
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile utility of felumin, a biological dye for the first fluorimetric determination of L-tetramisole drug through an “on–off fluorescence” strategy†
Ahmed Abdulhafez Hamad
Department of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Al-Azhar University, Assiut Branch, Assiut 71524, Egypt
First published on 21st March 2023
Abstract
L-Tetramisole is an anti-nematode and immunomodulating agent employed medically to treat diseases caused by worms. It is also used as an immune system-modifying agent in rheumatoid arthritis and as assistant therapy in treating cancers in the colorectal, head, and neck regions. Felumin is a safe food dye used in feed additives, flavors, biostaining, and recently, in analytical tracking. This is the first innovative spectrofluorimetric approach to L-Tetramisole drug analysis. It is both efficient and environmentally benign and was evaluated and validated in this investigation using a green, one-pot, and direct spectrofluorimetric technique. Instant complexes were created by combining L-Tetramisole and Felumin in an acidic solution. The fluorometric investigation was performed based on the off-effect strategy of L-Tetramisole on the emission amplitude of a biological dye (Felumin) at 557.5 nm. The linear range was from 0.1 to 1.7 μg mL−1, with sensitivity limits of 0.020 and 0.061 μg mL−1, respectively. Analytical modulation of L-Tetramisole–Felumin complexes was done for all system parameters. The system was found to meet ICH criteria. Moreover, this technique successfully recovered the substance in the prescribed medicinal dosage forms. The created fluorimetric technique was also successfully employed to track the drug of interest in human biofluids, which was a major accomplishment. The kinetics of the reaction system was also studied further. Finally, the proposed method's environmental friendliness was evaluated on the eco-scale.
Introduction and preface
Due to their high extinction indices, high quantum yields, and affinity for linking to biomolecules, there has been increased interest in using xanthene dyes and related compounds for labeling and monitoring purposes. Felumin (FLN, Fig. 1B) is a halogen-substituted xanthene that has been adapted as a laser dye, food dye, and even a clinical and pharmaceutical diagnostic. FLN is becoming well-established as an appealing reagent for the quantitative assessment of proteins in biological materials due to its preferential protein binding and intrinsic photoluminescence. Oxygen content, vitamin B6, and urine protein are only a few examples of the interactions reported using FLN as a probe.1 From an analytical viewpoint, xanthene-based biological dyes have also been used to determine proteins,2 and several pharmaceutical compounds.3–6 Structurally, the Felumin dye is 2,4,5,7-tetraiodo-fluorescein (Fig. 1B). The creation of an ion-association complex binding the dye to the fundamental chemicals is usually the strategy for these procedures. The complex formation causes a reduction in FLN's inherent fluorescence, and the analyte, which has a basic center, can be quantified using this indicator. Numerous medications have been chemically investigated using xanthene-based dyes such as Felumin and Eosin Y, for example, fluoroquinolones,4 antispasmodics,7 oncogenic agents,8 anthelmintic drugs,9,10 and antihypertensive drugs.11 L-Tetramisole is an anti-helminthic drug used to treat diseases caused by worms, rheumatoid arthritis, and colon, head, and neck malignancies. L-Tetramisole (Fig. 1A) is also known as (S)-(2)-2,3,5,6-tetrahydro-6-phenyl-imidazo-[2,1-b] thiazole according to IUPAC, and (2,3,5,6-tetrahydro-6-phenylimidazole [2,1-b] thiazole) according to USP.12 L-Tetramisole particularly activates the parasites' nicotinic acetylcholine receptors, which in turn promotes the parasites' immune system. In humans, L-Tetramisole can be used as an immunomodulatory.13 L-Tetramisole is frequently employed in the management and treatment of immunological and autoimmune disorders. To successfully treat the skin, L-Tetramisole hydrochloride liniments have been produced.14 L-Tetramisole has also been utilized as an adulterant in cocaine, generally at a concentration of 5% of the bulk substance, which piqued forensic attention.15 Steroid use can be minimized by L-Tetramisole, which is an immunoregulatory man-made imidazothiazole analog. When L-Tetramisole is given as the first alternative to steroids and after therapy failure with cyclophosphamide or ciclosporin, depressive episodes are lessened, and corticosteroid doses are minimized in patients with SSNS.16 L-Tetramisole 's plasma concentration (from a pharmacokinetic viewpoint) was found to be at its peak at 1.98 hours, and its half-life varied between 3.3 and 5.1 hours. It was found that after a single oral dose of 50–150 mg L-Tetramisole, peak plasma levels of the unmodified medication were attained roughly two hours after ingestion. This study found that L-Tetramisole has a 4 hour plasma half-life.17 With dosages of 2.5 mg and 5 mg kg−1 (according to another study), L-Tetramisole plasma concentrations peaked in 1.0 to 2.0 hours (with a tmax of 1.5 hours), were rapidly absorbed, and reached their peak at concentrations of 800 to 1600 ng mL−1.18 A novel therapeutic use for this anthelmintic medication has emerged since Renoux demonstrated its immunostimulatory properties.19 Herpes, leprosy, zona, rheumatoid arthritis, lupus, and many types of cancer have all been successfully treated with L-Tetramisole. L-Tetramisole–HCl is official in European,20 British,21 and United States Pharmacopeias.12 For its medicinal value, LTS was determined in artificial forms and biofluids. Many different approaches to evaluating LTS in raw or pharmaceutical formulations have been documented in the literature. However, to date, no fluorometric strategy has been reported for its estimation. HPLC was used to quantify LTS alone22,23 and in the presence of other drugs such as albendazole,24 heparin and pentobarbital,25 abamectin,26 and thiabendazole.27 Also, an electrophoretic technique28,29 and electroanalytical approaches30–32 have been applied for its determination. Spectroscopic techniques33–35 (including extractive spectrophotometric,34 and ultraviolet methods36), luminescence,37 and the atomic absorption spectrometric method38 have also been used. Despite its various advantages, there are no published spectrofluorimetric methods for LTS analysis, such as its simplicity of application, specificity, and elevated sensitivity. Because of this, the spectrofluorimetric analysis of L-Tetramisole is required. The spectrofluorimetric technique designed for this medicine achieves these objectives by shutting down the self-fluorescent dyes in the protogenic media. The proposed approach has been thoroughly validated under ICH requirements.39 It promptly and accurately determined LTS in bulk, pharmaceuticals, and human fluids.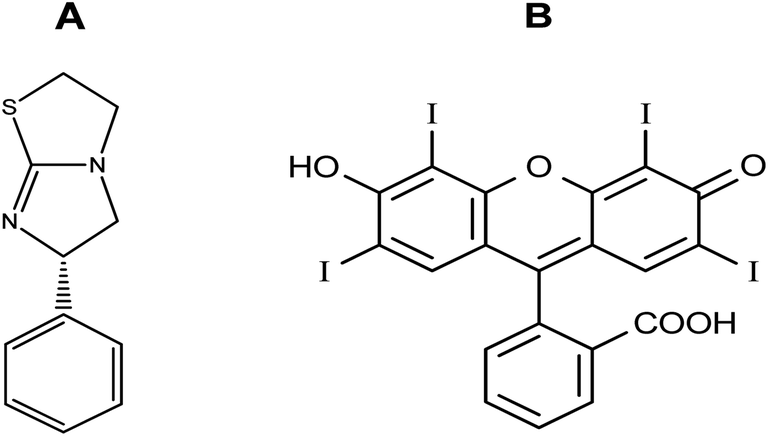 | ||
| Fig. 1 Molecular structures of L-Tetramisole (A) and Felumin (B). | ||
The LTS tertiary amino group can be protonated in acidic conditions. As a result, an ion-pair complex combining FLN and LTS can develop. The resulting “on–off fluorescence system” will be utilized to measure LTS via the LTS-induced suppression of the dye's internal emission. An easy and gentle method for drug testing might be achieved by using fluorescence quenching at a specific concentration of 0.1–1.7 μg mL−1. Reagents are inexpensive, and the apparatus is commonly present in most product control laboratories, rendering this a cost-effective technique.
