
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe proton conduction behavior of two 1D open-framework metal phosphates with similar crystal structures and different hydrogen bond networks†
Kai-ming Zhang ac,
Min-fang Jib,
Xue-Yi Zhoub,
Fang Xuanb,
Bo-yuan Duana,
Yuan Yuana,
Guang-xiang Liu*b,
Hai-bao Duan*b and
Hai-rong Zhao*b
ac,
Min-fang Jib,
Xue-Yi Zhoub,
Fang Xuanb,
Bo-yuan Duana,
Yuan Yuana,
Guang-xiang Liu*b,
Hai-bao Duan*b and
Hai-rong Zhao*b
aDepartment of Material Science and Engineering, Nanjing Institute of Technology, Nanjing 211167, P. R. China
bSchool of Environmental Science, Nanjing Xiaozhuang University, Nanjing 210009, P. R. E-mail: hairong_zhao111@163.com; Tel: +86 25 13914700426
cJiangsu Key Laboratory of Advanced Structural Materials and Application Technology, 1 Hongjing Road, Nanjing 211167, P. R. China
First published on 15th May 2023
Abstract
Two open-framework zinc phosphates [C3N2H12][Zn(HPO4)2] (1) and [C6N4H22]0.5[Zn(HPO4)2] (2) were synthesized via hydrothermal reaction and characterized by powder X-ray diffraction, thermogravimetric analysis and scanning electron microscopy. Both compounds have a similar crystal structure and macroscopic morphology. However, the difference in equilibrium cations, in which the propylene diamine is for 1 and the triethylenetetramine is for 2, results in a significant distinction in the dense hydrogen grid. The diprotonated propylene diamine molecule in 1 is more favorable for forming a hydrogen-bond network in three dimensions than in 2, in which the twisted triethylenetetramine forms a hydrogen bond grid with the inorganic framework only in two dimensions owing to its large steric effect. This distinction further leads to a disparity in the proton conductivity of both compounds. The proton conductivity of 1 can reach 1.00 × 10−3 S cm−1 under ambient conditions (303 K and 75% RH) and then increase to 1.11 × 10−2 S cm−1 at 333 K and 99% RH, which is the highest value among the open-framework metal phosphate proton conductors operated in the same conduction. In contrast, the proton conductivity of 2 is four orders of magnitude smaller than 1 at 303 K and 75% RH and two orders smaller than 1 at 333 K and 99% RH.
1. Introduction
Crystallized porous materials, such as coordination polymers, metal–organic frameworks (MOFs) and covalent–organic frameworks (COFs), which are considered to be new candidates for proton conduction materials because of their high surface area, tunable pore surface and designable framework, have been attracting considerable attention owing to their potential applications in electrochemical devices, such as fuel cells,1–7 sensing devices8,9 and super capacitors.Open-framework metal phosphates, composed of the anionic inorganic phosphate framework, which is formed by the metal ion and PO43−, HPO42−, H2PO4− or their complex substance, and organic amines or ammonium cations, are distinctive crystallize porous materials. They have similar designable structures to MOFs but have better thermal and aqueous stability owing to their robust inorganic units. Importantly, compared with MOFs, balanced organic amine cations (for example NH4+, enH22+, C3N2H122+ and Him+) and the hydrophilic groups PO4H/PO4H2 can directly provide an effective proton for proton transport. Moreover, the crystallinity of metal phosphates allows for the precise identification of their structural features to provide useful insights into proton-conduction pathways and mechanisms. Interestingly, most reported open-framework metal phosphates exhibit high proton conductivity. Yu reported JU103 (ref. 10) and JU102 (ref. 11) with the values of 3.59 × 10−3 S cm−1 (20 °C, 98% RH) and 1.19 × 10−3 S cm−1 (55 °C and 98% RH), respectively; Ren reported that compound (C2N2H10)0.5CoPO4 reached 2.05 × 10−3 S cm−1 (56 °C, 98% RH).12 Our group has also carried out several studies on the preparation of highly conductive metal phosphates, such as (C2H10N2)[Mn2(HPO4)3](H2O) reached 1.64 × 10−3 S cm−1 at 298 K and 99% RH,13 (C2H10N2)[Mn2(PO4)2]·2H2O reached 0.22 × 10−4 S cm−1 at 303 K and 99% RH (ref. 14) and (NH3(CH2)3NH3)2[Fe4(OH)3(HPO4)2(PO4)3].4H2O15 reached 0.8 × 10−3 S cm−1 at 317 K and 99% RH.
However, owing to the required balance of positive and negative charges in open-framework metal phosphate, its structural framework is fixed, which leads to its difficulty in increasing the proton conductivity by further modifying its structure using effective strategies, such as MOFs.16–24 For example (1) inclusion of functional guest molecules into the cavity of MOFs, such as acid molecules, imidazoles, and triazoles;25–31 (2) use of organic linkers bearing hydrophilic groups (including –COOH, –OH, –SO3H, and –PO3H2)32–38 as structural building units. Therefore, the most ways to improve the proton conductivity of open-framework metal phosphate are by optimizing the conductive environment at present, for example, increasing the temperature and relative humidity and changing the water to an ammonia environment. Compared to the regulation of the material's structure, these changes in the external environment are not the fundamental and optimal way to improve proton conductivity. Therefore, to increase the proton conductivity of open-framework metal phosphate, the reasonable design of its original structure based on a thorough understanding of its proton transport mechanism is particularly important and critical. However, research in this area is still very poorly explored.
