
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radiolytic degradation of dodecane substituted with common energetic functional groups†
Patricia L. Huestis ,
Nicholas Lease,
Chris E. Freye,
Daniel L. Huber,
Geoffrey W. Brown,
Daniel L. McDonald,
Tammie Nelson,
Christopher J. Snyder and
Virginia W. Manner*
,
Nicholas Lease,
Chris E. Freye,
Daniel L. Huber,
Geoffrey W. Brown,
Daniel L. McDonald,
Tammie Nelson,
Christopher J. Snyder and
Virginia W. Manner*
Los Alamos National Laboratory, USA. E-mail: vwmanner@lanl.gov
First published on 21st March 2023
Abstract
Explosives exist in and are expected to withstand a variety of harsh environments up to and including ionizing radiation, though little is known about the chemical consequences of exposing explosives to an ionizing radiation field. This study focused on the radiation-induced chemical changes to a variety of common energetic functional groups by utilizing a consistent molecular backbone. Dodecane was substituted with azide, nitro, nitrate ester, and nitramine functional groups and γ-irradiated with 60Co in order to study how the functional group degraded along with what the relative stability to ionizing radiation was. Chemical changes were assessed using a combination of analysis techniques including: nuclear magnetic resonance (NMR) spectroscopy, gas chromatography of both the condensed and gas phases, Raman spectroscopy, and Fourier transform infrared (FTIR) spectroscopy. Results revealed that much of the damage to the molecules was on the energetic functional group and often concentrated on the trigger linkage, also known as the weakest bond in the molecule. The general trend from most to least susceptible to radiolytic damage was found to be D–ONO2 → D–N3 → D–NHNO2 → D–NO2. These results also appear to be in line with the relative stability of these functional groups to things such as photolysis, thermolysis, and explosive insults.
1. Introduction
Explosives find use in many applications such as military,1 mining,2 medicine,3–5 and even space exploration.6–8 The variety of applications require explosives to be exposed to a large range of harsh conditions, one of which is ionizing radiation. Very little is known about how explosives are affected by ionizing radiation fields, and the logistical challenges associated with irradiating high explosives leads to little progress on that front. Additionally, the large number of explosives that are currently in use make it impractical to study the effects of radiation on every known explosive. Instead, a more generalized approach should be used to make broad predictions on the radiolytic stability for both currently used and not yet discovered explosives.Ionizing radiation is able to drive chemistry far from equilibrium through the creation of radicals and highly excited states. These reactive species lead to complicated chemistry as they react with the surrounding system, and the resultant products are not often easy to predict. Different structures, for instance, can display different tolerances to ionizing radiation even when the same atoms are present.9 Because explosives tend to utilize different backbone structures in addition to different energetic functional groups (FGs), it can be difficult to differentiate between backbone and FG resistance to ionizing radiation when comparing across multiple explosives. This study aims to understand the relative stability of commonly used energetic FGs themselves, so a common molecular backbone was used: dodecane.
Though many backbones could have in theory been used for this study, dodecane was a reasonable first choice due to the fact that it is commonly used in nuclear reprocessing10,11 and thus its stability to ionizing radiation has been studied.11–13 In addition, it is a low volatility liquid at ambient temperature and pressure and relatively cheap and easy to work with which make it a good candidate as the common backbone molecule for this study. Although several of the substituted dodecane molecules used in this study have been synthesized before, none were commercially available and thus were synthesized in-house. The energetic FGs chosen for this study were: azide (–N3), nitro (–NO2), nitrate ester (–ONO2), and nitramine (–NHNO2); the generalized molecular structure for these materials can be found in Fig. 1. These same molecules were also the focus of an earlier photolytic computational study.14 Although photolysis and radiolysis are different processes, it is still interesting to make broad comparisons across the different energy regimes.
 | ||
| Fig. 1 Generalized molecular structures of the materials utilized in this study. | ||
In this paper, the various substituted dodecanes were synthesized, irradiated with 60Co under vacuum, and then analyzed post radiolysis in order to compare with the unirradiated (control) material. Unsubstituted dodecane was also irradiated and analyzed in a similar manner to provide a benchmark system for radiation effects of the backbone structure. Analysis was completed using a suite of techniques. Nuclear magnetic resonance (NMR) spectroscopy, gas chromatography-time of flight mass spectrometry (GC-TOFMS), Raman spectroscopy, and Fourier transform infrared (FTIR) spectroscopy were used to analyze the condensed phase for differences resulting from irradiation. Radiolytically-produced gas was analyzed using gas chromatography-mass spectrometry (GC-MS) and the total gas produced during irradiation was used as a general benchmark for radiolytic activity.
In this manuscript, all details related to the synthesis, sample preparation and irradiation, and analysis are discussed in detail, including the synthesis of a compound used for proper identification of a suspected radiolytic degradation product that was not able to be purchased. The most important degradation products are identified and organized by compound. The results are then benchmarked against previous studies on the irradiation of alkanes and relevant explosives along with photolysis, thermolysis, and small-scale sensitivity measurements and computations in order to make broader statements about explosive degradation. Notably, this study is the first of its kind to investigate radiolytic degradation of targeted energetic FGs by combining the disparate fields of energetic material synthesis, trace analytical techniques, and high-energy radiation.
2. Methods and materials
Laboratory grade dodecane (D–H, D = C12H25) was purchased from Fisher Chemical and used as is. Analytical standard grade chemicals used for GC-TOFMS identification of octanol, octanal, decanal, dodecanol, and dodecene were purchased from Sigma Aldrich and used as is. All other compounds were synthesized in-house and had a purity >97% as measured with 13C- and 1H-NMR spectroscopy.2.1 Synthesis of dodecane-based compounds
Nitric acid (fuming), triflouroacetic anhydride were purchased from Acros chemical and used as received. Dodecanol, dodecylamine and 1-bromododecane were all purchased from Sigma Aldrich (reagent grade) and used as received. Deuterated solvents (chloroform d) were purchased from Cambridge Isotope Laboratories and used as received. 1H-NMR and 13C-NMR spectra were recorded on a 400 MHz Bruker spectrometer. NMR signals were referenced to residual solvent signals in the deuterated solvents. Synthesis reaction schemes can be found in Fig. S2 in the ESI.†![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1.65 (p, J = 7.36, 7.23, 2H, CH2–CH2–CN), 2.33 (t, J = 7.11, 2H, CH2–CN). 13C{1H}-NMR (100 MHz, CDCl3) δ 14.3, 17.3, 22.8, 25.5, 28.8, 28.9, 29.4, 29.6, 29.7, 32.0, 120.0.
N), 1.65 (p, J = 7.36, 7.23, 2H, CH2–CH2–CN), 2.33 (t, J = 7.11, 2H, CH2–CN). 13C{1H}-NMR (100 MHz, CDCl3) δ 14.3, 17.3, 22.8, 25.5, 28.8, 28.9, 29.4, 29.6, 29.7, 32.0, 120.0.2.2 Gamma irradiations
Materials for gamma irradiation were first placed into custom built irradiation vessels. Each vessel consisted of a glass ampoule hermetically bonded to a 1.33 in conflat flange (Accuglass) that was connected to a series of VCR fittings (Swagelok) to form a gas tight apparatus (see Fig. S1†). The final vessel had three “arms”: one contained a removable 50 cm3 gas bottle and was used for headspace gas analysis; one contained an MKS Baratron Capacitance Manometer; and the last one contained a metal seated valve (Swagelok) that was used to connect to the evacuation manifold before isolating the vessel. An 11.5 psi burst disk (Accuglass) was also connected as a pressure relief in the event of overpressurization. The total free space volume for the vessels before sample addition was approximately 105 cm3.After approximately 2–5 g of the materials were placed into the glass ampoule, the samples were evacuated to a final pressure of 25 hPa using the freeze–pump–thaw methodology. The vessels were then isolated using the valve, transported to the Gamma Irradiation Facility (GIF) at Sandia National Laboratories (SNL), and arranged in an arc around a 60Co source (γ-ray energies 1.17 and 1.33 MeV). The dose rate was measured with an ionization chamber and found to be ∼1 Gy s−1 with respect to CaF2. At this dose rate, the temperature during irradiation would not exceed 30 °C. Materials were irradiated for approximately 84 hours to achieve a total absorbed dose of 300 kGy-CaF2 before being transported back to LANL for analysis. The dose was kept consistent with respect to CaF2, and a simple conversion of the absorbed dose with respect to the various materials can be found in the ESI (Table S1†). The dose was chosen to induce a large enough change in the material that could be detected using the techniques chosen for analysis, and the dose rate was chosen for convenience with facility availability. No detectable amounts of solids were produced as a result of irradiation with the exception of dodecyl nitramine which was synthesized as a solid. Irradiations were completed twice on each material.
