaceuticals Ind., Cairo-Egypt. Applying their corresponding official methods34 revealed their potencies to be 100.45 ± 0.408% for ALF and 99.35 ± 1.706% for SOL. Commercially available Solitral capsules (BN: MPF18001SK), with labelled contents of 10 mg of ALF and 5 mg of SOL per capsule and manufactured by Sun Pharmaceutical Ind, Ltd., Mumbai-India, were purchased from a local Indian market.
Standard solutions
Stock standard solutions of ALF and SOL were separately prepared in ethanol with concentrations of 1.0 mg mL−1 for the HPTLC/densitometric method and 4.0 mg mL−1 for the HPTLC/smartphone method. The prepared stock solutions were stored at 4 °C and found to be stable for three weeks.
Construction of illumination chamber
For the HPTLC/smartphone method, a customized illumination chamber was designed as shown in Fig. 1a, with dimensions of 30 cm length, 15 cm width and 15 cm height. The building material was medium-density fibre (MDF) boards, an inexpensive, widely available and easily tailored material, with a thickness of 5 mm. In the middle of the upper housing part, a rectangular window was drilled to fit the rear camera of the selected smartphone model (Redmi Note 8, Xiaomi, Beijing-China). The plate was inserted from the removable bottom lid and fitted using a static paper clip to keep the plate adequately in the camera field of view (Fig. 1b). The chamber was also equipped with two illumination sources: a 254 nm UV lamp (Philips, model: G11T5, 11 W, 21 cm length) and a daylight LED strip (cold white, 7 W), each plugged into an electrical outlet rather than a battery source to avoid fluctuation in the light intensity (Fig. 1b).
 |
| | Fig. 1 Structural design of the (A) exterior and (B) interior of the smartphone-based illumination chamber. | |
Chromatographic conditions
HPTLC silica gel 60 F254 aluminium sheets (Merck, Darmstadt-Germany) were utilized as the stationary phase for chromatographic separation. First, the plates were washed overnight in methanol and then activated by heating in an oven at 80 °C for 15 minutes. A Camag Linomat-5 autosampler (Muttenz-Switzerland) applied a sample aliquot, 10 μL for the HPTLC/densitometric method and 25 μL for the smartphone method, to the 20 cm × 10 cm plates at speed of 150 nL s−1 in the form of compact 4 mm spots. A mobile phase composed of ethyl acetate, ethanol and ammonia (2.0:8.0:0.5 by volume) was accurately prepared, transferred to a twin trough glass jar and left for 30 minutes for saturation and phase equilibrium. After that, the plates were inserted in the glass jar, allowing the development of the mobile phase in a linear ascending manner over a distance of 8.0 cm. The plates were then raised and left to dry in a vertical position.
Calibration curve and plate detection
HPTLC/densitometric method. From the ALF and SOL stock solutions (each 1.0 mg mL−1), serial aliquots equivalent to 0.2–8.0 mg and 0.1–6.0 mg, respectively, were withdrawn and transferred into two separate series of 10 mL volumetric flasks. Ethanol was then added to the mark as a diluent. Chromatographic procedures were conducted as previously mentioned and the developed plates were quantified densitometrically using a Camag TLC scanner (model 3, S/N 1302319) run with winCATS® software. The scanner was set to reflectance measuring mode with a measuring wavelength of 215.0 nm, slit dimension of 4 × 0.45 mm and scanning speed of 20 mm s−1. The purity of the resolved peaks was also assessed via the winCATS® spectral correlation tool.
HPTLC/smartphone method. Aliquots equivalent to 0.8–12 mg were accurately transferred from the ALF and SOL stock solutions (each 4.0 mg mL−1) into two individual sets of 10 mL volumetric flasks. The volume was then completed to the mark with ethanol and the previously mentioned chromatographic procedures were followed. Each plate included a total of fourteen samples with about 1 cm spacing to guarantee the absence of spot overlap. The developed plates were divided into two equal parts along the direction of mobile phase development so that every developed track was adequately fitted in the camera's field of view. Each part was immersed in Dragendorff's reagent33,35,36 for 30 seconds, rinsed with a small amount of distilled water and left until complete dryness. After that, the plates were fixed on the bottom lid of the illumination chamber using the paper clip and illuminated under the daylight LED strip. Before image acquisition, the lamp was allowed to warm up for about 5 minutes to stabilize the light intensity and limit the variability among measurements. The smartphone was placed on the upper housing part where the 48 MP wide rear camera with f/1.8 aperture was facing the plate. Different camera settings were optimized and finally adjusted to shutter speed of 1/250 s, ISO sensitivity of 100, autofocus and daylight white balance along with switching off the camera flash. All images were transferred to ImageJ software (NIH, Maryland-USA) on a laptop, opened as RGB colour images and analysed via the “Gels” function. First, sample tracks were identified by drawing equally sized rectangles using the “rectangular selection” tool followed by selecting the “Plot Lanes” option from the Gels menu. Peaks corresponding to each spot were generated and their areas were then manipulated using the “Straight” and “Magic Wand” tools. The different procedures of the proposed HPTLC/smartphone method are schematically illustrated in Fig. S2.†
Application to pharmaceutical formulations analysis
Ten Solitral capsules were evacuated and their contents were ground into fine powder using a hand mortar. For the HPTLC/densitometric method, a weighed quantity claiming to contain 10 mg ALF and 5 mg SOL was thoroughly transferred into a conical flask and sonicated in 30 mL ethanol for 30 minutes. The contents were then filtered through Whatman® (grade 1) filter paper and quantitatively transferred into a 50 mL volumetric flask to obtain a final solution of 0.2 mg per mL ALF and 0.1 mg per mL SOL. A sample volume of 10 μL was applied. Similarly, an assay of the dosage forms via the HPTLC/smartphone method was conducted, using a 25 mL volumetric flask and applying a sample volume of 25 μL. The subsequent procedures were followed as previously explained under each method to determine the concentration of the two cited drugs.