Moreover, open-framework metal phosphates or other crystallize porous materials generally exhibit excellent conductive behavior when high temperatures and high humidity coexist. However, it is also very important for proton-conducting materials to exhibit high proton conductivity in the medium-low temperature and humidity regions to maintain high conductivity over a wide temperature and humidity range, while research on high-proton conductive materials under relatively medium-low temperature and low humidity is still relatively lacking.28,35,39–42
Herein, to further understand the proton conduction mechanism of open-framework metal phosphates and to prepare materials that can exhibit high conductivity even at medium-low temperature and low humidity, we synthesized two metal phosphate compounds [C3N2H12][Zn(HPO4)2] (1) and [C6N4H22]0.5[Zn(HPO4)2] (2) with similar crystal structures, both showing 1D ladder-like chain structure of the inorganic skeleton and the organic cations interspersed in the inorganic chains. The results demonstrate that the effective hydrogen bond grid plays a key role in proton transport. For 2, the twisted structure of the large organic cationic triethylenetetramine resulted in the formation of hydrogen bond fragments only in the two-dimensional plane owing to its large steric hindrance, whereas 1 formed dense hydrogen bond interactions in the three-dimensional space. As anticipated, the value of proton conductivity for 1 reached 1.00 × 10−3 S cm−1 at low 75% RH and 303 K and then achieved 10−2 S cm−1 at 99% RH and 333 K, compared to the Nafion membrane; while for 2, only 1.04 × 10−7 S cm−1 at low 75% RH and 298 K. To the best of our knowledge, 1 serves as the highest proton conductivity at low humidity in porous phosphate compounds. This encouraging result offers more opportunities for real-world conductive materials in fuel cells. It gives us more confidence to further investigate the relationship between proton conductivity and hydrogen bond network and organic amine cations to find a class of metal phosphate compounds that exhibit high electrical conductivity at low humidity.
2. Experimental section
2.1 Chemicals and reagents
All reagents and chemicals were purchased from commercial sources and used without further purification. Compounds 1 and 2 were synthesized according to the method reported in the literature.43,44 The detailed preparation process is as follows.2.2 Synthesis of [C3N2H12][Zn(HPO4)2] (1)
0.5480 g Zn(ac)2·2H2O was dispersed in 4.5 mL H2O, 0.22 mL hydrochloric acid (35%) and 0.81 mL phosphoric acid (aq, 85 wt%). 1.26 mL of DAP was dropwise added to the mixture, which was homogenized for 30 min. The mixture solution was transferred and sealed in a 25 mL Parr Teflon lined stainless steel autoclave and heated at 438 K for 24 hours. The white plate crystals of 1 were obtained.2.3 Synthesis of [C6N4H22]0.5[Zn(HPO4)2] (2)
0.3255 g zinc oxide was dispersed in 7.2 mL H2O, 0.7 mL hydrochloric acid (35%) and 2.6978 g phosphoric acid (aq, 85 wt%). 1.19 mL of TETA was dropwise added to the mixture, which was homogenized for 30 min. The mixture solution was transferred and sealed in a 25 mL Parr Teflon lined stainless steel autoclave and heated at 353 K for 50 hours. The white plate crystals of 2 were obtained.2.4 Material characterizations
Powder X-ray diffraction (PXRD) data were collected using a Bruker D8 diffractometer with Cu Kα radiation (λ = 1.5418 Å) in the degree range of 5–50° (298 K). TGA experiments were performed using a STA449 F3 thermogravimetric analyzer in the temperature range of 298–1073 K (25–800 °C) at a warming rate of 10 °C min−1 under a nitrogen atmosphere, and the polycrystalline samples were placed in an Al2O3 crucible. The morphologies of the sample were observed using a Hitachi SU8010 scanning electron microscope (SEM).2.5 Conductivity measurements
