
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring novel derivatives of isatin-based Schiff bases as multi-target agents: design, synthesis, in vitro biological evaluation, and in silico ADMET analysis with molecular modeling simulations†
Ashraf S. Hassan *a,
Nesrin M. Morsya,
Wael M. Aboulthanab and
Ahmed Ragab*c
*a,
Nesrin M. Morsya,
Wael M. Aboulthanab and
Ahmed Ragab*c
aOrganometallic and Organometalloid Chemistry Department, National Research Centre, Dokki 12622, Cairo, Egypt. E-mail: ashraf_salmoon@yahoo.com; as.el-salmoon@nrc.sci.eg
bBiochemistry Department, Biotechnology Research Institute, National Research Centre, Dokki 12622, Cairo, Egypt
cChemistry Department, Faculty of Science (Boys), Al-Azhar University, Nasr City, Cairo 11884, Egypt. E-mail: ahmed_ragab7@ymail.com; Ahmed_ragab@azhar.edu.eg
First published on 20th March 2023
Abstract
Recently, scientists developed a powerful strategy called “one drug-multiple targets” to discover vital and unique therapies to fight the most challenging diseases. Novel derivatives of isatin-based Schiff bases 2–7 have been synthesized by the reaction of 3-hydrazino-isatin (1) with aryl aldehydes, hetero-aryl aldehydes, and dialdehydes. The structure of the synthesized derivatives was proved by physical and spectral analysis. Additionally, in vitro biological studies were performed, including antioxidant, anti-diabetic, anti-Alzheimer, and anti-arthritic activities. The four derivatives 3b, 5a, 5b, and 5c possess the highest activities. Among the four potent derivatives, compound 5a exhibited the highest antioxidant (TAC = 68.02 ± 0.15 mg gallic acid per g; IRP = 50.39 ± 0.11) and scavenging activities (ABTS = 53.98 ± 0.12% and DPPH = 8.65 ± 0.02 μg mL−1). Furthermore, compound 5a exhibited an α-amylase inhibitory percentage of 57.64 ± 0.13% near the acarbose (ACA = 69.11 ± 0.15%) and displayed inhibitor activity of the acetylcholinesterase (AChE) enzyme = 36.38 ± 0.08%. Moreover, our work extended to determining the anti-arthritic effect, and compound 5a revealed good inhibitor activities with very close values for proteinase denaturation (PDI) = 39.59 ± 0.09% and proteinase inhibition (PI) = 36.39 ± 0.08%, compared to diclofenac sodium PDI = 49.33 ± 0.11% and PI = 41.88 ± 0.09%. Additionally, the quantum chemical calculations, including HOMO, LUMO, and energy band gap were determined, and in silico ADMET properties were predicted, and their probability was recorded. Finally, molecular docking simulations were performed inside α-amylase and acetylcholinesterase enzymes.