Content uniformity testing
The above procedures for quantifying the ALF and SOL in Solitral® capsules were followed using only one capsule. Ten capsules were individually analysed by each method and the uniformity of their contents was examined as per USP guidelines.37
Cleaning validation
Swabbing protocol for sample extraction. Square stainless-steel plates (10 cm × 10 cm) were spiked with known amounts of ALF and SOL to simulate manufacturing equipment surfaces covered with drug residues. This was achieved by using micropipettes to add 500 μL, in the form of small droplets all over the stainless-steel plate, from standard mixture solutions containing ALF and SOL at various concentration levels in ethanol (0.6/0.3 mg mL−1, 1.2/0.6 mg mL−1 and 2.4/1.2 mg mL−1). The stainless-steel plates were allowed to dry until complete evaporation of the solvent, leaving drug residues. A swabbing protocol was then adopted using an Alpha® polyester swab (model: TX714A, Texwipe, North Carolina, USA). First, the swab was wetted with ethanol and the excess diluent was squeezed. Sampling began by swabbing the surface with the first side of the swab in unidirectional, horizontal, parallel and overlapping strokes. The swab was flipped to the other side where the same sampling technique was applied but in a perpendicular direction. The head of the swabs was then cut and transferred into a clean 25 mL screw-cap test tube containing 7 mL ethanol. The procedure was repeated with a second swab at 45° to enhance extraction recovery and the second swab head was added to the same screw-cap test tube. After sonication for 10 minutes, the solution was quantitatively transferred to a 10 mL measuring flask and the volume was adjusted to the mark with ethanol.
Analytical method. The chromatographic conditions and scanning procedures of the HPTLC/densitometric method were tracked as previously explained to determine the residual content of the two cited drugs using a volume of 20 μL. The recovery efficacy of the adopted swabbing technique was evaluated by comparing the obtained concentrations with the nominal ones.
Results and discussion
Development and optimization of HPTLC separation
One of the most challenging issues during development of a new analytical method is controlling its impacts on health, safety and the environment without affecting its desired performance.28,38,39 Adopting the 3R green philosophy (reuse, replace, reduce), the sustainability extent of HPTLC could be greatly promoted through either substituting hazardous solvents with eco-friendlier ones or reducing their consumption.19,20
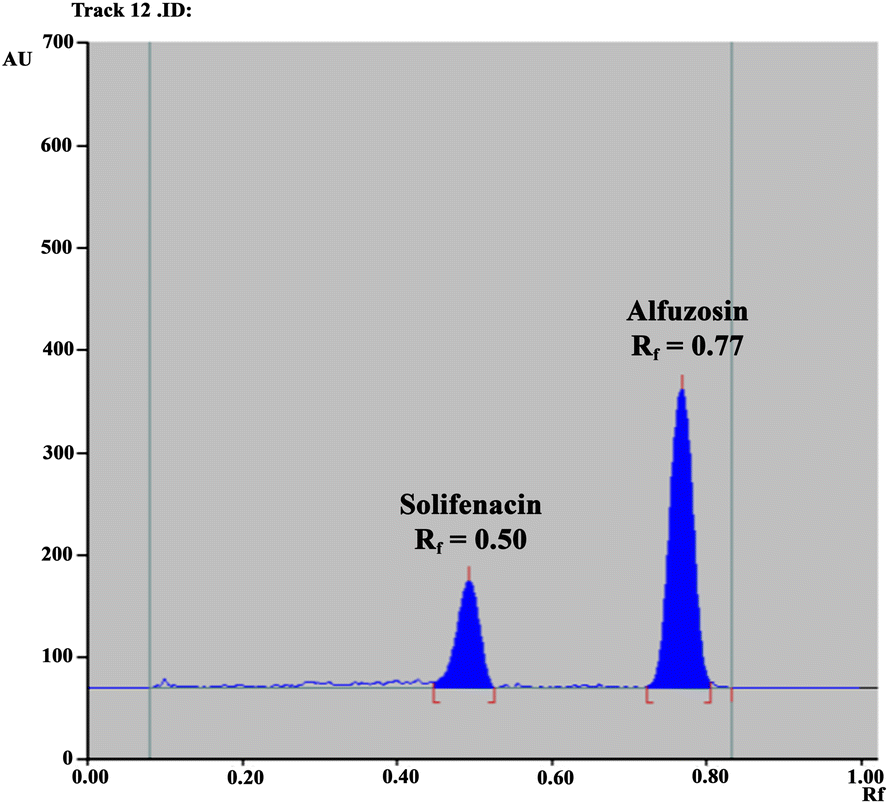
Consequently, our efforts were focused on excluding any reported hazardous solvents used for ALF and SOL separation, like methanol, acetonitrile, chloroform, dichloromethane and toluene, and resorting to greener ones as per the GlaxoSmithKline (GSK) solvent sustainability guide.40 Of these solvents, n-butanol, isopropanol, ethanol, ethyl acetate and acetone were examined in various ratios for efficient separation of the studied drug mixture. Preliminary trials revealed the promising capability of an ethyl acetate and ethanol mixture to separate the two drugs, but in form of streaks rather than compact spots. This tailing dilemma was resolved by exploring the chemical structures of the studied drugs, as demonstrated in Fig. S1A,† where the existence of basic functional groups suggested the addition of a basic reagent such as ammonia or triethylamine.41 A few drops of ammonia were found to possess a better enhancement effect on peak shape than triethylamine and optimum chromatographic parameters were attained with a mixture of ethyl acetate, ethanol and ammonia at a volumetric ratio of 2.0:8.0:0.5. The two drugs were successfully resolved with retardation factor (Rf) values of 0.77 for ALF and 0.50 for SOL (Fig. 2). Table 1 summarizes the calculated system suitability parameters42 where the values were within the acceptable ranges.37
