
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of a lipase/reduced graphene oxide/metal–organic framework electrode using a central composite design-response surface methodology approach†
Nur Aina Izzati Mohd Mokhtar a,
Siti Efliza Asharibc and
Ruzniza Mohd Zawawi*a
a,
Siti Efliza Asharibc and
Ruzniza Mohd Zawawi*a
aDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, 43400 UPM, Serdang, Selangor, Malaysia. E-mail: ruzniza@upm.edu.my
bIntegrated Chemical BioPhysics Research, Faculty of Science, Universiti Putra Malaysia, 43400 UPM, Serdang, Selangor, Malaysia
cCentre of Foundation Studies for Agricultural Sciences, Universiti Putra Malaysia, 43400 UPM, Serdang, Selangor, Malaysia
First published on 3rd May 2023
Abstract
Lipase has been gaining attention as the recognition element in electrochemical biosensors. Lipase immobilization is important to maintain its stability while providing excellent conductivity. In this study, a lipase electrochemical biosensor immobilized on a copper-centred metal–organic framework integrated with reduced graphene oxide (lipase/rGO/Cu-MOF) was synthesized by a facile method at room temperature. Response surface methodology (RSM) via central composite design (CCD) was used to optimize the synthesis parameters, which are rGO weight, ultrasonication time, and lipase concentration, to maximize the current response for the detection of p-nitrophenyl acetate (p-NPA). The results of the analysis of variance (ANOVA) showed that all three parameters were significant, while the interaction between the ultrasonication time and lipase concentration was the only significant interaction with a p-value of less than 0.05. The optimized electrode with parameters of 1 mg of rGO, 30 min ultrasonication time, and 30 mg mL−1 lipase exhibited the highest current response of 116.93 μA using cyclic voltammetry (CV) and had a residual standard error (RSE) of less than 2% in validation, indicating that the model is suitable to be used. It was characterized by X-ray diffraction (XRD), field-emission scanning electron microscopy (FESEM), and Fourier transform infrared spectroscopy (FTIR), where the integration of the composite was observed. Immobilization using ultrasonication altered the lipase's secondary structure, but reduced its unorderly coils. The electrochemical and thermal analysis showed that the combination of Cu-MOF with rGO enhanced the electrochemical conductivity and thermostability.
Introduction
Lipase (EC 3.1.1.3) is one of the hydrolase enzymes widely used in biocatalysts and organic reactions owing to its non-toxicity and unique chemoselectivity, regioselectivity, and stereoselectivity.1 It has also attracted attention as the recognition element in electrochemical biosensors to detect analytes such as triglycerides, pesticides, and fungicides.2–5 The use of an enzyme in biosensors is still limited due to its unstable nature and short lifetime. It is prone to denaturation, biofouling, and electrode passivation, resulting in low selectivity. The difficulty in recovering and reusing the lipase and its tendency to agglomerate easily make the industrial application challenging.1,6 Hence, enzyme immobilization is utilized to reduce these disadvantages. It can enhance the lipase's stability and resistance to environmental changes and is practical in industry as it can reduce cost and is reusable.7A highly porous material with a large surface area, such as a metal–organic framework (MOF), is advantageous for this purpose.8 A metal biomolecule framework (MBioF), copper aspartate MOF (Cu-MOF), has been used as the lipase immobilization matrix in an electrochemical biosensor due to its properties.9 It is also beneficial owing to its biocompatibility as it employs amino acid as the ligand.10 However, MOFs possess poor electroconductivity and catalytic activity, which is a problem in electrochemical sensors.11 The integration with reduced graphene oxide (rGO) with superior electrochemical and thermal conductivity can overcome these limitations.12 It can be done through the presence of oxygen vacancies and chemically active sites which enable modification of MOFs.13 The integration of rGO with the MOF (rGO/MOF) has been reported to exhibit excellent electroconductivity to be utilized in electrochemical sensors owing to their synergistic effects.14
The solvothermal/hydrothermal method is the commonly employed method for rGO/MOF synthesis. However, it requires harsh experimental conditions and a long reaction time, and has difficulties in scaling up for industrial applications.15,16 In contrast, the ultrasonication method is faster, easier to use, and does not require high temperature and pressure. It operates by cavitation and collapse of bubbles in the aqueous media, which leads to high pressure variation that can increase the chemical reactivity of the composite.17 Thus, ultrasonication is used for the synthesis of rGO/MOF.
To the best of the authors' knowledge, rGO/MOF composite has never been used as a lipase immobilization matrix in biosensors. Herein, the parameters for the synthesis of a lipase biosensor immobilized on rGO/Cu-MOF (lipase/rGO/Cu-MOF) were studied and optimized using response surface methodology (RSM). It is used for the detection of p-nitrophenyl acetate (p-NPA). p-nitrophenyl esters like p-NPA and p-nitrophenyl palmitate (p-NPP) are widely used to assess lipase activity and can also be used as the model pesticide due to their similar detection mechanism with organophosphorus pesticide (OPP) containing p-nitrophenol groups such as methyl paraoxon, methyl parathion, parathion, and ethyl paraoxon.5,18,19 They have also been utilized as the substrate to study the inhibition of pesticides, such as malathion and carbendazim, using lipase biosensors.3,20 RSM can reduce the time and cost compared to the one-factor-at-a-time approach. Plus, it can provide information on the significance of the synthesis parameters and the interactions between them, which influence the outcome greatly.21
Experimental
Chemicals
All the chemicals used are of analytical grade without further purification. Copper(II) nitrate trihydrate (Cu(NO3)2·3H2O), L-aspartic acid, acetic acid (glacial), and ethanol 95% (denatured) were acquired from Chemiz. Chitosan (low molecular weight), rGO, Burkholderia cepacia lipase, and p-NPA were obtained from Sigma Aldrich. The p-NPA was dissolved in 0.1 M phosphate buffer solution (PBS) pH 7.0, which was prepared by mixing potassium dihydrogen phosphate (KH2PO4) and dipotassium phosphate (K2HPO4), both were purchased from Chemiz. [Fe(CN6)]3−/4− was prepared by mixing potassium ferrocyanide (K4[Fe(CN6)]·3H2O) and potassium hexacyanoferrate(III) (K3[Fe(CN6)]) in 0.1 M potassium chloride solution (KCl), which were obtained from Friendemann Schmidt and Chemiz, respectively. Ultrapure 18.2 M Ω cm deionized water (Sartorius) and distilled water (BioPure) were used throughout the experiment.Synthesis of Cu-MOF and rGO/Cu-MOF
The synthesis of Cu-MOF was carried out following the reference with slight modifications.22 1.0872 g of Cu(NO3)2·3H2O was dissolved in 6 mL of ethanol. The solution was mixed with 30 mL of distilled water containing 0.24 g of NaOH and 0.3993 g of L-aspartic acid and allowed to stand for 24 hours. The resulting deep blue solution was filtered and washed six times with a mixture of distilled water and ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) before being vacuum dried at 60 °C for 8 hours. For the synthesis of rGO/Cu-MOF, 1 mg of Cu-MOF and various amounts of rGO were ultrasonicated in 10 mL of ethanol using an ultrasonic water bath at room temperature. Then, the rGO/Cu-MOF was filtered and washed with ethanol followed by deionized water. The composites were dried at ambient temperature.
:1) before being vacuum dried at 60 °C for 8 hours. For the synthesis of rGO/Cu-MOF, 1 mg of Cu-MOF and various amounts of rGO were ultrasonicated in 10 mL of ethanol using an ultrasonic water bath at room temperature. Then, the rGO/Cu-MOF was filtered and washed with ethanol followed by deionized water. The composites were dried at ambient temperature.