1. Introduction
People with diseases have many serious complications that affect many body areas.1 Diabetes mellitus is a disorder in which the sugar (glucose) level in the blood is elevated. Diabetes mellitus leads to numerous complications, including atherosclerosis which leads to heart attacks and strokes, and blood vessel damage of the eye, then loss of vision (diabetic retinopathy). The other diabetes mellitus complications are damage to the liver, kidney, nerves, and food disorders. Also, gangrene (tissue death), bone infection (osteomyelitis), and reduced immunity against pneumonia and influenza.2 Alzheimer's disease is a progressive neurological disorder characterized by degeneration, atrophy, and death of brain cells.3 Alzheimer's disease is caused by several factors, such as infections, cytotoxicity of amyloid β-protein, increasing age (aging), and genetic characteristics.4,5 Alzheimer's disease has complications such as depression, hallucinosis (psychiatric disturbances), sleep disturbance, difficulty speaking, pneumonia, constipation or diarrhea, malnutrition or dehydration, and dental problems,6–8 in addition to cognitive and functional decline.9 Aging is marked by a progressive loss of human cells and tissues because of genetic and environmental factors and oxidative damage.10 The free radicals (oxidative damage) are responsible for miscellaneous diseases like cancer, malaria, atherosclerosis, inflammatory diseases, Alzheimer's disease, and diabetes complications.11,12 Age-related complications are hearing loss, osteoarthritis, diabetes mellitus, depression, Alzheimer's disease, and cardiovascular disease.13Recently, studies, modern research, and bioinformatics analysis proved the close relationship between Alzheimer's disease, diabetes mellitus, oxidative damage, aging, and cardiovascular disease14–16 (Fig. 1). Numerous drugs on the market target a single disease; therefore, these drugs are working on one target “one drug-one target”. A patient with various diseases suffers a problem when taking more than one drug at the same time or near times between doses. This problem may be causes interference for some drugs that don't work effectively against disease and, in some cases, don't work with each other (antagonistic properties appear) and in some cases may don't work completely. Consequently, research cooperation continues between scientists for the chemical studies