The hydrated proton conductivities of the compound pellets and composite membranes were measured using a CHI 660D electrochemical workstation and a conventional three-electrode method. The ac frequencies ranged from 102 to 106 Hz with 5 mV signal amplitude. The powdered polycrystalline pellets (with 0.7 mm thickness and 1.33 cm2 area) prepared under 10 MPa pressure were sandwiched by two copper electrodes used for proton conductivity measurement. The conductivity (σ) of the sample was calculated using the following equation: σ = L/RS, where L and S represent the thickness and cross-sectional area of the sample, respectively, and R represents the resistance value obtained from the impedance spectra.3. Results and discussion
The crystal structures of compounds 1 and 2 were first described, and then some differences between their structures were analyzed in detail to gain insight into conductive behavior. Compound 1 crystallized in the orthorhombic space group P212121. Its asymmetric unit contains one Zn2+ ion (labeled Zn(1)), two crystallographically independent HPO42− ions (labeled P(1) and P(2)), and one protonated diction of 1,3-diaminopropane. As shown in Fig. 1, the Zn2+ showed tetrahedral coordination by four oxygen atoms, which all come from HPO42− ions. The ZnO4 and HP(2)O4 were connected into four-membered rings, and then these four-membered rings were arranged around the a axis, forming a ladder-like chain. Moreover, the HP(1)O4 tetrahedra are grafted onto the one-dimensional chain, which acts as a pendant. The action amine molecule, diprotonated DAP molecule, sits in the middle of these channels and interacts with the framework through hydrogen bonds.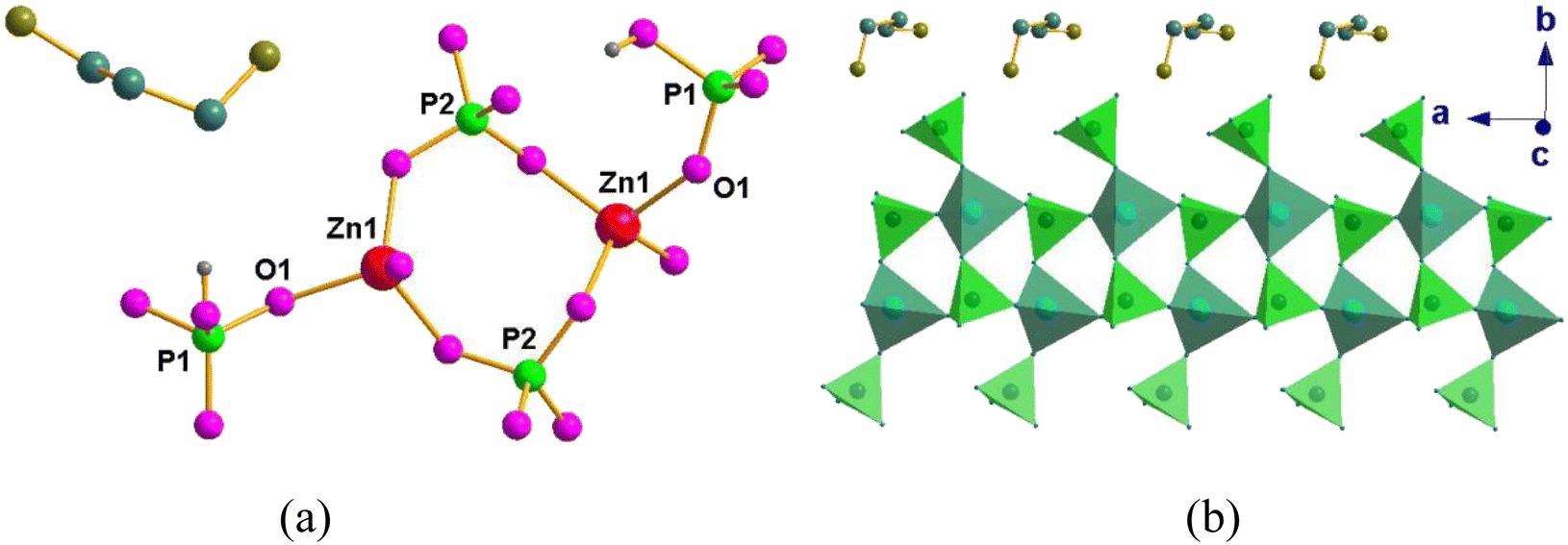 | ||
| Fig. 1 (a) The asymmetric unit of 1 and (b) polyhedral view of 1 along the [100] axis showing a one-dimensional chain. The amine molecules are packed along the chains. | ||
Compound 2 crystallized in the monoclinic space group P21/c; its asymmetric unit is similar to that of 1, which also contains one Zn2+ ion, two crystallographically independent HPO42− ions and one quadruply protonated triethylenetetramine. As shown in Fig. 2, the Zn2+ ions of compound 2 have similar coordination structures to 1, and then the ZnO4 and HP(2)O4 tetrahedra were also connected into a one-dimensional ladder-like chain, in which the quadruply triethylenetetramine molecule sits in the middle of these channels and interacts with the framework through hydrogen bonds.
 | ||
| Fig. 2 (a) The asymmetric unit of 2 and (b) polyhedral view of 2 along the [100] axis showing a one-dimensional chain. The amine molecules are packed along the chains. | ||
Obviously, both compounds have a similar asymmetric unit and stacking structure. However, there is a great difference in the dense hydrogen bonds between the two compounds. As shown in Fig. 3(a), the nitrogen atom of the DAP with the oxygen of the phosphate has intense O⋯N distances for compound 1, such as dO8⋯N1 = 2.965 Å, dO6⋯N1 = 2.938 Å, dO5⋯N1 = 2.818 Å, dO5⋯N2 = 2.853 Å, dO7⋯N2 = 2.881 Å, and dO4⋯N2 = 2.966 Å. These close distances extend in three dimensions, resulting in a dense hydrogen grid between the inorganic framework and the organic cations. Fig. 3(b) shows that the nitrogen atom of the triethylenetetramine with the oxygen of the phosphate has closer O⋯N distances, such as dO8⋯N1 = 2.891 Å, dO5⋯N1 = 2.645 Å, dO5⋯N2 = 2.681 Å, and dO6⋯N2 = 2.688 Å. Unfortunately, the twisted triethylenetetramine formed a hydrogen bond grid with the inorganic framework only in the ac plane owing to its large steric effect.
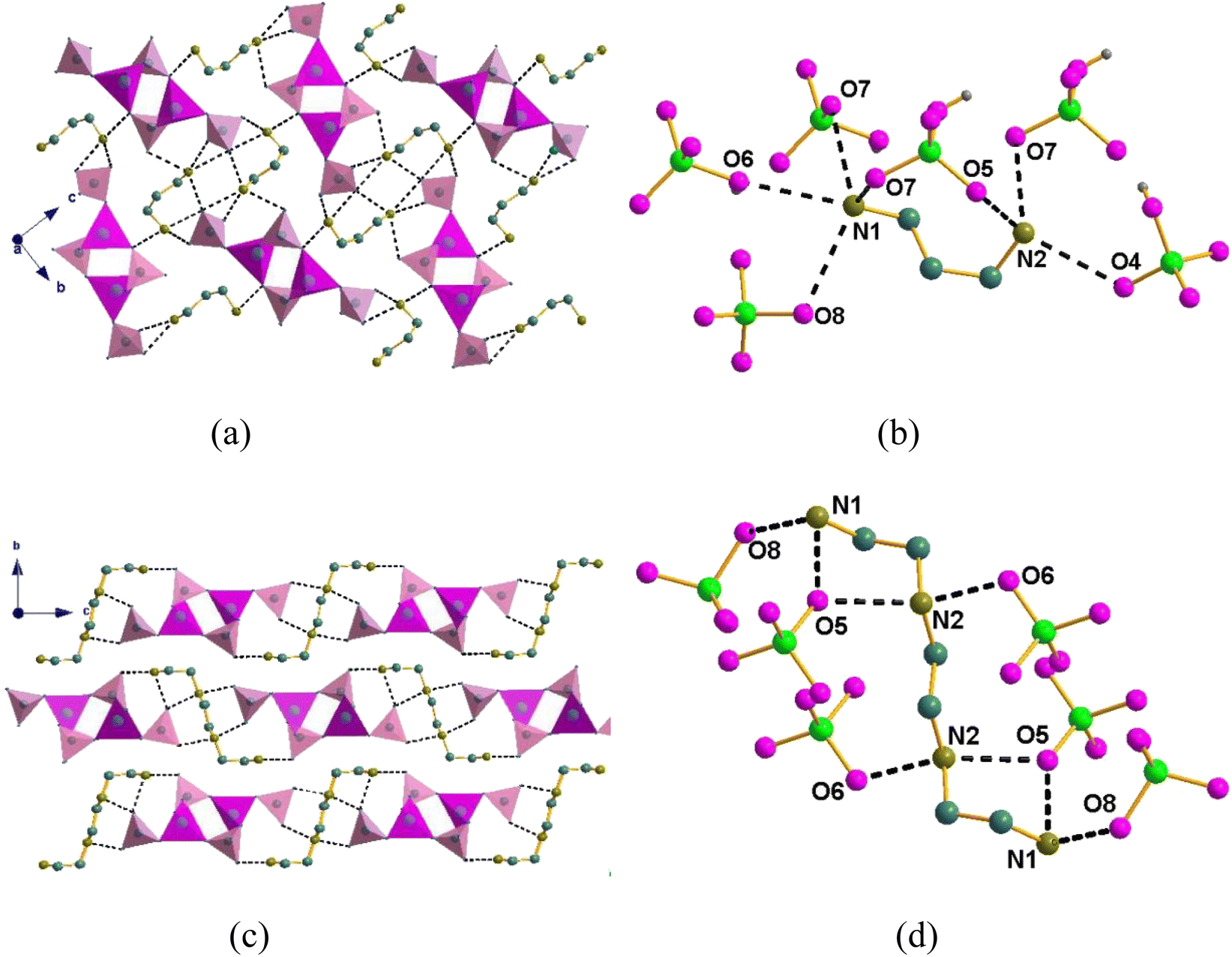 | ||
| Fig. 3 H-bond interactions between the guests and the inorganic [Zn(HPO4)2]∞ chains in the crystals of (a) and (b) 1 and (c) and (d) 2. | ||
The experiment PXRD and FTIR of 1 and 2 are shown in Fig. 4 and S3,† respectively. The PXRD curves are consistent with those of the simulations, indicating that they have high purity (Fig. 4). Scanning electron microscopy (SEM) characterized the morphology of the obtained samples (Fig. 5) and demonstrated that the two compounds show a regular rod-like structure with similar diameter and length, which further indicates that they not only have a similar crystal structure but also have the same macroscopic morphology. As shown in Fig. S2,† the TG curves of compounds 1 and 2 showed similar one-step decomposition thermogravimetric behavior. They started weight loss at 500 K and 527 K for 1 and 2, respectively, which corresponded to the decomposition of charge-compensating cations occupied within the inorganic chains. The mass loss (ca. 29.5% and 29.3%) is also close to the calculated value (29.6%). The TG results demonstrated that 1 and 2 were thermally stable below ca. 500 K.