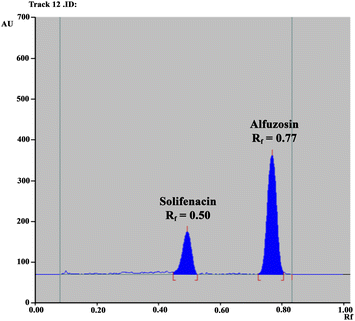 |
| | Fig. 2 HPTLC chromatogram of the resolved mixture of ALF (2.0 μg per band) and SOL (1.0 μg per band) using a mobile system composed of ethyl acetate, ethanol and ammonia (2.0:8.0:0.5, by volume) with detection at 215.0 nm. | |
Table 1 System suitability parameters of the proposed HPTLC method for determination of alfuzosin and solifenacin
| Parameters |
SOL |
ALF |
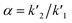 , where k′ is the capacity factor k′ = (1 − Rf)/Rf. Rs = [2(z2 − z1)]/(w1 + w2), where z is migration distance and w is peak width at the baseline. T = W0.05/2f, where W0.05 is peak width at 5% of height and f is distance between peak maximum and leading edge. N = 16(z/w)2, where z is the migration length of the spot and w is the spot width. , where k′ is the capacity factor k′ = (1 − Rf)/Rf. Rs = [2(z2 − z1)]/(w1 + w2), where z is migration distance and w is peak width at the baseline. T = W0.05/2f, where W0.05 is peak width at 5% of height and f is distance between peak maximum and leading edge. N = 16(z/w)2, where z is the migration length of the spot and w is the spot width. |
| Retardation factor (Rf) |
0.50 |
0.77 |
| Capacity factor (k′) |
1.00 |
0.30 |
| Selectivity factor (α)a |
3.33 |
| Resolution (Rs)b |
3.42 |
| Tailing factor (T)c |
0.85 |
0.91 |
| Theoretical plate number (N)d |
1600 |
6745 |
| Height equivalent to theoretical plate (HETP) [mm] |
0.050 |
0.012 |
Development of densitometric quantification
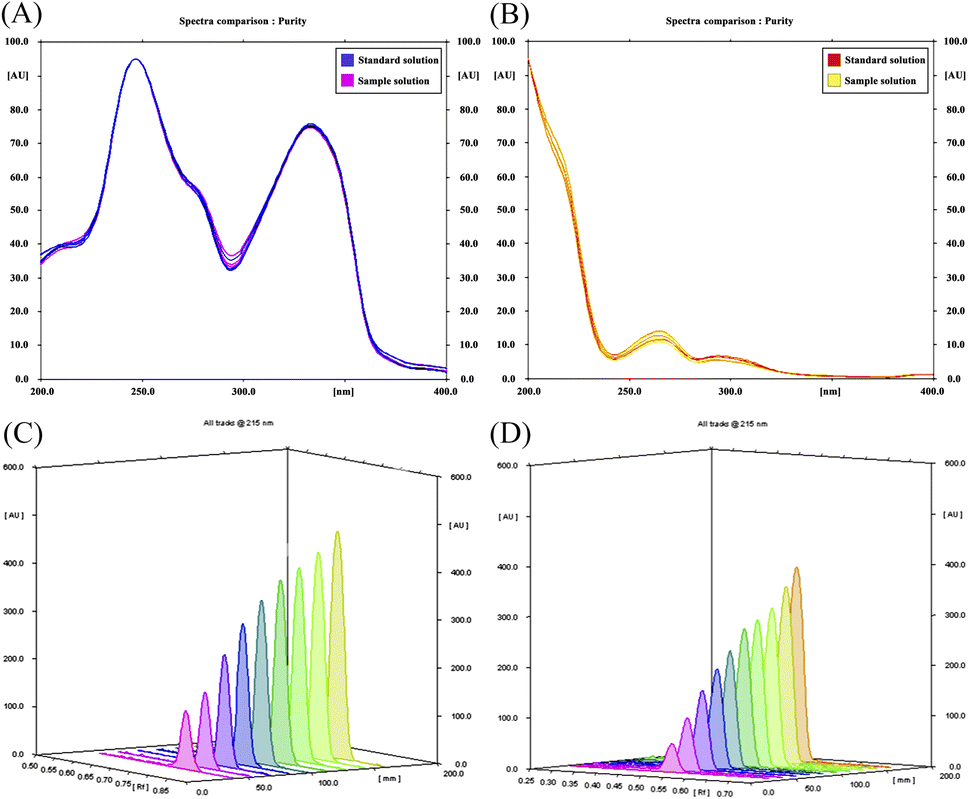
The scanning wavelength in the HPTLC/densitometric method was optimized via recording spectrodensitograms of each drug after the chromatographic process. As demonstrated in Fig. 3A and B, SOL has poor absorptivity and sensitivity at higher wavelengths in addition to being the minor component in the pharmaceutical dosage form ratio. As a result, the selection criteria were chosen in favour of SOL rather than ALF and a wavelength of 215.0 nm was found to achieve minimum background noise and the highest densitometric response for SOL without greatly affecting the sensitivity towards ALF.43
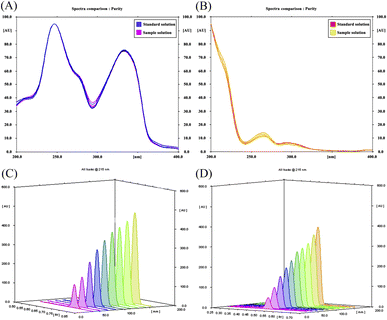 |
| | Fig. 3 Superimposed spectrodensitograms for the standard and sample tablet solutions of (A) ALF and (B) SOL for wavelength selection and peak purity assessment. (C and D) Scanning 3D profiles in range of 0.2–8.0 μg per band for ALF and 0.1–6.0 μg per band for SOL, respectively. | |
Development of HPTLC/smartphone method
The tremendous revolution in the built-in digital cameras of smartphones has opened new horizons for their applicability in different analytical techniques.44,45 In this work, a HPTLC/smartphone method was proposed, excluding any sophisticated instruments or arduous work, where the developed plates were captured by smartphone camera and rapidly processed to extract chromatographic results. To improve the method's accuracy and reproducibility, various key construction and shooting parameters were investigated and optimized as discussed below.