Lipase immobilization on rGO/Cu-MOF and electrode fabrication
Electrodes were fabricated following the reference with slight modifications.9 300 μL of rGO/Cu-MOF solution was mixed with 100 μL of Burkholderia cepacia lipase (BCL) solution prepared in 0.1 M PBS (pH 7.0). The solution was ultrasonicated in an ultrasonic water bath for 10 min to immobilize the enzyme. Then, 100 μL of a chitosan solution (6 mg mL−1) prepared in acetic acid (1% v/v) was added to the mixture and shaken for about three minutes. Chitosan was chosen as the film-forming agent to fabricate the electrode. 5 μL of the resulting composite was drop-casted onto the surface of a screen-printed carbon electrode (SPCE) and dried before analysis. The procedures for the synthesis and fabrication of the biosensor were visualized in Fig. 1.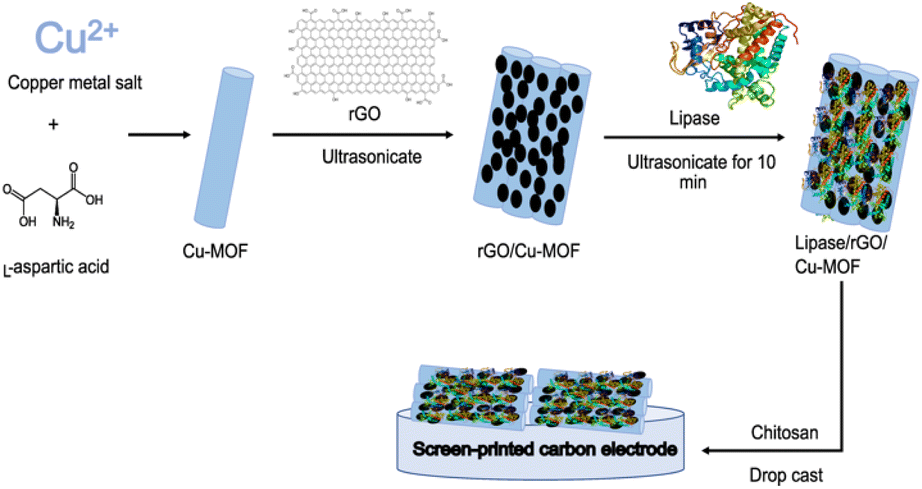 | ||
| Fig. 1 Schematic diagram of the synthesis of lipase/rGO/Cu-MOF biosensor. | ||
Electrode optimization using RSM
There are several steps for optimization using RSM. They consist of screening the significant factors and the interactions, generating the coefficient equation and appropriate model via mathematical modelling, predicting the optimized parameters, and validating the model.23,24 The chosen parameters were rGO weight (mg), ultrasonication time of rGO/Cu-MOF synthesis (min), and lipase concentration (mg mL−1), which were denoted as A, B, and C, respectively. The levels for each parameter used are shown in Table 1.| Symbol | Independent variables (unit) | Range | ||
|---|---|---|---|---|
| Low (−1) | Middle (0) | High (+1) | ||
| A | rGO weight (mg) | 1 | 1.5 | 2.0 |
| B | Ultrasonication time (min) | 30 | 45 | 60 |
| C | Lipase concentration (mg mL−1) | 10 | 20 | 30 |
A face-centred central composite design (CCD) was developed using Design Expert 13.0 (Stat Ease Inc., Minnesota, United States of America). 20 sets of experimental runs were generated, including six central points, six axial points, and eight factorial points. The star points were located at the centre of each face of the factorial space, hence α = ±1. The second-order polynomial equation was shown in eqn (1),
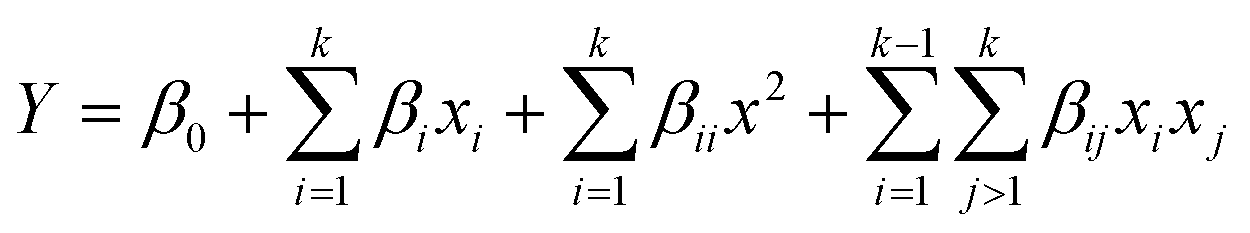 | (1) |
The modified electrode was subjected to electrochemical analysis using cyclic voltammetry (CV) with 750 μM p-NPA in 0.1 M PBS (pH 7) as the analyte. The oxidation peak of p-NPA was chosen as the dependent parameter for statistical analysis. The analysis of variance (ANOVA) was used for model generation, prediction, the significance of the model and parameters, and the optimization of parameters. The p-value <0.05 was chosen to indicate the significance of the data. The optimization was performed via numerical and graphical optimization by choosing the levels for each parameter that had the highest predicted current response and desirability.
After the optimization, the model was validated by running five experiments with randomly selected levels. The residual standard error (RSE) was calculated using eqn (2) to evaluate the validity of the model.
 | (2) |
Electrochemical analysis
Electrochemical analysis was carried out using Metrohm Multi Autolab M101 and Nova software 2.1.4 (Barendrecht, Netherlands). SPCE was used as it integrates the three-electrode system in a miniaturized electrode. CV was used to measure the current response of p-NPA detection at a potential range of −1.5 V to 1.0 V, a start and stop potential at 0 V, and a scan rate of 100 mV s−1. The potential was negatively scanned from 0 V to −1.5 V before being scanned to the positive potential until 1.0 V. The electrode was subjected to ten cycles, with the highest oxidation peak selected as the output for RSM optimization. The behaviour of the modified electrodes was evaluated with CV using 5 mM [Fe(CN6)]3−/4− in 0.1 M KCl at a potential range of −0.4 V to 0.7 V and a scan rate of 100 mV s−1. The electrochemical impedance spectroscopy (EIS) was carried out using the FRA32 impedance potentiostat module. A sinusoidal potential modulation of ±10 mV amplitude in the 104 to 10−2 Hz frequency range, with the number of frequencies of 10 per decade, was superimposed onto the formal potential of the redox couple [Fe(CN6)]3−/4−. It was operated under an open circuit potential (OCP) with a 60 s duration and 0.1 s interval time. Before analysis, the electrolytes were purged with nitrogen gas for 5–10 min to avoid the interferences caused by the oxygen.Characterization of the composite
The structural, morphological, and crystallographic analysis of the composite were studied using the JEOL JSM-7600F field-emission scanning electron microscope (FESEM) (Tokyo, Japan) and Shimadzu XRD-6000 X-ray diffractometer (XRD) at a 2θ range from 2° to 60° using Cu Kα radiation with a wavelength of 1.54 Å (Kyoto, Japan). Thermogravimetric analysis (TGA) (Mettler Toledo, Ohio, United States of America) was carried out under a nitrogen gas atmosphere with a heating rate of 10 °C min−1 from 50 °C to 800 °C to evaluate the thermal stability of the composite. The chemical structure and interaction within the materials were examined using a Fourier transform infrared spectroscopy (FTIR) with attenuated total reflection (ATR) (Bruker Invenio, United States of America). Diamond crystal was used for L-aspartic acid, Cu-MOF, and free lipase. On the other hand, germanium crystal was employed for carbon-based materials, which are rGO, rGO/Cu-MOF, and lipase/rGO/Cu-MOF. The scanning was done with a range from 4000 cm−1 to 400 cm−1, 16 sample and background scans, and a resolution of 4 cm−1. Peak deconvolution was done for free lipase and lipase/rGO/Cu-MOF spectrum from 1700 cm−1 to 1600 cm−1 using OriginPro software to assess the secondary structure of the protein. Peak integration was carried out to calculate the percentage area for the secondary structure. The total peak area was calculated by adding up all the peak areas. The percentage area of the peak was calculated using eqn (3).25
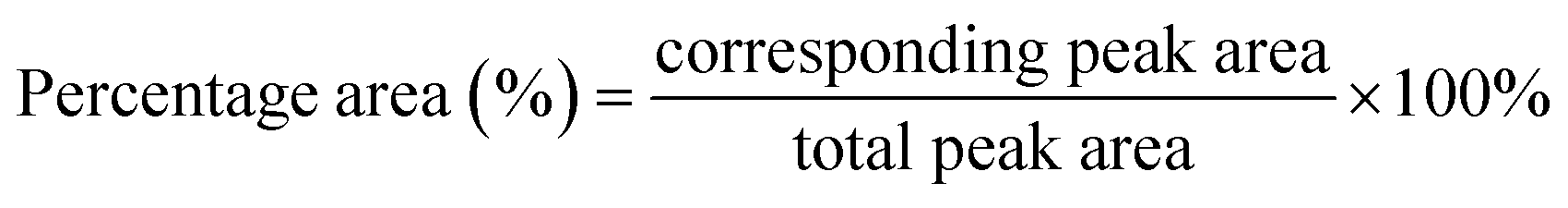 | (3) |
Results and discussion
Electrochemical detection of p-NPA
The detection of p-NPA using lipase involved hydrolysis, irreversible reduction, and reversible redox processes. These resulted in three peaks shown using cyclic voltammetry (CV), which were two cathodic peaks and one anodic peak, as shown in Fig. S1.† The reactions were explained by Pohanka5 and the schematic diagram for the reactions involved is shown in Scheme S1.† The product of lipase hydrolysis, p-nitrophenol, was reduced to p-(hydroxylamino)phenol, resulting in the first cathodic peak at −1.00 V (reaction (1)). The oxidation peak at 0.07 V and another reduction peak at −0.10 V were the results of a redox process of p-(hydroxylamino)phenol and p-nitrosophenol (reaction (2)). The mechanism is similar to that of detection of OPP containing p-nitrophenol groups using lipase, where the OPP is hydrolyzed to form p-nitrophenol that undergoes reversible redox process.9,19 In this study, the anodic peak was chosen as the dependent parameter for RSM optimization.Model fitting and statistical analysis