 | ||
| Fig. 4 PXRD patterns of (a) 1 and (b) 2 for the simulated and synthesized compounds. | ||
 | ||
| Fig. 5 SEM images of (a) compound 1 and (b) compound 2. | ||
The conducting performance of the samples was first measured at a fixed temperature of 303 K with a humidity range of 75–99% RH using ac impedance spectroscopy. The unregular arc in the high frequency was observed corresponding to the boundary resistance and a spike in the low-frequency range related to the electrode contribution, respectively. As shown in Fig. S4,† both 1 and 2 have strong humidity-dependent proton conductivity; specifically, proton conductivity increases with humidity. Of the two compounds, compound 1 exhibited higher proton conductivity at low humidity. Its proton conductivity reached 1.00 × 10−3 S cm−1 at 75% RH, which is higher than almost all reported open-framework metal phosphates, such as [HIm]2[Zn3(HPO3)4]28 (2.2 × 10−4 S cm−1, 77% RH and 298 K), which is also superior to most other types of crystallize porous materials, such as (Me2NH2)2(H3O)[GdL2]·8H2O35 (2.35 × 10−4 S cm−1, 60% RH and 298 K) and {R3N(CH2CO2H)}[MCr(ox)3]·nH2O41 (8 × 10−5 S cm−1, 60% RH and 298 K). Then, the values increased to 2.54 × 10−3 S cm−1 at 85% RH and achieved a maximum value of 8.33 × 10−3 S cm−1 at 99% RH.
However, for 2, its proton conductivity is only 1.04 × 10−7 S cm−1 at 75% RH and then reaches 2.87 × 10−6 S cm−1 at 85% RH and 1.96 × 10−4 S cm−1 at 99% RH. In contrast, the proton conductivity of 2 is far less than that of 1 under the same conditions, but the gap narrowed with increasing humidity, which is four orders of magnitude difference at 75% RH and two orders of magnitude difference, while the proton conductivity increases to 99% RH. This indicates that the effective hydrogen bonding network between the proton-hoping sites (–HPO4) and DAP for 1 is favorable for proton conduction under a low humidity environment by analyzing the structure of both compounds. The increasing proton conductivity with humidity of 1 and 2 shows that water molecules also play a role in proton conduction, which promotes effective proton transport because water molecules form proton carriers in the form of H2O–H3O+ for proton hopping.
To gain insight into the influence of the structure of the compounds on proton conduction, we measured the water sorption isotherms of 1 and 2 at 298 K. The results show that the amount of water sorption for 1 is considerably higher than that for 2, as shown in Fig. 6. This further indicates that a dense hydrogen bond grid in the chain is essential for the high conductivity of the compound.
 | ||
| Fig. 6 Water-adsorption/desorption isotherm of 1 and 2 at 298 K. | ||
Given the high proton conductivity of compound 1 at low RH, we further explored the variation in its proton conductivity with temperature under the same low RH condition. The temperature dependence of compound 1 was tested at fixed humidity of 75% RH, and the resultant curves in Fig. S5† show that the proton conductivity increased as the temperature increased. The values showed 1.00 × 10−3 S cm−1, 1.32 × 10−3 S cm−1, 1.64 × 10−3 S cm−1 and 1.97 × 10−3 S cm−1 at 303 K, 313 K, 323 K and 333 K, respectively. This indicates that compound 1 can exhibit high proton conductivity in a wide range of medium-low temperatures, and the extent of conductivity change is not large, which is conducive for stable proton transport in its practical application. Furthermore, we measured the temperature dependence of proton conductivity at 85% RH and 99% RH (Fig. 7 and S5†). At 85% RH, the values showed 2.54 × 10−3 S cm−1, 2.99 × 10−3 S cm−1, 3.21 × 10−3 S cm−1 and 3.43 × 10−3 S cm−1 at 303 K, 313 K, 323 K and 333 K, respectively; then, at 99% RH, the conductivity increased from 6.99 × 10−3 S cm−1 to 9.66 × 10−3 S cm−1 and reached the highest value of 1.11 × 10−2 S cm−1 at 333 K. This remarkable proton conduction of 1 (1.11 × 10−2 S cm−1) is comparable to Nafion as well as several of the highest performing metal phosphate materials under the same conditions.