Designing and optimization of illumination chamber. Since any external light or stray shadows from the surrounding environment might interfere with the analysis process, it was essential to control these variables through designing a tightly closed illumination chamber so that HPTLC plates would be exposed to uniform and consistent illumination throughout the whole analysis process (Fig. 1). The chamber also served to securely orient the smartphone camera and the plate at the same position and distance during image acquisition. Two main construction parameters were thoroughly optimized. The first was the vertical distance from the HPTLC plate to the smartphone camera; sharp and wide photos were captured at 15 cm. Increasing this distance slightly disturbed the image sharpness and its resolution, while capturing at a nearer distance reduced the camera shooting range and required cutting the plate into more parts. The second parameter was centering the position of the two illumination sources with reference to the plate to ensure complete brightness of the plate and the absence of any light reflection zone on the plate (Fig. 1).
Selection of illumination mode. The constructed illuminated chamber was equipped with two light sources: a 254 nm UV lamp and a daylight LED strip to offer multiple modes; consequently, it could be employed for further analysis of various compounds. Preliminary trials revealed that the low absorptivity of SOL at 254 nm hindered its direct quantification using the UV-illumination mode (Fig. 4A). To overcome this limitation, several TLC staining reagents were tried to achieve stable, intensely coloured spots for both studied drugs without fading with time.2,33 Those reagents included iodine crystals, Dragendorff's reagent, ninhydrin, anisaldehyde and phosphotungstic acid. Although the literature reported the utilization of iodine crystals as a universal visualizing agent producing brown spots,6,45 the intensity of the coloured spots was found to be highly variable, very sensitive to time and rapidly diminishing. Satisfactory and rapid results were obtained upon immersing the developed plate in Dragendorff's reagent for 30 seconds, with intense orange spots achieved for ALF and SOL.35,36 This may be attributed to the abundance of nitrogenous groups in the chemical structures of ALF and SOL (Fig. S1A†). The plates were then rinsed with a small amount of distilled water to remove the excess reagent and left until complete dryness. Quantification was performed under the daylight-illumination mode using the constructed chamber (Fig. 4B).
 |
| | Fig. 4 Images of the assembled smartphone HPTLC analyser under (A) 254 nm UV lamp and (B) daylight lamp after visualization using Dragendorff's stain. | |
Optimization of image acquisition and analysis. The shooting parameters of the smartphone camera under daylight-illumination mode were manually adjusted by switching to pro-mode rather than auto-mode to standardize the camera exposure among images.4 The shooting parameters adjusted were the shutter speed, ISO sensitivity and white balance. The influence of the different levels for each parameter was monitored by capturing the same plate and comparing the obtained peak areas. First, the shutter speed was examined in the range of 1/600 s to 1/10 s. Extending the shutter speed permitted extensive light to enter to camera lens and the image to become brighter with a limitation of increasing background noise.4 A shutter speed of 1/250 s achieved maximum peak area with minimum noise. ISO sensitivity was another key parameter controlling image brightness and its sharpness. Its effect was assessed in gradual increments from 100 to 800, and optimum results were attained at a value of 100. Finally, the white balance was optimized to adjust the colour temperature of pictures under LED illumination. Although varying the white balance slightly influenced the peak reading, a daylight white balance was found to be best in comparison to the tungsten or fluorescent ones.The images were subsequently opened in the ImageJ desktop software. Sample tracks were identified and the software automatically detected the signals over the background for each track. Colour intensities of the spots were converted into analytical signals in the form of peak areas.
Validation of the proposed methods
The capability of the proposed methods for reliable quantification of ALF and SOL was validated in accordance with ICH guidelines.46
Accuracy. To assure the methods' accuracy, ALF and SOL were assessed in triplicate at five concentration levels: 0.3, 0.5, 1.0, 3.0 and 5.0 μg per band for the HPTLC/densitometric method and 5.0, 10.0, 15.0, 20.0 and 25.0 μg per band for the HPTLC/smartphone method (Fig. S4†). Acceptable mean recoveries were achieved in the range of 98–102%, highlighting the good accuracy of the two proposed methods (Table 2).
Precision. First, the variability extent in the scanning performance of each method was assessed via intra-plate precision repeatability. This was performed by triplicate scanning of the same spots corresponding to three different concentration levels (0.3, 1.0 and 5.0 μg per band for the HPTLC/densitometric method and 5.0, 15.0 and 25.0 μg per band for the smartphone method, Fig. S5†). Intermediate precision was conducted by analysing the same concentrations either on two successive days or on three different plates (Fig. S6†). Relative standard deviations (RSD%) for each method were calculated and found to be less than two (Table 2). In addition, the obtained RSD% values of the smartphone-based measurements were in line with those of the benchtop densitometer.