The calculated predicted values were compared with the actual values of the current response for each run, as shown in Table S1,† where both values did not differ greatly with each other. The results of ANOVA are shown in Table 2. The calculated F-value of the selected model, which was 57.91, showed that this model is significant. Furthermore, the p-value, which was less than 0.0001, strengthened this evaluation. Conversely, the lack-of-fit of this model is deemed insignificant based on its F-value of 0.84, which is relative to the pure error. The p-value of 0.5719 indicated that 57.19% chances of the high F-value of lack-of-fit was due to the noise. Thus, the insignificant lack-of-fit is necessary to show that this model can be used.| Source | Sum of squares | df | Mean square | F-Value | p-Value | |
|---|---|---|---|---|---|---|
| Model | 561.42 | 9 | 62.38 | 57.91 | <0.0001 | Significant |
| A | 10.36 | 1 | 10.36 | 9.62 | 0.0112 | |
| B | 5.66 | 1 | 5.66 | 5.25 | 0.0449 | |
| C | 262.96 | 1 | 262.96 | 244.13 | <0.0001 | |
| AB | 0.0018 | 1 | 0.0018 | 0.0017 | 0.9682 | |
| AC | 4.62 | 1 | 4.62 | 4.29 | 0.0652 | |
| BC | 24.57 | 1 | 24.57 | 22.81 | 0.0008 | |
| A2 | 25.32 | 1 | 25.32 | 23.50 | 0.0007 | |
| B2 | 5.21 | 1 | 5.21 | 4.83 | 0.0526 | |
| C2 | 91.05 | 1 | 91.05 | 84.53 | <0.0001 | |
| Residual | 10.77 | 10 | 1.08 | |||
| Lack of fit | 4.93 | 5 | 0.9855 | 0.8432 | 0.5719 | Not significant |
| Pure error | 5.84 | 5 | 1.17 | |||
| Cor total | 572.19 | 19 |
The coefficient of determination, R2, explains the overall efficiency and adequacy of the model, where the value nearing one is encouraged. The R2 value of 0.9812 as shown in Table S2† indicated that 98.12% of this model can be explained through the independent parameters. The adjusted and predicted R2, which were 0.9642 and 0.9235 respectively, showed small differences of 0.0507, indicating that this model agrees with each other. There were also small differences observed between R2 and adjusted R2, which implied that the model contained mostly statistically significant factors. The model exhibited a high adequate precision of 25.8022 meaning a high signal-to-noise ratio. A high ratio of 4 is required for the model to be used to navigate the design space.
Based on the statistical analysis, a quadratic model was proposed. The quadratic model equation based on the coded factor is shown in eqn (4), where A, B, and C indicated the rGO weight, ultrasonication time, and lipase concentration, respectively. It included all linear, quadratic and interaction terms that are either significant or not. The positive and negative signs indicated synergistic and antagonistic effects, respectively.
| Current = 100.55 − 1.02A − 0.752B + 5.13C − 0.015AB − 0.76AC − 1.75BC + 3.03A2 − 1.38B2 + 5.75C2 | (4) |
From the ANOVA result in Table 2, the significance of the independent parameters and the interaction between the parameters were also evaluated. Significant values were found in the model terms for all three independent parameters based on the p-value of less than 0.05, indicating that these three parameters are important in the synthesis of lipase/rGO/Cu-MOF electrodes. However, only the interaction between BC was deemed significant compared to AB and AC. The two interactions had p-value greater than 0.05, indicating no mutual effects shown through the interactions. The quadratic terms A2 and C2 were also considered significant, while B2 was insignificant. These insignificant factors are still required in the model to support the hierarchy. If there are too many insignificant terms in the model, a model reduction can be applied to improve the model.
Diagnostics
The diagnostics plots showed the accuracy and reliability of the model chosen. As depicted in Fig. S2a,† the actual responses were almost linear to the predicted response line. Thus, this model can accurately predict the outcome and the linear regression fit can be used for model analysis. In Fig. S2b,† the externally studentized residuals response versus the predicted response plot showed that the data were mainly circulated at the mean point of the response variables. The plot of studentized residuals response versus run number in Fig. S2c† exhibited random scattering of data within the limit with no apparent trends. This implied that the data had good distribution and no constant errors. Therefore, no model transformation is needed and there is no data ignored. This can also be strengthened by looking into the plot of the difference in fits (DFFITS) as a function of the run number in Fig. S2d,† where no outliers were observed. Thus, this model is appropriate for the optimization design.Effects of individual parameters
Apart from ANOVA, the graph of each independent parameter can be observed to determine the significance of the parameters. In Fig. S3a,† the maximum current response for rGO weight (A) was obtained when 1 mg of rGO was used, which was the minimum level. The current response decreased when the amount of rGO increased to 1.5 mg. However, the current response increased slightly when the amount of rGO was further increased up to 2 mg. A higher amount of rGO may lead to agglomeration due to the interlayer effect of the van der Waals interaction26 that can reduce the surface area for lipase immobilization. Thus, it can be concluded that a minimum amount of rGO is required to obtain the highest current response.Fig. S3b† shows the effect of ultrasonication time (B) on the current response. The trend for ultrasonication time showed a decrease in the current response with increasing ultrasonication time. This may be due to the ultrasonication treatment exceeding the optimum time, which may lead to a rise in the metal oxide concentration of the Cu-MOF due to excess turbulence.27 It can contribute to structural collapse and a decrease in production yield and reactivity of the rGO/Cu-MOF composite, therefore lowering the current response.
The effect of lipase concentration (C) was the most significant compared to A and B based on the statistical analysis, with a p-value of less than 0.0001. Fig. S3c† shows that the current response decreased slightly when the lipase concentration increased to 15 mg mL−1. However, the current response drastically improved upon the increase to 30 mg mL−1. This observation is consistent with the previous research, which the maximum level of lipase concentration generated the highest current response.9,28 Low enzyme concentration caused the enzyme molecules to be distributed randomly. This led to ineffective interaction with the acoustic cavitation generated by the ultrasonication. As the enzyme concentration increased, the distribution became more orderly and the effective interaction with the cavitation bubbles increased during lipase immobilization.29 Plus, a high enzyme concentration provided a higher reaction rate for the hydrolysis of p-NPA, generating more electroactive species that enhanced the peak current response.30
Effects of the interactions between the parameters