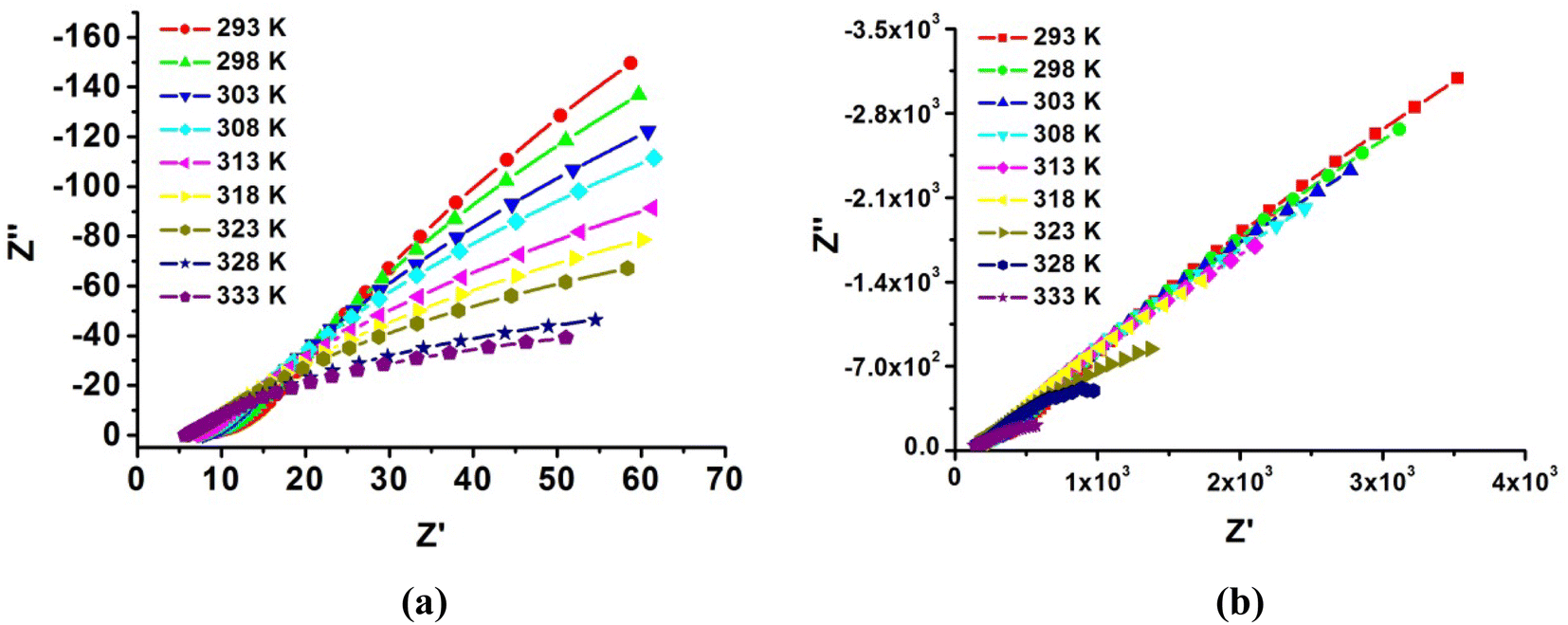 | ||
| Fig. 7 Temperature-dependent of Nyquist plots of (a) compound 1 and (b) compound 2 at 99% RH. | ||
Similarly, it can also be clearly observed that the temperature dependence on the proton conductivity of compound 2 is proportional to the increasing temperature. The conductivity increased from 1.74 × 10−4 S cm−1 (298 K) to 2.82 × 10−4 S cm−1 (313 K) and reached 4.60 × 10−4 S cm−1 at 333 K and 99% RH. Undoubtedly, the value of 2 at any temperature point is lower than 1.
To investigate the proton transfer mechanism, the activation energy (Ea) of 1 and 2 were obtained from the Arrhenius equation in the temperature range of 293–333 K:
 | (1) |
It can be induced that the activation energy is 0.24 eV (75% RH), 0.15 eV (85% RH) and 0.14 eV (99% RH) for 1, demonstrating that 1 followed the Grothuss mechanism (Fig. 8 and S6†). The activation energy of 2 is 0.23 eV at 99% RH, indicating that the proton transfer mechanism of 2 also belongs to the Grothuss mechanism at this conduction. The calculated activation energy, together with the structural difference between both compounds, is helpful in obtaining insight into the proton pathways. For 1, the three-dimensional hydrogen bond network can absorb more water molecules to construct intense hydrogen bond networks in the chains for proton hopping, resulting in a lower Ea and Grotthuss mechanism in proton conduction. However, at high RH, 2 can also absorb more water molecules into the inter-chain spaces; therefore, 2 preferred to form efficient proton-hopping pathways rather than the migration of the cations owing to its large hindrance, resulting in the Grotthuss mechanism.