NOTE: 60Co is a high energy gamma emitter that can cause serious harm or death. Experiments using 60Co should only be conducted in an approved facility with trained personnel.
2.3 Experimental investigations
Following the synthesis of suitably pure materials (as determined by NMR spectroscopy), the samples were irradiated and analyzed using a variety of analytical techniques. By using materials with similar purity, the expectation was that the effects of impurities in the samples would be comparable. For all analytical techniques completed on the condensed phase, the focus was simply on the differences between the irradiated and unirradiated materials; impurities in the starting material revealed using more sensitive analytical techniques were not evaluated. Both irradiated and unirradiated materials were analyzed at the same time to ensure the most direct comparison between the samples.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with an inlet temperature of 250 °C. Ultra-high purity helium (Grade 5, 99.999%) was used as the carrier gas at a constant flow of 2.0 mL min−1. The oven was held at 40 °C for 1.5 min and ramped at 10 °C min−1 to 250 °C where it was held for 1 min. 6 replicates of each sample were collected and the resulting data was exported as a .csv file and then imported into Matlab 2021a for visualization and subsequent evaluation using Fisher ratio analysis.18 Standards of dodecanenitrile, dodecyl amine, dodecanol, dodecanal, dodecene, octanol, and octanal were also run.
:1 with an inlet temperature of 250 °C. Ultra-high purity helium (Grade 5, 99.999%) was used as the carrier gas at a constant flow of 2.0 mL min−1. The oven was held at 40 °C for 1.5 min and ramped at 10 °C min−1 to 250 °C where it was held for 1 min. 6 replicates of each sample were collected and the resulting data was exported as a .csv file and then imported into Matlab 2021a for visualization and subsequent evaluation using Fisher ratio analysis.18 Standards of dodecanenitrile, dodecyl amine, dodecanol, dodecanal, dodecene, octanol, and octanal were also run.After the data was imported into Matlab, the chromatograms were baseline corrected using in-house software, smoothed using the Savitzky–Golay function native to Matlab, and then normalized to the total ion current. A tile size of 20 data points (4 seconds) was used. A S/N threshold of 3 was applied and the Fisher ratios were calculated at each tile for each m/z. The average Fisher ratio for each tile and grid scheme was calculated by taking the average Fisher ratio of the top ten m/z Fisher ratios with the requirement that there be at least 3 m/z present in the tile above the S/N threshold. If a tile contained at least 3 m/z ratios above the threshold but fewer than 10 m/z, then average Fisher ratio was calculated using those m/zs (e.g. if there were only 6 m/zs above the S/N threshold, then the average Fisher ratio was calculated with those 6 m/zs). The redundant hits were removed by using a “pinning and clustering” method with a cluster window of 15 data points (3 seconds).18,19 Finally, a statistical threshold was obtained using a combinatorial null distribution. After a chemical change was discovered using Fisher ratio analysis, the resulting spectra was matched to the NIST database to obtain identification and match value where possible.
Hydrogen was calibrated using certified ppm-level standards (Airgas). A semi-quantitative method was used to provide mole percent (mol%) level estimates of unknown gas-phase species detected by GC-MS in each sample. Chromatographic peak areas were calculated for all unknown compounds with a library match probability greater than eighty percent. Peak areas of each unknown compound that passed the library match criteria were divided by the total peak area sum for all compounds to obtain a percent-level estimate. Semi-quantitative estimates of unknown compounds were tabulated and reported for each sample. It is important to note that quantified hydrogen was accounted for in each sample prior to calculating an estimated concentration for unknown compounds. The estimated uncertainty for the mol% calculation is 10% of the calculated value.
3. Results
In the case of all condensed phase analyses, direct comparisons were made between the irradiated and unirradiated samples. For this study, we chose a single radiation dose, 300 kGy-CaF2, in replicate. This allowed us to benchmark the relative stability of the various functional groups without the added complexity of various doses, which would be more appropriate for a follow-on mechanistic study. The initial state of the materials, i.e. synthesis impurities, is not considered in the analysis; purity was determined using 1H-NMR, and anything above 97% was considered pure enough for this study. Both irradiated and unirradiated samples were analyzed at the same time to provide the best comparison and to ensure that any observed differences could be attributed to irradiation. Less sensitive analyses were completed using Raman and FTIR spectroscopy. No differences were seen for any of the samples, and thus the results are not discussed in the main text. However, all Raman and FTIR spectra can be found in the ESI (Fig. S13–S22†). Headspace gas analysis revealed many different products that were identified using a database or, in the case of hydrogen, with a standard. The total ion chromatographs (TICs) and tables containing the identified products can be found in the SI. This section will only focus on the most relevant results. A full list of GC-MS results can be found in the ESI (Tables S5–S9 and Fig. S23–S27†). The rest of the data used in this paper can also be found in the ESI.†3.1 Dodecane (D–H)
Unsubstituted dodecane was irradiated to understand how the backbone molecule degraded as well as provide a reference point for the derivatized dodecane materials, and the summary of the results from chemical analyses on the solid phase can be found in Fig. 2. GC-TOFMS coupled with Fisher ratio analysis on the condensed phase revealed a total of 25 statistically significant compounds (e.g. hits), though most of these hits had a Fisher ratio below 100 indicating that the chemical changes were minor; this was the case for all the samples analyzed (see Fig. S28–S35 and Tables S10–S13†). This result matches previous results where the limit of discovery is often below the limit of identification.18,20 Of the 25 hits, only dodecene was able to be identified through matching of the NIST database as well as confirmation by a dodecene standard, though the exact isomer of dodecene is not known and the hit is likely a mixture of several isomers. The formation of dodecene requires the removal of two hydrogen atoms and the formation of a carbon–carbon double bond. Many others have also found evidence of the formation of carbon double bonds,12,13,21 so this result is not surprising. For the other hits, the most significant m/z was 43, 57, and 71, which correspond with alkane derivatives. These are likely chain scission products, which again has been seen in previous dodecane irradiations.12,22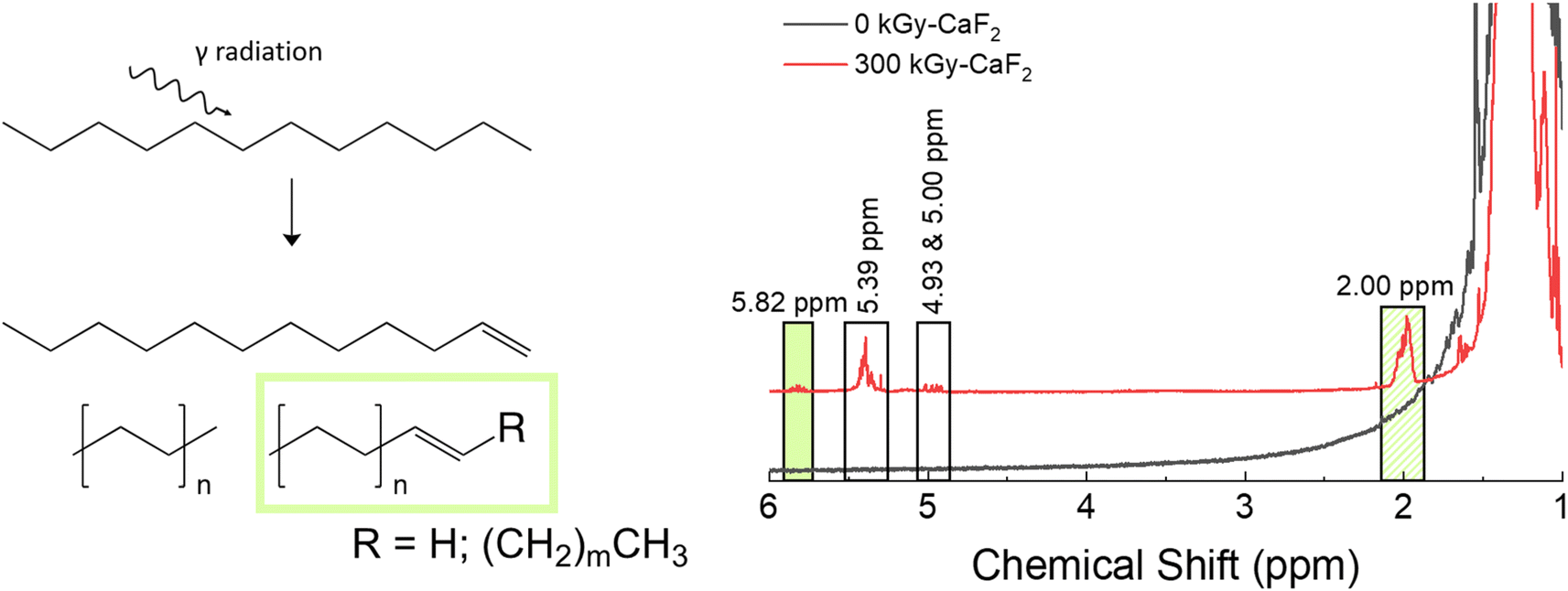 | ||
| Fig. 2 Summary of results for condensed phase analysis of irradiated D–H. Left: Various products identified with GC-TOFMS. Right: Various products identified with 1H-NMR spectroscopy. | ||
Chain scissions resulting in smaller alkanes yield NMR peaks that fall within the broad dodecane peaks at ∼2 ppm, so 1H-NMR spectroscopy was not useful for identifying these products and thus no quantification could be made for them. Instead, a few new peaks were seen in the 1H-NMR spectra: a multiplet at 2.00, a doublet of sextets as 4.93, a doublet of quintets at 5.00, a multiplet at 5.39, and a multiplet at 5.82 ppm. Spiking the irradiated sample with dodecene revealed that the multiplet at 5.82 ppm and at least some of the multiplet at 2.00 ppm are due to alkenes. Integrations of all peaks with respect to the large characteristic dodecane multiplets indicative of dodecane yielded an approximate decomposition of 0.1%.
