
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDysoticans F–H: three unprecedented dimeric cadinanes from Dysoxylum parasiticum (Osbeck) Kosterm. stem bark†
Al Arofatus Naini ab,
Tri Mayantiac,
Rani Maharaniac,
Sofa Fajriahd,
Kazuya Kabayamae,
Atsushi Shimoyamae,
Yoshiyuki Manabee,
Koichi Fukasee,
Sirriporn Jungsuttiwongf and
Unang Supratman*ab
ab,
Tri Mayantiac,
Rani Maharaniac,
Sofa Fajriahd,
Kazuya Kabayamae,
Atsushi Shimoyamae,
Yoshiyuki Manabee,
Koichi Fukasee,
Sirriporn Jungsuttiwongf and
Unang Supratman*ab
aDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jatinangor 45363, Sumedang, West Java, Indonesia. E-mail: unang.supratman@unpad.ac.id
bCentral Laboratory, Universitas Padjadjaran, Jatinangor 45363, Sumedang, West Java, Indonesia
cStudy Centre of Natural Product Chemistry and Synthesis, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jatinangor 45363, Sumedang, West Java, Indonesia
dResearch Center for Chemistry, National Research and Innovation Agency (BRIN), Kawasan PUSPIPTEK Serpong, Tangerang Selatan, 15314, Indonesia
eDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
fDepartment of Chemistry, Faculty of Science, Ubon Ratchathani University, Ubon Ratchathani, 34190, Thailand
First published on 22nd March 2023
Abstract
An asymmetrical true-dimeric cadinane via ketonic bridge [C-15/C-3′], dysotican F (1), two symmetrical pseudo-cadinane dimers through an O-ether linkage [C-3/C-3′], dysoticans G (2) and H (3), as well as three known sesquiterpenoids 4–6 were obtained from the stem bark of Dysoxylum parasiticum (Osbeck) Kosterm. (Meliaceae). Their structures were determined by spectroscopic and quantum chemical calculations of 13C NMR shifts using a GIAO method and electronic circular dichroism (ECD) using a TDDFT method. A possible biogenetic pathway for 1–3 beginning from the known compounds (i–ii) was proposed. Cytotoxic evaluation showed that 2 as a new lead compound is the most potent against the MCF-7 and HeLa cell lines with IC50 values of 12.07 ± 0.17 μM and 9.29 ± 0.33 μM, while 1 has moderate inhibition with IC50 values of 31.59 ± 0.34 μM and 27.93 ± 0.25 μM. Furthermore, 3 is a selective inhibitor against the HeLa cell growth with an IC50 value of 39.72 ± 0.18 μM. A brief structure–activity relationship analysis of all isolated compounds 1–6 was also provided, including comparison with the coexisting molecules in the previous report.
Introduction
Sesquiterpenoid dimers, are a group of naturally occurring metabolites that usually have C30 cores derived from two units of sesquiterpenoids, which may possess homo- or heterodimeric skeletons. Based on structural features and coupling patterns of the two sesquiterpenoid units, they are assigned into three major classes, namely true-disesquiterpenoids (linked via C–C bonds), pseudo-disesquiterpenoids (linked via C–O bonds), and mero-disesquiterpenoids (hybridized to both sesquiterpenoids and other constituents via C–C bods).1 More than 400 unprecedented sesquiterpenoid dimers have been discovered and extensively characterized, demonstrating the “majesty” of nature in producing fascinating compounds.2 The diverse architectures and the intriguing pharmacological profile of sesquiterpenoid dimers attracted the attention of natural product chemists, synthetic chemists, and pharmacologists.3 Furthermore, the genus Dysoxylum (Meliaceae) comprises approximately 200 species distributed in Australia, New Zealand, India, Indonesia, and Southeast Asia.4,5 Its previous phytochemical studies led to the reports of an array of products, including sesquiterpenoids,6,7 diterpenoids,8 triterpenoids as the major compound,5 steroids,9 and alkaloids,10 which exhibit diverse biological effects.Dysoxylum parasiticum (Osbeck) Kosterm. which is known as the fragrant tree or “Majegau” (http://powo.science.kew.org/taxon/578268-1/) is widely cultivated in West Java and Bali. The leaves have been reported to yield three dimers11 and two trimers of sesquiterpene phenols12 with activity against HL-60 cell line, as well as one bicoumarin derivative.13 Furthermore, the stem bark produced two undescribed sesquiterpenoids, namely 10β-11-dihydroxy-1β-hydroperoxide-4αH,5αH,7βH-guaiane, 10β-hydroxy-4α,4β-dimethyl-eudesm-3-one, as well as three known sesquiterpenoids with moderate anticancer activity against MCF-7 breast cancer (IC50 12.17–33.46 μM).7 Two undescribed sesquiterpenoids (dysoticans A and B), three undescribed sesquiterpenoid dimers (dysoticans C–E), and six analogs, showing IC50 values ranging from 22.15 to 45.14 μM against MCF-7 and HeLa cell lines, were also yielded.14 In the continuous search for novel bioactive metabolites from the genus Dysoxylum, Dysoxylum parasiticum (Osbeck) Kosterm. collected from West Java Province of Indonesia was chemically investigated. Dysotican F (1), the first asymmetrical true-dimeric cadinanes via ketonic bridge, and dysoticans G and H (2–3), two symmetrical pseudo-cadinane dimers through O-ether linkage, as well as three known sesquiterpenoid compounds, 4–6, were isolated and identified by diverse method. Finally, the isolation, structure determination, and biological evaluation of these molecules were described.