and biological applications of the nitrogen heterocycle compounds in the hope of finding a new drug with multi-biologically target “one drug-multiple target”. This is one of the efficient strategies for discovering novel categories of therapeutics or the development of drugs to face and fight against the most challenging diseases, especially after the Covid-19 pandemic and appearing numerous healthy problems complicated.17–22
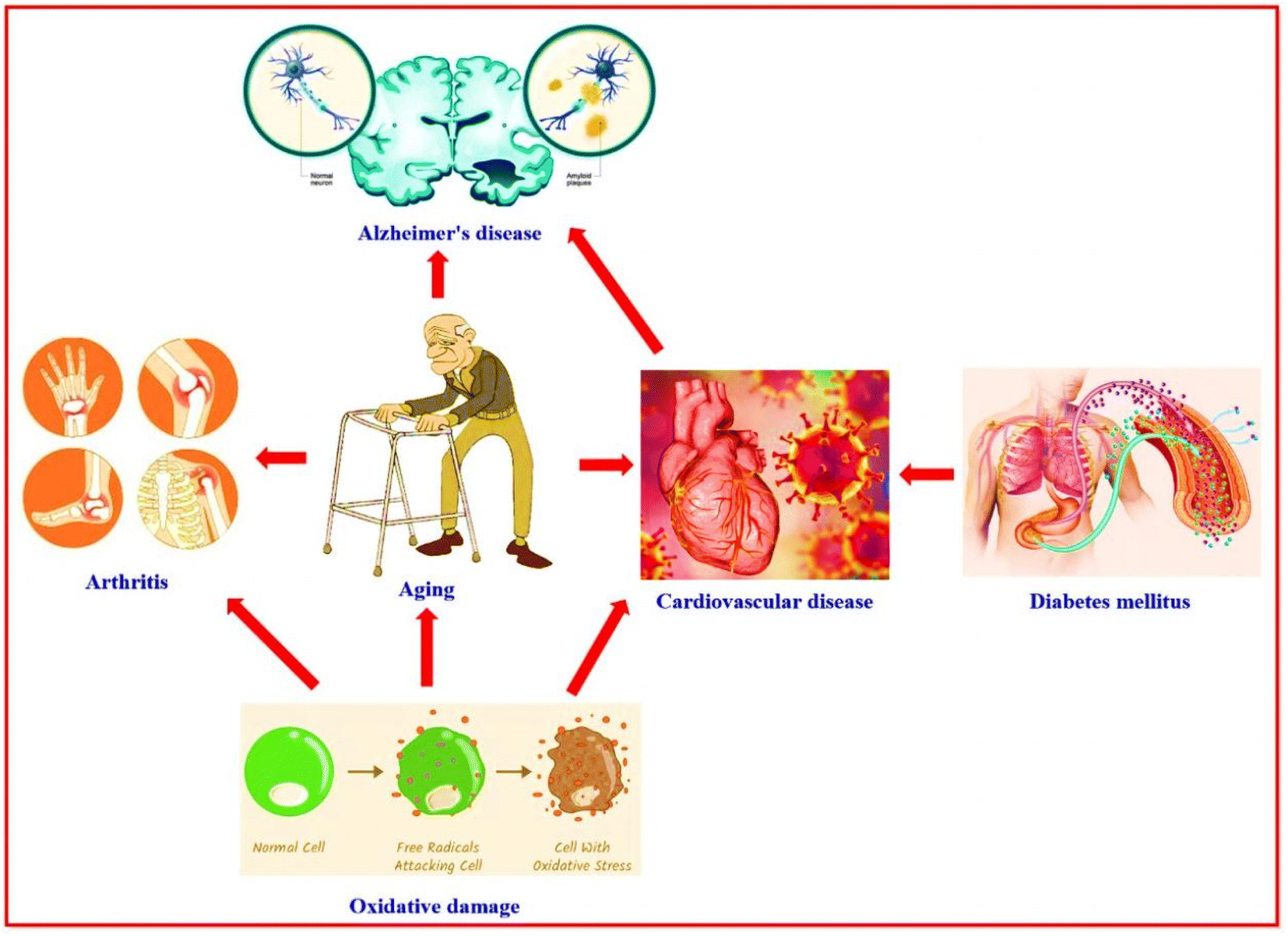 | ||
| Fig. 1 Illustration of the relationship between diseases. | ||
In recent decades, the isatin (2,3-dioxindole) (I) molecule has attracted the concern of scientists for its potent biological and therapeutic applications.23–25 Also, isatin derivatives have numerous distinct biological activities. Ethyl 4-chlorobenzoyl hydrazono-2-oxoindolin-1-yl acetate II is biologically active as a powerful in vitro inhibitor for an α-amylase enzyme (anti-diabetic agent).26 The isatin thiosemicarbazone derivative III acts as an inhibitor against acetylcholinesterase enzyme in vitro (anti-Alzheimer agent).27 Additionally, 2-oxoindolin-3-ylidene-pyrrolidine-3-carbohydrazide derivative IV possessed a high ability to iron reducing power (antioxidant agent).28 Moreover, the morpholinosulfonyl-2-oxoindolin-3-ylidene compound V showed a broad-spectrum and potent anticancer activity against HepG-2, HCT-116, and MCF-7 cell lines.29 Besides, the derivative 1H-pyrazol-4-yl-5-(morpholinosulfonyl)indolin-2-one VI expositions antibacterial activity and is a potent inhibitor of S. aureus DNA gyrase.30 Additionally, some clinical drugs incorporate isatin moiety as nintedanib®, which acts as a tyrosine kinases inhibitor and a lung cancer treatment,31 methisazone® that act as an mRNA inhibitor, protein synthesis, and an antiviral drug;32 and sunitinib® is act as receptor tyrosine kinase (RTK) inhibitor and renal cell carcinoma (RCC) therapy33 (Fig. 2). On the other hand, the Schiff bases linked to a heterocyclic scaffold displayed interesting and numerous biological activities. Schiff base associated with 1,2,4-triazole VII displayed highly antibacterial activity against Halomonas salina and inhibitor activity against the two enzymes butyrylcholinesterase and acetylcholinesterase (enzymes of anti-Alzheimer disease).34 Further, the Schiff base tethered pyrazole scaffold VIII demonstrated an inhibitory impact in vitro against B. subtilis DNA gyrase and dihydrofolate reductase (enzymes of bacterial infection)35 (Fig. 2).