The total radiolytic gas yield, also known as the G-value with traditional units of molecules produced per 100 eV of energy absorbed, was found to be 4.2 molecules/100 eV, roughly 29% lower than the previously measured yield for de-aerated dodecane.11 The exact reasoning for this is not known but could be due to the scavenging of chemistry-inducing precursors (i.e. radicals) due to impurities in the starting material. Although Fisher Chemical did not give a numerical value for the purity of the material, it was likely lower than the 99+% pure material utilized by LaVerne and Kleemola. The yield measured in this study matched better with the aerated dodecane yield measured by LaVerne and Kleemola, so it is also possible that the samples were not evacuated to a high enough vacuum level. GC-MS analysis on the headspace gas revealed the formation of many species, including some that support the incomplete evacuation (see Table S6†). Approximately 64 mol% was hydrogen gas, while a further 16 mol% was identified as volatile alkanes such as isobutane, pentane, heptane, etc. that were formed due to chain scission as discussed above. Also seen was roughly 8 mol% of volatile alkenes, which suggests a significant amount of the degradation resulted in the formation of carbon double bonds. Overall, these results indicate that multiple degradation events were likely occurring on a dodecane molecule, though it is certainly possible that some amount of the degradation seen in the gas phase occurred due to gas phase radiolysis. This phenomenon would be the case for all irradiated samples.
3.2 Dodecyl azide (D–N3)
A summary of the chemical analyses on the condensed phase of irradiated D–N3 can be found in Fig. 3. Trace analysis identification with GC-TOFMS and Fisher ratio analysis resulted in 64 hits above the statistical threshold. From these hits, numerous degradation products were identified: dodecene, dodecyl amine, octyl azide, octanenitrile, nonanenitrile, decanenitrile, undecanenitrile, and dodecanenitrile. The identities of dodecene, dodecyl amine, and dodecanenitrile were confirmed with standards. The presence of these compounds indicates scission of part of the FG (nitriles and amine), removal of the entire FG (dodecene), and chain scission (octyl azide) as well as a combination of FG scissions and chain scissions. The identities of the remaining hits are not known.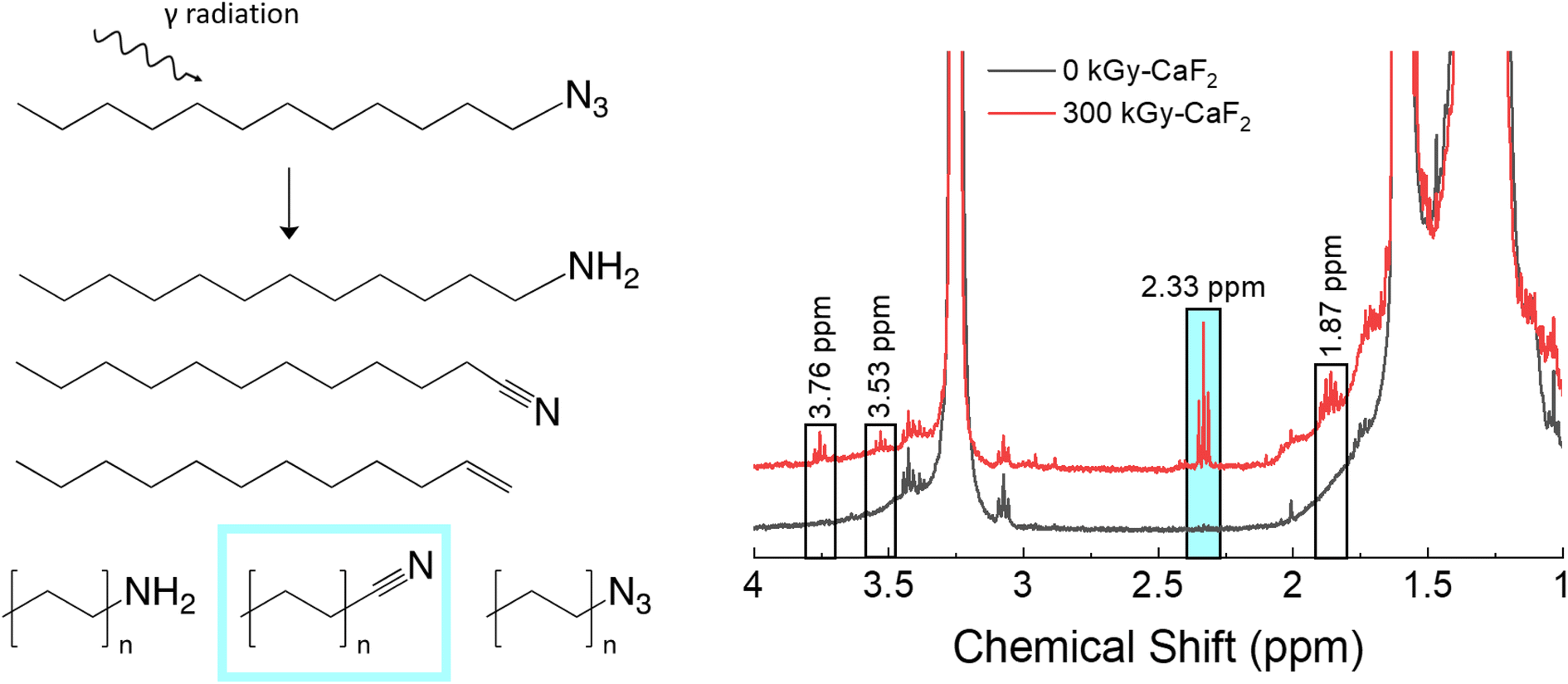 | ||
| Fig. 3 Summary of condensed phase results for irradiated D–N3. Left: Various products as identified by GC-TOFMS. Right: Various peaks as identified by 1H-NMR spectroscopy. | ||
For the unirradiated D–N3, the main identifying NMR signal is a triplet at 3.25 ppm. This triplet is due to the CH2 adjacent to the N3, and thus significant shifts of this triplet are likely due to changes in the N3 FG. Quantitative analysis with 1H-NMR spectroscopy revealed several small signals in the baseline: a quintet at 1.86 ppm and triplets at 2.33, 3.53, and 3.76 ppm. Due to the abundance of nitrile species revealed using GC-TOFMS, the 1H-NMR spectrum of dodecanenitrile was compared to the irradiated D–N3 spectrum. The triplet at 2.33 ppm was confirmed to be the CH2 adjacent to the nitrile. Although shorter alkane chains may give measurable peak shifts in 1H-NMR spectra, samples containing octanal, decanal, and dodecanal were indistinguishable from one another (see Table S5†). As a result, it was deemed not possible to distinguish the various alkane nitrile species identified using GC-TOFMS. This fact is presumed to be the same for all similar species identified using 1H-NMR spectroscopy in this study. Integration of the nitrile triplet with respect to the azide triplet gives an estimated decomposition of approximately 1.9%. Both dodecene and dodecyl amine were analyzed using 1H-NMR spectroscopy, but none of the peaks were visible in the irradiated D–N3 sample. The identity of the quintet at 1.86 ppm is therefore not known. The 3.53 and 3.76 ppm triplets can be assumed to be related to the 3.25 ppm triplet (i.e. corresponding to a change in the FG), though they do not appear to be a nitrile or an amine. In this case, the 3.53 ppm triplet integrates to about a 0.3% degradation while the 3.76 ppm signal integrates to approximately 0.4% degradation.