Experimental
Apparatus
All fluorescence measurements were performed using an FS-2 (Korea) SCINCO spectrometer and a light source lamp (150 W Xe-arc). A ThermoFisher scientific centrifuge was used for clinical samples (PICO 21, Germany). The SONICOR SC-101TH bath sonicator is an additional component. The pH of the working medium was adjusted using an AD11P pH meter (Adwa, Romania).Materials and reagents
L-Tetramisole HCl was obtained from the EIPICO company and was used without additional treatment. Felumin dye reagent was purchased from Market Harborough, Leicestershire, UK, and dissolved in doubly distilled water. Katrex® 40 mg per tablets and syrup were purchased from the local pharmacy.Buffer solutions and solvents
Chemicals such as acetone, acetonitrile, alcohol, methanol, and dimethylformamide were purchased from the El Nasr Company for Intermediate Chemicals in Cairo, Egypt. El Nasr also provided DMSO and other chemicals, including 1,2-dioxane, isopropanol, HCl, and NaOH.Acids of 0.04 M concentration were found in the Britton–Robinson solution (pH 2.0–12.0). Acids include boric, phosphoric, and acetic acids. The pH of this combination was altered by adding 0.2 M NaOH to a beaker.40
There were two master solutions in the Teorell–Stenhagen control solution, each of which spans the pH range from 2.0–12.0. The solution contained phosphoric, citric, hydrochloric acids, and sodium hydroxide (A). The second master solution is HCl (0.1 M) (B). It was possible to attain the operational pH when the two solutions were combined in the correct proportions.
Here, 0.2 M citric acid and 0.01 M Na2HPO4 were mixed to make a McIlvaine buffer solution with a pH of 2.2–8.0 6.
An acetate pH-controlling solution with a concentration of 0.2 M was prepared from acetic acid and sodium acetate solutions.
Non-purified analytical or pharmaceutical-grade compounds were used in their unprocessed original form. Every day, a new solution was created.
Human body fluids
Drug-free urine and plasma samples were taken from healthy adults and stored in a freezer. The Committee on Scientific Research Ethics approved the collection of biological samples from human beings at the Al-Azhar University Hospital, Assiut Branch. The Al-Azhar University Faculty of Pharmacy Ethical Committee in the Assiut Branch, Egypt accepted and approved the technique (ZA-AS/PH/5/C/2022). In all cases, authorization forms were signed by each party. Following the Declaration of Helsinki recommendations, the experimentation, including human specimens, was executed.Working solutions
For a standard solution of LTS, a volumetric flask of 100 mL was charged with 10 mg of LTS, dispersed in an adequate amount of water, and then topped up with the same liquid. The stock solution was dispersed in water to the desired volume to create the standard operating solution. The solutions were kept at 4 °C in the refrigerator to preserve their chemical stability.The processing steps
An aliquot of LTS in calibrated 10.0 mL flasks in the 1.0–17.0 μg mL−1 range was utilized for the fluorometric experiment. Each flask was pre-mixed with 1.5 mL of acetate-regulating solution and 1.2 mL of FLN solution. It was then replenished with double-distilled water and matured for another 6 minutes before being analyzed. Aside from the drug solution, the blank reagent was tested to the same standard. Endogenous FLN fluorescence intensity was determined using a fluorimetric technique at 557 nm, then correlated to the LTS dose in μg mL−1.Tablet and suspension analysis
Crushed Katrex® tablets and syrup were transferred using 250 mL calibration volumetric jars. Each flask contained 60 mL of water and was sonicated for 0.5 hours. The same liquid was used to complete the solution, with the first portion of the filter discarded. The final doses of 0.5, 1.0, and 1.5 μg mL−1 were produced by dispersing the solvents further.Clinical sample analysis
At the same time, blank testing was performed. Calibration was established in plasma by tracking the drug-loaded plasma's signal to dose levels. The subject plasma samples, with varying drug doses present in the operating range, were used to estimate the levels of LTS.
Comparisons were made between the fluorescence amplitudes of treated plasma samples and those obtained from a control drug of equal dosage. The absolute recovery was determined.
Molar ratio calculation method
The stoichiometry of the LTS–FLN reaction was predicted using continuous variation charting (Job's technique42) as part of the present design. Different complementary ratios of the analyte and dye were introduced into 10 mL calibrated jars to give a volume of 1.0 mL of LTS and FLN (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0, 0.1:0.9, …, 0.9:0.1, 1.0:0). The general procedure was followed. Blanks were also prepared and measured at the same time. The mole fraction of the tested medication was plotted against the corrected emission intensity (ΔF).