The interactions between the parameters were shown through the 3D surface graphs and the 2D contour shapes, where the shapes indicate the type of interactions that could happen at different levels. For the interaction between AB and BC, they were observed to have saddle points, where the minimum or maximum points cannot be seen in the centre point. The AB interaction, which is the interaction between the rGO weight and ultrasonication time of the rGO/Cu-MOF synthesis, was the least significant according to its high p-value, which was 0.9682. The graph in Fig. 2a and b also showed that there was no apparent minimum or maximum value at the centre point. Hence, no visible trend can be deduced from the graph. However, the contour shape showed that the maximum current response can be obtained by utilizing a minimum amount of rGO and ultrasonication time. This is to prevent rGO agglomeration and the collapse of Cu-MOF structure that can interfere with lipase immobilization.26,27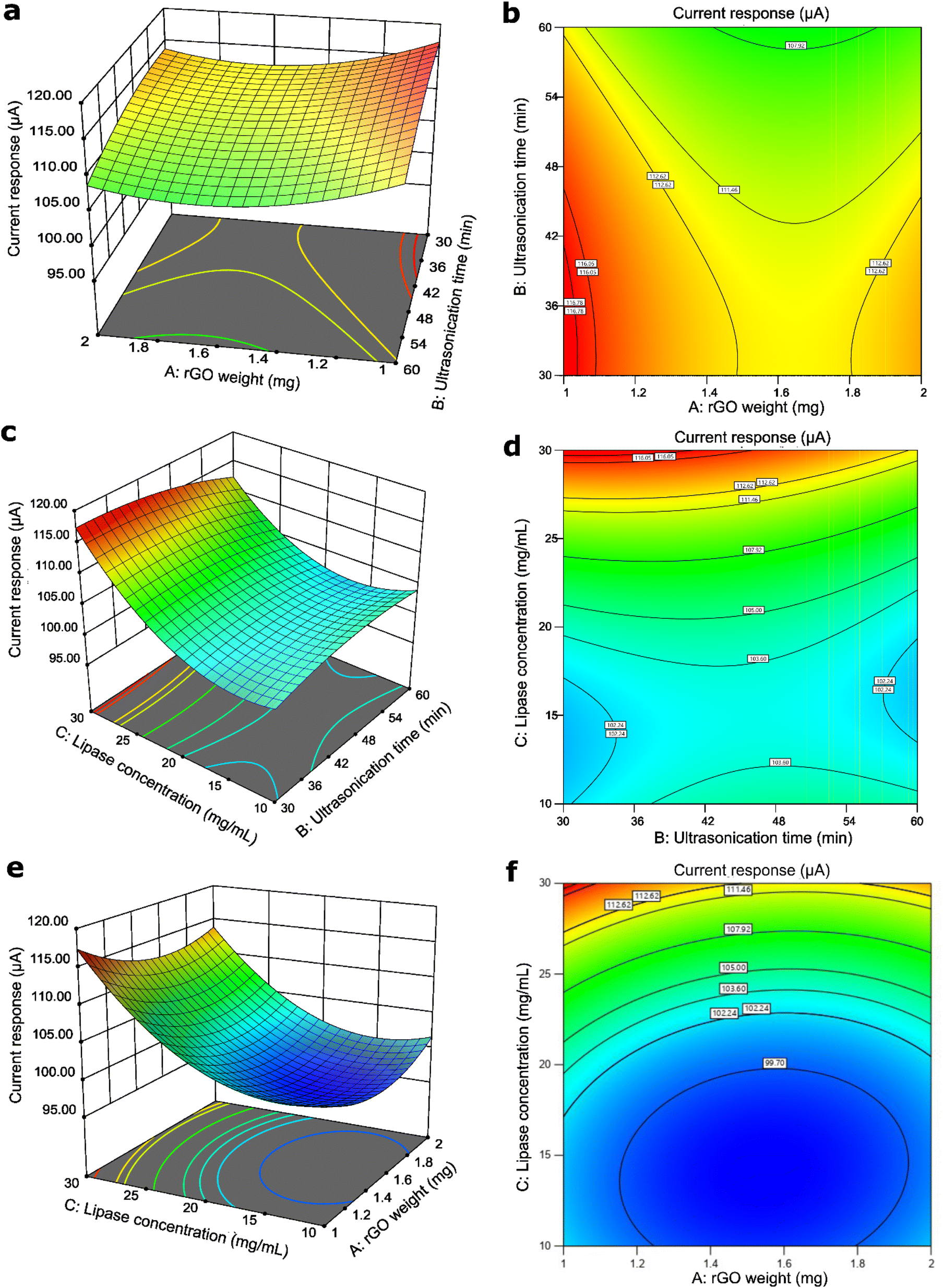 | ||
| Fig. 2 The 3D graph and 2D contour shape of the interaction between the rGO weight and the ultrasonication time (a) and (b), the ultrasonication time and the lipase concentration (c) and (d), and the rGO weight and the lipase concentration (e) and (f). | ||
Conversely, BC has a p-value of 0.0008, which was deemed as the most statistically significant interaction compared to other interactions. However, it also had a saddle point where the centre point did not show any minimum or maximum value. Despite no visible trend observed at the centre point of the graph in Fig. 2c and d, it was noted that the maximum current response was generated at the lowest ultrasonication time and highest lipase concentration. Low ultrasonication time is required to retain a stable rGO/Cu-MOF structure for lipase immobilization.27 Immobilizing high concentration of lipase increased the enzymatic activity, resulting in high electroactive species that translated to a high current response.30
The interaction between rGO weight and lipase concentration, AC, showed a different trend from the previous two interactions. It was observed that the graph in Fig. 2e and f exhibited a minimum trend where the centre point has the minimum value of the generated current response. It can also be seen that the maximum current response was generated with the lowest rGO weight and the highest lipase concentration. A low rGO weight is needed to avoid rGO agglomeration,26 which can reduce the surface area for lipase immobilization. A high lipase concentration increased the reaction rate of p-NPA hydrolysis for an enhanced enzymatic activity.30
Optimization and validation
Numerical and graphical optimization were utilized to predict the optimized conditions for the synthesis of lipase/rGO/Cu-MOF with maximum current response. The constraints were set for each parameter within the range of minimum and maximum, and the goal for the current response was set to maximum, as shown in Table S3.† The optimum conditions were 1 mg of rGO, 30 min ultrasonication, and 30 mg mL−1 lipase. The experimental value for these parameters was 116.93 ± 0.28 μA. Under these conditions, a current response of 117.353 μA was predicted. This was the highest current response predicted with a desirability of 1.00. The point prediction also predicted the same outcome with the optimized conditions. The optimum parameters were used to synthesize the electrode.To validate the model, five experiments with randomized conditions were selected to predict the outcome and compare it with the actual current response. For each parameter, the conditions were within the range that maximizes the current response. It was calculated that all responses had a residual standard error (RSE) of less than 2%, indicating that the model is fit to use. The results of the validation are shown in Table S4.†
Characterization
Subsequently, the composite was characterized based on the optimum parameters. The morphology and crystallinity of the rGO/Cu-MOF immobilization matrix was studied using XRD and FESEM. As shown in Fig. S4,† the Cu-MOF exhibited peaks at 2θ = 5.56°, 11.1°, 14.72°, 16.68°, 19.86°, 25.62°, and 26.6°, consistent with those in the literature.9 The peak intensity showed that it was a highly crystalline material, which can facilitate enzyme immobilization and orientation toward a preferred direction.31 The detailed information on the Cu-MOF structure is unattainable due to its nanosize under 200 nm, which makes the analysis using single crystal X-ray diffraction (SCXRD) difficult.32 Plus, it is challenging to synthesize large crystals of Cu-MOF.33 However, it did not show any peak corresponding to other phases, such as copper oxide, indicating the synthesized Cu-MOF was in a pure state.34 On the other hand, rGO exhibited an amorphous structure with no visible sharp peak. After integration with rGO, the peak intensity of Cu-MOF decreased significantly, which may be due to the shifting in atomic position. However, there was no significant difference in the peak positions. This showed that the integration with rGO did not cause severe structural damage to the Cu-MOF. This is important, as the stability of the composite can affect lipase immobilization.The FESEM images of Cu-MOF in Fig. 3a exhibited a rod-like shape with several micrometers in length and a diameter of 80 to 180 nm, which is slightly smaller than the reported literature.10,22,34–36 This might be due to the slow crystallization process of the amino acid-based MOF.37 This structure is the most promising among one-dimensional nanostructures owing to its high surface-to-volume ratio and web-like configuration,34 which are advantageous for rGO attachment and lipase immobilization. The presence of wrinkled sheets displayed in Fig. 3b indicated the integration of rGO with the Cu-MOF. The wrinkled structure can enhance charge transport on the electrode surface as it shortens the ionic diffusion path and improves its electroconductivity.38 Agglomeration of Cu-MOF was exhibited after integration using ultrasonication. Although it was reported that ultrasonication reduced the MOF size,39 it may cause agglomeration due to the molecular collisions of the Cu-MOF.
 | ||
| Fig. 3 The FESEM images of Cu-MOF (a) and rGO/Cu-MOF (b) in 10000× magnification. | ||
The FTIR spectrum of L-aspartic acid, Cu-MOF, rGO, and rGO/Cu-MOF are displayed in Fig. 4a. The spectrum of Cu-MOF and L-aspartic acid, which is the Cu-MOF linker, were compared to study the formation of the Cu-MOF. A small broad peak from 3159 cm−1 to 2873 cm−1 in the L-aspartic acid spectrum and a large broad peak from 3569 cm−1 to 2857 cm−1 in the Cu-MOF are peaks corresponding to the overlapping bands of N–H stretching and O–H stretching. Apart from that, the L-aspartic acid spectrum showed peaks at 1689 cm−1, 1648 cm−1, and 1595 cm−1, which were assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, N–H stretching, and N–H bending, respectively. Two peaks at 1139 cm−1 and 1118 cm−1 were related to NH2 rocking mode.10 All the peaks were corresponding to the presence of carboxyl and amine groups in L-aspartic acid.
O stretching, N–H stretching, and N–H bending, respectively. Two peaks at 1139 cm−1 and 1118 cm−1 were related to NH2 rocking mode.10 All the peaks were corresponding to the presence of carboxyl and amine groups in L-aspartic acid.