The radiolytic gas yield of all species for D–N3 was found to be 18.6 molecules/100 eV. Of this gas, approximately 4 mol% was hydrogen gas, 49 mol% was volatile alkanes, and 3 mol% was volatile alkenes. Interestingly, there was also a 16 mol% contribution due to cyclopentane and a 1 mol% contribution due to cyclopentene, both of which are ring structures and not seen in large quantities in the headspace gas of irradiated dodecane. The N2 content was not able to be assessed with this technique, though N2 is thought to be a large fraction of the headspace gas. The ratio of alkanes to alkenes was also significantly different from dodecane as was the mol% of hydrogen gas. Overall, these results suggest that a significant portion of the degradation occurred on the energetic FG, and that the degradation seen with D–N3 is not simply a combination of the degradation of the dodecane backbone and the degradation of the azide energetic FG.
3.3 Dodecyl nitro (D–NO2)
A summary of the condensed phase chemical analyses performed on D–NO2 is shown in Fig. 4. GC-TOFMS analysis resulted in a total of 80 hits above the statistical threshold, but only 3 products were able to be identified: dodecene, dodecane, and hexyl nitro. Both dodecene and dodecane indicate a removal of the nitro energetic FG while the hexyl nitro indicates a chain scission. It should be noted that the match value for the hexyl nitro was not very high (849) and no other nitrated alkane was found in the NIST database making the identification tentative, though the product is positively identified as a product of chain scission due to the characteristic m/z signals. It could therefore be a longer chain nitro alkane, but the conclusion is still the same regardless of the chain length.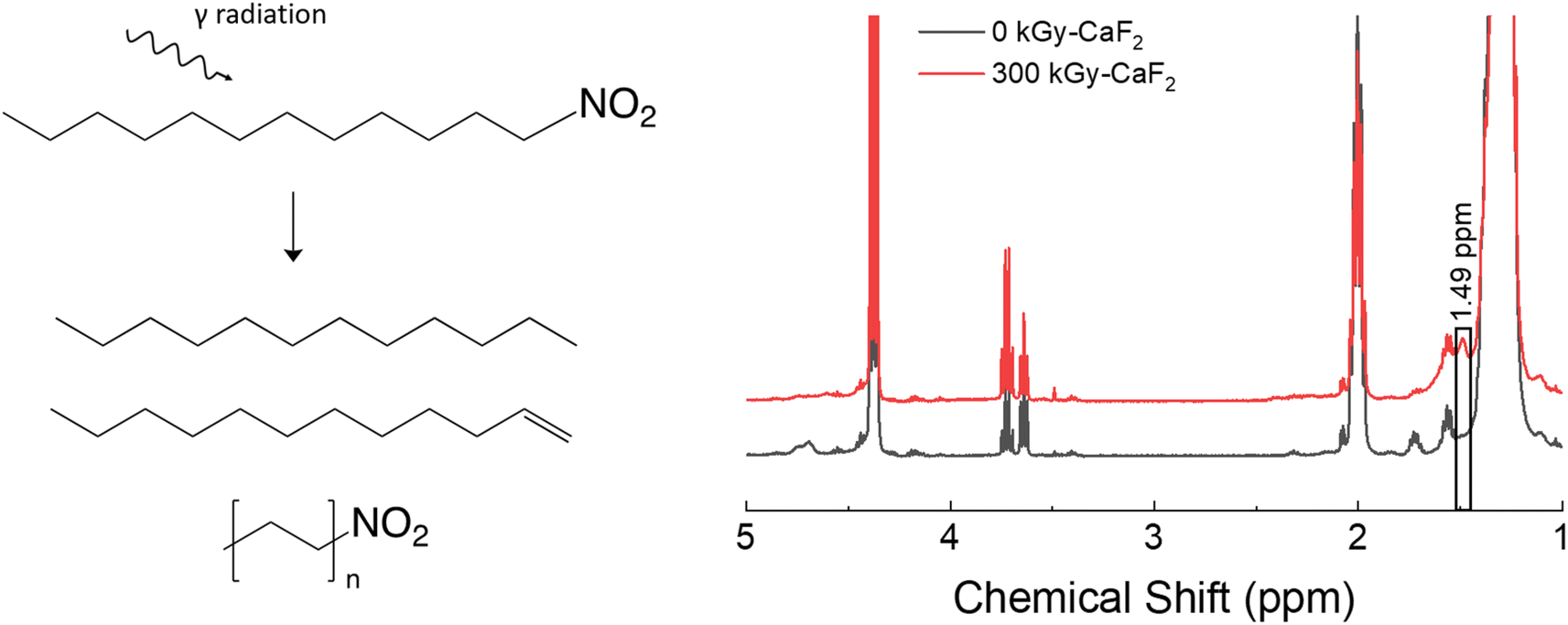 | ||
| Fig. 4 Summary of results for the condensed phase analysis of irradiated D–NO2. Left: Various products identified by GC-TOFMS. Right: 1H-NMR spectra for both samples. | ||
1H-NMR spectroscopy did not reveal meaningful changes. Of the identified hits on GC-TOFMS, only dodecene would result in a visible change to the 1H-NMR spectra. However, no peaks belonging to dodecene could be discerned from the baseline; a broad signal at 1.49 ppm was the only visible change, and the identity of this peak is not known. Given the lack of meaningful results, integration of the quantity of degradation products was not possible with D–NO2.
Gas analysis of the radiolytically-produced headspace gas gave a total yield of 3.8 molecules/100 eV, the lowest yield of any of the samples. GC-MS identified 22 mol% was hydrogen gas while 6 mol% was identified as volatile alkenes and 3 mol% was identified as volatile alkanes. The largest contributor was nitrous oxide at 39 mol%, followed by an amine at 24 mol%. Nitrous oxide has also been seen as a major component in the gas phase of irradiated explosives23–25 containing the nitro energetic FG, so this result was expected. Overall, these results indicate that D–NO2 is a relatively stable molecule to irradiation when compared to the other molecules used in this study, though degradation of the nitro energetic FG is a large contributor to the degradation gas products observed.