Specificity. The specificity of the proposed methods towards ALF and SOL was evaluated by their ability to analyse laboratory-prepared mixtures containing various concentrations along their linearity ranges (Fig. S7†). Good mean recoveries were obtained, revealing the selective determination of ALF and SOL without pronounced interference from each other (Table 2). This work also assured method specificity by checking the purity of the obtained peaks in the HPTLC/densitometric method.43 For each drug peak, UV-absorption spectra were recorded at three levels: peak start slope (s), peak maximum (m) and peak end slope (e) (Fig. 3A and B). The correlation between those spectra were then manipulated using the winCATS® spectral correlation tool, giving two correlation coefficients (rs,m and rm,e). Their values were found to surpass 0.9990, a good indication of the peaks' homogeneity as well as the absence of any masked co-eluting components.47,48 Moreover, the similar identity of the standard and sample of each drug was guaranteed through the good correlation and superimposition of their UV spectra, as illustrated in Fig. 3A and B.
Robustness. The developed HPTLC chromatographic conditions were reassessed after deliberate alteration in the ethanol and ethyl acetate ratio with a value of ±0.1. No remarkable variation was detected in system suitability parameters, as reflected by low RSD% values (Table 2). The robustness of the densitometric measurements was additionally evaluated by a slight change in the selected wavelength (±1 nm). For the HPTLC/smartphone method, the impact of minor changes in shooting parameters, including shutter speed and ISO sensitivity, was investigated. Table 2 displays the satisfactory values of RSD% for both methods, reflecting their good robustness.
Application of the two proposed methods for assaying Solitral capsules
ALF and SOL were successfully determined in the ratio of their marketed pharmaceutical formulation without any noticeable contribution from tablet excipients. The percentage values of found amount ± RSD% are summarized in Table 3. These satisfactory results provide insight into the eligibility of the proposed HPTLC/smartphone method as a greener and faster candidate for drug assaying at quality control laboratories, particularly in developing countries.
Table 3 Quantitative estimation of alfuzosin and solifenacin in Solitral capsules (BN: MPF18001SK) and in cleaning validation samples
| Application |
Solitral capsules |
Cleaning validation |
| Found % ± RSDa |
Nominal concentration (μg per band) |
Recovery % ± RSDb |
| HPTLC/densitometric method |
HPTLC/smartphone method |
HPTLC/densitometric method |
| Average of five determinations of tablet dosage form solution. Average of three determinations. |
| ALF |
99.42 ± 1.385 |
99.84 ± 0.898 |
0.6 |
98.36 ± 0.485 |
| 1.2 |
98.53 ± 0.824 |
| 2.4 |
98.02 ± 1.138 |
| SOL |
100.53 ± 1.597 |
100.29 ± 1.798 |
0.3 |
97.89 ± 0.324 |
| 0.6 |
98.52 ± 0.652 |
| 1.2 |
98.21 ± 0.725 |
Content uniformity testing
Regulatory and pharmacopeial compendia emphasize performing content uniformity testing as one of multiple quality control tests to guarantee the potency of a manufactured drug product. Multiple dosage units were randomly selected from the production batch followed by their individual quantifications.49 However, such analysis consumes much time, effort and resources, raising the need for fast, cost-effective and reliable analytical methods with little impact on the environment. Herein, the proposed HPTLC/smartphone method offers a sustainable and onsite analytical platform to monitor the content uniformity of the marketed pharmaceutical formulation. The procedures and processes were conducted as per USP guidelines50 and the acceptance value (AV) was found using the following equation.
AV = |M − ![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) | + k × SD | + k × SD |
Ten capsules were individually assayed and the obtained means of their recovery percents () were found to be in the range of 98.50–101.50% (Table S1†). In this case, the reference value (M) equals the mean () and thus AV was the multiplication product of the acceptability constant (k) and the sample standard deviation (SD). Since a sample size of ten was analyzed, the value of k was set at 2.4 (i.e. AV = 2.4 × SD). The calculated AV for ALF and SOL in Solitral capsules was smaller than the maximum allowed level, assuring adequate content uniformity of the capsules (Table S1†). Moreover, the functionality of the proposed HPTLC/smartphone method was confirmed by comparing these results with those of the conventional HPTLC/densitometric method and similar values were obtained (Table S1†).
Cleaning validation
Establishing cleaning limits. In the pharmaceutical industry, successive production of different drug products on the same manufacturing equipment must be accompanied by highly stringent cleaning procedures to eschew any contamination from previous drug residues.51 This is monitored by establishing the maximum allowable carryover (MACO) limit, which is the maximum acceptable amount of the previous drug that can be transferred to the following products.16 The instrument is considered clean if the previous drug residues are found below this limit. The principle of the MACO limit is that the patient should not receive the previous drug in an amount exceeding 0.1% of its therapeutic daily dose.12 Thus, the adopted calculation formula is based on the dosage criteria that consider the therapeutic daily doses of the previous (TDDa) and next (TDDb) products, the minimum batch size of the next product (MBS), the total surface area of all equipment and the safety factor (Sf, usually 100 for oral dosage form) as per the following equation.
The acceptance limit for residue per surface area is calculated using the following equation.
In our work, the MACO limit and further calculations were performed at a worst-case scenario of large TDDb and equipment surface area as well as a small batch size to assess the sensitivity of the proposed HPTLC/densitometric method to quantify trace amounts of the studied drugs.
18,52 The acceptance limits were calculated to be 6.67 μg cm
−2 for ALF and 3.33 μg cm
−2 for SOL.