Results and discussion
Compound 1 was isolated as a colourless oil, and its molecular formula, C30H47O4, was established by positive HR-ESI-TOFMS (m/z 471.3479 [M + H]+, calcd for 471.3474), referencing eight degrees of unsaturation. It showed a maximum UV absorption at 246 and 315 nm in methanol, and its infrared spectrum exhibited a broad and sharp band at 3431 cm−1 and 1650 cm−1, respectively, as well as a paired bands at 1464 and 1374 cm−1, which are characteristic of a hydroxy group, ketone conjugation system, and gem-dimethyl group. The 1H NMR spectra of 1 showed six secondary methyls [δH 0.75 (d = 7 Hz, H-13); 0.77 (d = 7 Hz, H-13′); 0.90 (d = 7 Hz, H-14′); 0.91 (d = 7 Hz, H-12); 0.95 (d = 7 Hz, H-14); 0.99 (d = 7 Hz, H-12′)], one tertiary methyl [δH 1.29 (s, H-15′)], two oxygenated methines [δH 3.82 (m, H-2); 4.80 (brs, H-5′)], and one olefinic hydrogen [δH 5.70 (brs, H-3)]. Also, the 13C NMR and DEPT results as well as HMQC spectra, indicated that 30 carbons were assigned to a ketone carbonyl at δC 196.9 (C-15′), six olefinic carbons, including one methine at δC 102.0 (C-3) and five tertiary carbons [δC 110.0 (C-6′); 131.9 (C-6); 137.5 (C-1′); 143.7 (C-1); 157.5 (C-4)], 23 sp3 carbon atoms, involving two oxygen-bearing system methines [δC 70.9 (C-5′); 73.8 (C-2)] and one nonprotonated at δC 88.7 (C-4′), seven methines [δC 25.5 (C-10′); 28.7 (C-11); 30.7 (C-11′); 37.8 (C-10); 39.3 (C-7′); 42.0 (C-7); 55.3 (C-3′)], five methylenes [δC 18.0 (C-8); 20.9 (C-8′); 26.7 (C-9′); 30.6 (C-9); 35.9 (C-2′)], and seven methyls [δC 16.8 (C-13); 18.8 (C-15′); 19.6 (C-14); 19.6 (C-13′); 20.5 (C-14′); 21.8 (C-12′)], representing four double-bond equivalents. The remaining degrees of unsaturation were due to the presence of four rings in the molecule 1. These characteristics, considering the structure types from Dysoxylum genus, indicated that 1 could be a sesquiterpene dimer, and its architecture was constructed mainly by 2D NMR data (Fig. 1). A spin system inferred from 1H–1H COSY correlations (Fig. 2), between H-14/H-10/H-9/H-8/H-7/H-11, H-12/H-11/H-13 and H-14′/H-10′/H-9′/H-8′/H-7′/H-11′, H-12′/H-11′/H-13′ of units A and B, respectively, in addition to HMBC correlations (Fig. 2), from H-14 to C-1, C-9, C-10, H-14′ to C-1′, C-9′, C-10′, H-12/H-13 to C-7, C-11, H-12′/H-13′ to C-7′, C-11′, showed the moiety of secondary methyl at C-10/C-10′ and the isopropyl at C-7/C-7′ in the structure of compound 1. HMBC correlations of H-8/H-11 to C-6 and H-8′/H-11′ to C-6′ confirmed that the skeleton of the two units in 1 belongs to sesquiterpenoid cadinene-type with two pairs of double bonds positioned at C-1/C-6 and C-1′/C-6′. Subsequently, an α,β-ketone at C-3/C-4/C-15 of A unit was established by correlations of H-2/H-5 to C-3, H-5 to C-15, as well as the cross-peak of H-2/H-3 in 1H–1H COSY. Meanwhile, the attachment of three hydroxyls at C-2 in the A unit and C-4′, C-5′ in the B unit was evident by correlations of (H-10 to C-2, H-2 to C-1, C-10), (H-15′ to C-3′, C-4′), and (H-15′ to C-5′). Units A and B were connected by the direct carbon–carbon bond between C-15 and C-5′ based on the 1H–1H COSY key correlation of H-2/H-3′ and the apparent HMBC correlations from H-2 to C-6, C-10, H-3′ to C-4, and H-2′/H-3′ to C-15 to form a ketonic bridge linking the two units in 1, thereby deducing the planar structure of 1.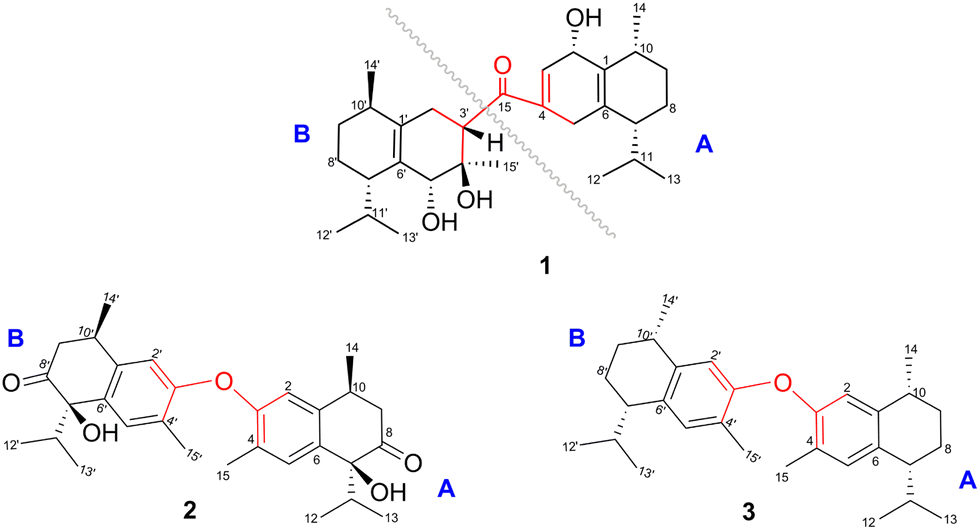 | ||
| Fig. 1 Chemical structures of compounds 1–3. | ||
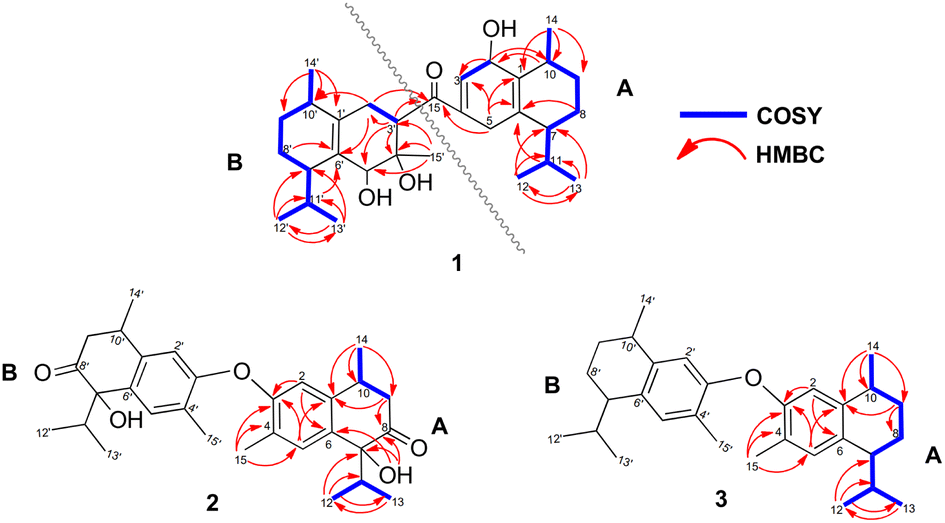 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of compounds 1–3. | ||
The relative configurations of the chiral centers in 1 were assigned by the basis of the detailed analysis of its Nuclear Over Hauser Effect Spectroscopy (NOESY) spectrum, as shown in Fig. 3. Since the two monomers of 1 connected through a flexible segment, the configuration was determined separately. The correlations of H-10(β-oriented) to H-2 and H-2/H-10 to H-7 suggested that the relative configurations of hydroxyl at C-2, isopropyl at C-7, and CH3-14 at C-10 in the A unit were an α-orientation. Furthermore, NOE interactions of H-7′ to H-13′, H-13′ to H-11′, H-11′/H-13′ to H-5′, and H-5′ to H-3′, indicated the α-orientations of isopropyl at C-7′ and hydroxyl at C-5′ as well as β-orientation of hydrogen methine at C-3′. The β-orientation of CH3-14′ was supported by NOESY cross-peak (Fig. 3) from H-14′ to H-3′(β-oriented), but the absence of correlation between CH3-15′ and these β protons in the B unit indicate that CH3-15′ was an α-configuration. In order to further establish the relative configuration of 1 as deduced above based on the spectral analysis, the (2S,7R,10R,3′S,4′R,5′R,7′S,10′R)-1 configuration was performed according to NMR chemical shift calculations using gauge independent atomic orbital (GIAO) method at PCM/mPW1PW91/6-311+G(d,p) level using chloroform as a solution. As a results (Fig. 4), the calculated 13C NMR chemical shifts of (2S,7R,10R,3′S,4′R,5′R,7′S,10′R)-1 were in strong agreement with the experimental one (Table 1). The evidence of an excellent correlation with R-coefficient of 0.9996 and a low standard deviation (STDEV) of 0.98 ppm, the relative configuration of 1 was determined. Finally, its absolute configuration was also supported by a combination of electronic circular dichroism (ECD) calculations based on the time-dependent density functional theory (TDDFT) method at the B3LYP/6-31G(d,p) level using the Gaussian 09 program with the ECD spectra experimentally collected in methanol, as shown in Fig. 5. Therefore, compound 1 was defined as a true-dimeric cadinanes core featuring a ketonic bridge at C-15/C-3′ via direct C–C bond, and it was named as dysotican F.