3.4 Dodecyl nitrate ester (D–ONO2)
A summary of the results from the condensed phase chemical analyses completed on the D–ONO2 system is shown in Fig. 5. Trace analysis with GC-TOFMS revealed a total of 57 hits above the statistical threshold, and several were able to be identified: octanal, decanal, undecanal, dodecanol, undecanol, heptyl nitrate ester, octyl nitrate ester, and nonyl nitrate ester. The aldehydes and alcohols indicate a scission along the O–NO2 bond of the nitrate ester where the NO2 is released but the O remains with the parent molecule. This bond is referred to as the trigger linkage, or weakest bond, for the nitrate ester FG26 and it is thought to be the first bond to break during thermal and explosive decomposition.27,28 Interestingly, GC-TOFMS did not identify products indicative of the removal of the entire nitrate ester energetic FG (scission of the C–ONO2 bond). Shorter chain nitrate esters were seen, indicating chain scission was also a significant degradation product.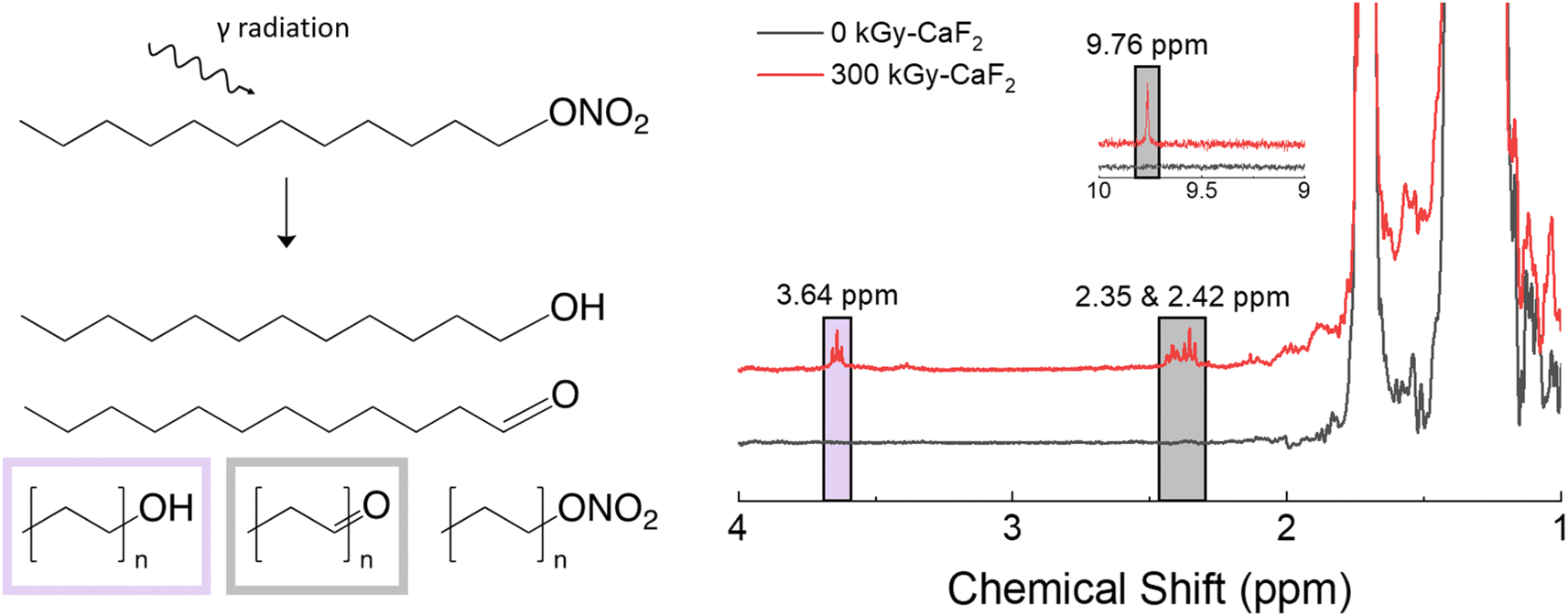 | ||
| Fig. 5 Summary of condensed phase chemical analysis results for irradiated D–ONO2. Left: Various products identified with GC-TOFMS. Right: Products identified with 1H-NMR spectroscopy. | ||
1H-NMR spectroscopy showed the formation of several new species: triplets at 2.35, 2.42, 3.64, and a small singlet at 9.76 ppm. The CH2 group next to the nitrate ester FG has a characteristic triplet at 4.47 ppm, so significant shifts in that triplet are likely due to changes in the nitrate ester FG. Based on the GC-TOFMS results, samples were spiked with octanol and octanal to discern the location of the alcohol and aldehyde peaks, respectively. The triplet at 3.64 ppm was identified as an alcohol signal through spiking with an independently prepared sample and integrated to 0.8% of the parent triplet at 4.47 ppm. Similarly, the triplet peaks at 2.35 and 2.42 ppm as well as the singlet at 9.76 ppm were identified as aldehyde signals and integrated to ∼2.2% of the parent triplet.
The total radiolytic gas yield of D–ONO2 was measured at 20.6 molecules/100 eV, the largest yield of the materials used in this study. GC-MS revealed 3 mol% of that gas was hydrogen gas, 2 mol% was volatile alkenes, and 4 mol% was volatile alkanes. A further 12 mol% was due to volatile hydrocarbons containing at least oxygen, of which 8 mol% contained aldehyde and 1 mol% contained alcohol groups. Lastly, 1 mol% was due to volatile nitrate ester-containing hydrocarbons, and 75 mol% was due to nitrous oxide. The relative ratios of backbone degradation to FG degradation suggests that a large majority of the radiolytic degradation of D–ONO2 occurs on the energetic FG. Overall, these results indicate that D–ONO2 is particularly susceptible to radiolytic degradation, and most of that degradation occurs on the nitrate ester FG. Furthermore, the degradation tends to occur along the trigger linkage and leads to the formation of alcohol and aldehyde FGs, though aldehyde formation is more common.
3.5 Dodecyl nitramine (D–NHNO2)
D–NHNO2 was a solid at ambient temperature and pressure and did not vaporize below 250 °C, so GC-TOFMS could not be completed on the material. Surprisingly, the 1H-NMR spectra (shown in Fig. 6) resulted in the broad peak at 7.94 ppm (NHNO2) shifting to 7.77 ppm following irradiation rather than visible changes in the baseline. Addition of acid to the NMR sample of unirradiated D–NHNO2 resulted in shifts in the opposite direction, suggesting that the change is not due to the formation of an acid from the release of H+. The only insightful data that could be collected was the radiolytically-produced gas. The total gas yield was calculated to be 16.6 molecules/100 eV, and GC-MS revealed 2 mol% was due to hydrogen gas, <1 mol% was due to volatile alkenes, and 3 mol% was due to volatile alkanes. A further 40 mol% was due to partial degradation of the nitramine FG, of which 37 mol% was identified as an amine. 26 mol% was attributed to nitrous oxide, and 19 mol% was attributed to oxygen gas. From the gas analysis, it appears that the energetic FG is once again more susceptible to radiolytic degradation than the backbone molecule is.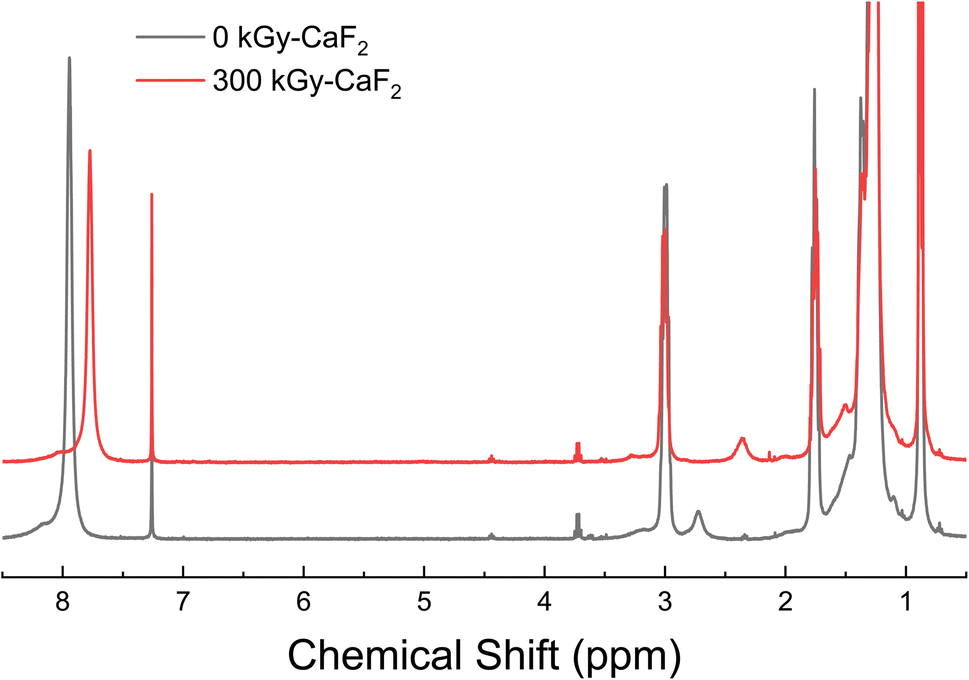 | ||
| Fig. 6 1H-NMR spectra for control (bottom black line) and irradiated (top red line) D–NHNO2. | ||
4. Discussion
The purpose of this study was to assess the relative radiation stability of various energetic FGs by utilizing a common molecular backbone. To that degree, the level of degradation to the backbone molecule is not entirely important, though the ratio of energetic FG degradation and backbone degradation can give some interesting insights. In the following section, we will discuss comparisons of gas production for dodecane versus the substituted dodecanes in order to estimate how the energy is localized on the overall molecule as well as the energetic functional group.Radiolytic gas yields are often used as an indication of the radiolytic activity of a material. In the case of these materials, many gas species were created and identified. If only the total G-value is considered, then the stability of the studied substituted molecules from most to least susceptible to radiation is: D–ONO2 → D–N3 → D–NHNO2 → D–NO2. To the extent that 1H-NMR spectroscopy results could be collected, the integrated amount of decomposition products relative to the parent peaks show a similar trend to the total as yield analysis, as shown in Table 1; the uncertainties listed are systematic uncertainties that were propagated through for the G-value calculation. Interestingly, the order of the results matches well with both photolytic molecular dynamics simulations of these molecules at 8 eV14 as well as the general trend for the sub-shock impact sensitivity of various explosives.31–33 Although a more limited study, these results also provide an explanation why, for a previous study exploring radiolytic damage to a variety of explosives,25 the only explosive that was found to degrade with low-level radiation in large enough quantities to be detectable using 1H-NMR spectroscopy was pentaerythritol tetranitrate (PETN), a nitrate ester-based explosive. The total gas yields, however, are a measure of how radiolytically active the entire molecule is rather than just the energetic FG. Based on the amount of data able to be collected for these systems, the total gas yield is the only value to give results across the entire substitution series and thus is the only true means of comparison. It is important to note that this simplistic analysis will convolute factors such as the different phase of D–NHNO2 (solid) with respect to the other materials (liquid) and the synthesis impurities of the various materials, both of which could have an effect on the results. However, as the general trend seen in these results closely follows the trend seen in photolytic simulations and sub-shock impact sensitivity tests, the total G-value seems to be an overall valid approach to ranking the relative radiolytic susceptibility of the investigated energetic FGs.