Swabbing recovery and sample assay. The high sensitivity of the proposed densitometric method the over smartphone-based one was used, for the first time, to determine drug residues during cleaning validation. The recovery of the sampling techniques was optimized to assure a high extent of drug extraction from the stainless-steel surfaces. First, enhancement in the extraction efficacy was attained by re-swabbing the surface with the other swab side in a perpendicular direction. Moreover, higher recovery values (>90%) were observed upon adopting a double swabbing protocol at an angle of 45°. The recovery results of the extracted samples from the simulated manufacturing surfaces are summarized in Table 3, highlighting the potential of the proposed HPTLC/densitometric method to monitor the residual content of ALF and SOL at the levels of the acceptable allowed limits.
Statistical comparison between the proposed and official methods
To guarantee the performance of the two proposed HPTLC methods for determining ALF and SOL, the results were statistically compared with those obtained upon conducting the corresponding official method for each drug.34 The Student's t-test and the F-test revealed a lack of pronounced difference between the proposed and official methods upon adjusting the p-value at 0.05 (Table S2†).
Methods evaluation and greenness profile assessment
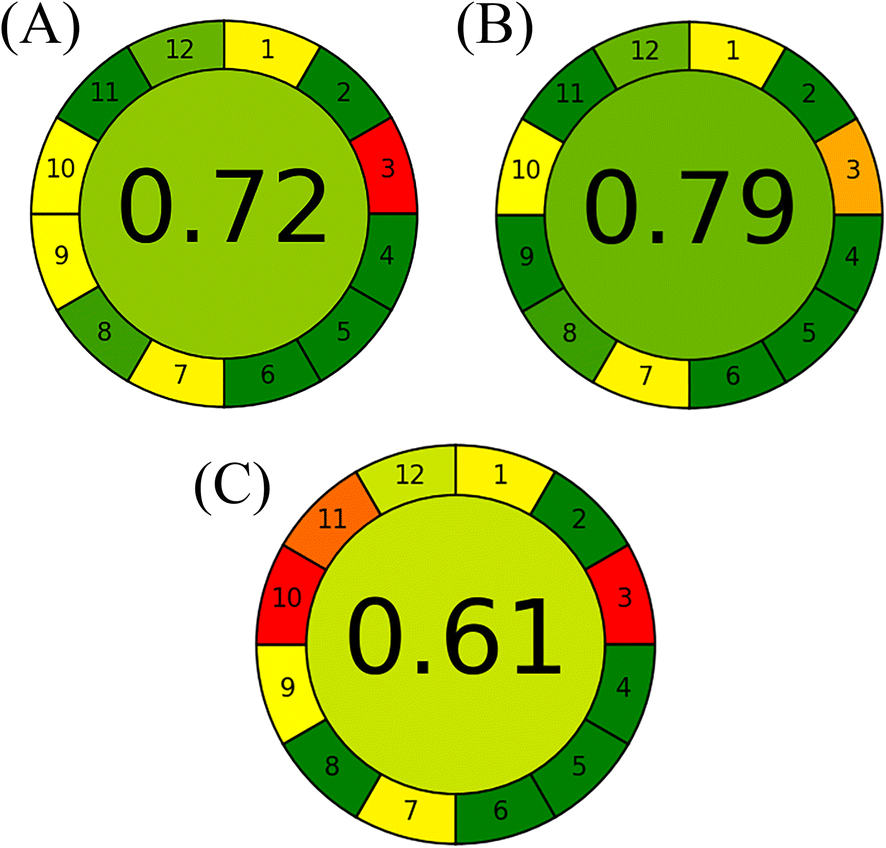
A comprehensive comparative study was conducted to illustrate the additional advantages of the proposed HPTLC methods over previously reported ones in terms of analytical performance and method sustainability. Although the reported spectrophotometric methods were based on distilled water as a solvent and were greener,26 the performance of the proposed HPTLC approaches was superior in allowing determination of several samples in one run as well as being easier and faster without requiring several manipulation steps to resolve the two drugs. In addition, our proposed methods excelled over the reported HPLC methods,27–29,31 as multiple samples could be simultaneously analysed with the lowest energy, mobile phase consumption and waste per sample. In the view of green analytical chemistry, the proposed methods were superior or relatively similar to such HPLC methods in utilizing ethanol and ethyl acetate as greener and bio-based organic solvents instead of the hazardous acetonitrile, as endorsed by the GSK solvent sustainability guide.26,29,40 Finally, the sustainability of the whole analytical method consisting of the two proposed methods and the reported HPTLC one30 was appraised by the state-of-the-art Analytical Greenness (AGREE) metric.53 It provides a simple and quantitative benchmark for the greenness profile assessment of analytical methods through free downloadable software.54 The findings are diagrammatically summarized in a circular pictogram with an overall assessment score. Methods with a higher score are considered greener. As demonstrated in Fig. 5, the independence of the proposed HPTLC/smartphone from the high energy-consumption detector and the facility of its at-line detection promoted it as the most sustainable method, followed by the proposed HPTLC/densitometric one, while the reported method had the lowest score owing to the hazardous solvents in its mobile phase composition.