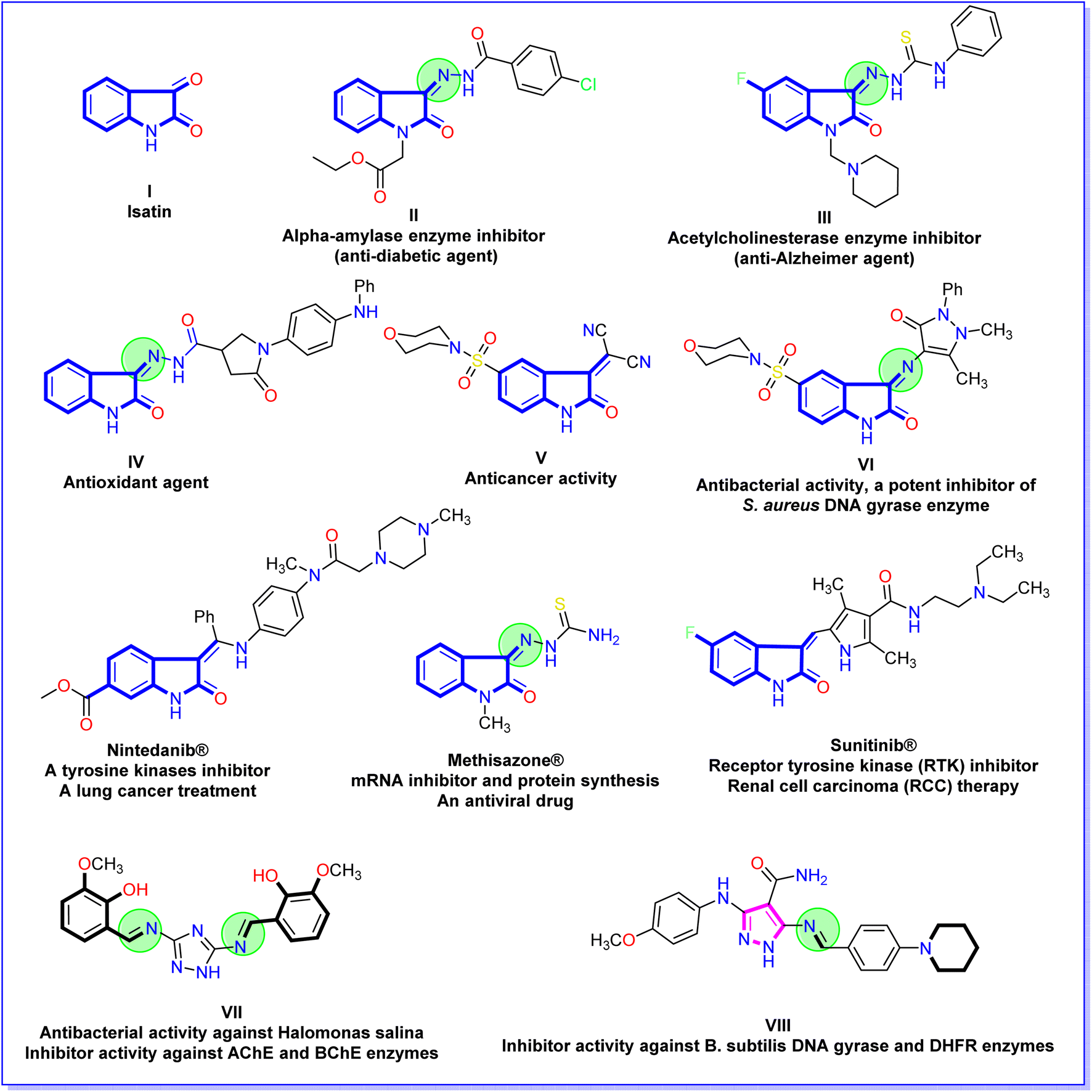 | ||
| Fig. 2 Chemical structures of bioactive isatin derivatives, isatin drugs, and Schiff bases associated with heterocyclic scaffold. | ||
In view of our scientific team targets in medicinal chemistry based on bioactive heterocyclic compounds36–42 and the previous-mentioned facts about the relationship between various diseases, applications of isatin derivatives, and various activities of Schiff bases tethered with a heterocyclic scaffold. Hence, the present study aims to explore isatin-based Schiff bases (azomethine) as multi-target agents. Consequently, we synthesized a new series of isatin-based azomethine derivatives 2a and 2b, isatin-azomethine-aryl azo 3a–d, isatin-azomethine-heterocyclic moiety 4, 5a–c, and 6, and bis-Schiff bases based on isatin 7a and 7b. All the synthesized isatin-based Schiff bases structures were confirmed and evaluated for their antioxidant, anti-diabetic, anti-Alzheimer, and anti-arthritic activities. Finally, the structure–activity relationship study, the Quantum chemical calculations, the molecular docking simulation, and in silico ADMET properties of the most potent isatin-based Schiff bases (azomethine) 3b and 5a–c were predicted and studied.