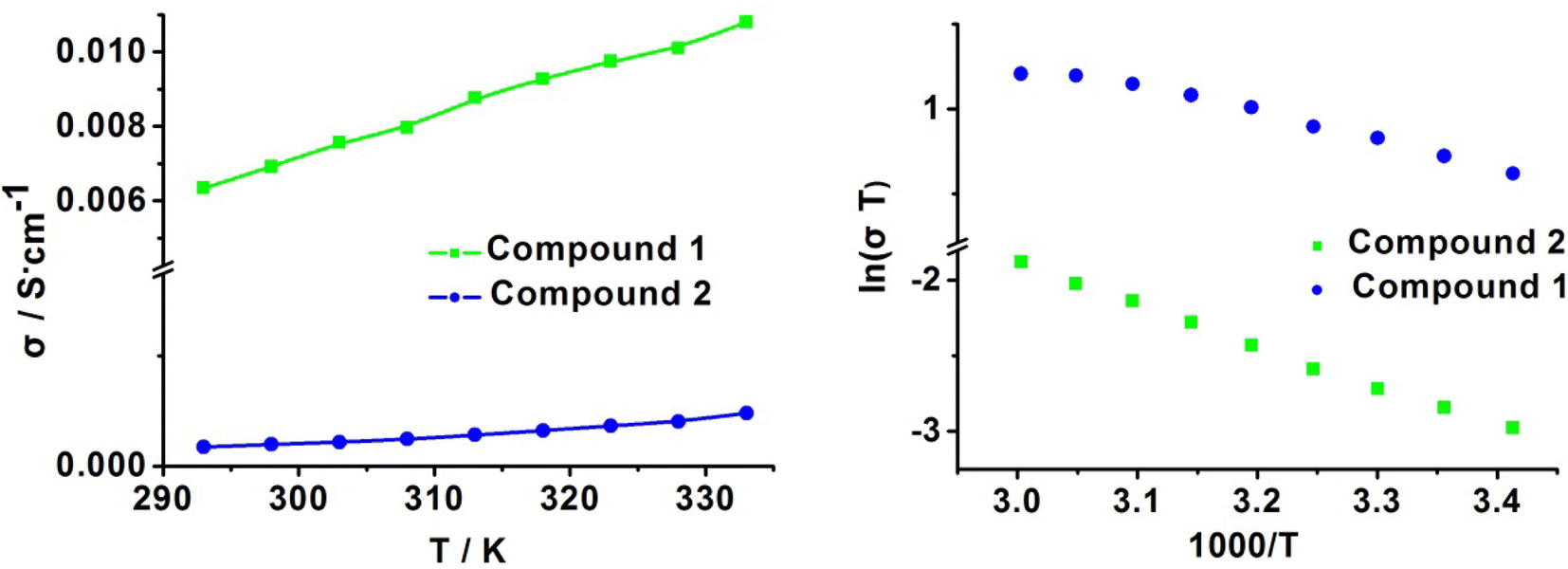 | ||
| Fig. 8 The corresponding conductivity in the form of (a) σ vs. T and (b) ln(σT) vs. 1000/T at 99% RH for 1 and 2. | ||
These results demonstrate that the intense hydrogen bond networks play an important role in the proton conducting of open-framework metal phosphates and that the appropriate size of the organic amine cations helps to form denser hydrogen bonds. For example, the diprotonated DAP molecules in 1 are more favorable for forming a dense hydrogen-bond network in three dimensions than in compound 2, in which the twisted triethylenetetramine formed a hydrogen bond grid with the inorganic framework only in two dimensions owing to its large steric effect. Compared with the previously prepared 1D compound (C4H12N2)1.5[Fe2(OH)(H2PO4)(HPO4)2(PO4)]·H2O (3), its larger size piperazine leads to the formation of weaker hydrogen bonds, which also makes the proton conductivity of 3 (2.61 × 10−4 S cm−1 at 99% RH and 293 K) lower than 2.
Moreover, in addition to organic equilibrium cations, we pay attention to the important role of inorganic frameworks in the construction of hydrogen bond grids of open-framework metal phosphates. For example, the two previously prepared 2D open-framework metal phosphates (C2H10N2)[Mn2(HPO4)3(H2O)] (4) and (C2H10N2)[Mn2(PO4)2]·2H2O (5) have the same organic equilibrium cations but various inorganic frameworks. The more hydrogen bonding sites of the inorganic framework in 4 resulted in proton conductivity (1.65 × 10−3 S cm−1), which was much higher than 5 (1.14 × 10−5 S cm−1) under the same conditions (293 K and 99% RH). Therefore, the design of an inorganic framework with abundant hydrogen bonding sites and matching suitable small molecular equilibrium cations can form dense hydrogen bonds, which is helpful for obtaining highly conductive open-framework metal phosphates. Further studies to design and prepare open-framework metal phosphates with high proton conductivity based on the above two aspects of analysis are in progress and will be reported in due course.
4. Conclusion
In summary, we fabricated two 1D zinc phosphates [C3N2H12][Zn(HPO4)2] (1) and [C6N4H22]0.5[Zn(HPO4)2] (2) with similar crystal structures. For 1, the pendant phosphoate groups of the inorganic framework and the 1,3-diaminopropane formed an intense hydrogen bond network for proton conducting, which exhibited high proton conductivity of 1.00 × 10−3 S cm−1 at 75% RH (303 K) and then up to 1.11 × 10−2 S cm−1 at 99% RH (333 K). For 2, the pendant phosphoate groups and the triethylenetetramine formed only hydrogen bond fragments in the planes owing to the larger steric hindrance of cations, which gives the result that the values only reached 10−4 S cm−1 at 99% RH. These detailed investigations demonstrate that the suitable size of the organic cations in 1 is helpful for the formation of efficient hydrogen bond grids and then provide efficient proton-conductive pathways in a wide humidity range. This study provides the promise of selecting appropriate organic amine for constructing reasonable proton conducting pathways in metal phosphates to improve conductivity. Our group will further study the relationship between the structure of the compound and proton conductivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Foundation of the Jiangsu Education Committee (20KJB150006), Major Project of Nanjing Institute of Technology Innovation Fund (CKJA202001) for financial support. The present work was supported by the outstanding scientific and technological innovation team in colleges and universities of Jiangsu province (202211276140H).References
- K.-D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Transport in Proton Conductors for Fuel-Cell Applications: Simulations, Elementary Reactions, and Phenomenology, Chem. Rev., 2004, 104, 4637–4678 CrossRef CAS PubMed.
- B. C. H. Steele and A. Heinzel, Materials for Fuel-Cell Technologies, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- M. Inukai, S. Horike, T. Itakura, R. Shinozaki, N. Ogiwara, D. Umeyama, S. Nagarkar, Y. Nishiyama, M. Malon, A. Hayashi, T. Ohhara, R. Kiyanagi and S. Kitagawa, Encapsulating Mobile Proton Carriers into Structural Defects in Coordination Polymer Crystals: High Anhydrous Proton Conduction and Fuel Cell Application, J. Am. Chem. Soc., 2016, 138, 8505–8511 CrossRef CAS PubMed.
- G. Shen, J. Liu, H. B. Wu, P. Xu, F. Liu, C. Tongsh, K. Jiao, J. Li, M. Liu, M. Cai, J. P. Lemmon, G. Soloveichik, H. Li, J. Zhu and Y. Lu, Multi-functional anodes boost the transient power and durability of proton exchange membrane fuel cells, Nat. Commun., 2020, 11, 1191–1202 CrossRef CAS PubMed.