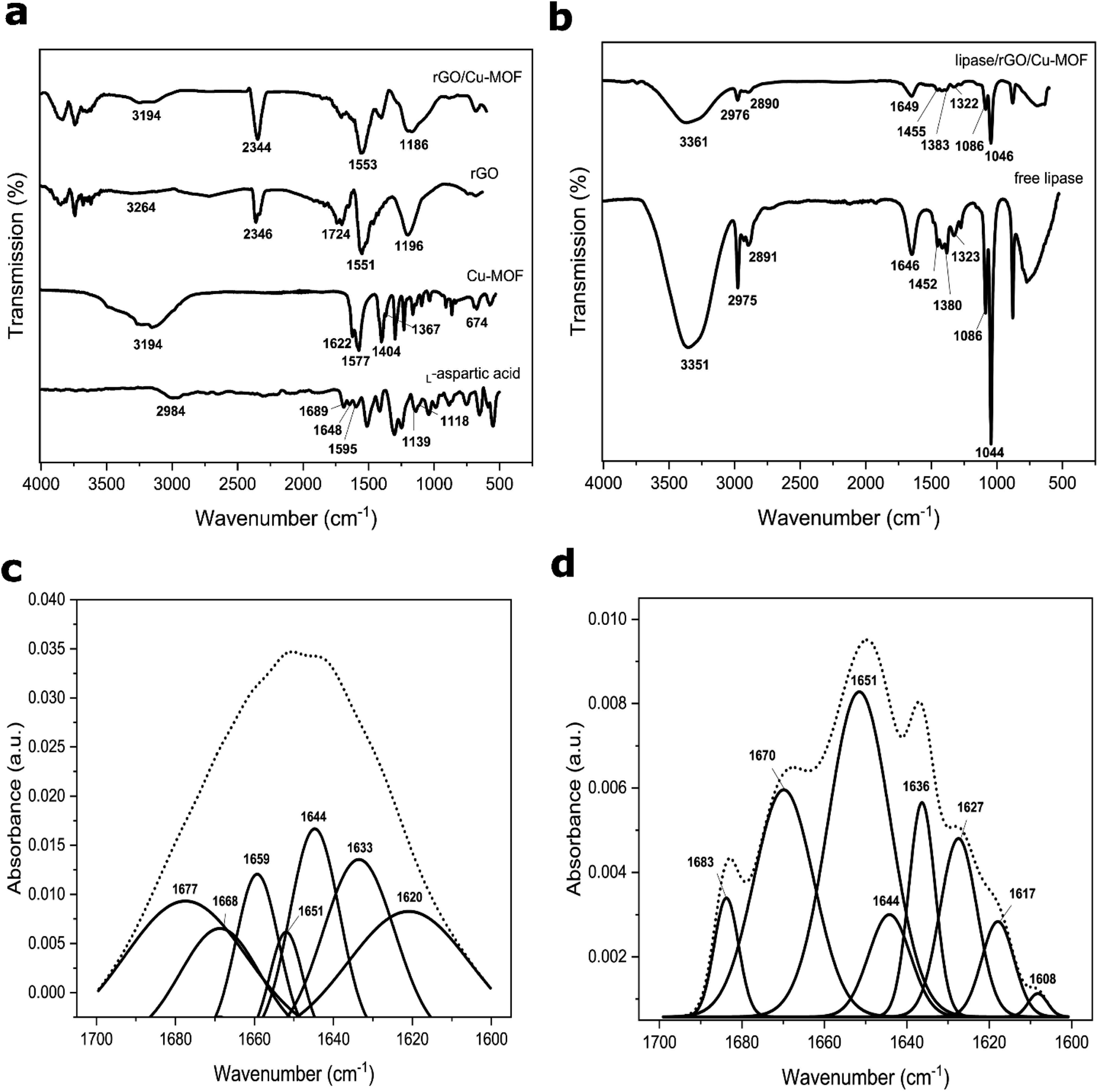 | ||
| Fig. 4 FTIR spectrum of L-aspartic acid, Cu-MOF, rGO, rGO/Cu-MOF (a), free lipase, and lipase/rGO/Cu-MOF (b). Spectrum deconvolution of amide I for free lipase (c) and lipase/rGO/Cu-MOF (d) from 1700 cm−1 to 1600 cm−1. | ||
In the Cu-MOF, the asymmetric and symmetric COO− stretching were recorded at 1622/1577 cm−1 and 1400/1367 cm−1, respectively. Furthermore, a small peak located at 674 cm−1 was related to the Cu–O bond.34 The blue shifting of the CO stretching peak and the presence of a Cu–O peak in the Cu-MOF spectrum indicated the formation of Cu-MOF via the binding of the copper ion with the carboxyl group of L-aspartic acid. This finding is consistent with the previous literature.10,33,34 There is also a possibility of the copper ion coordination with the amine group due to the shifting of the overlapping bands of the N–H stretch and O–H stretch.34 Thus, the Cu-MOF can be formed via a coordination bond between the copper ion with the carboxyl group only, or with the carboxyl and amine groups of the L-aspartic acid.
In the rGO spectrum, a sharp peak that appeared at 1551 cm−1 was attributed to the CC of the graphene structure. A broad peak from 3461 cm−1 to 2981 cm−1 with extremely low intensity was recorded, indicating the low amount of oxygenated functional groups. Two peaks at 1724 cm−1 and 1196 cm−1 were attributed to the CO stretching and C–O stretching of the functional groups. The integration of Cu-MOF with rGO occurred possibly via hydrogen bonding due to the reduction in peak intensity corresponding to the O–H and N–H stretching in rGO/Cu-MOF compared to Cu-MOF. An almost diminished peak related to the CO stretching of rGO and a blue shifting of the C–O stretching peak indicated the possible interaction between the oxygenated functional groups of rGO with the carboxyl and amine groups of Cu-MOF. A sharp peak at 1553 cm−1 was related to the CC stretching of the rGO, showing that the sp2 structure retained upon integration. The presence of peaks at 2346 cm−1 and 2344 cm−1 in rGO and rGO/Cu-MOF respectively were due to CO2 contamination.
The spectrum of free lipase and lipase/rGO/Cu-MOF are shown in Fig. 4b. A broad peak from 3500 cm−1 to 3200 cm−1 in both free and immobilized lipases was assigned to the carboxyl, hydroxyl, and amine groups of amino acids in lipase. The two peaks at 2975 cm−1 and 2891 cm−1 in free lipase were related to the alkane C–H stretch. The peak at 1646 cm−1 was attributed to the amide I linkage corresponding to the CO stretch and C–N stretch. Two peaks at 1455 cm−1 and 1383 cm−1 corresponded to the asymmetric and symmetric CH3 bending of the protein's methyl groups. A peak at 1322 cm−1 was related to the O–H bending, whereas two sharp peaks at 1086 cm−1 and 1044 cm−1 were attributed to the C–O stretching. A reduction in peak intensity was observed for all the peaks corresponding to the lipase upon immobilization. The decrease in peak intensity and no apparent shifting observed after immobilization showed that the lipase retained its structure using the physical adsorption method. The ultrasonication provided a facile and rapid immobilization method compared to the simple stirring method, which can take up hours. It can be done via π–π interaction, hydrogen bonding, hydrophobic interaction, or electrostatic interaction.
The amide I peak from 1700 cm−1 to 1600 cm−1 is attributed to the α-helix and β-sheet structures of the enzyme. They are the important backbone for the secondary structure of the protein. Spectrum deconvolution was done on the amide I linkage peak to assess the effect of the lipase immobilization via ultrasonication towards the secondary structure of the lipase before and after immobilization. The peaks were assigned based on the literature.25,40 In Fig. 4c, the peaks of free lipase at 1651 cm−1 and 1659 cm−1 were related to the α-helices, whereas the peaks at 1620 cm−1 and 1633 cm−1 were corresponding to β-sheets, with a peak attributed to β-turns at 1677 cm−1. The presence of α-helices and β-sheets confirmed the secondary structure of the free lipase. A peak at 1668 cm−1 was assigned to the turns and bands, while a peak at 1644 cm−1 was attributed to the unordered random coils of lipase.
On the other hand, the lipase/rGO/Cu-MOF exhibited only a peak attributed to α-helix at 1651 cm−1, as shown in Fig. 4d. The peaks corresponding to β-sheets were at 1608 cm−1, 1617 cm−1, 1627 cm−1, and 1636 cm−1, with 1670 cm−1 corresponding to the β-turns. A peak related to the turns and bands was recorded at 1683 cm−1, whereas a smaller peak at 1644 cm−1 was attributed to unordered coils.