:1.0, 0.1:0.9, …, 0.9:0.1, 1.0:0). The general procedure was followed. Blanks were also prepared and measured at the same time. The mole fraction of the tested medication was plotted against the corrected emission intensity (ΔF).
Results and discussion
The spectrum of the present system
FLN (a xanthene dye example) was employed in the developed system for analyzing the suggested drug. Because FLN has an intrinsic fluorescence at 557.5 nm that may be quantitatively lowered by LTS, it is possible to quantify the drug using the fluorescence detection response (Fig. 2).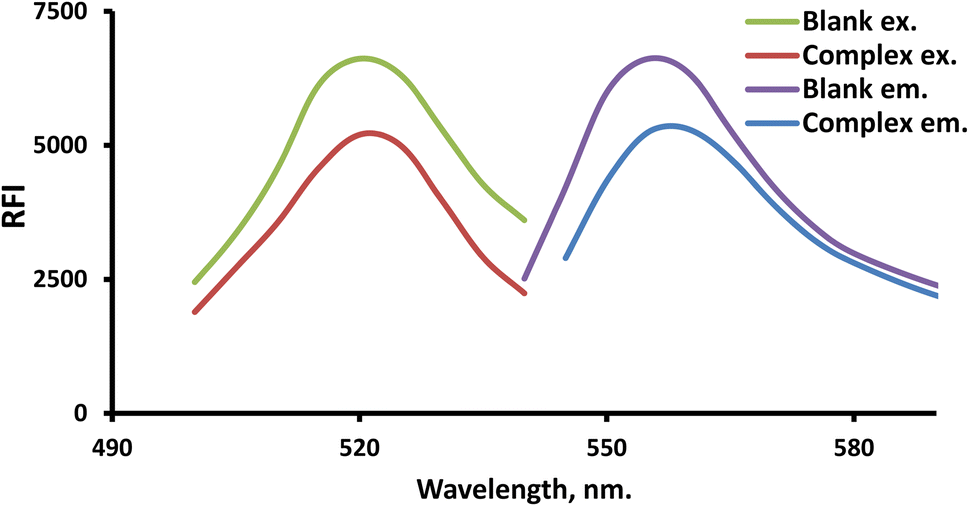 | ||
| Fig. 2 The scanned spectra of the resulting LTS–FLN ion-paired complex and the blank. | ||
Control of the experiment's parameters
To achieve the greatest possible scores for this specific spectroscopic application, it was necessary to investigate and correct any reaction factors that might have an impact on the system's signal.pH and buffer volume effects
The pH scale of 2.0–6.0 was used to study the LTS–FLN binary complex, which has a crucial function in forming this complex. The sample pH had a significant impact on the LTS–FLN complex. The planned system had the highest RFI values in the pH range of 3.0–4.2. The RFI values dropped when the pH was shifted outside of this range (Fig. 3).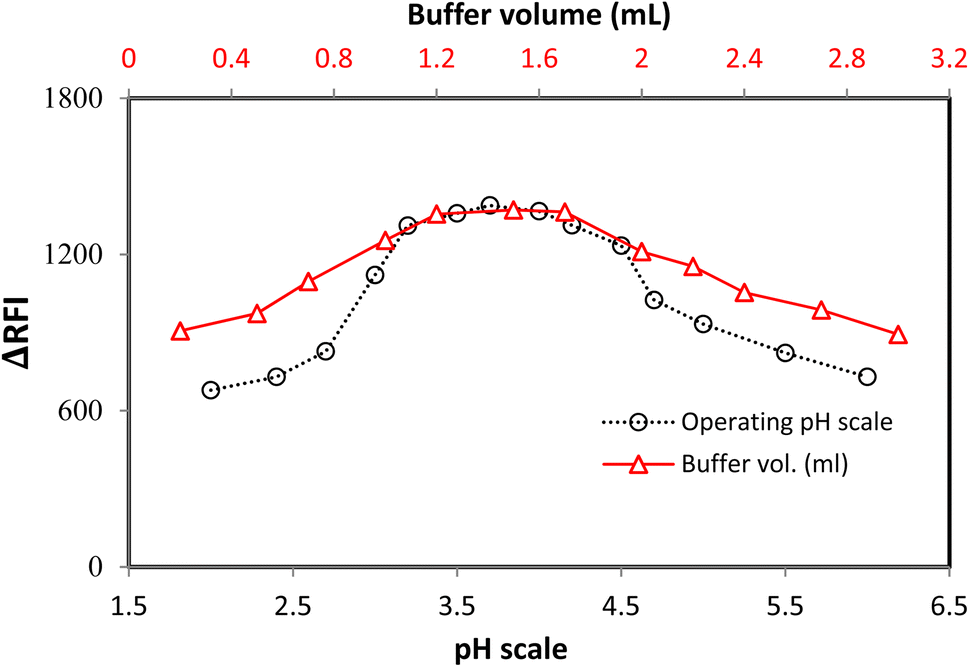 | ||
| Fig. 3 The impact of pH and buffer volumes on the system response. | ||
The buffer volume significantly impacted the growth of the LTS–FLN complex, and the optimal pH level was found to be 3.7 in this investigation. The acetate buffer solution was employed in quantities ranging from 0.2 to 3.0 mL to examine the LTS–FLN binary complex. For the chosen spectroscopic technique, a buffer volume of 1.2–1.7 mL produced the strongest responses (i.e., the greatest drop in dye emission). The response values were lower when the volume was outside of this range. To keep the pH stable, the buffer needs to be limited; if the buffer is too large, the anionic dye will compete with the positive component of the buffer for coupling, preventing the complexation process. Due to these considerations, the experiment was best carried out at a volume of 1.5 mL (Fig. 3).