| Molecule | 1H-NMR decomposition (%) [does not include chain scissions] | G-value (molecules/100 eV) [total gas yield] |
|---|---|---|
| a Note: D = C12H25.b Meaningful data was not able to be obtained from the 1H-NMR spectra of D–NHNO2. | ||
| D–H | 0.1 | 4.2 ± 0.2 |
| D–N3 | 2.6 | 18.6 ± 0.8 |
| D–NO2 | Below detection limit | 3.8 ± 0.2 |
| D–ONO2 | 3.0 | 20.6 ± 0.9 |
| D–NHNO2 | Inconclusiveb | 16.6 ± 0.7 |
For the dodecane backbone, most of the degradation involves the removal of H atoms which forms both hydrogen gas as well as various alkenes. This result has been seen in previous studies, as well.11,22,29,30 Carbon–carbon bonds do also break, as evidenced by the presence of shorter chain alkanes and alkenes, though to a lesser degree. All the substituted dodecane samples showed both alkanes and alkenes being formed, though these were often at a much lower rate than degradation to the energetic FG. Degradation to the dodecane backbone was approximated by the amount of hydrogen gas produced during irradiation; C–C bond breaks were left out of the analysis due to the uncertainty of which side of the molecule the break occurred on and how or when the FG was involved. As a result, the best gas species for tracking where the damage is occurring was determined to be hydrogen. Hydrogen is a major component of the dodecane backbone and is not present in any FG except for the nitramine. Therefore, although some amount of H2 gas in irradiated D–NHNO2 could come from the nitramine FG, most of it likely comes from the dodecane backbone, and all hydrogen from the other materials must come from the backbone. Hydrogen gas is therefore considered to be evidence of alkane backbone degradation.
Using hydrogen gas as a comparison, the substituted dodecane materials produced only 12–31% the amount of hydrogen gas that pure dodecane produced. This result suggests that the addition of an energetic FG actually reduces the damage to the dodecane backbone structure even though the overall degradation to the molecule generally increases. There is a difference in the energy directly absorbed by each functional group (see ESI†), however the degradation trend suggests that these differences are small in comparison to other factors such as energy transport. One possible explanation for the above observation is exciton migration. During γ-radiolysis, both ionizations and excitations are created throughout the entire system. Excitations can lead to the formation of excitons that operate as an energy transfer mechanism and have been found to be an important mode of energy transport in alkanes.34 In fact, n-alkanes are considered to be efficient molecular exciton transporters even including the transport of excitons between molecules.29,30,35 Scission of the C–H bond in alkanes is the most likely degradation pathway,11,22 and this process is thought to occur quickly.22 C–C excitons, on the other hand, would be free to travel along and beyond the length of the molecule, assuming the molecule maintains the alkane characteristics. In the case of the substituted dodecane molecules, however, the energetic FG would likely prevent the exciton from moving to another molecule by acting as an exciton trap. The excitation energy would therefore be located on the energetic FG where degradation could occur. Exciton migration and localization on the energetic FG was also seen with non-adiabatic excited state molecular dynamics (NEXMD) calculation performed on the same molecules.14 By concentrating the energy on the energetic FGs, less degradation of the dodecane backbone could occur and thus most of the H2 gas measured must be due to direct interactions with the C–H bonds of the dodecane molecule. Another possible explanation for the decrease in the hydrogen gas G-value for the substituted dodecane molecules with respect to pure dodecane involve the general delocalization of the molecular exciton in alkanes rather than localized transport.36 Electron paramagnetic resonance (EPR) spectroscopy has revealed that the n-alkane cations are delocalized along the entire length of the alkane chain.37 In this instance, the exciton would simply need to couple to the energetic FG to induce damage on it. In either explanation, however, energy does appear to localize on the energetic FG regardless of where the energy is initially deposited, and this localization results in a larger fraction of damage on the FG than on the molecular backbone.