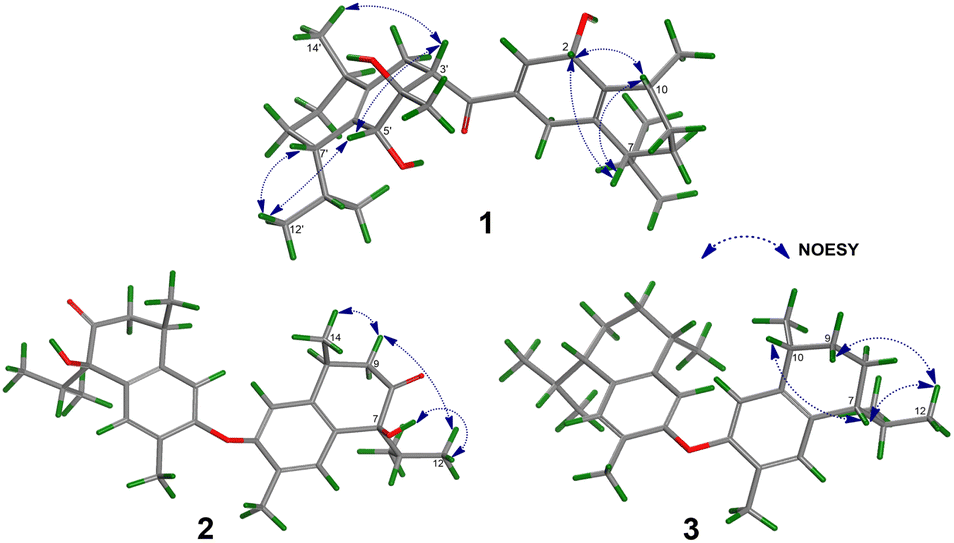 | ||
| Fig. 3 Key NOESY correlations of compounds 1–3. | ||
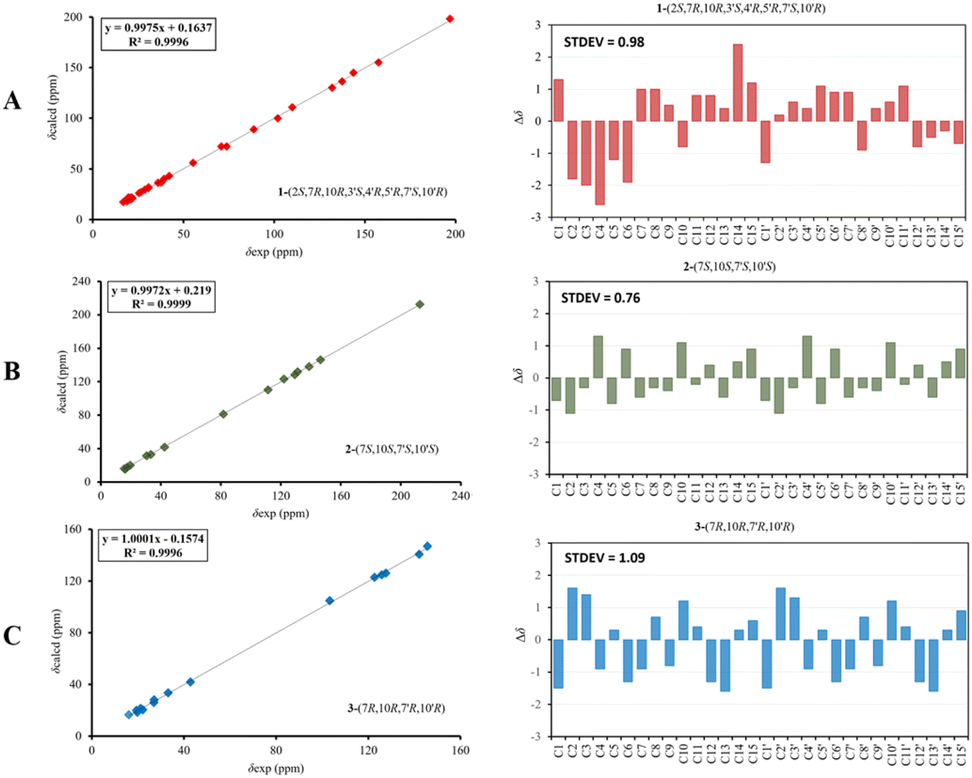 | ||
| Fig. 4 Regression analysis and individual deviations between experimental and calculated 13C NMR of 1 (A), 2 (B), and 3 (C). | ||
| 1 | |||||
|---|---|---|---|---|---|
| No. | Unit A | No. | Unit B | ||
| δH (J in Hz) | δC | δH (J in Hz) | δC | ||
| a Overlapped signal. | |||||
| 1 | — | 143.7, s | 1′ | — | 137.5, s |
| 2 | 3.82 (m) | 73.8, d | 2a′ | 1.99 (m) | 35.9, t |
| 2b′ | 2.90 (m) | ||||
| 3 | 5.70 (brs) | 102.0, d | 3a′ | 2.79 (m) | 55.3, d |
| 3b′ | |||||
| 4 | — | 157.5, s | 4′ | — | 88.7, s |
| 5 | 2.05a (m) | 37.7, t | 5′ | 4.80 (s) | 70.9, d |
| 6 | — | 131.9, s | 6′ | — | 110.0, s |
| 7 | 2.29 (m) | 42.0, d | 7′ | 2.57 (m) | 39.3, d |
| 8a | 1.47 (m) | 18.0, t | 8a′ | 1.55 (m) | 20.9, t |
| 8b | 1.69a (m) | 8b′ | 1.60 (m) | ||
| 9a | 1.67a (m) | 30.6, t | 9a′ | 1.34 (m) | 26.7, t |
| 9b | 1.68a (m) | 9b′ | 1.64 (m) | ||
| 10 | 2.01a (m) | 37.8, d | 10′ | 2.87 (m) | 25.5, d |
| 11 | 2.25 (m) | 28.7, d | 11′ | 2.23 (m) | 30.7, d |
| 12 | 0.91 (d = 7) | 21.1, q | 12′ | 0.99 (d = 7) | 21.8, q |
| 13 | 0.75 (d = 7) | 16.8, q | 13′ | 0.77 (d = 7) | 19.6, q |
| 14 | 0.95 (d = 7) | 19.6, q | 14′ | 0.90 (d = 7) | 20.5, q |
| 15 | — | 196.9, s | 15′ | 1.29 (s) | 18.8, q |
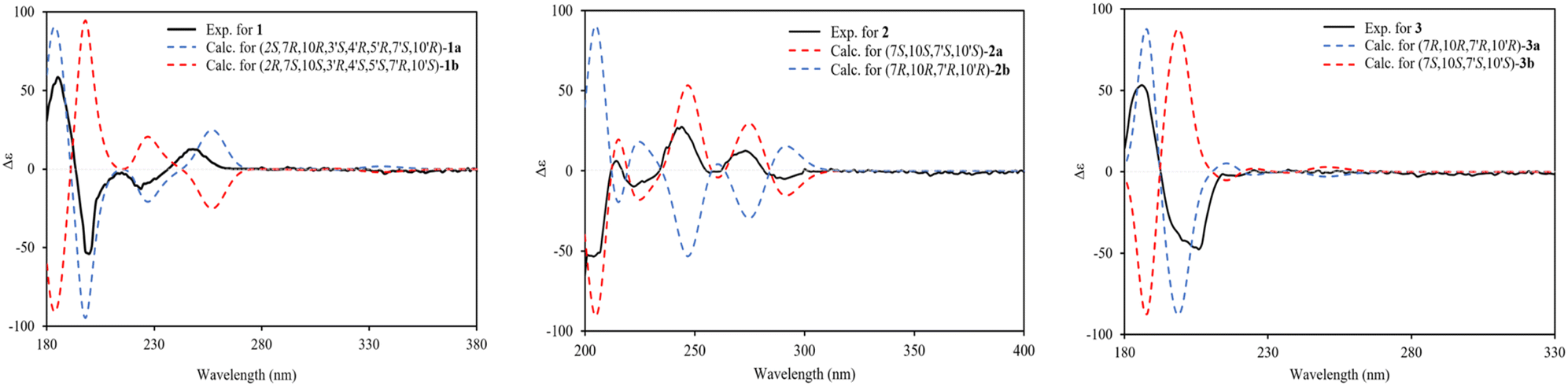 | ||
| Fig. 5 Experimental and calculated ECD spectra of compounds 1–3. | ||
Compound 2 was obtained as a pale brown, while a sodium adduct quasimolecular ion [M + Na]+ at m/z 501.2620 suggested a molecular formula of C30H38O5 (calcd for C30H38O5Na, 501.2617), indicating 12 degrees of unsaturation. The UV spectrum in methanol, showing an absorption band of about 189 nm, was indicative of a benzene group. The IR spectrum of 2 displayed characteristic of OH (3398 cm−1), carbonyl (1710 cm−1), olefinic/aromatic (1605 and 1513 cm−1), and gem-dimethyl (1441 and 1375 cm−1). Furthermore, the 1H NMR data (Table 2) of 2 showed typical signals assigned to a sesquiterpene–calamenene derivatives, including the presence of one deshielded tertiary methyl at δH 2.23 (s, H-15), three