The impact of the pH-controlling solution type
Several buffer types were evaluated in 1.5 mL volumes to get the best results from the applied procedure. All buffers studied (McIlvaine, acetate, Teorell–Stanhagen, and Britton–Robinson) produced similar values, with the acetate buffer producing considerably greater signals and more tolerable results, according to the testing; therefore, the acetate was used as the buffer for this procedure (ESI 1†).The impact of FLN volume and reaction time
A variety of FLN reagent concentrations were tested to achieve the desired results. The strongest fluorescence quenching was achieved with an FLN solution at a concentration of 0.015% w/v in 1.2 mL. Low FLN concentration led to a lower response as shown in ESI Fig. 2,† as the reaction was not complete at that concentration. Because FLN self-agglomeration led to lesser dye responses, larger dye concentrations resulted in weaker responses.8 This could also be related to the self-aggregation of FLN molecules into dimers when a very large amount is present.10Fast and efficient LTS–FLN synthesis took place at room temperature when reactant solution interactions were established. After ten minutes, all measurements were carried out so that the complex components could face each other and ensure that the complex formation would continue progressing.
The effect of dispersing liquid
Alcohols (ethyl, methyl, and propyl), acetone, dioxane, and distilled water are only a few of the dispersing solvents that were tested (Fig. 4). When distilled water was employed as the dispersion liquid, fluorescence suppression scores were at their maximum. A possible explanation for this study's low measurement scores using organic media is that the solvents may have a detrimental influence on the complex. Fluorescence signals can be modified by specific solvents, which can destabilize the complicated system. As a result, breaking short-chain solvents like ethyl and methyl alcohols in an aqueous environment disrupts the complicated formation process. At high levels of alcohol, the complexation process is impaired, and the system may be substantially damaged.43 Pure water was discovered to be the best final solvent for dilution, which was fortunate. Water has a higher dielectric constant (80.2) and a higher polarity score (9) as compared to the other solvents (ESI Table 1†). Because most of the system's components are dissolved in water, it is also likely that the system will be completely soluble. The system's formation may be hindered by the low miscibility of various organic solvents with variable dielectric constants.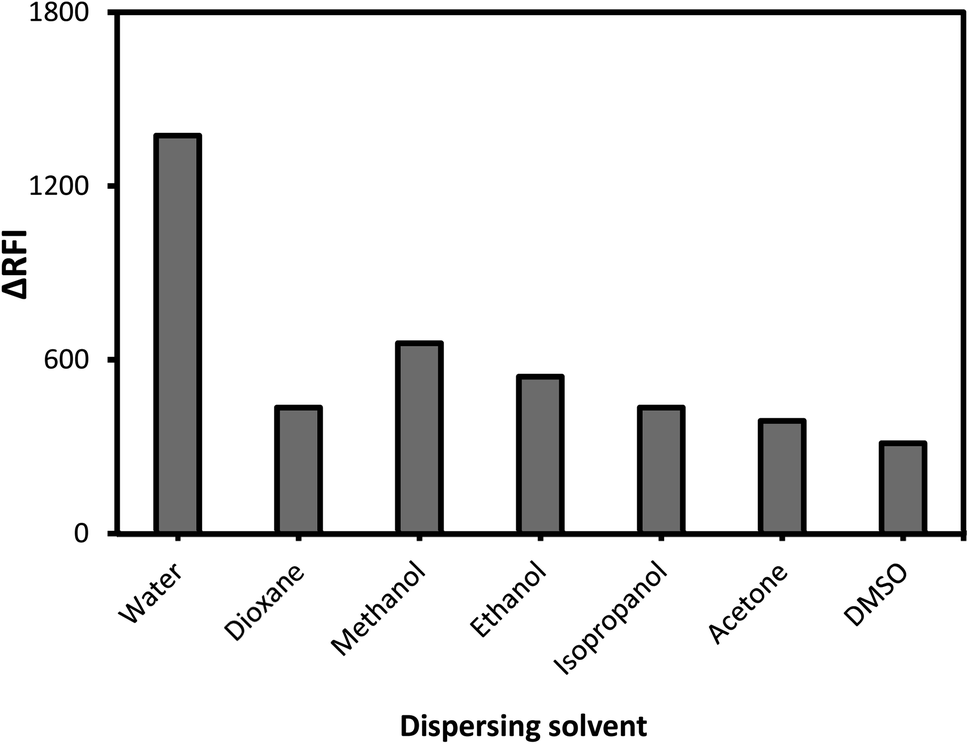 | ||
| Fig. 4 The impact of dispersion liquids on the developed association complex. | ||
The mechanism and strategy for the complexation process
FLN, fluorescein, and eosin's maximum intrinsic emission were found to be inhibited by many nitrogen-containing drugs.9,10 When the LTS analyte (which possesses a basic nitrogenous core) is combined with the FLN reagent in moderately acidic environments, an ion-pair complex is created, leading to the formation of a binary compound (Fig. 5). | ||
| Fig. 5 The mechanism of the ion pair complex and the hypothesized pathway between LTS and the FLN dye. | ||
The FLN molecule can take one of the following forms, depending on the pH of the solution:
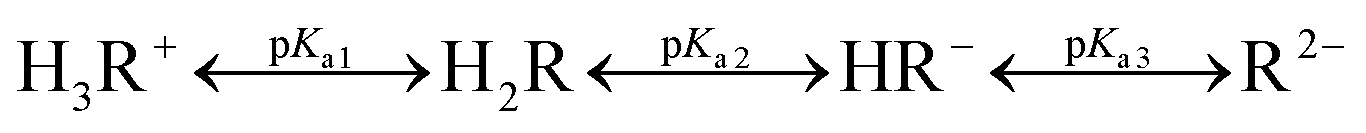
The ratio of the included reactants
Job's plot was used to determine the ion-pair complex's molar ratio. Equal doses of the drug and the dye were coupled in cascade molar ratios to keep the overall molarity constant. As stated in the experimental stages, analysis was carried out in a similar manner. A correlation between a drug's mole fraction and its corresponding response was depicted in Job's plots. According to the plots, a mole fraction of roughly 0.5 produced the best results. This score advocated the development of a drug: reagent blending in the ratio of 1:1. Using this ratio, the presence of one tertiary amino group in the medication that might be ion-linked to one dye molecule was confirmed and supported (ESI Fig. 3†).
Stern–Volmer formula and quenching kinetic study
To understand the quenching mechanism of FLN generated by LTS, the Stern–Volmer formula was used.46| f0/f = 1 + kSV[M] = 1 + kqτ0[Q] | (1) |
The fluorophore's lifespan in the excited state (typically within 0.089 nanoseconds (ns)47 or 89 picoseconds (ps)48), the Stern–Volmer dynamic suppressing constant (KSV), and the bimolecular suppressing rate constant (kq) all play a role in this equation. FLN and FLN-LTS have fluorescence intensities of F0 and F, respectively.