The exact location of the degradation on the energetic FG is also worth noting. For D–N3, GC-TOFMS results indicate that the azide FG is broken both at the C–N3 and the CN–N2 bonds. The N–N bond is the trigger linkage for the azide FG26,38 and is considered to be the most susceptible bond to be broken. GC-TOFMS results for D–NO2 indicate only the removal of the NO2 FG; the C–N bond is the trigger linkage for the nitro FG.26 This result is consistent with the observation that the irradiation of trinitrotoluene (TNT) only revealed the formation of dinitrotoluene23 which would be formed by the removal of a single NO2 group from the molecular structure. As mentioned earlier in the text, D–ONO2 was found to degrade only along the O–NO2 bond which is the trigger linkage for the nitrate ester FG. This bond has also been found to be particularly susceptible to damage in radiolytic,23,25 thermal,39–41 and explosive insult42,43 degradation events. No instances were observed with the loss of –ONO2; instead, the damage to the nitrate ester FG appears to be exclusively through the N–O bond. Though condensed phase analysis for D–NHNO2 were unsuccessful, the gas phase analysis reveals at least a partial degradation of the nitramine FG. It is unclear where exactly the bond is breaking for these products, though radiolytic degradation with X-rays of the nitramine-containing explosive RDX suggests the most likely bond to break is the N–N bond44 which is the trigger linkage for the nitramine FG.26
It is interesting to compare the above results with the 8 eV results found in the photolytic NEXMD calculations completed on the same molecules.14 Of course, radiolysis and photolysis are not the same process, but a significant portion of the primary decomposition in alkanes can be attributed to low-energy electronically excited states generated through absorption of scattered radiation.22 Additionally, the photolytic NEXMD calculations allow for a look at the fast time scales for degradation involving the S1 excited state, an insight that is unachievable for the radiolysis experiments in this study. The photolytic NEXMD calculations were also completed on the same molecules utilized in this study, which allows for a more direct comparison between the computational results and these experimental results. In the 8 eV computations involving the substituted dodecane molecules, the exciton was initially localized on the dodecane backbone where it then quickly relaxed to the S1 excited state and moved to the energetic FG to undergo degradation.14 For the D–N3 molecule, the degradation would mostly involve the scission of the N–N bond, but contributions involving the scission of the C–N3 as well as degradation to the dodecane backbone were also seen. For the D–NO2 molecule, only the C–NO2 bond was broken. The D–ONO2 molecule underwent scission of the O–NO2 bond but would sometimes also include some degradation to the dodecane backbone, and the D–NHNO2 molecule would either break at the N–N bond or it would remove a hydrogen ion from the nitramine; the ion nature was simply due to how the calculations were completed. In all cases, the primary degradation pathways were in line with what has been seen in this experimental investigation. Obviously, higher energy radiation results in additional degradation pathways, but much of the degradation can be explained by lower energy excitations. This result also follows for what is currently known about the radiolysis of explosives: most of the degradation occurs not on the carbon molecular structure supporting the energetic FGs, but rather on the energetic FGs themselves, and particularly along the trigger linkage.23,25,44
5. Conclusions
In order to study the effects of ionizing radiation on various commonly used energetic functional groups, a selection of molecules were synthesized with a common molecular backbone and irradiated with γ-rays using a 60Co source. The studied materials included: dodecyl azide, dodecyl nitro, dodecyl nitrate ester, and dodecyl nitramine. These materials were irradiated under vacuum and analyzed post-radiolysis which was directly compared to the unirradiated materials. Trace analyses revealed the formation of several species not originally present in the sample that were indicative of chain scission, trigger linkage scission, removal of the entire energetic functional group in the case of some materials, and a combination of all of the above. Analysis of the radiolytically-produced gas revealed that a significant portion of the damage took place on the energetic functional group, specifically along the trigger linkage. A cursory ranking of the radiolytic susceptibility was determined using the total gas G-value and was found to be D–ONO2 → D–N3 → D–NHNO2 → D–NO2 when ranked from most to least susceptible. These results have some caveats, but do follow the same trend that photolytic calculations and sub-shock impact tests show which lends credence to this approach.Author contributions
Patricia L. Huestis – conceptualization, investigation, writing-original draft, visualization. Nicholas Lease – investigation, resources, writing-review & editing. Chris E. Freye – investigation, writing – review & editing. Daniel L. Huber – investigation, writing-review & editing. Geoff Brown – supervision. Daniel L. McDonald – resources. Tammie Nelson – supervision, writing-review & editing. Chris Snyder – resources. Virginia W. Manner – conceptualization, writing-review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Joe Lichthardt for his help with irradiation vessel design, Taylor Dehner for her help with FTIR and Raman spectra collection, and Don Hanson, Maryla Wasiolek, and Keven McRunnels for their help in facilitating the irradiations. The authors also thank Brooke Baumgarten, Jeremy Moore, and Andy Cook for their incredibly helpful and insightful discussion throughout the project. This work was supported by the US Department of Energy through the Los Alamos National Laboratory. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (Contract No. 89233218CNA000001). Lastly, the authors would like to thank the LANL Aging and Lifetimes Program for funding, as well as Sheldon Larson and Charles Hills for their helpful discussion during this project. This contribution is LA-UR-23-21456 from Los Alamos National Laboratory.References
- J. Pichtel, Distribution and Fate of Military Explosives and Propellants in Soil: A Review, Appl. Environ. Soil Sci., 2012, 2012, 617236, DOI:10.1155/2012/617236.
- N. Lease, N. J. Burnside, G. W. Brown, J. P. Lichthardt, M. C. Campbell, R. T. Buckley, J. F. Kramer, K. M. Parrack, S. P. Anthony, H. Tian, S. K. Sjue, D. N. Preston and V. W. Manner, The Role of Pentaerythritol Tetranitrate (PETN) Aging in Determining Detonator Firing Characteristics, Propellants, Explos., Pyrotech., 2021, 46, 26–38, DOI:10.1002/prep.202000181.
- A. M. Sarquis, L. M. Sennet, M. Canepa Kittredge, K. W. Kittredge and M. S. Sokol, Investigating the Stability of Benzoyl Peroxide in Over-the-Counter Acne Medications, J. Chem. Educ., 2008, 85, 1655, DOI:10.1021/ed085p1655.
- S. Steven, M. Oelze, M. Brandt, E. Ullmann, S. Kröller-Schön, T. Heeren, L. P. Tran, S. Daub, M. Dib, D. Stalleicken, P. Wenzel, T. Münzel and A. Daiber, Pentaerythritol Tetranitrate In Vivo Treatment Improves Oxidative Stress and Vascular Dysfunction by Suppression of Endothelin-1 Signaling in Monocrotaline-Induced Pulmonary Hypertension, Oxid. Med. Cell. Longev., 2017, 2017, 4353462, DOI:10.1155/2017/4353462.
- T. Groten, T. Lehmann, E. Schleußner, U. Pecks, C. von Kaisenberg, M. Condic, S. Verlohren, D. Schlembach, C. Neukoelln, G. Seliger, S. Seeger, A. Tauscher, M. Komar, K. O. Kagan, U. Friebe-Hoffmann, C. Hübener, L. de Vries and for the P. S. Group, Does Pentaerytrithyltetranitrate Reduce Fetal Growth Restriction in Pregnancies Complicated by Uterine Mal-Perfusion? Study Protocol of the PETN-Study: A Randomized Controlled Multicenter-Trial, BMC Pregnancy Childbirth, 2019, 19, 336, DOI:10.1186/s12884-019-2456-7.
- E. R. Lake, S. J. Thompson and V. W. Drexelius, A Study of the Role of Pyrotechnic Systems on the Space Shuttle Program, Report NASA CR-2292, St. Louis, MO, 1973.
- M. R. Manaa, A. R. Mitchell, R. G. Garza, P. F. Pagoria and B. E. Watkins, Flash Ignition and Initiation of Explosives−Nanotubes Mixture, J. Am. Chem. Soc., 2005, 127, 13786–13787, DOI:10.1021/ja0547127.
- L. Glavier, A. Nicollet, F. Jouot, B. Martin, J. Barberon, L. Renaud and C. Rossi, Nanothermite/RDX-Based Miniature Device for Impact Ignition of High Explosives, Propellants, Explos., Pyrotech., 2017, 42, 308–317, DOI:10.1002/prep.201600154.
- J. A. LaVerne, M. S. Driscoll and M. Al-Sheikhly, Radiation Stability of Lignocellulosic Material Components, Radiat. Phys. Chem., 2020, 171, 108716, DOI:10.1016/j.radphyschem.2020.108716.
- J. N. Mathur, M. S. Murali and K. L. Nash, Actinide Partitioning - A Review, Solvent Extr. Ion Exch., 2001, 19, 357–390, DOI:10.1081/SEI-100103276.
- J. A. LaVerne and S. R. Kleemola, Hydrogen Production in the Radiolysis of Dodecane and Hexane, Solvent Extr. Ion Exch., 2017, 35, 210–220, DOI:10.1080/07366299.2017.1312887.
- H. A. Dewhurst, Radiation Chemistry of Organic Compounds. I. n-Alkane Liquids, J. Phys. Chem., 1957, 61, 1466–1471, DOI:10.1021/j150557a004.
- J. P. Holland, J. F. Merklin and J. Razvi, The Radiolysis of Dodecane-Tributylphosphate Solutions, Nucl. Instrum. Methods, 1978, 153, 589–593, DOI:10.1016/0029-554X(78)91011-X.
- T. Nelson, P. L. Huestis and V. W. Manner, Modeling Photolytic Decomposition of Energetically Functionalized Dodecanes, J. Phys. Chem. A, 2022, 126, 7094–7101, DOI:10.1021/acs.jpca.2c0340.
- S. Y. Baek, Y. W. Kim, S. H. Yoo, K. Chung, N. K. Kim and J. S. Kim, Synthesis and Rust Preventing Properties of Dodecyl Succinate Derivatives Containing Triazole Groups, Ind. Eng. Chem. Res., 2012, 51, 9669–9678, DOI:10.1021/ie300316f.
- K. J. Hock, J. Grimmer, D. Göbel, G. G. T. Gasaya, J. Roos, I. V Maucher, B. Kühn, J. Fettel, T. J. Maier and G. Manolikakes, Modular Regiospecific Synthesis of Nitrated Fatty Acids, Synthesis (Stuttg), 2017, 49, 615–636 CAS.