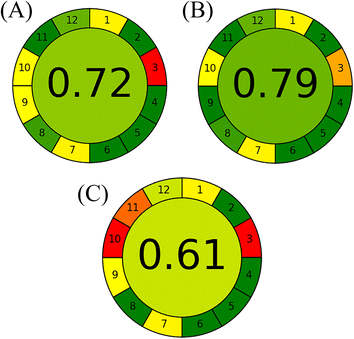 |
| | Fig. 5 Greenness profile evaluation of (A) proposed HPTLC/densitometric method, (B) proposed HPTLC/smartphone method and (C) reported HPTLC method for simultaneous determination of ALF and SOL using AGREE metric. | |
Conclusion
This work introduced new and eco-friendly HPTLC methods for determining ALF and SOL in their binary mixture. Its sustainability was increased by swapping hazardous solvents for greener ones such as ethyl acetate and ethanol. Two quantification methods were developed: the first used conventional densitometry while the second was a smartphone-based method that allowed fast quantification of the studied drug in daylight-illumination mode after a simple visualization step. The smartphone method was integrated with the license-free software ImageJ for image analysis and a constructed multi-illumination chamber for light uniformity. This work described a comprehensive evaluation of the analytical performance and greenness profile of the HPTLC/smartphone method compared to conventional benchtop densitometric scanners for drug analysis. The comparative study revealed the superiority of the densitometric method in determining lower drug concentrations; however, the proposed HPTLC/smartphone method is a promising alternative for more sustainable, onsite drug detection with minimal image processing. The results shone a light on not only the successful applicability of smartphones for drug assaying and content uniformity testing in quality control laboratories but also the capability of HPTLC for tracing drug residues during cleaning validation.
Author contributions
Mina Wadie: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, visualization. Ezzat M. Abdel-Moety: writing – review & editing, supervision. Mamdouh R. Rezk: conceptualization, methodology, resources, writing – review & editing, supervision. Hoda M. Marzouk: conceptualization, methodology, writing – review & editing, supervision.
Conflicts of interest
There are no conflicts to declare.
References
- N. Mac Fhionnlaoich, S. Ibsen, L. A. Serrano, A. Taylor, R. Qi and S. Guldin, J. Chem. Educ., 2018, 95, 2191–2196 CrossRef CAS.
- M. E. Sowers, R. Ambrose, E. Bethea, C. Harmon and D. Jenkins, J. Chromatogr. A, 2022, 1669, 462942 CrossRef CAS PubMed.
- F. Tosato, T. R. Rosa, C. L. M. Morais, A. O. Maldaner, R. S. Ortiz, P. R. Filgueiras, K. M. Gomes Lima and W. Romão, Anal. Methods, 2016, 8, 7632–7637 RSC.
- M.-T. Liu, J. Zhao and S.-P. Li, J. Chromatogr. A, 2021, 1637, 461826 CrossRef CAS PubMed.
- S. M. Sibug-Torres, I. D. Padolina, P. Cruz, F. C. Garcia, M. J. Garrovillas, M. R. Yabillo and E. P. Enriquez, Anal. Methods, 2019, 11, 721–732 RSC.
- A. G. Gad, Y. M. Fayez, K. M. Kelani and A. M. Mahmoud, RSC Adv., 2021, 11, 19196–19202 RSC.
- H. Yu, H. M. Le, E. Kaale, K. D. Long, T. Layloff, S. S. Lumetta and B. T. Cunningham, J. Pharm. Biomed. Anal., 2016, 125, 85–93 CrossRef CAS PubMed.
- L. Pieszczek and M. Daszykowski, Talanta, 2021, 221, 121599 CrossRef CAS PubMed.
- ImageJ Software, U. S. National Institutes of Health, Bethesda, Maryland-USA, https://imagej.nih.gov/ij/, accessed February, 2023 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- L. Chen, S. Wang, H. Yuan, J. Yang, M. Meng and Z.-l. Zhan, JPC – Journal of Planar Chromatography – Modern TLC, 2022, 35, 403–410 CrossRef CAS.
- World Health Organization (WHO), Appendix 3: Cleaning validation, Annex 3: Good manufacturing practices: guidelines on validation, 2006 Search PubMed.
- European Commission, EudraLex Volume 4: Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 16 Certification by a Qualified Person and Batch Release, European Commission, Brussels, Belgium, 2015 Search PubMed.
- Food and Drug Administration (FDA) Guidelines, Validation of Cleaning Processes (7/93), Guide to inspections validation of cleaning processes, 2014 Search PubMed.
- M. A. Akl, M. A. Ahmed and A. Ramadan, J. Pharm. Biomed. Anal., 2011, 55, 247–252 CrossRef CAS PubMed.
- Active Pharmaceutical Ingredients Committee (APIC), Guidance on aspects of cleaning validation in active pharmaceutical ingredient plants, 2014 Search PubMed.
- M. a. J. Nozal, J. L. Bernal, L. Toribio, M. a. T. Martín and F. J. Diez, J. Chromatogr. A, 2001, 919, 87–93 CrossRef CAS PubMed.
- N. Dubey, N. Dubey, M. Mandhanya and D. Kumar Jain, J. Pharm. Anal., 2012, 2, 478–483 CrossRef PubMed.
- M. A. Tantawy, S. A. Weshahy, M. Wadie and M. R. Rezk, Biomed. Chromatogr., 2020, 34, e4850 CrossRef CAS PubMed.
- D. Milojkovic-Opsenica and F. Andric, in Green chromatographic techniques: Separation and purification of organic and inorganic analytes, ed. D. Inamuddin and A. Mohammad, Springer, Dordrecht-Netherlands, 2014, ch. 4, pp. 81–101 Search PubMed.
- M. a. J. Nozal, J. L. Bernal, L. Toribio, M. a. T. Martín and F. J. Diez, J. Pharm. Biomed. Anal., 2002, 30, 285–291 CrossRef CAS PubMed.
- M. B. Boca, Z. Apostolides and E. Pretorius, J. Pharm. Biomed. Anal., 2005, 37, 461–468 CrossRef CAS PubMed.
- S. Fekete, J. Fekete and K. Ganzler, J. Pharm. Biomed. Anal., 2009, 49, 833–838 CrossRef CAS PubMed.
- Z. Slivová and R. Opatřilová, J. Spectrosc., 2015, 2015, 1–8 CrossRef.