The quenching rate constant kq = KSV/τ0 confirmed the formation of the complex
| kq = kSV/τ0, | (2) |
On plotting F0/F for FLN and LTS at 35, 45, and 55 °C (308, 318, and 328 K), the linear regressions were obtained.
The results and the dose of interest had an excellent straight-line relationship, as shown in ESI Fig. 4.† The creation of a ground state complex was responsible for quenching the fluorescence of FLN. For dynamic quenching, the maximum scattering collision constant of the quencher was 6 × 109 M−1 s−1.49
According to the study's findings, LTS suppression mechanisms are not expected to be dampened by dynamic collisions but rather by the complexation process. Dynamic and static quenching methods can both use the Stern–Volmer equation. However, the Stern–Volmer slope (KSV) for static quenching processes is reliant on the concentration of FLN, but the KSV for dynamic quenching processes is not. This shows that the suppression is the outcome of a complicated mechanism rather than a dynamic process. The fluorescence data were examined using a modified Stern–Volmer equation:50
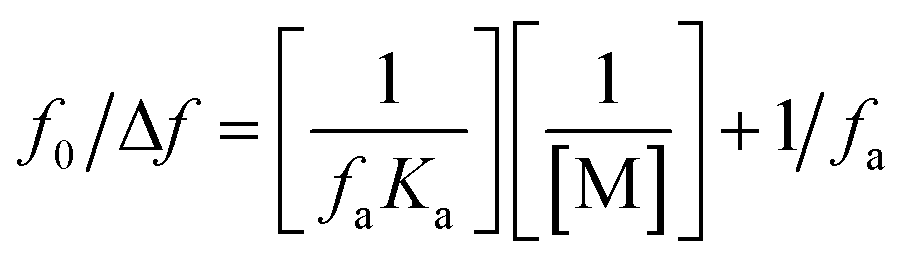 | (3) |
The suppression constant (Ka) is the amount of initial fluorescence that can be quenched by the quencher (fa). ESI Fig. 5† shows a straight-line relationship between F0/ΔF and the inverted quencher dose; (1/[M]). A static quenching process was confirmed by the data, which demonstrated a linear relationship between the drug and dye.
Evaluation of the binding constant and site
Treatment-related drug stability and safety are closely linked to the therapeutic agent's binding capacity. The complexed drug–protein is a great example of how drug–protein binding can be studied. Binding constant values were calculated using fluorescence intensity data. The equilibrium between bound and nonbound particles can be calculated using the equation below.9
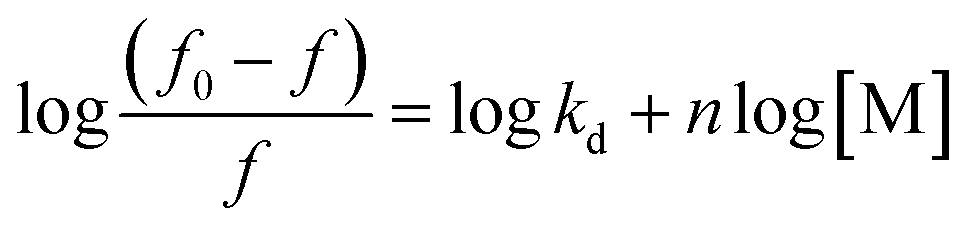 | (4) |
For a reaction in a static quenching mode, the linking constant (K) and the number of linking sites (n) are identical to those of the reaction in eqn (1), so the graph in ESI Fig. 6† shows that [(f0 − f)/f] is plotted against log[M] and the Y-axis intercept is equal to logK. The value of n is close to one (0.94) when the drug interacts with FLN, indicating that the drug has one binding site for the FLN. To estimate the drug's speed and ease of transport from circulation to the intended receptor, FLN binding is the most significant component.’ Binding constants of approximately 7.071 × 104 L mol−1 are frequent in reversible protein binding.51 As a result, the K value indicates a moderate binding between LTS and FLN, pointing to the creation of an LTS–FLN complex that is reversible.
Binding forces and thermodynamic parameters
Biomolecular interactions can occur via hydrophobic, van der Waals, or electrostatic forces between a big biological molecule and a small organic one. There are many different sorts of interactions that can take place in protein association processes, and each one has its own set of thermodynamic parameters (ΔH and ΔS), as detailed by Ross and Subramanian.52 When both ΔH and ΔS are greater than zero, hydrophobic contact is the dominant force. If ΔH and ΔS are smaller than zero, the major forces are van der Waals and hydrogen-bonding interactions. Electrostatic forces take dominance when ΔH is negative, and ΔS is positive. If the temperature changes only slightly, the enthalpy change (ΔH) can be taken for granted. The van't Hoff equation is used here:|
lnKT = −ΔH/RT + ΔS/R
| (5) |
The gas constant R, the linking constant KT, and the temperature T (in kelvin scale) were used to derive the thermodynamic characteristics of the LTS–FLN binding.
LTS–FLN binding thermodynamic parameters were calculated using the van't Hoff equation, and the repercussions of this conclusion were studied.
The slope of the van't Hoff plot is used to compute the change in enthalpy (ΔH). The free energy change (G) can be calculated using the formula shown below:
| ΔG° = ΔH° − TΔS° | (6) |
At three different temperatures, the binding constants were utilized to derive the thermodynamic criterion using a linear van't Hoff plot (ESI Fig. 7†).
Using the van't Hoff equation, lnK and the inverse of T (1/T) were plotted on a straight line. It is clear that LTS binding to FLN occurs spontaneously, as evidenced by the negative free energy (ΔG) and decrease in entropy (ΔS). This binding is an endothermic reaction because of the positive enthalpy change (ΔH) indicated by the increasing K values at higher temperatures.
Using the formula ΔG° = −2.303RTlogKd, where R denotes gas constant (8.314 J K−1 mol−1), T: temperature (in Kelvin scale), and Kd signifies the linking constant, one may calculate Gibb's free energy (ΔG°), which was −24.76 kJ mol−1 for the reaction of interest. At room temperature, the system's signal was both immediate and practical because of this significant negative free energy value.