- B. Inci, I. Lieberwirth, W. Steffen, M. Mezger, R. Graf, K. Landfester and K. B. Wagener, Decreasing the Alkyl Branch Frequency in Precision Polyethylene: Effect of Alkyl Branch Size on Nanoscale Morphology, Macromolecules, 2012, 45, 3367–3376, DOI:10.1021/ma3002577.
- C. E. Freye, P. R. Bowden, M. T. Greenfield and B. C. Tappan, Non-Targeted Discovery-Based Analysis for Gas Chromatography with Mass Spectrometry: A Comparison of Peak Table, Tile, and Pixel-Based Fisher Ratio Analysis, Talanta, 2020, 211, 120668, DOI:10.1016/j.talanta.2019.120668.
- B. A. Parsons, L. C. Marney, W. C. Siegler, J. C. Hoggard, B. W. Wright and R. E. Synovec, Tile-Based Fisher Ratio Analysis of Comprehensive Two-Dimensional Gas Chromatography Time-of-Flight Mass Spectrometry (GC × GC-TOFMS) Data Using a Null Distribution Approach, Anal. Chem., 2015, 87, 3812–3819, DOI:10.1021/ac504472s.
- C. E. Freye, N. R. Moore and R. E. Synovec, Enhancing the Chemical Selectivity in Discovery-Based Analysis with Tandem Ionization Time-of-Flight Mass Spectrometry Detection for Comprehensive Two-Dimensional Gas Chromatography, J. Chromatogr. A, 2018, 1537, 99–108, DOI:10.1016/j.chroma.2018.01.008.
- H. Ikeda and A. Suzuki, Radiolysis of N-Dodecane and Its Physical Property Change Based on the Dose in One Pass through a Reference HA Column, J. Nucl. Sci. Technol., 1998, 35, 697–704, DOI:10.1080/18811248.1998.9733930.
- L. Wojnarovitz, in Charg. Part. Phot. Interact. with Matter, ed. A. Mozumder and Y. Hatano, CRC Press, Boca Raton, 2003, pp. 371–408. doi: DOI:10.1201/9780203913284-12.
- M. J. Urizar, E. D. Loughran and L. C. Smith, The Effects of Nuclear Radiation on Organic Explosives, Report TID-12491, Los Alamos, New Mexico, 1960, doi: DOI:10.2172/4027488.
- T. C. Castorina, J. Haberman and A. F. Smetana, Radiation-Enhanced Surface Activity of an Organic Explosive, γ-HMX, Int. J. Appl. Radiat. Isot., 1968, 19, 495–503, DOI:10.1016/0020-708X(68)90001-X.
- P. L. Huestis, J. A. Stull, J. P. Lichthardt, M. A. Wasiolek, L. Montano-Martinez and V. W. Manner, Effects of Low-Level Gamma Radiation on Common Nitroaromatic, Nitramine, and Nitrate Ester Explosives, ACS Omega, 2022, 7, 2842–2849, DOI:10.1021/acsomega.1c05703.
- P. Politzer, J. S. Murray and P. Lane, Computational Determination of Effects of Electric Fields upon “Trigger Linkages” of Prototypical Energetic Molecules, Int. J. Quantum Chem., 2009, 109, 534–539, DOI:10.1002/qua.21800.
- J. Joy, D. Danovich and S. Shaik, Nature of the Trigger Linkage in Explosive Materials Is a Charge-Shift Bond, J. Org. Chem., 2021, 86, 15588–15596, DOI:10.1021/acs.joc.1c02066.
- J. S. Murray, M. C. Concha and P. Politzer, Links between Surface Electrostatic Potentials of Energetic Molecules, Impact Sensitivities and C–NO2/N–NO2 Bond Dissociation Energies, Mol. Phys., 2009, 107, 89–97, DOI:10.1080/00268970902744375.
- R. H. Partridge, Excitation Energy Transfer in Alkanes. II. Experimental Demonstration, J. Chem. Phys., 1970, 52, 2491–2500, DOI:10.1063/1.1673333.
- R. H. Partridge, Excitation Energy Transfer in Alkanes. III. Radiation Chemistry of Alkane Polymers, J. Chem. Phys., 1970, 52, 2501–2510, DOI:10.1063/1.1673334.
- S. Zeman and M. Jungová, Sensitivity and Performance of Energetic Materials, Propellants, Explos., Pyrotech., 2016, 41, 426–451, DOI:10.1002/prep.201500351.
- M. J. Cawkwell and V. W. Manner, Ranking the Drop-Weight Impact Sensitivity of Common Explosives Using Arrhenius Chemical Rates Computed from Quantum Molecular Dynamics Simulations, J. Phys. Chem. A, 2020, 124, 74–81, DOI:10.1021/acs.jpca.9b10808.
- N. Lease, L. M. Kay, G. W. Brown, D. E. Chavez, D. Robbins, E. F. C. Byrd, G. H. Imler, D. A. Parrish and V. W. Manner, Synthesis of Erythritol Tetranitrate Derivatives: Functional Group Tuning of Explosive Sensitivity, J. Org. Chem., 2020, 85, 4619–4626, DOI:10.1021/acs.joc.9b03344.
- T. Miyazaki, The Migration of the Exciton in the Radiolysis of Alkanes, Bull. Chem. Soc. Jpn., 1973, 46, 329–330, DOI:10.1246/bcsj.46.329.
- R. H. Partridge, Excitation Energy Transfer in Alkanes. I. Exciton Model, J. Chem. Phys., 1970, 52, 2485–2490, DOI:10.1063/1.1673332.
- A. R. Cook, in Poster Int. Conf. Ioniz. Process., vol. 2022, 2022 Search PubMed.
- K. Toriyama, K. Nunome and M. Iwasaki, σ-Delocalized Radical Cations [H(CH2)NH]+ of Primary Alkanes: ESR Evidence, J. Phys. Chem., 1981, 85, 2149–2152, DOI:10.1021/j150615a001.
- A. L. Shoaf and C. A. Bayse, The Effect of Nitro Groups on N2 Extrusion from Aromatic Azide-Based Energetic Materials, New J. Chem., 2019, 43, 15326–15334, 10.1039/c9nj03220g.
- D. M. Chambers, C. L. Brackett and O. D. Sparkman, Perspectives on Pentaerythritol Tetranitrate (PETN) Decomposition, Report UCRL-ID-148956, Lawrence Livermore National Laboratory, Livermore, CA, 2002.
- B. D. Roos and T. B. Brill, Thermal Decomposition of Energetic Materials 82. Correlations of Gaseous Products with the Composition of Aliphatic Nitrate Esters, Combust. Flame, 2002, 128, 181–190, DOI:10.1016/S0010-2180(01)00343-1.
- V. W. Manner, L. Smilowitz, C. E. Freye, A. H. Cleveland, G. W. Brown, N. Suvorova and H. Tian, Chemical Evaluation and Performance Characterization of Pentaerythritol Tetranitrate (PETN) under Melt Conditions, ACS Mater. Au, 2022, 2, 464–473, DOI:10.1021/acsmaterialsau.2c00022.
- H. Brust, A. van Asten, M. Koeberg, J. Dalmolen, A. van der Heijden and P. Schoenmakers, Accurate Quantitation of Pentaerythritol Tetranitrate and Its Degradation Products Using Liquid Chromatography–Atmospheric Pressure Chemical Ionization–Mass Spectrometry, J. Chromatogr. A, 2014, 1338, 111–116, DOI:10.1016/j.chroma.2014.02.067.
- A. Basch, Y. Margalit, S. Abramovich-Bar, Y. Bamberger, D. Daphna, T. Tamiri and S. Zitrin, Decomposition Products of Petn in Post Explosion Analysis, J. Energ. Mater., 1986, 4, 77–91, DOI:10.1080/07370658608011334.
- B. C. Beard, X-ray radiation decomposition of cyclo-1,3,5-trimethylene-2,4,6-trinitramine (RDX), at low temperature: Initial reaction steps, Propellants, Explos. Pyrotech., 1991, 16, 81–87, DOI:10.1002/prep.19910160208.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00998j |
| This journal is © The Royal Society of Chemistry 2023 |