2. Results and discussion
2.1. Chemistry
A new series of isatin-based Schiff bases 2a, 2b, 3a–d, 4, 5a–c, 6, 7a, and 7b were synthesized based on a reaction of 3-hydrazonoindolin-2-one (1) that was previously prepared43 with different formyl derivatives, and the structures of the newly designed compounds are outlined in Schemes 1–3. Our research was designed by reaction of 3-hydrazonoindolin-2-one (1) with different formyl cores as series 1 containing aromatic formyl containing aliphatic or azo (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) group (Scheme 1), series 2 involved heterocyclic formyl based on pyrazole or furane core (Scheme 2), and series 3 including bis-formyl derivatives (Scheme 3).
N) group (Scheme 1), series 2 involved heterocyclic formyl based on pyrazole or furane core (Scheme 2), and series 3 including bis-formyl derivatives (Scheme 3).
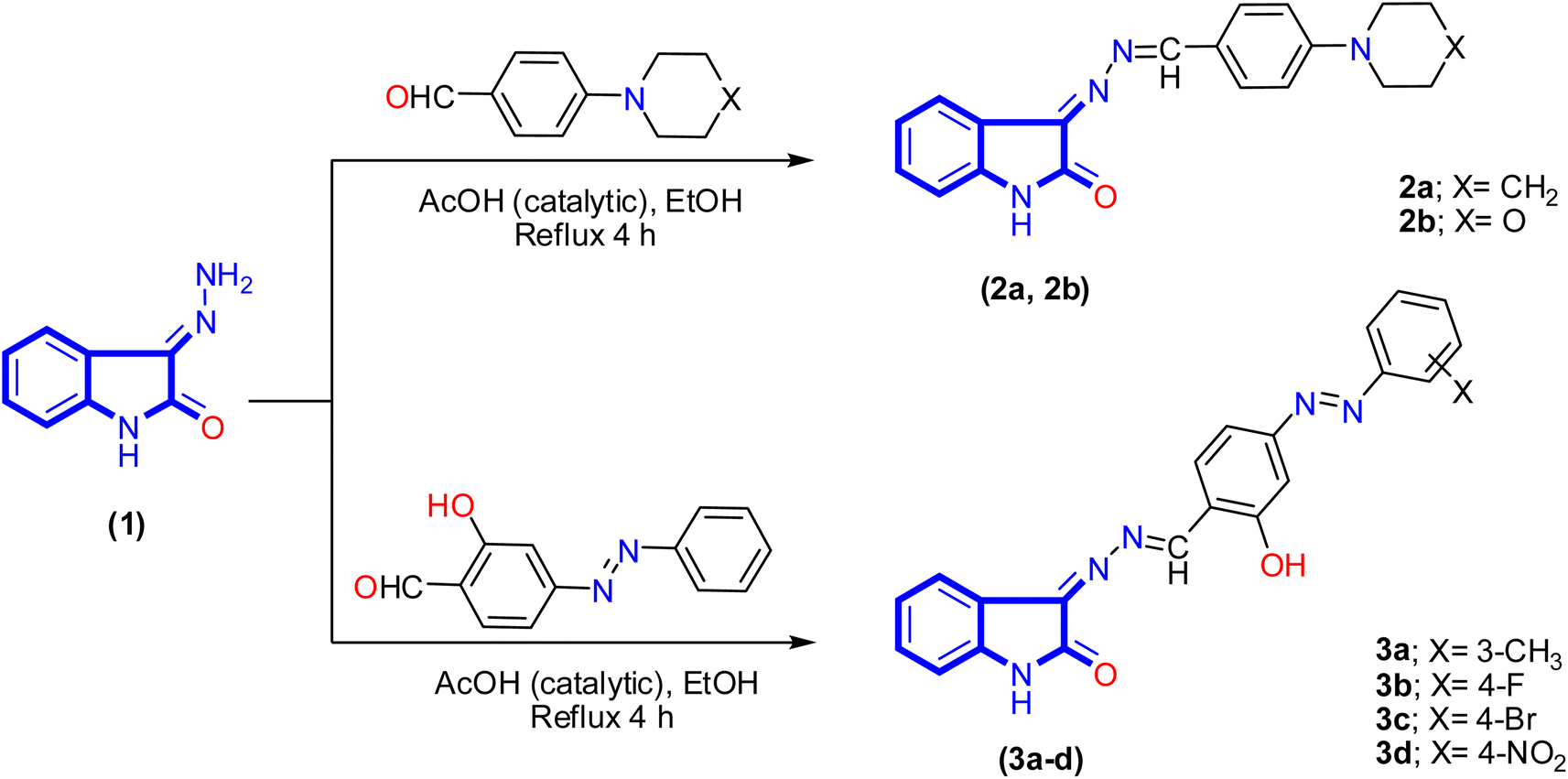 | ||
| Scheme 1 Synthesis of isatin-based azomethine 2a, 2b, and isatin-azomethine-arylazo 3a–d. | ||
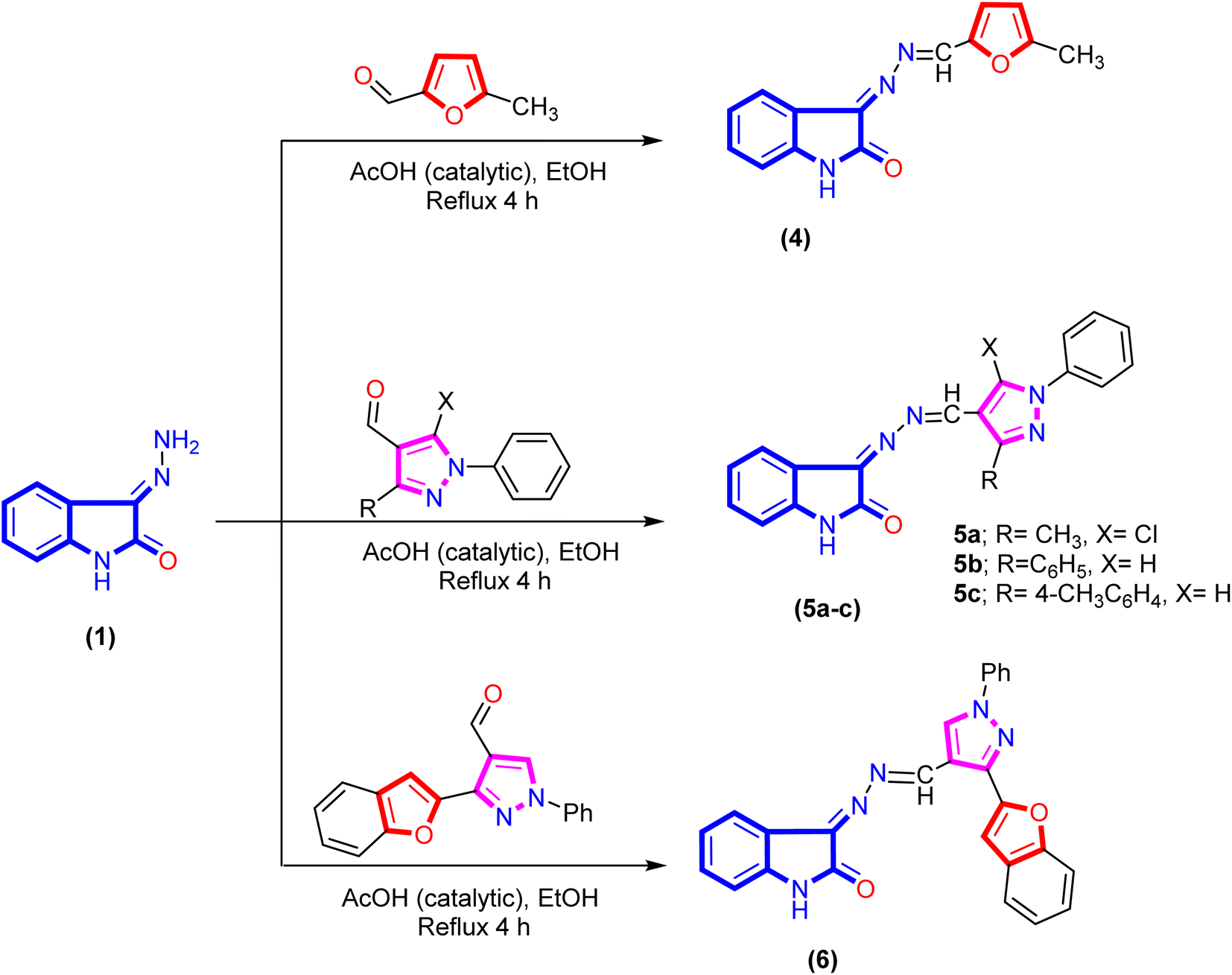 | ||
| Scheme 2 Synthesis of isatin-azomethine-heterocyclic 4, 5a–c, and 6. | ||
 | ||
| Scheme 3 Synthesis of bis-Schiff bases based on isatin 7a and 7b. | ||
Firstly, the isatin-based azomethine derivatives 2a, 2b, and isatin-azomethine-arylazo derivatives 3a–d were synthesized via the reaction of 3-hydrazonoindolin-2-one (1) with aryl aldehydes (namely: 4-(piperidin-1-yl)benzaldehyde, 