Complex confirmation
FTIR spectroscopy was used to verify and characterize the functional groups in the crude drug sample and the drug-dye-produced product. The LTS's FTIR spectra corroborated the amino group's absence in the resultant IR spectrum. This proved that a novel product could be developed from multiple complex parts. To verify the complex formation, we compared the spectra of the drug, the dye, and the product. The new product showed improved features that are unrelated to LTS. As shown in ESI Fig. 8,† the dye's –OH group stretching band in the spectrum area between 3200 and 3500 cm−1 and the –OH group bending band at 1310–1390 cm−1 were significantly reduced in their measured responses due to the complexation process, confirming the formation of the new compound.The absence of an amino peak in the compound suggested that the amino group in LTS was either absent or bound to the dye, FLN, in some other fashion (ESI Fig. 8†).
By comparing the LTS spectrum with the product spectrum after complexation with LTS and FLN, we were able to confirm the formation of the complex. Additionally, many spectroscopic properties of LTS were lost in the synthesis of the complex, and the resulting product had novel functionality. Because the –N– peak in the LTS–FLN complex was no longer visible, it was determined that the amino groups in the LTS molecule were either lost or had been incorporated into a new bond with FLN.
Method vetting and validation
The constructed spectroscopic system was tested in accordance with the necessary criteria.39 Several parameters, including linearity and range, precision, accuracy, and flexibility, were tested as part of the validation process.Sensitivity and linearity
Several LTS solutions were analyzed using the spectrofluorimetric technique. The LTS concentration-dependent calibration graph was created by plotting FI scores against the LTS dose in μg mL−1. From 0.1 to 1.7 μg mL−1, the new approach demonstrated a linear response. Besides that, the technique was evaluated using a linear decline analysis, and the results are shown in Table 1.| Parameter | Value |
|---|---|
| Linear range (μg mL−1) | 0.1–1.7 |
| Slope | 1.029 |
| Intercept | 338.425 |
| Standard deviation of the intercept | 6.345 |
| Determination coefficient (r2) | 0.9996 |
| Limit of quantitation (μg mL−1) | 0.061 |
| Limit of detection (μg mL−1) | 0.020 |
The method's sensitivity was estimated using LOQ and LOD formulas.
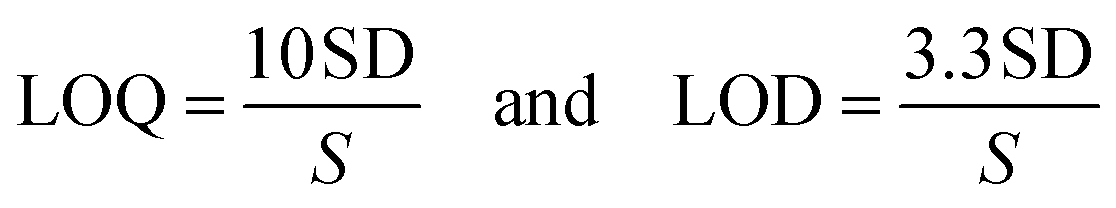
The sensing response of Felumin dye with L-Tetramisole in the present article was also compared with different techniques listed in the literature reports (ESI Table 2†).
Precision and accuracy
The accuracy of the spectroscopic technique was evaluated at three dose levels (0.5, 1.0, and 1.5 μg mL−1). Accuracy can be assessed using two metrics: recovery percentage and relative error. Table 2 shows that the procedure has a high degree of accuracy.As an additional step, the proposed technique was employed to evaluate the inter-and intra-day precision of the current system at dose levels of 0.5 to 1.5 μg mL−1, respectively. The RSD value was utilized to evaluate the spectroscopic technique's precision. Table 3 shows that the current approach has good precision, with RSD values under 2% (at both levels).
The system adaptation testing (robustness)
System parameters like pH, FLN solution volume, and reaction rate were examined to see if the suggested spectroscopic system could handle the light modification of these objects. It was possible to evaluate the method's robustness based on these metrics. The tiny modifications had no major impact on the fluorescence quenching, supporting the suggested spectrofluorimetric application's tolerance (ESI Table 3†).Interference and selectivity
Various additions of prescription medicine were tested to see if the anticipated method's selectivity for the cited dosage form was adequate.In the presence of pharmaceutical additives and LTS, the suggested approach was applied to analyze the solution samples. ESI Table 4† reveals that the excipients investigated had no significant impact on the outcomes of the procedure.
The spectroscopic approach described in the literature examined the pharmaceutical LTS tablets and syrup available under the brand name Katrex®. The published spectrofluorimetric method53 was used to test the identical dose formulations. Equilibration of the spectrophotometric technique with the designed system was achieved using the t and F-statistical tests. Since the obtained t- and F-values at a confidence level of 95% were smaller than the documented scores, there was no apparent difference in accuracy or precision between the described method and the suggested spectroscopic system (Table 4).
Experiments were done to evaluate the efficacy of the two prescription forms of LTS, Katrex®. This method outperformed others regarding sensitivity, convenience of use, saving time, the use of a “green” liquid, and detection levels. High recovery rates and a lack of interference from prescription package additives allow the system to be ideal for QC labs when testing dosage forms and tests that use LTS.
The results of the selectivity and interference experiments were obtained in the presence of different cations, anions, and small molecules to test their effects on FLN (ESI Table 5†). The experimental findings showed no significant interference, hence the high selectivity of the proposed system, which may be attributed to the lack of target moieties required for the complexation reaction.
Clinical sample-loaded-drug analysis (in vitro analysis)
The spectroscopic system was used to measure LTS concentrations in clinical samples. According to the method, a standard LTS solution was added to three replicated plasma or urine samples before they were tested. ESI Table 6† shows that the method has a recovery rate of around 100% for assessing LTS levels in human blood and urine.LTS in vivo study
Tests on healthy human volunteers (37 years old and weighing 60 kg) confirmed the method's suitability by assessing their plasma LTS concentrations. A single Katrex (5 mg kg−1 LTS of body weight) tablet was administered orally to the healthy participant who supplied written consent. Blood samples were obtained from the subject and analyzed. Because of the method's high sensitivity, it was used to determine the concentrations of unaltered medication in human plasma. After the ingestion of 300 mg of LTS by healthy individuals, their plasma samples were collected. The RFI of the genuine samples was identical to the RFI of the spiked samples, and no interference was found.The LTS concentrations were 1.6 μg mL−1 in all genuine plasma samples. According to the current investigation, the concentration of LTS in real plasma after 1.5 hours was about 1.483 μg mL−1, and the percentage recovery (92%) was identical to those obtained with published methods.18 LTS's Cmax and Tmax were similar to those obtained using the previously referenced method. As a result, the fluorescence approach proposed here could provide a simple alternative for accurately quantifying LTS in biological samples. Results show that the system developed for clinical and pharmacokinetic research of LTS is convenient for this application.