secondary methyls [δH 0.80 (d = 7 Hz, H-13); 0.83 (d = 7 Hz, H-12); 1.32 (d = 7 Hz, H-14)], two aromatic protons [δH 6.68 (s, H-2); 7.27 (s, H-5)], and one exchangeable proton at δH 3.88 (1H, brs). In contrast to its molecular formula, comprehensive analysis of the 13C NMR and HMQC spectra only exhibited 15 carbons, including one carbonyl of ketone at δC 212.6 (C-8), two sp2 methines [δC 111.5 (C-2); 129.3 (C-5)], four quaternary sp2 carbons [δC 122.0 (C-4); 131.1 (C-6); 138.8 (C-1); 146.4 (C-3)], two sp3 methines [δC 30.4 (C-10); 33.2 (C-11)], one sp3 methylene at δC 42.4 (C-9), and four sp3 methyls [δC 15.5 (C-15); 16.1 (C-13); 17.4 (C-12); 19.6 (C-14)] (Table 2). These molecular pieces accounted for four degrees of unsaturation out of 12, leaving eight DBEs in 2. Therefore, compound 2 was a symmetrical dimeric cadinanes of calamenene derivative. In the HMBC spectrum, the correlations from: H-12/H-13 to C-7 and C-11; H-14 to C-1, C-9, and C-10; H-15 to C-3, C-4, and C-5; H-5 to C-1 and C-3; H-2 to C-3 and C-4 were consistent with a calamenene compound, having a structure that consist of benzene in ring A. Furthermore, the correlations of the exchangeable proton at δH 3.88 to C-6, C-7, and C-8, as well as between H-9 to C-8 allowed the attachment of hydroxyl at C-7 and carbonyl ketone at C-8. The observed cross-peaks between H-9/H-10/H-14 and H-12/H-11/H-13 showed the fragments in compound 2 through the 1H–1H COSY-connected proton pairs. A comparison between the 1H and 13C NMR signals of 2 with those of 1,6-dihydroxy-1-isopropyl-4,7-dimethyl-3,4-dihydronaphthalen-2(1H)-one,15 indicates that they were structurally related. However, unlike the former, the latter showed a lower chemical shift of C-3 (δC 146.4) compared to the known one (δC 156.1), which proved that the hydroxyl at C-3 forms an ether bond to bridge the units A and B via C-3/C-3′. The connectivity through an ether bridge was also evident from the molecular weight to satisfy the requirement for the remaining oxygen atom in compound 2. Therefore, the planar structure of 2 was assumed to be a symmetrical pseudo-dimeric cadinanes featuring an ether linkage at C-3/C-3′.