4-morpholinobenzaldehyde, and 2-hydroxy-4-((4-substituted-phenyl)azo)benzaldehyde) in ethanol (solvent)/glacial acetic acid (catalytic) under reflux conditions, where the product obtained after cooling (Scheme 1). Spectral data and elemental analysis confirmed the structures of the synthesized isatin-based azomethine derivatives 2a, 2b, and 3a–d.
The IR spectrum of 3-((4-morpholino/pipridino-benzylidene)hydrazono)indolin-2-one 2a and 2b displayed various bands related to functional groups as NH, carbonyl (CO), and azomethine (CN) groups ranging between 3171–3193, 1723–1725, and 1605–1611 cm−1, respectively. Similarly, the azo-aryl-Schiff bases 3a–d exhibited bands ranging between 3429–3440, 3159–3200, 1715–1726, and 1618–1620 cm−1 assigned to OH, NH, CO, and CN groups, respectively. The 1H NMR spectrum analysis of compound 2b appeared the morpholinyl moiety as two signals with the integration of four for each single and displayed as multiplet between δ 3.27–3.29 ppm and singlet signal at δ 3.71 ppm. Besides, two singlet signals at δ 8.58 and 10.75 ppm correspond to azomethine and NH proton of the isatin derivative. The aromatic protons (8 protons) range between δ 6.86–8.11 ppm appearing as four doublets signals for six protons and two triplet signals for two protons. Moreover, the 13C NMR spectrum of compound 2b revealed two signals at δ 47.42 and 66.40 ppm for the four carbon atoms of morpholinyl moiety, the aromatic and azomethine carbon atoms range from