In vivo studies revealed that the intake of Katrex tablets at the dose of 5 mg kg−1 of body weight had a mean peak plasma recovery % of 92.74 after 1.5 hours of administration, which was close to previously published data.18 Thus, the proposed method is precise and accurate, with good recovery and relative standard deviations (ESI Table 7†).
System eco-friendliness assessment
An analyst's primary responsibility is to protect nature and humanity from the chemical and pharmaceutical industries' hazardous radicals and organic waste.41,54 Efforts to improve green chemistry must continue. The analytical eco-scale rank55 and the Environmental Quality Methods level56 are two recent concepts. The greenness of the system was evaluated using the eco-scale. As a result of the “ultimate green analysis,” an analytical eco-scale evaluation resulted in a penalty point being deducted from a possible 100. The higher the rate (number), the more green the analysis is.57–59 Energy consumption per sample was less than 0.1 kWh because no extraction or heating was required. Using an aqueous medium for all of the procedure's steps60 contributed to the procedure's high eco-scale score of 95 (ESI Table 8†). As a result, our approach was deemed environmentally friendly.Conclusion
In this study, a new spectrofluorimetric method for detecting LTS was established. The electrostatic attraction was used to assess LTS concentrations between 0.1 and 1.7 μg mL−1 in an acidic solution. Felumin, a less hazardous reagent, is also a better option. The procedure's major advantage is environmental safety because the material was immersed in water, and the resultant ion-linked complex could be easily detected in watery environments. For the current system, LTS and the protonated hydroxyl group of FLN form an ion-associated complex. Due to the extraction process, the analysis time was shortened, and the costs were decreased, making the technique simple and rapid. Water is the reaction fluid since it is the least harmful to the environment. The approaches described here are environmentally friendly. No volatile solvents were used in this study. The described method successfully detected medication in its dose forms, in blood, and urine, all without interference from the matrices. The process was rated highly on the eco-scale for greenness, which indicated that it was ecologically friendly. Therefore, this procedure can be utilized in research laboratories, clinical trials, and pharmaceutical manufacturing to ensure the quality of this treatment.Conflicts of interest
There are no conflicts to declare.References
- L. Jin, T. Wang, C. Cui, H. Wu, H. Ren and M. Wei, Dyes Pigm., 2014, 111, 39–44 CrossRef CAS.
- A. M. O. Azevedo, C. Sousa, S. S. M. Rodrigues, M. Chen, C. E. Ayala, R. L. Pérez, J. L. M. Santos, I. M. Warner and M. L. M. F. S. Saraiva, Dyes Pigm., 2022, 110635, DOI:10.1016/j.dyepig.2022.110635.
- S. M. Derayea, A. A. Hamad, D. M. Nagy, D. A. Nour-Eldeen, H. R. H. Ali and R. Ali, J. Mol. Liq., 2018, 272, 337–343 CrossRef CAS.
- J. Wang, Z. Liu, J. Liu, S. Liu and W. Shen, Spectrochim. Acta, Part A, 2008, 69, 956–963 CrossRef PubMed.
- A. A. Hamad, Talanta Open, 2022, 6, 100156 CrossRef.
- A. A. Hamad, RSC Adv., 2022, 12, 26566–26574 RSC.
- S. M. Derayea, Anal. Methods, 2014, 6, 2270–2275 RSC.
- A. A. Hamad, R. Ali and S. M. Derayea, RSC Adv., 2022, 12, 7413–7421 RSC.
- A. A. Hamad, R. Ali, H. R. H. Ali, D. M. Nagy and S. M. Derayea, RSC Adv., 2018, 8, 5373–5381 RSC.
- S. M. Derayea, A. A. Hamad, R. Ali and H. R. H. Ali, Microchem. J., 2019, 149, 104024 CrossRef CAS.
- S. M. Derayea, H. F. Askal, O. H. Abdel-Megeed and M. A. El Hamd, J. Appl. Pharm. Sci., 2012, 2, 84–89 Search PubMed.
- C. USP, Natl. Formul., 2008, 14, 1519 Search PubMed.
- H. Wang, J. Zhang, C. Gao, Y. Zhu, C. Wang and W. Zheng, Eur. J. Pharmacol., 2007, 577, 162–169 CrossRef CAS PubMed.
- L. Tong, L. Ding, Y. Li, Z. Wang, J. Wang, Y. Liu, L. Yang and A. Wen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 299–303 CrossRef CAS PubMed.
- S. Schneider and F. Meys, Forensic Sci. Int., 2011, 212, 242–246 CrossRef CAS PubMed.
- A. Kreeftmeijer-Vegter, T. P. Dorlo, M. Gruppen, A. De Boer and P. De Vries, Br. J. Clin. Pharmacol., 2015, 80, 242–252 CrossRef CAS PubMed.
- C. Hess, N. Ritke, K. Sydow, L. M. Mehling, H. Ruehs, B. Madea and F. Musshoff, Drug Test. Anal., 2014, 6, 1049–1054 CrossRef CAS PubMed.
- M. Luyckx, F. Rousseau, M. Cazin, C. Brunet, J. Cazin, J. Haguenoer, B. Devulder, I. Lesieur, D. Lesieur and P. Gosselin, Eur. J. Drug Metab. Pharmacokinet., 1982, 7, 247–254 CrossRef CAS PubMed.
- G. Renoux, C. R. Acad. Sci., 1971, 272, 349–350 CAS.
- E. P. Commission and E. D. f. t. Q. o. Medicines and Healthcare, European pharmacopoeia, Council of Europe, 2010 Search PubMed.
- B. Pharmacopoeia, Social Services and Public Safety, 2011 Search PubMed.
- P. Ravisankar and G. D. Rao, Asian J. Pharm. Clin. Res., 2013, 6, 169–173 Search PubMed.