| No. | 2 | 3 | ||
|---|---|---|---|---|
| Units A and B | ||||
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1/1′ | — | 138.8, s | — | 127.6, s |
| 2/2′ | 6.68 (s) | 111.5, d | 6.27 (s) | 103.3, d |
| 3/3′ | — | 146.4, s | — | 145.7, s |
| 4/4′ | — | 122.0, s | — | 125.8, s |
| 5/5′ | 7.27 (s) | 129.3, d | 6.38 (s) | 122.7, d |
| 6/6′ | — | 131.1, s | — | 142.1, s |
| 7/7′ | — | 81.7, s | 2.37 (td = 6.0, 3.0) | 42.8, d |
| 8a/8a′ | 212.6, s | 1.70 (m) | ||
| 8b/8b′ | — | 1.80 (m) | 19.4, t | |
| 9a/9a′ | 2.90 (dd = 6.9, 17.0) | 42.4, t | 1.42 (m) | |
| 9b/9b′ | 2.95 (dd = 9.0, 17.1) | 1.92 (m) | 26.9, t | |
| 10/10′ | 3.43 (m) | 30.4, d | 3.13 (m) | 27.0, d |
| 11/11′ | 2.24 (q = 5.1) | 33.2, d | 1.90 (m) | 33.1, d |
| 12/12′ | 0.83 (d = 7.0) | 17.4, q | 0.78 (d = 7.0) | 19.8, q |
| 13/13′ | 0.80 (d = 7.0) | 16.1, q | 0.91 (d = 6.8) | 22.1, q |
| 14/14′ | 1.32 (d = 7.0) | 19.6, q | 1.18 (d = 6.9) | 21.2, q |
| 15/15′ | 2.23 (s) | 15.5, q | 2.08 (s) | 16.1, q |
In the NOESY spectrum, the correlations of H-12 with OH-7/H-9b and H-9b with H-14 indicated that they are spatially close, with OH-7 and CH3-14 being assigned arbitrarily as β-oriented, while isopropyl at C-7 was an α-orientation. To obtain evidence for the structural and stereochemical assignment of 2, the 13C NMR chemical shift calculation (in CDCl3) on (7S,10S,7′S,10′S)-2 configuration was carried out. Comparison of the calculated and the experimental NMR shifts yield the convincing linear correlation coefficients (R2 = 0.9999) as well as the standard deviation (STDEV) of 0.76 ppm, approving that (7S,10S,7′S,10′S)-2 is the correct structure. Furthermore, calculations of two possible absolute configurations, namely (7S,10S,7′S,10′S)-2 (2a) and (7R,10R,7′R,10′R)-2 (2b) were performed using TDDFT method. As a result, the calculation of ECD spectrum for (7S,10S,7′S,10′S)-2 (2a) was in good agreement with experimental value of 2, as shown in Fig. 5. The complete structure of 2 with a novel symmetrical pseudo-dimeric cadinanes bearing an O-ether bridge skeleton was identified and named as dysotican G.
Compound 3 was isolated as a pale yellow. The molecular formula C30H42O with 10 degrees of unsaturation was established by the HR-ESI-TOFMS positive ion at m/z 441.3137 ([M + Na]+, calcd for 441.3133). The 1H and 13C NMR data in Table 2 clarified that compound 3 was also a symmetrical dimeric of cadinenes from calamenene derivative, and its core structure was identical to 2. A careful analysis of 1D NMR data revealed the most significant differences between 2 and 3 were that the quaternary oxygenated carbon [δC 81.7 (C-7)] and the C-8 ketone carbonyl [δC 212.6 (C-8)] in 2 were replaced by a methine [δC 42.8 (C-7)] and methylene carbon [δC 19.4 (C-8)] in 3. This arrangement was supported by HMBC correlations from [δH 0.78 (d = 7 Hz, H-12)] and [δH 0.91 (d = 7 Hz, H-13)] to [δC 33.1 (C-11); 42.8 (C-7)], from [δH 6.38 (s, H-5)] to C-7, as well as a spin system deduced from 1H–1H COSY correlations between [H-12, δH 0.78/(H-11), δH 1.90/H-13, δH 0.91/H-7, δH 2.37/H-8, δH 1.70; δH 1.80/H-9, δH 1.42; δH 1.92/H-10, δH 3.13/H-14, δH 1.18]. Subsequently, the HMBC correlations from δH 2.08 (s, H-15) to C-3 (δC 145.7), C-4 (δC 125.8), C-5 (δC 122.7) and δH 6.38 (s, H-5) to C-7 (δC 42.8), together with a remaining oxygen atom from its molecular formula, enabled the linkage bond of C-3-O-C-3′ between the two symmetrical monomer units (A and B) in 3. Therefore, compound 3 was determined to contain a symmetrical cadinane dimer bridged by an ether bond via C-3/C-3′. The pairwise NOESY correlations of H-12 to H-9b, H-12/H-9b to H-7, and H-7/H-10 indicated that they were co-facial and assigned as β-oriented (Fig. 3), referencing with the known 2,15-dihydroxycalamenene in previous study,14 which the chemical shift of C-7 (δC 43.2) and C-14 (δC 21.0) in the known one compared to 3 [C-7 (δC 42.8) and C-14 (δC 21.2)] was strongly similar. Therefore, the isopropyl located at C-7/C-7′ and secondary methyl at C-10/C-10′ in these four chiral centers were established as α-oriented. The absolute configuration of 3 as (7R,10R,7′R,10′R) was established based the high linear correlation coefficient (R2) and the low standard deviation (STDEV) (Fig. 4), as well as the signs of cotton effects in the ECD curves (Fig. 5). Consequently, the structure of 3 was unequivocally established as unprecedented symmetrical pseudo-cadinane dimer and named as dysotican H.
Based on the structural features, a presumable biogenesis pathway for 1–3 was outlined (Scheme 1). Compound 1 represented an unprecedented asymmetrical true-cadinene dimeric that possessed direct C–C bridged fusion of a ketonic group at C-15 to C-3′. As described in Scheme 1, it was proposed from the co-occurring dysotican A (i) isolated from the plant.14 Compound (i) underwent dehydration to form a double bond at C-2/C-3 and double bond shift at C-4/C-5 to C-3/C-4, which could successively trigger the hydration of sp2 (C-2/C-3) to provide hydroxyl at C-2. The dehydration at C-6 involving the H-1 yielded a pair of sp2 at C-1/C-6, followed by a strong reduction of aldehyde at C-15 to generate unit A (a). Furthermore, the unit B (b) was achieved from i by a series of chemical reactions similar to a, including a strong reduction of an aldehyde as well as dehydration to form a methyl group at C15′ and a double bond pair at (C-1′/C-6′). Hydration at C-4′ followed by 1,2-hydroxyl shift from C-3′ to C-4′ can occur to build the final construct of b. Finally, compound 1 could be produced from a free radical coupling by releasing hydrogen at C-15 of a and C-3′ of b to generate a direct C–C bond, in which the C-15 underwent further oxidation to produce a ketonic bridge. Meanwhile, (−)-hydroxycalemenene (ii) was proposed as the primary precursor of the symmetrical cadinane dimers of 2 and 3. The bridging diphenyl ether bond of 3 was formed through the dehydration of (ii) catalyzed by a hydronium ion in the hydroxyl site, generating compound 3. Moreover, the epimerization of C-10/C-10′ as well as the strong oxidation of C-7,8/C-7′,8′ might occur to transform into compound 2.