Peak integration was carried out for the deconvoluted peaks of free and immobilized lipase to observe if there are any secondary structure changes upon immobilization. The results are shown in Table S5,† where the contents are studied based on the percentage area. The β-sheets content was higher than α-helices in free lipase, with a considerably high content of unordered coils. However, the β-sheets content decreased significantly upon immobilization, with α-helix constituting the highest percentage of the secondary structure. This may be attributed to lipase immobilization via ultrasonication. A decrease in β-sheets and an increase in β-turns and unordered coils were reported after enzyme encapsulation41 and ultrasonication42 caused by the unfolding of β-sheets. However, the unordered coils content was reduced greatly after lipase immobilization in this study. This may indicate interactions between the lipase and the immobilization matrix. The results showed that immobilization using ultrasonication can alter the lipase's secondary structure, but it is majorly preserved due to the high content of the orderly structure.
The thermal stability of Cu-MOF (Fig. 5a) and rGO/Cu-MOF (Fig. 5b) was evaluated by TGA and derivative thermogravimetric analysis (DTG). The first weight loss at about 56 °C to 120 °C for both materials were attributed to the removal of solvent molecules and adsorbed gas within the pores.43,44 The weight loss at around 122 °C in the rGO/Cu-MOF was due to the loss of oxygen-containing functional groups of rGO.45 The small reduction of weight indicated a low amount of oxygenated functional groups in the rGO.46 The reduction of weight at about 230 °C and 232 °C in Cu-MOF and rGO/Cu-MOF was attributed to the decomposition of the functional groups of Cu-MOF to copper oxide,47 causing the structure to collapse and become amorphous. The temperature was extended in rGO/Cu-MOF to around 517 °C due to the presence of carbon structure in rGO. This also contributed to lower weight loss compared to Cu-MOF.11 This is caused by the large aspect ratio and interfacial contact area of rGO that can lower thermal resistance.12 Cu-MOF retained 37% of the weight at 385 °C, whereas rGO/Cu-MOF still has 59% of the weight left even after heating at 517 °C. A higher pyrolysis temperature indicates a more stable material, where in this case rGO helped in increasing the thermostability of Cu-MOF.
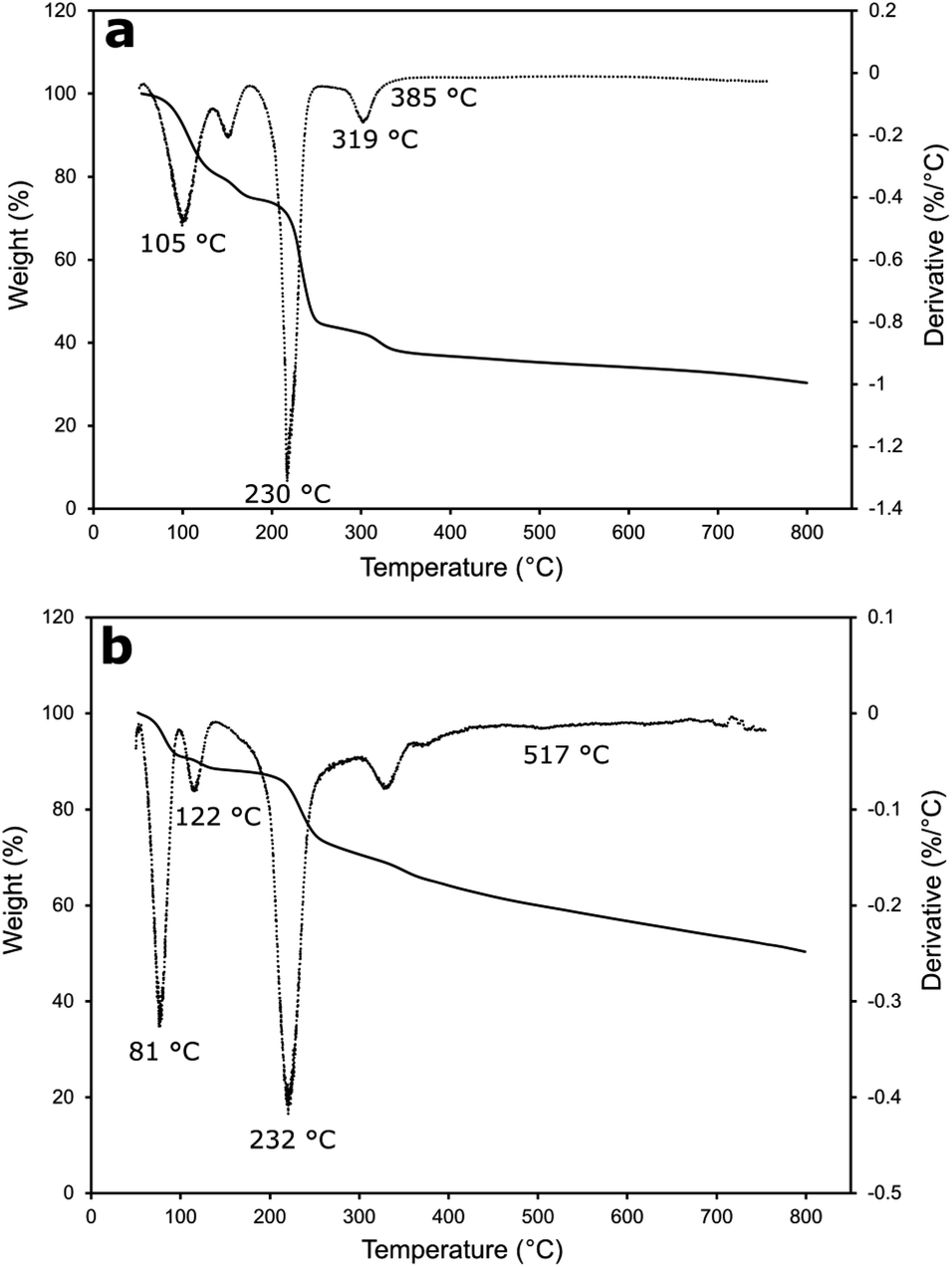 | ||
| Fig. 5 TGA (solid line) and DTG (dotted line) curves of Cu-MOF (a) and rGO/Cu-MOF (b). | ||
Cyclic voltammetry (CV) was used to evaluate the electrochemical behaviour of the optimized lipase/rGO/Cu-MOF electrode with other non-enzymatic electrodes using 5 mM [Fe(CN6)]3−/4− in 0.1 M KCl solution. The results of the analysis are demonstrated in Fig. 6a. The current responses for the electrodes in ascending order are as follows; Cu-MOF/SPCE < lipase/rGO/Cu-MOF/SPCE < bare SPCE < rGO/Cu-MOF/SPCE. The Cu-MOF/SPCE exhibited the lowest current response (ipa = 65.17 μA) demonstrating its low electroconductivity. This could be due to the weak electron transfer between the copper center and the amino acid ligand.48 However, the current response increased by 1.78-fold upon integration with rGO. The restoration of sp2 structure in rGO upon reduction from graphene oxide reduced the defects and sp3 structure of carbon–oxygen that hindered electron transport within the structure.49 The current response for lipase/rGO/Cu-MOF/SPCE (ipa = 78.62 μA) was lower than that of rGO/Cu-MOF/SPCE (ipa = 116.21 μA) due to the steric hindrance of the lipase, resulted in partial blockage of interfacial electrons.2 However, the current response was higher than that of Cu-MOF/SPCE due to the immobilization within the rGO/Cu-MOF matrix.