- Chemaxon, Marvin Software for drawing, displaying and characterizing chemical structures, https://www.chemaxon.com, accessed February, 2023 Search PubMed.
- M. A. Tantawy, S. A. Weshahy, M. Wadie and M. R. Rezk, Curr. Pharm. Anal., 2021, 17, 1093–1103 CAS.
- H. N. Mistri, A. G. Jangid, A. Pudage, D. M. Rathod and P. S. Shrivastav, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 876, 236–244 CrossRef CAS PubMed.
- E. M. Abdel-Moety, M. R. Rezk, M. Wadie and M. A. Tantawy, Microchem. J., 2021, 160, 105711 CrossRef CAS.
- M. Wadie, E. M. Abdel-Moety, M. R. Rezk and M. A. Tantawy, Microchem. J., 2021, 165, 106095 CrossRef CAS.
- M. A. Tantawy, S. A. Weshahy, M. Wadie and M. R. Rezk, Microchem. J., 2020, 157, 104905 CrossRef CAS.
- M. A. Tantawy, S. A. Weshahy, M. Wadie and M. R. Rezk, Anal. Methods, 2020, 12, 3368–3375 RSC.
- M. Wadie, H. M. Marzouk, M. R. Rezk, E. M. Abdel-Moety and M. A. Tantawy, Anal. Chim. Acta, 2022, 1200, 339599 CrossRef CAS PubMed.
- L. Cai, Current Protocols Essential Laboratory Techniques, 2014, vol. 8, pp. 6.3.1–6.3.18 Search PubMed.
- British Pharmacopoeia, The Stationary Office, London-UK, 2019, vol. II Search PubMed.
- S. S. Tanzey, A. V. Mossine, A. R. Sowa, J. Torres, A. F. Brooks, M. S. Sanford and P. J. H. Scott, Anal. Methods, 2020, 12, 5004–5009 RSC.
- F. A. Mohamed, G. A. Saleh, S. R. El-Shaboury and A. H. Rageh, Chromatographia, 2008, 68, 365–374 CrossRef CAS.
- US Pharmacopeia 41, The United States Pharmacopial Convention, Rockville-USA, Asian edn, 2019 Search PubMed.
- M. R. Rezk, H. H. Monir and H. M. Marzouk, Microchem. J., 2019, 146, 828–834 CrossRef CAS.
- E. M. Abdel-Moety, N. A. Elragehy, N. Y. Hassan and M. R. Rezk, J. Chromatogr. Sci., 2010, 48, 624–630 CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- J. P. Taygerly, L. M. Miller, A. Yee and E. A. Peterson, Green Chem., 2012, 14, 3020–3025 RSC.
- P. S. Variyar, S. Chatterjee and A. Sharma, Fundamentals and theory of HPTLC-based separation, Springer, London-UK, 2011 Search PubMed.
- I. I. Hewala, M. M. Bedair and S. M. Shousha, Talanta, 2012, 88, 623–630 CrossRef CAS PubMed.
- M. Rezazadeh, S. Seidi, M. Lid, S. Pedersen-Bjergaard and Y. Yamini, TrAC, Trends Anal. Chem., 2019, 118, 548–555 CrossRef CAS.
- M. M. Ibrahim, K. M. Kelani, N. K. Ramadan and E. S. Elzanfaly, ACS Omega, 2022, 7, 23815–23820 CrossRef CAS PubMed.
- International Council for Harmonisation (ICH) Guidelines, Validation of Analytical Procedures: Text and Methodology, Q2 (R1), 2005 Search PubMed.
- A. F. El-Yazbi and R. M. Youssef, Anal. Methods, 2015, 7, 7590–7595 RSC.
- M. R. Rezk, E. M. Abdel-Moety, M. Wadie and M. A. Tantawy, J. Sep. Sci., 2020, 44, 530–538 CrossRef PubMed.
- B. Banfai, K. Ganzler and S. Kemeny, J. Chromatogr. A, 2007, 1156, 206–212 CrossRef CAS PubMed.
- US Pharmacopeia, Uniformity of dosage units, 2011, ch. 905 Search PubMed.
- G. L. Fourman and M. Mullen, Pharm. Technol., 1993, 17, 54–58 Search PubMed.
- N. Kumar, D. Sangeetha and P. Balakrishna, Pharm. Methods, 2011, 2, 161–166 CrossRef PubMed.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS PubMed.
- Y. Rostom, M. Wadie, M. R. Rezk, H. M. Marzouk and E. M. Abdel-Moety, Spectrochim. Acta, Part A, 2022, 273, 121063 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Ezzat M. Abdel-Moety,
Mamdouh R. Rezk and
Hoda M. Marzouk
*,
Ezzat M. Abdel-Moety,
Mamdouh R. Rezk and
Hoda M. Marzouk
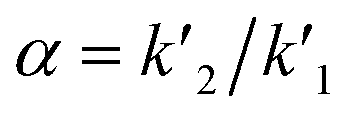 , where k′ is the capacity factor k′ = (1 − Rf)/Rf.b Rs = [2(z2 − z1)]/(w1 + w2), where z is migration distance and w is peak width at the baseline.c T = W0.05/2f, where W0.05 is peak width at 5% of height and f is distance between peak maximum and leading edge.d N = 16(z/w)2, where z is the migration length of the spot and w is the spot width.
, where k′ is the capacity factor k′ = (1 − Rf)/Rf.b Rs = [2(z2 − z1)]/(w1 + w2), where z is migration distance and w is peak width at the baseline.c T = W0.05/2f, where W0.05 is peak width at 5% of height and f is distance between peak maximum and leading edge.d N = 16(z/w)2, where z is the migration length of the spot and w is the spot width.