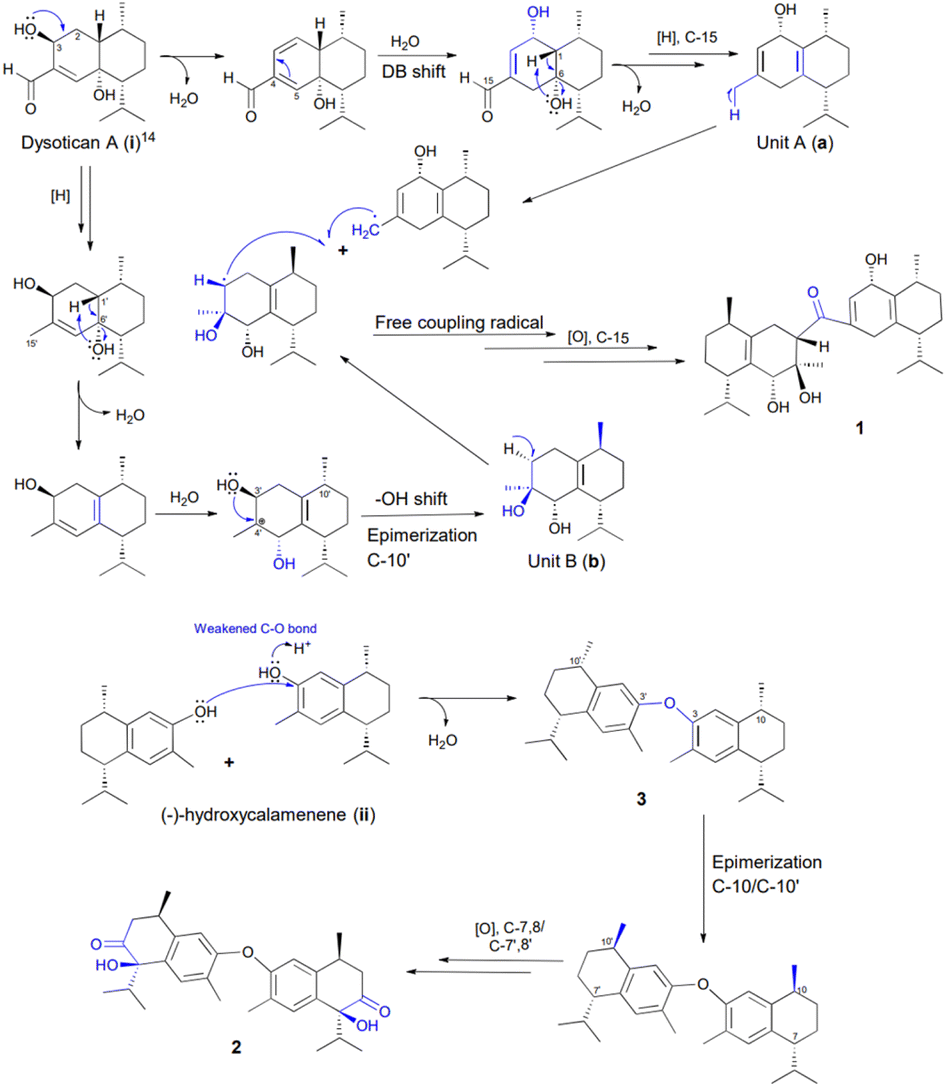 | ||
| Scheme 1 Plausible biogenetic pathway of compounds 1–3. | ||
The cytotoxic activities of 1–6 against the breast cancer MCF-7 and the cervical cancer HeLa cell lines were evaluated by the Resazurin (PrestoBlue) assay to obtain their IC50 values (Table 3). Among these compounds, the symmetrical pseudo-cadinane dimer 2 showed remarkable cytotoxicity against these two cell lines with IC50 values of 12.07 ± 0.17 μM and 9.29 ± 0.33 μM, for each, and exhibited less cytotoxicity than that of cisplatin. Meanwhile, another unprecedented symmetrical pseudo-cadinane dimer 3 displayed selective inhibitory effect against the HeLa cell line with an IC50 value of 39.72 ± 0.18 μM. The asymmetrical true-cadinane dimer 1 showed a moderate activity with IC50 values of 31.59 ± 0.34 μM and 27.93 ± 0.25 μM, respectively.
| Compound | MCF-7 | HeLa |
|---|---|---|
| a Data were reported as the mean ± SD; n = 3 independent experiments.b Positive control. | ||
| 1 | 31.59 ± 0.34 | 27.93 ± 0.25 |
| 2 | 12.07 ± 0.17 | 9.29 ± 0.33 |
| 3 | >100 ± 0.27 | 39.72 ± 0.18 |
| 4 | >100 ± 0.21 | 92.78 ± 0.15 |
| 5 | >100 ± 0.35 | >100 ± 0.31 |
| 6 | 79.83 ± 0.28 | 85.31 ± 0.24 |
| Cisplatinb | 53.00 ± 0.02 | 16.00 ± 0.01 |
A brief structure–activity relationship (SAR) analyses revealed that the oxygen substituent group at C-7/C-7′ and C-8/C-8′ as well as the epimerization at C-10/C-10′ of the symmetrical pseudo-cadinane dimer 2 can directly affect its cytotoxic activity, especially against the MCF-7 cell line compared to that of 3. Furthermore, hydroxyl shift from C-6 to C-10 and epimerization of C-7/C-10 by 6 reduces its inhibitory activity against the two cell lines by two-fold compared to that of i,14 while the hydroxyl movement from C-2 to C-3 of 4 and epimerization of hydroxyl at C-7 of 5 showed no obvious effects against these two cell lines compared to their analogs schiffnerone B and eudesm-4(15)-ene-1β,6α-diol, for each, in our previous report.14
Conclusions
In summary, one unprecedented true-cadinane dimer via C–C bond bearing a ketonic bridge at C-15/C-3′, dysotican F (1), and two symmetrical pseudo-cadinane dimers through C–O bond featuring an ether linkage at C-3/C-3′ scaffold, dysoticans G-H (2–3), were characterized from the n-hexane extract of Dysoxylum parasiticum (Osbeck) Kosterm. stem bark. The results will enrich the chemical diversity of sesquiterpenoid dimers from the genus Dysoxylum and also stimulate the interest in chemical synthesis in order to duplicate the efficiency and the selectivity of natural compounds. Furthermore, compound 2 was discovered to be the most potent of a new class at inhibiting growth of the MCF-7 and HeLa cell lines', which could be further developed into lead compound for drug discovery.Experimental
General experimental procedures
Optical rotations were performed using an ATAGO AP-300 automatic polarimeter. UV and IR were obtained using PerkinElmer Lambda 35 UV/VIS Double Beam and Everest ATR Thermos scientific FTIR spectrometers. ECD were recorded on a JASCO J-720 CD spectrometer. Furthermore, high-resolution mass spectra (HR-MS) were determined on a Waters Xevo Q-TOF direct probe/MS system, using ESI+ mode and microchannel plates MCPs detector (Milford, MA, USA). JEOL ECZ and Bruker Topspin spectrometers at 500 MHz for 1H and 125 MHz for 13C NMR were used to measure NMR spectra, using TMS as an internal standard. The reference to the solvent of CDCl3 were assigned in δ (ppm) with signals (δH 7.26, δC 77.16). Additionally, column chromatography (CC) was subjected using silica gel 60 (Merck, 70–230 and 230–400 mesh) and octa desylsilane (Fuji sylisia chemical LTD, Chromatorex® C18 DM1020 M, 100–200 mesh). Thin-layer chromatography (TLC) precoated silica gel 60 F254 (Merck) and RP-18 F254s plates (Merck) were used to guide the separation process while UV light at 254 and 365 nm was employed for detection before spraying with 10% H2SO4 in ethanol.Plant materials