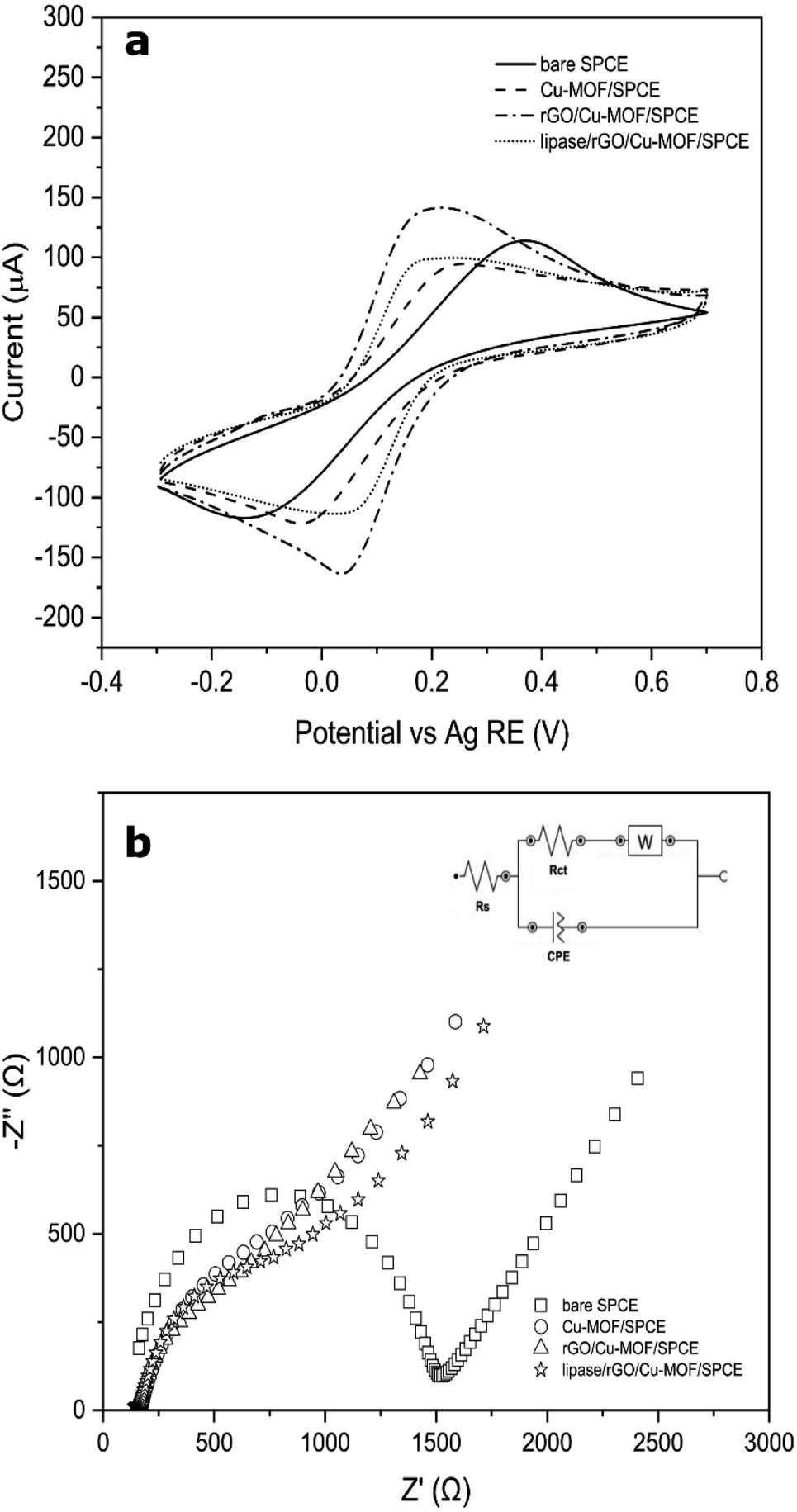 | ||
| Fig. 6 Cyclic voltammograms of bare SPCE, Cu-MOF/SPCE, rGO/Cu-MOF/SPCE, and lipase/rGO/Cu-MOF/SPCE in 0.1 M KCl containing 5.0 mM [Fe(CN6)]3−/4− at scan rate of 100 mV s−1 (a) and Nyquist plots of bare SPCE, Cu-MOF/SPCE, rGO/Cu-MOF/SPCE, and lipase/rGO/Cu-MOF/SPCE in 0.1 M KCl solution containing 5.0 mM of [Fe(CN)6]3−/4− (b) inset shows the Randles equivalent circuit model for the electrodes. | ||
The rGO/Cu-MOF/SPCE also exhibited low peak-to-peak separation (ΔEp = 158.7 mV) compared to Cu-MOF (ΔEp = 266.5 mV) and bare SPCE (ΔEp = 515.2 mV). The enhancement of the kinetic transfer of the electrode is due to the synergistic effects of Cu-MOF and rGO in improving the surface area and conductivity.14 Interestingly, an even lower ΔEp was shown by lipase/rGO/Cu-MOF/SPCE (ΔEp = 124.5 mV), demonstrating an enhanced kinetic transfer with reversibility behavior. This observation can provide insights on the electrochemical behavior of the lipase upon immobilization on the rGO/Cu-MOF.
The electrochemical processes that occurred at the electrode–electrolyte interface for the modified electrodes were evaluated by EIS. The analysis was carried out in an N2-saturated 0.1 M KCl solution containing 5 mM of [Fe(CN)6]3−/4−. The plots were fitted and visualized using the Randles equivalent circuit model, which consists of the electrolyte resistance (Rs), charge transfer resistance (Rct), Warburg impedance (W), and constant phase elements (CPE). As shown in Fig. 6b, bare SPCE exhibited the largest semicircle diameter (Rct = 1350 Ω), implying a slow charge transfer occurred on the electrode surface. Upon electrode modification, the semicircle diameter decreased in the following order: Cu-MOF/SPCE > lipase/rGO/Cu-MOF/SPCE > rGO/Cu-MOF/SPCE, with the Rct values of 786 Ω, 765 Ω, and 580 Ω, respectively. The Cu-MOF/SPCE demonstrated a high resistance on the electrode surface due to its low energy charge-transport pathways within the framework, which led to its low electroconductivity.48 However, the integration of the rGO reduced the resistivity of the Cu-MOF/SPCE. This indicates that the incorporation of rGO into Cu-MOF helps in reducing the impedance of the electrode and enhance the charge transfer kinetic. On the other hand, the high resistivity of the lipase/rGO/Cu-MOF/SPCE was due to the partial blockage of the interfacial electrons by the lipase.2 The Nyquist plots of the electrodes were in agreement with the CVs obtained in Fig. 6a.
Conclusions
The lipase/rGO/Cu-MOF electrode was successfully optimized in this study using RSM. The CCD was able to generate a quadratic model that can be used to evaluate the significance of the three synthesis parameters based on the statistical data. Interestingly, the highest current response was generated when the rGO weight and the ultrasonication time were set at the minimum level, whereas the lipase concentration was at the maximum. Only the interaction between the ultrasonication time of rGO/Cu-MOF synthesis and lipase concentration was found to be significant based on the p-value. This is to ensure the stability of the rGO/Cu-MOF structure for lipase immobilization to obtain an enhanced enzymatic activity in the detection of p-NPA. The ultrasonication provided a rapid and facile integration of rGO with the Cu-MOF and successful lipase immobilization via physical adsorption compared to other time-consuming methods. The rGO/Cu-MOF showed improved electrochemical and thermal stability and has a high potential to be used as lipase immobilization matrix in electrochemical biosensors.Author contributions
Nur Aina Izzati Mohd Mokhtar: methodology, investigation, data curation, formal analysis, visualization, writing – original draft. Ruzniza Mohd Zawawi: conceptualization, methodology, formal analysis, validation, resources, supervision, funding acquisition, writing – review and editing. Siti Efliza Ashari: methodology, formal analysis, validation, writing – review and editing.Conflicts of interest
The authors have no conflicts of interest to declare that are relevant of this article.Acknowledgements
The authors thank the Ministry of Higher Education Malaysia for funding this study. Grant no.: FRGS/1/2020/STG04/UPM/02/7. Nur Aina Izzati Mohd Mokhtar would like to thank the Public Service Department of Malaysia for scholarship (BYDPA). Special thanks to Department of Chemistry, Faculty of Science and Institute of Bioscience, Universiti Putra Malaysia for the facilities provided.References
- F. Rafiee and M. Rezaee, Int. J. Biol. Macromol., 2021, 179, 170–195, DOI:10.1016/j.ijbiomac.2021.02.198.
- Y. Cheng, B. Ma, C. P. Tan, O. M. Lai, W. Panpipat, L. Z. Cheong and C. Shen, Sens. Actuators, B, 2020, 321, 128477, DOI:10.1016/j.snb.2020.128477.
- A. de Moura Barboza, A. Brunca Da Silva, E. Mendonça Da Silva, W. Pietro De Souza, M. A. Soares, L. Gomes De Vasconcelos, A. J. Terezo and M. Castilho, Anal. Methods, 2019, 11, 5388–5397, 10.1039/c9ay01600g.
- M. Iyer, I. Shreshtha, H. Baradia and S. Chattopadhyay, Biotechnol. Genet. Eng. Rev., 2022, 38, 87–110, DOI:10.1080/02648725.2022.2050499.
- M. Pohanka, Molecules, 2019, 24, 616, DOI:10.3390/molecules24030616.
- G. Rocchitta, A. Spanu, S. Babudieri, G. Latte, G. Madeddu, G. Galleri, S. Nuvoli, P. Bagella, M. I. Demartis, V. F. R. Manetti and P. A. Serra, Sensors, 2016, 16, 780, DOI:10.3390/s16060780.
- D. M. Liu, J. Chen and Y. P. Shi, TrAC, Trends Anal. Chem., 2018, 102, 332–342, DOI:10.1016/j.trac.2018.03.011.