- T. F. Vandamme, M. Demoustier and B. Rollmann, Eur. J. Drug Metab. Pharmacokinet., 1995, 20, 145–149 CrossRef CAS PubMed.
- S. Sowjanya and C. Devadasu, Int. J. Anal. Chem., 2018, 2018, 5746305 CAS.
- J. J. García, M. J. Diez, M. Sierra and M. T. Terán, J. Liq. Chromatogr., 1990, 13, 743–749 CrossRef.
- P. Sari, M. Razzak and I. G. Tucker, J. Liq. Chromatogr. Relat. Technol., 2004, 27, 351–364 CrossRef CAS.
- B. Wyhowski de Bukanski, J.-M. Degroodt and H. Beernaert, Z. Lebensm.-Unters. Forsch., 1991, 193, 545–547 CrossRef CAS PubMed.
- L. Xu, F. Luan, F. Hu, H. Liu and Y. Gao, Anal. Methods, 2013, 5, 762–766 RSC.
- L. Xu, F. Luan, L. Wang, H. Liu and Y. Gao, J. AOAC Int., 2014, 97, 128–132 CrossRef CAS PubMed.
- B. C. Lourencao, R. A. Medeiros, S. S. Thomasi, A. G. Ferreira, R. C. Rocha-Filho and O. Fatibello-Filho, Sens. Actuators, B, 2016, 222, 181–189 CrossRef CAS.
- R. Elaryan, S. toubar, A. Ashour and M. Elshahed, Electroanalysis, 2022, 1450–1462 CrossRef.
- N. Zubenya, Z. Kormosh, D. Saribekova and S. Sukharev, Mediterr. J. Chem., 2017, 6, 7–14 CrossRef CAS.
- U. Shah, T. Talaviya and A. Gajjar, Int. J. Pharm. Chem. Anal., 2015, 2, 108–112 Search PubMed.
- J. Misquith, P. P. Prabhu, E. Subrahmanyam and A. Shabaraya, Int. J. PharmTech Res., 2012, 4, 1215–1220 CAS.
- A. M. El-Didamony, Spectrochim. Acta, Part A, 2008, 69, 770–775 CrossRef PubMed.
- S. M. Syed, Z. Farooqui, K. Gawale and D. Hamde, Int. J. Pharm. Res. Appl., 2020, 5, 97–101 Search PubMed.
- Y. Xiao, J. Li and C. Fu, Luminescence, 2014, 29, 183–187 CrossRef CAS PubMed.
- S. Khalil and N. Borham, J. Pharm. Biomed. Anal., 2000, 22, 235–240 CrossRef CAS PubMed.
- M. E. Swartz and I. S. Krull, Analytical method development and validation, CRC Press, 2018 Search PubMed.
- A. A. Hamad, Y. F. Hassan, W. E. Eltoukhi, S. M. Derayea, M. A. S. Abourehab and B. S. Mohammed, Luminescence, 2023, 38, 166–175 CrossRef CAS PubMed.
- S. M. Derayea, R. Ali and A. A. Hamad, Arabian J. Chem., 2020, 13, 8026–8038 CrossRef CAS.
- C. Y. Huang, in Methods in enzymology, Elsevier, 1982, vol. 87, pp. 509–525 Search PubMed.
- S. Shiao, V. Chhabra, A. Patist, M. Free, P. Huibers, A. Gregory, S. Patel and D. Shah, Adv. Colloid Interface Sci., 1998, 74, 1–29 CrossRef CAS.
- D. Snigur, M. Fizer, A. Chebotarev, O. Lukianova and O. Zhukovetska, Dyes Pigm., 2022, 198, 110028 CrossRef CAS.
- L. Yu, Z. Liu, X. Hu, L. Kong and S. Liu, Microchim. Acta, 2010, 169, 375–382 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, 2006 Search PubMed.
- J. P. Eichorst, K. W. Teng and R. M. Clegg, in Fluorescence Spectroscopy and Microscopy, Springer, 2014, pp. 97–112 Search PubMed.
- N. Boens, W. Qin, N. Basarić, J. Hofkens, M. Ameloot, J. Pouget, J.-P. Lefevre, B. Valeur, E. Gratton and M. VandeVen, Anal. Chem., 2007, 79, 2137–2149 CrossRef CAS PubMed.
- M. Szabelski, D. Ilijev, P. Sarkar, R. Luchowski, Z. Gryczynski, P. Kapusta, R. Erdmann and I. Gryczynski, Appl. Spectrosc., 2009, 63, 363–368 CrossRef CAS PubMed.
- S. Lehrer, Biochemistry, 1971, 10, 3254–3263 CrossRef CAS PubMed.
- L. N. Jattinagoudar, S. T. Nandibewoor and S. A. Chimatadar, J. Biomol. Struct. Dyn., 2017, 35, 1200–1214 CrossRef CAS PubMed.
- M. Hema and S. Arivoli, Int. J. Phys. Sci., 2007, 2, 10–17 Search PubMed.
- P. Polawar, U. Shivhare, K. Bhusari and V. Mathur, Res. J. Pharm. Technol., 2008, 1, 539–540 Search PubMed.
- A. A. Hamad and S. M. Derayea, Spectrochim. Acta, Part A, 2023, 293, 122460 CrossRef CAS PubMed.
- A. Gałuszka, Z. M. Migaszewski, P. Konieczka and J. Namieśnik, TrAC, Trends Anal. Chem., 2012, 37, 61–72 CrossRef.
- M. De La Guardia and S. Armenta, Green analytical chemistry: theory and practice, Elsevier, 2010 Search PubMed.
- S. M. Derayea, H. Madian, E. Samir, A. A. Hamad and K. M. B. El-Din, Spectrochim. Acta, Part A, 2022, 273, 121024 CrossRef CAS PubMed.
- A. A. Hamad, Talanta Open, 2023, 7, 100179 CrossRef.
- S. M. Derayea, D. M. Nagy, K. M. B. El-Din, T. Z. Attia, E. Samir, A. A. Mohamed and A. A. Hamad, RSC Adv., 2022, 12, 17607–17616 RSC.
- A. Abdulhafez Hamad, Spectrochim. Acta, Part A, 2023, 288, 122187 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra08183k |
| This journal is © The Royal Society of Chemistry 2023 |