The plant material was collected in August 2019 from Bogor Botanical Garden, West Java Province, Indonesia (latitude −6.597629, longitude 106.799568, elevation 277 m) and identified by Mr Harto of the Bogoriense Herbarium where the voucher specimen (III. F.79) was deposited.Extraction and isolation
The air-dried and powdered stem barks (5.50 kg) was extracted with MeOH at room temperature (3 × 12 L, 24 h each) to afford a crude extract (481.7 g), which was suspended in water, followed by sequential partitioning with n-hexane, EtOAc, and n-butanol. The organic layer was evaporated under reduced pressure to obtain crude extracts of n-hexane (153.9 g), EtOAc (65.2 g), and n-butanol (48.6 g), respectively. A total of eight fractions were separated from 150 g of the extract using vacuum liquid chromatography over silica gel eluted with a gradient of n-hexane–EtOAc (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–0:10, 10% increments) (Fr.A–Fr.L). Using a silica gel column with a gradient elution of n-hexane–EtOAc (10:0–7:3, step 5%), Fr.D (40.90 g) was separated into nine subfractions (D1–D9). Fr.D.6 (5 g) was passed through a CC (70–230 mesh) eluted with a gradient of n-hexane–EtOAc (10:0–6:4, 2%, v/v) to generate seven fractions (D.6.a–D.6.g). Fr.D.6.b (900 mg) was separated into six fractions (Fr.D.6.b.1–Fr.D.6.b.6) through silica gel (230–400 mesh) column chromatography (CC) with n-hexane–EtOAc (8:2) as solvents. Fr.D.6.b.6 (50 mg) was subjected to an open ODS column (MeOH–H2O, 8:2) to produce compounds 1 (3.3 mg) and 4 (4.0 mg). Fr.D.7 (5 g) was subjected to silica gel (70–230 mesh) CC using a gradient elution of n-hexane–EtOAc (10:0–5:5, 2.5%, v/v) to generate eight subfractions (Fr.D.7.a–Fr.D.7.h). Furthermore, Fr.D.7.b (300 mg) was separated over silica gel (230–400 mesh) with the eluent n-hexane–EtOAc (10:1) and purified by ODS CC (MeOH–H2O, 8:2) to obtain compounds 2 (3.0 mg) and 3 (6.0 mg). Finally, Fr.D.7.d (450 mg) was chromatographed by silica gel CC (230–400 mesh) and then purified by a flash ODS CC (MeOH:H2O, 5:5) to yield compounds 5 (4.0 mg) and 6 (5.0 mg).
ε) 246 (4.42), 315 (3.21) nm; ECD (c 0.29 mM, MeOH) λmax (Δε): 186 (−57.2), 199 (−53.5), 233 (−11.1), 243 (+10.2) nm; IR (KBr): 3431, 2957, 2927, 1650, 1464, 1374, 1221, 1035 cm−1; 1H and 13C NMR data are presented in Table 1; HR-ESI-QTOFMS m/z 471.3479 [M + H]+ (calcd for C30H47O4, 471.3474).ε) 189 (4.14), 207 (3.39), 249 (3.53) nm; ECD (c 0.17 mM, MeOH) λmax (Δε) 203 (−53.0), 213 (+9.2), 221 (−9.7), 243 (+26.2), 275 (+11.3), 293 (−4.3) nm; IR (KBr): 3398, 2938, 2915, 1710, 1605, 1513, 1441, 1375, 1205, 1105 cm−1; 1H and 13C NMR data are presented in Table 2; HR-ESI-QTOFMS m/z 501.2620 [M + Na]+ (calcd for C30H38O5Na, 501.2617).ε) 197 (4.01), 218 (3.46), 247 (2.66) nm; ECD (c 0.72 mM, MeOH) λmax (Δε) 185 (+59.9), 206 (−47.6) nm; IR (KBr): 2977, 2925, 1604, 1502, 1425, 1319, 1219, 1011 cm−1; 1H and 13C NMR data are presented in Table 2; HR-ESI-QTOFMS m/z 441.3137 [M + Na]+ (calcd for C30H42ONa, 441.3133).
:0–0:10, 10% increments) (Fr.A–Fr.L). Using a silica gel column with a gradient elution of n-hexane–EtOAc (10:0–7:3, step 5%), Fr.D (40.90 g) was separated into nine subfractions (D1–D9). Fr.D.6 (5 g) was passed through a CC (70–230 mesh) eluted with a gradient of n-hexane–EtOAc (10:0–6:4, 2%, v/v) to generate seven fractions (D.6.a–D.6.g). Fr.D.6.b (900 mg) was separated into six fractions (Fr.D.6.b.1–Fr.D.6.b.6) through silica gel (230–400 mesh) column chromatography (CC) with n-hexane–EtOAc (8:2) as solvents. Fr.D.6.b.6 (50 mg) was subjected to an open ODS column (MeOH–H2O, 8:2) to produce compounds 1 (3.3 mg) and 4 (4.0 mg). Fr.D.7 (5 g) was subjected to silica gel (70–230 mesh) CC using a gradient elution of n-hexane–EtOAc (10:0–5:5, 2.5%, v/v) to generate eight subfractions (Fr.D.7.a–Fr.D.7.h). Furthermore, Fr.D.7.b (300 mg) was separated over silica gel (230–400 mesh) with the eluent n-hexane–EtOAc (10:1) and purified by ODS CC (MeOH–H2O, 8:2) to obtain compounds 2 (3.0 mg) and 3 (6.0 mg). Finally, Fr.D.7.d (450 mg) was chromatographed by silica gel CC (230–400 mesh) and then purified by a flash ODS CC (MeOH:H2O, 5:5) to yield compounds 5 (4.0 mg) and 6 (5.0 mg).