- Y. Hu, L. Dai, D. Liu, W. Du and Y. Wang, Renewable Sustainable Energy Rev., 2018, 91, 793–801, DOI:10.1016/j.rser.2018.04.103.
- Z. Wang, B. Ma, C. Shen and L. Z. Cheong, Talanta, 2019, 197, 356–362, DOI:10.1016/j.talanta.2019.01.052.
- G. Gizer, M. Sahiner, Y. Yildirim, S. Demirci, M. Can and N. Sahiner, Curr. Res. Green Sustainable Chem., 2021, 4, 100110, DOI:10.1016/j.crgsc.2021.100110.
- S. Rani, S. Kapoor, B. Sharma, S. Kumar, R. Malhotra and N. Dilbaghi, J. Alloys Compd., 2020, 816, 152509, DOI:10.1016/j.jallcom.2019.152509.
- A. T. Smith, A. M. LaChance, S. Zeng, B. Liu and L. Sun, Nano Mater. Sci., 2019, 1, 31–47, DOI:10.1016/j.nanoms.2019.02.004.
- H. Yu, W. Guo, X. Lu, H. Xu, Q. Yang, J. Tan and W. Zhang, Food Control, 2021, 127, 108117, DOI:10.1016/j.foodcont.2021.108117.
- N. A. I. M. Mokhtar, R. M. Zawawi, W. M. Khairul and N. A. Yusof, Environ. Chem. Lett., 2022, 3099–3131, DOI:10.1007/s10311-022-01403-2.
- S. Hao, L. Yuling and J. Yang, J. Mol. Struct., 2022, 1251, DOI:10.1016/j.molstruc.2021.132046.
- D. Sud and G. Kaur, Polyhedron, 2021, 193, 114897, DOI:10.1016/j.poly.2020.114897.
- N. A. Khan, Z. Hasan and S. H. Jhung, Coord. Chem. Rev., 2018, 376, 20–45, DOI:10.1016/j.ccr.2018.07.016.
- R. Syeed-León, M. Sandoval-Barramtes, H. Trimiño-Vásquez, L. Villegas-Peñaranda and G. Rodríguez-Rodríguez, Uniciencia, 2020, 34, 31–43, DOI:10.15359/ru.34-2.2.
- L. Qiu, P. Lv, C. Zhao, X. Feng, G. Fang, J. Liu and S. Wang, Sens. Actuators, B, 2019, 286, 386–393, DOI:10.1016/j.snb.2019.02.007.
- K. G. Reddy, G. Madhavi and B. E. Kumara Swamy, J. Mol. Liq., 2014, 198, 181–186, DOI:10.1016/j.molliq.2014.06.019.
- S. Nazarpour, R. Hajian and M. H. Sabzvari, Microchem. J., 2020, 154, 104634, DOI:10.1016/j.microc.2020.104634.
- C. Zhao, C. Shen and W. Han, RSC Adv., 2015, 5, 20386–20389, 10.1039/C4RA16416D.
- G. E. de Benedetto, S. Di Masi, A. Pennetta and C. Malitesta, Biosensors, 2019, 9, 26, DOI:10.3390/bios9010026.
- S. Bhattacharya, Response Surface Methodology in Engineering Science, 2021, DOI:10.5772/intechopen.95835.
- H. A. Alhazmi, M. Al Bratty, A. M. Meraya, A. Najmi, M. S. Alam, S. A. Javed and W. Ahsan, Acta Biochim. Pol., 2021, 68, 99–107, DOI:10.18388/abp.2020_5462.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279, DOI:10.1038/nmat4170.
- A. Dastbaz, J. Karimi-Sabet and M. A. Moosavian, Int. J. Hydrogen Energy, 2019, 44, 26444–26458, DOI:10.1016/j.ijhydene.2019.08.116.
- B. Ma, L. Z. Cheong, X. Weng, C. P. Tan and C. Shen, Electrochim. Acta, 2018, 283, 509–516, DOI:10.1016/j.electacta.2018.06.17.
- S. H. Jadhav and P. R. Gogate, Ind. Eng. Chem., 2014, 53, 1377–1385, DOI:10.1021/ie403419e.
- P. K. Robinson, Essays Biochem., 2015, 59, 1–41, DOI:10.1042/BSE0590001.
- X. Wang, P. C. Lan and S. Ma, ACS Cent. Sci., 2020, 6, 1497–1506, DOI:10.1021/acscentsci.0c00687.
- B. Shan, J. B. James, M. R. Armstrong, E. C. Close, P. A. Letham, K. Nikkhah, Y. S. Lin and B. Mu, J. Phys. Chem. C, 2018, 122, 2200–2206, DOI:10.1021/acs.jpcc.7b11012.
- G. Khandelwal, M. K. Ediriweera, N. Kumari, N. P. Maria Joseph Raj, S. K. Cho and S. J. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 18887–18896, DOI:10.1021/acsami.1c03075.
- S. Nami-Ana, S. Nasresfahani, J. Tashkhourian, M. Shamsipur, Z. Zagarpour and M. H. Sheikhi, ACS Appl. Mater. Interfaces, 2021, 13, 39791–39805, DOI:10.1021/acsami.1c07116.
- I. Imaz, M. Rubio-Martínez, W. J. Saletra, D. B. Amabilino and D. Maspoch, J. Am. Chem. Soc., 2009, 131, 18222–18223, DOI:10.1021/ja908721t.
- L. Lei, D. Song, L. Fan, B. Liu, M. He, X. Sun, W. Xu, K. Tao, H. Huang and Y. Li, Microchim. Acta, 2022, 189, 1–11, DOI:10.1007/s00604-021-05160-x.
- S. Wang, M. Wahiduzzaman, L. Davis, A. Tissot, W. Shepard, J. Marrot, C. Martineau-Corcos, D. Hamdane, G. Maurin, S. Devautour-Vinot and C. Serre, Nat. Commun., 2018, 9, 1–8, DOI:10.1038/s41467-018-07414-4.
- M. Saraf, R. Rajak and S. M. Mobin, J. Mater. Chem. A, 2016, 4, 16432–16445, 10.1039/c6ta06470a.
- E. Moradi, R. Rahimi, V. Safarifard and S. Azari, Proceedings, 2020, 41, 31, DOI:10.3390/ecsoc-23-06482.
- P. Sedlacek, E. Slaninova, V. Enev, M. Koller, J. Nebesarova, I. Marova, K. Hrubanova, V. Krzyzanek, O. Samek and S. Obruca, Appl. Microbiol. Biotechnol., 2019, 103, 1905–1917, DOI:10.1007/s00253-018-09584-z.
- M. I. Burgos, A. Ochoa and M. A. Perillo, Biochem. Biophys. Res. Commun., 2019, 508, 270–274, DOI:10.1016/j.bbrc.2018.11.077.
- S. S. Nadar and V. K. Rathod, World J. Microbiol. Biotechnol., 2017, 33, 1–12, DOI:10.1007/s11274-017-2322-6.
- E. Akbarzadeh, H. Z. Soheili, M. Hosseinifard and M. R. Gholami, Mater. Res. Bull., 2020, 121, 110621, DOI:10.1016/j.materresbull.2019.110621.
- J. O. Olowoyo, U. Saini, M. Kumar, H. Valdés, H. Singh, M. O. Omorogie, J. O. Babalola, A. V. Vorontsov, U. Kumar and P. G. Smirniotis, J. CO2 Util., 2020, 42, 101300, DOI:10.1016/j.jcou.2020.101300.
- M. Coros, F. Pogacean, A. Turza, M. Dan, C. Berghian-Grosan, I. O. Pana and S. Pruneanu, Phys. E, 2020, 119, 113971, DOI:10.1016/j.physe.2020.113971.
- G. G. Gebreegziabher, A. S. Asemahegne, D. W. Ayele, M. Dhakshnamoorthy and A. Kumar, Mater. Today Chem., 2019, 12, 233–239, DOI:10.1016/j.mtchem.2019.02.003.
- Y. Sun, Y. Li, N. Wang, Q. Q. Xu, L. Xu and M. Lin, Electroanalysis, 2018, 30, 474–478, DOI:10.1002/elan.201700629.
- S. K. Bhardwaj, N. Bhardwaj, R. Kaur, J. Mehta, A. L. Sharma, K. H. Kim and A. Deep, J. Mater. Chem. A, 2018, 6, 14992–15009, 10.1039/c8ta04220a.
- D. Shahdeo, A. Roberts, N. Abbineni and S. Gandhi, Compr. Anal. Chem., 2020, 91, 175–199, DOI:10.1016/bs.coac.2020.08.007.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra01060k |
| This journal is © The Royal Society of Chemistry 2023 |