- H. W. Zhang and P. K. Shen, Recent Development of Polymer Electrolyte Membranes for Fuel Cells, Chem. Rev., 2012, 112, 2780–2832 CrossRef CAS PubMed.
- H. Yuan, H. F. Dai, X. Z. Wei and P. W. Ming, Model-based observers for internal states estimation and control of proton exchange membrane fuel cell system: A review, J. Power Sources, 2020, 468, 228376 CrossRef CAS.
- J. Zhang, B. Hu, X. Deng, C. Li, Y. S. Wu, C. Zhou, D. Z. Zhang, L. Ge, W. Zhou and Z. P. Shao, Perovskite-Carbon Joint Substrate for Practical Application in Proton Exchange Membrane Fuel Cells under Low-Humidity/High- Temperature Conditions, ACS Appl. Mater. Interfaces, 2022, 14, 30872–30880 CrossRef CAS PubMed.
- M. Dogru, M. Handloser, F. Auras, T. Kunz, D. Medina, A. Hartschuh, P. Knochel and T. Bein, A Photoconductive Thienothiophene-Based Covalent Organic Framework Showing Charge Transfer Towards Included Fullerene, Angew. Chem., 2013, 52, 2920–2924 CrossRef CAS PubMed.
- D. Gopalakrishnan and W. R. Dichtel, Direct Detection of RDX Vapor Using a Conjugated Polymer Network, J. Am. Chem. Soc., 2013, 135, 8357–8362 CrossRef CAS PubMed.
- Y. J. Sun, Y. Yan, Y. Y. Wang, Y. Li, J. Y. Li and J. H. Yu, High proton conduction in a new alkali metal-templated open-framework aluminophosphate, Chem. Commun., 2015, 51, 9317–9319 RSC.
- Y. Mu, Y. Wang, Y. Li, J. Li and J. H. Yu, Organotemplate-free synthesis of an open-framework magnesium aluminophosphate with proton conduction properties, Chem. Commun., 2015, 51, 2149–2151 RSC.
- M. Wang, H. B. Luo, S. X. Liu, Y. Zou, Z. F. Tian, L. Li, J. L. Liu and X. M. Ren, Water assisted high proton conductance in a highly thermally stable and superior water-stable open-framework cobalt phosphate, Dalton Trans., 2016, 45, 19466–19472 RSC.
- H. R. Zhao, C. Xue, C. P. Li, K. M. Zhang, H. B. Luo, S. X. Liu and X. M. Ren, A Two-Dimensional Inorganic-Organic Hybrid Solid of Manganese(II) Hydrogenophosphate Showing High Proton Conductivity at Room Temperature, Inorg. Chem., 2016, 55, 8971–8975 CrossRef CAS PubMed.
- K. M. Zhang, F. Y. He, H. B. Duan and H. R. Zhao, An Alkali Metal Ion-Exchanged Metal-Phosphate (C2H10N2)xNa1-x[Mn2(PO4)2] with High Proton Conductivity of 10-2 S.cm-1, Inorg. Chem., 2019, 58, 6639–6646 CrossRef CAS PubMed.
- H. R. Zhao, Y. Jia, Y. Gu, F. Y. He, K. M. Zhang, Z. F. Tian and J. L. Liu, A 3D open-framework iron hydrogenophosphate showing high proton conductance under water and aqua-ammonia vapor, RSC Adv., 2020, 10, 9046–9051 RSC.
- X. Chen and G. Li, Proton conductive Zr-based MOFs, Inorg. Chem. Front., 2020, 7, 3765–3784 RSC.
- D. W. Lim and H. Kitagawa, Proton Transport in Metal-Organic Frameworks, Chem. Rev., 2020, 120, 8416–8467 CrossRef CAS PubMed.
- Y. X. Ye, L. S. Gong, S. C. Xiang, Z. J. Zhang and B. L. Chen, Metal-Organic Frameworks as a Versatile Platform for Proton Conductors, Adv. Mater., 2020, 32, 1907090 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Proton Conduction in Metal-Organic Frameworks and Related Modularly Built Porous Solids, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- X. Y. Dong, J. H. Wang, S. S. Liu, Z. Han, Q. J. Tang, F. F. Li and S. Q. Zang, Synergy between Isomorphous Acid and Basic Metal-Organic Frameworks for Anhydrous Proton Conduction of Low-Cost Hybrid Membranes at High Temperatures, ACS Appl. Mater. Interfaces, 2018, 10, 38209–38216 CrossRef CAS PubMed.
- S. Horike, D. Umeyama and S. Kitagawa, Ion Conductivity and Transport by Porous Coordination Polymers and Metal-Organic Frameworks, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS.
- S. Chand, S. M. Elahi, A. Pal and C. D. Madhab, Metal Organic Frameworks and Other Crystalline Materials for Ultrahigh Superprotonic Conductivities of 10-2 S.cm-1 or Higher, Chem. - Eur. J., 2019, 25, 6259–6269 CrossRef CAS.
- X. Meng, H. N. Wang, S. Y. Song and H. J. Zhang, Proton-conducting crystalline porous materials, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
- A. L. Li, Q. Gao, J. Xu and X. H. Bu, Proton-conductive metalorganic frameworks: Recent advances and perspectives, Coord. Chem. Rev., 2017, 344, 54–82 CrossRef CAS.
- C. R. Kayaramkodath, I. Rajith, D. V. Sairam, P. Joseph, C. W. Vivek, K. V. R. Goudappagouda, K. Sreekumar and S. B. Sukumaran, Imidazole-Linked Crystalline Two-Dimensional Polymer with Ultrahigh Proton-Conductivity, J. Am. Chem. Soc., 2019, 141, 14950–14954 CrossRef PubMed.
- F. M. Zhang, L. Z. Dong, J. S. Qin, W. Guan, J. Liu, S. L. Li, M. Lu, Y. Q. Lan, Z. M. Su and H. C. Zhou, Effect of Imidazole Arrangements on Proton-Conductivity in Metal-Organic Frameworks, J. Am. Chem. Soc., 2017, 139, 6183–6189 CrossRef CAS PubMed.
- Q. Qiao, H. J. Wang, C. P. Li, X. Z. Wang and X. M. Ren, Improving proton conduction of the Prussian blue analogue Cu3[Co(CN)6]2·nH2O at low humidity by forming hydrogel composites, Inorg. Chem. Front., 2021, 8, 2305–2314 RSC.