ε) 246 (4.42), 315 (3.21) nm; ECD (c 0.29 mM, MeOH) λmax (Δε): 186 (−57.2), 199 (−53.5), 233 (−11.1), 243 (+10.2) nm; IR (KBr): 3431, 2957, 2927, 1650, 1464, 1374, 1221, 1035 cm−1; 1H and 13C NMR data are presented in Table 1; HR-ESI-QTOFMS m/z 471.3479 [M + H]+ (calcd for C30H47O4, 471.3474).ε) 189 (4.14), 207 (3.39), 249 (3.53) nm; ECD (c 0.17 mM, MeOH) λmax (Δε) 203 (−53.0), 213 (+9.2), 221 (−9.7), 243 (+26.2), 275 (+11.3), 293 (−4.3) nm; IR (KBr): 3398, 2938, 2915, 1710, 1605, 1513, 1441, 1375, 1205, 1105 cm−1; 1H and 13C NMR data are presented in Table 2; HR-ESI-QTOFMS m/z 501.2620 [M + Na]+ (calcd for C30H38O5Na, 501.2617).ε) 197 (4.01), 218 (3.46), 247 (2.66) nm; ECD (c 0.72 mM, MeOH) λmax (Δε) 185 (+59.9), 206 (−47.6) nm; IR (KBr): 2977, 2925, 1604, 1502, 1425, 1319, 1219, 1011 cm−1; 1H and 13C NMR data are presented in Table 2; HR-ESI-QTOFMS m/z 441.3137 [M + Na]+ (calcd for C30H42ONa, 441.3133).Computational methods
Molecular Merck force field (MMFF) was used to perform the conformational analyses via random searching in Spartan 14.0 (Wavefunction Inc., Irvine, CA, USA) software. The resulting conformers were optimized using DFT method at the B3LYP/6-31G (d,p) level with PCM in MeOH by the Gaussian 09 software package.16 ECD calculations were then performed using TD-DFT at the B3LYP/6-31G (d,p) basis set with the rotary strengths of 120 excited states. Subsequently, the results were processed with SpecDis 1.64 program. For the 13C NMR chemical shifts calculation, Gauge-Independent Atomic Orbital (GIAO) was performed and accomplished by DFT at the mPW1PW91/6-311+G(d,p) level in CDCl3 using the PCM solvent model. The outcome was analysed by subtracting the isotopic shifts for TMS calculated with the same methods. Linear correlation coefficients (R2) and standard deviation (STDEV) were calculated to evaluate the results.Cytotoxicity assays
For this analysis, the MCF-7 and HeLa cells were cultured in 5% CO2 at 37 °C using the RPMI-1640 medium which supplemented by 10% fetal bovine serum and antibiotics. The cells were then seeded into 96-well plates at an initial cell density of approximately 3 × 104 cells per cm3 and incubated for 24 h. The compounds were prepared by dissolving in 2% aqueous DMSO and added at the required concentrations (500, 250, 125, 62.5, 31.25, 15.63, 7.81, and 3.91 μM) and incubated for 48 h. The measurement of the viability cell was conducted by adding 10 μL of PrestoBlue™ and incubation for 1–2 h. Afterwards, the optical density was measured by a micro plate reader at 570 nm. The IC50 values were then obtained by substituting the absorbance of negative control (2% aqueous DMSO) in the plotted graph of the tested compound concentrations. The test was performed in triplicate and cisplatin as the positive control.Conflicts of interest
The authors confirmed that this study has no conflict of interest.Acknowledgements
The authors are grateful to the Directorate of Ministry of Research, Technology/National Research and Innovation Agency, Indonesia for rendering financial support with contract no. 1207/UN6.3.1/PT.00/2021 under the PMDSU and PKPI program 2022, as well as Universitas Padjadjaran, for the Academic Leadership Grant, No. 1959/UN6.3.1/PT.00/2022 provided by Unang Supratman.Notes and references
- Z. J. Zhan, Y. M. Ying, L. F. Ma and W. G. Shan, Natural disesquiterpenoids, Nat. Prod. Rep., 2011, 28, 594–629 RSC.
- Y. H. Ma, X. X. Dou and X. H. Tian, Natural Disesquiterpenoids: An Overview of Their Chemical Structures, Pharmacological Activities, and Biosynthetic Pathways, Phytochem. Rev., 2020, 19, 983–1043 CrossRef CAS.
- J. J. Qin, W. Wang and R. W. Zhang, Novel natural product therapeutics targeting both inflammation and cancer, Chin. J. Nat. Med., 2017, 15, 401–416 CAS.
- V. Laksmi, K. Pandey and S. K. Agarwal, Bioactivity of the compounds in genus Dysoxylum, Acta Ecol. Sin., 2009, 29, 30–44 CrossRef.
- A. A. Naini, T. Mayanti and U. Supratman, Triterpenoids from Dysoxylum genus and their biological activities, Arch. Pharmacal Res., 2022, 1–27 Search PubMed.
- B. J. Xie, S. P. Yang and J. M. Yue, Terpenoids from Dysoxylum densiflorum, Phytochemistry, 2008, 69, 2993–2997 CrossRef CAS PubMed.
- A. A. Naini, T. Mayanti, D. Harneti, R. Maharani, A. Safari, A. T. Hidayat, K. Farabi, R. Lesmana, U. Supratman and Y. Shiono, Cytotoxic sesquiterpenoids from Dysoxylum parasiticum (Osbeck) Kosterm. stem bark, Phytochem. Lett., 2022, 47, 102–106 CrossRef CAS.
- J. Gu, S. Y. Qian, Y. L Zhao, G. G. Cheng, D. B. Hu, B. H. Zhang, Y. Li, Y. P. Liu and X. D. Luo, Prenyleudesmanes, rare natural diterpenoids from Dysoxylum densiflorum, Tetrahedron, 2014, 70, 1375–1382 CrossRef CAS.
- H. J. Yan, J. S. Wang and L. Y. Kong, Cytotoxic steroids from the leaves of Dysoxylum binectariferum, Steroids, 2014, 86, 26–31 CrossRef CAS PubMed.
- V. Mahajan, N. Sharma, S. Kumar, V. Bhardwaj, A. Ali, R. K. Khajuria, Y. S. Bedi, R. A. Vishwakarma and S. G. Gandhi, Production of rohitukine in leaves and seeds of Dysoxylum binectariferum: an alternate renewable resource, Pharm. Biol., 2015, 53, 446–450 CrossRef CAS PubMed.
- F. F. Sofian, A. Subarnas, M. Hakozaki, S. Uesugi, T. Koseki and Y. Shiono, Bidysoxyphenols A–C., dimeric sesquiterpene phenols from the leaves of Dysoxylum parasiticum (Osbeck) Kosterm, Fitoterapia, 2022, 158, 105157 CrossRef CAS PubMed.
- F. F. Sofian, A. Subarnas, M. Hakozaki, S. Uesugi, T. Koseki and Y. Shiono, Tridysoxyphenols A and B, two new trimeric sesquiterpene phenols from Dysoxylum parasiticum leaves, Phytochem. Lett., 2022, 50, 134–140 CrossRef CAS.
- F. F. Sofian, A. Subarnas, T. Koseki and Y. Shiono, Structure elucidation of a new bicoumarin derivative from the leaves of Dysoxylum parasiticum (Osbeck) Kosterm, Magn. Reson. Chem., 2022, 60, 857–863 CrossRef CAS PubMed.
- A. A. Naini, T. Mayanti, D. Harneti, R. Maharani, K. Farabi, T. Herlina, U. Supratman, S. Fajriah, H. Kuncoro, M. N. Azmi, Y. Shiono and S. Jungsuttiwong, Sesquiterpenoids and sesquiterpenoid dimers from the stem bark of Dysoxylum parasiticum (osbeck) kosterm, Phytochemistry, 2023, 205, 113477 CrossRef CAS PubMed.
- B. Luo, L. M. Dong, Q. L. Xu, X. Zhang, Q. Zhang, W. B Liu and J. W. Tan, A new monoterpene and a new sesquiterpene from the roots of Ageratina Adenophora, Phytochem. Lett., 2018, 24, 67–70 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, J. E. Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01085f |
| This journal is © The Royal Society of Chemistry 2023 |