- R. Shankar, E. Jakhar, A. Dubey, P. Chauhan, P. K. Tiwari and S. Basu, Studies on the Genesis and Proton Conductivity of Imidazole-Based Linear and Open-Framework Zinc Phosphites, Inorg. Chem., 2021, 60, 6569–6575 CrossRef CAS PubMed.
- R. Li, S. H. Wang, X. X. Chen, J. Lu, Z. H. Fu, Y. Li, G. Xu, F. K. Zheng and G. C. Guo, Highly Anisotropic and Water Molecule-Dependent Proton Conductivity in a 2D Homochiral Copper(II) Metal-Organic Framework, Chem. Mater., 2017, 29, 2321–2331 CrossRef CAS.
- S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, One-Dimensional Imidazole Aggregate in Aluminium Porous Coordination Polymers with High Proton Conductivity, Nat. Mater., 2009, 8, 831–836 CrossRef CAS PubMed.
- S. Horike, D. Umeyama, M. Inukai, T. Itakura and S. Kitagawa, Coordination -Network-Based Ionic Plastic Crystal for Anhydrous Proton Conductivity, J. Am. Chem. Soc., 2012, 134, 7612–7615 CrossRef CAS PubMed.
- X. M. Li, Y. M. Wang, Y. Mu, J. Liu, L. Zeng and Y.-Q. Lan, Superprotonic Conductivity of a Functionalized Metal-Organic Framework at Ambient Conditions, ACS Appl. Mater. Interface, 2022, 14, 9264–9271 CrossRef CAS PubMed.
- W. J. Phang, H. Jo, W. R. Lee, J. H. Song, K. Yoo, B. Kim and C. S. Hong, Superprotonic Conductivity of a UiO-66 Framework Functionalized with Sulfonic Acid Groups by Facile Postsynthetic Oxidation, Angew. Chem., Int. Ed., 2015, 54, 5142–5146 CrossRef CAS PubMed.
- Z. N. Que, Y. X. Ye, Y. S. Yang, F. H. Xiang, S. M. Chen, J. L. Huang, Y. B. Li, C. L. Liu, S. C. Xiang and Z. J. Zhang, Solvent-Assisted Modification to Enhance Proton Conductivity and Water Stability in Metal Phosphonates, Inorg. Chem., 2020, 59, 3518–3522 CrossRef CAS PubMed.
- S. L. Zhang, Y. X. Xie, M. R. Yang and D. R. Zhu, Porosity regulation of metal-organic frameworks for high proton conductivity by rational ligand design: mono-versus disulfonyl-4,4’-biphenyldicarboxylic acid, Inorg. Chem. Front., 2022, 9, 1134–1142 RSC.
- B. G. Montse, R. S. Inés, M. P. C. Rosario, X. Konstantinos, V. Didier, S. Norbert, L. G. Mar, d. R. Carmen, R. L. Enrique, C. Aurelio, D. D. Konstantinos and O. P. Pascual, Layered Lanthanide Sulfophosphonates and Their Proton Conduction Properties in Membrane Electrode Assemblies, Chem. Mater., 2019, 31, 9625–9634 CrossRef.
- P. Bhanja, A. Palui, S. Chatterjee, Y. V. Kaneti, J. Na, Y. Sugahara, A. Bhaumik and Y. Yamauchi, Crystalline Porous Organic Polymer Bearing -SO3H Functionality for High Proton Conductivity, ACS Sustainable Chem. Eng., 2020, 8, 2423–2432 CrossRef CAS.
- M. Subhabrata, D. Joyashish, S. Chandani, S. Rudraditya, O. Basu and S. K. Das, Designing UiO-66-Based Superprotonic Conductor with the Highest Metal-Organic Framework Based Proton Conductivity, ACS Appl. Mater. Interfaces, 2019, 11, 13423–13432 CrossRef PubMed.
- R. Wang, X. Y. Dong, H. Xu, R. B. Pei, M. L. Ma, S. Q. Zang, H. W. Hou and C. W. M. Thomas, Super water-stable europium-organic framework: guests inducing low humidity proton conduction and sensing of metal ions, Chem. Commun., 2014, 50, 9153–9156 RSC.
- J. Z. Li, H. Wu, L. Cao, X. Y. He, B. B. Shi, Y. Li, M. Z. Xu and Z. Y. Jiang, Enhanced Proton Conductivity of Sulfonated Polysulfone Membranes under Low Humidity via the Incorporation of Multifunctional Graphene Oxide, ACS Appl. Nano Mater., 2019, 2, 4734–4743 CrossRef CAS.
- M. Sadakiyo, H. Ōkawa, A. Shigematsu, M. Ohba, T. Yamada and H. Kitagawa, Promotion of Low-Humidity Proton Conduction by Controlling Hydrophilicity in Layered Metal-Organic Frameworks, J. Am. Chem. Soc., 2012, 134, 5472–5475 CrossRef CAS PubMed.
- H. A. Patel, N. Mansor, S. Gadipelli, D. J. L. Brett and Z. X. Guo, Superacidity in Nafion/MOF Hybrid Membranes Retains Water at Low Humidity to Enhance Proton Conduction for Fuel Cells, ACS Appl. Mater. Interface, 2016, 8, 30687–30691 CrossRef CAS PubMed.
- W. T. A. Harrison, Z. Bircsak, L. Hannooman and Z. H. Zhang, Two New 1,3-Diammonium-Propane Zinc Hydrogen Phosphates: H3N(CH2)3NH3.Zn2(HPO4)2 (H2PO4)2, with 12-Ring Layers, and H3N(CH2)3NH3.Zn(HPO4)2, with 4-Ring Ladders, J. Solid State Chem., 1998, 136, 93–102 CrossRef CAS.
- A. Choudhury, S. Neeraj, S. Natarajana and C. N. R. Rao, Transformations of the low-dimensional zinc phosphates to complex open-framework structures. Part 2:{one-dimensional ladder to two and three-dimensional structures, J. Mater. Chem., 2001, 11, 1537–1546 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01130e |
| This journal is © The Royal Society of Chemistry 2023 |