δ 111.18 to 164.17 ppm, and carbonyl carbon at δ 165.54 ppm. In the same way, the 1H NMR spectrum analysis of salicylaldimine derivatives 3a–d showed significant signals at δ 8.81–8.87, 10.90–10.97, and 11.03–12.56 ppm attributed to azomethine proton, phenolic hydroxyl group, and NH of isatin derivative, respectively. Moreover, in the case of salicylaldimine derivative 3a the signal that appeared at δ 2.42 ppm confirmed three protons presented in the methyl group. All the above-mentioned spectroscopic data confirmed the structure of the proposed compounds.
Furthermore, refluxing 3-hydrazonoindolin-2-one (1) with hetero-aryl aldehydes under the same reaction conditions afforded the corresponding 3-(((hetero-aryl)methylene)hydrazono)-indolin-2-one derivatives 4, 5a–c, and 6 (Scheme 2). Additionally, the Schiff base of isatin hybrid heterocyclic core structures 4–6 were confirmed by spectral data. The IR spectrum of compound 5c exhibited the absence of forked bands for the amino group (corresponding to hydrazone) and the presence of other stretching bands at 3202, 1727, and 1615 cm−1 attributed to NH, CO, and CN groups, respectively. The 1H NMR spectrum of compound 4 exhibited three significant signals at δ 2.38, 8.53, and 10.87 ppm, which are assignable to methyl protons, azomethine proton, and NH proton, respectively. In addition, an increasing number of aromatic protons with two doublet signals at δ 6.50 and 7.30 ppm with coupling constant (J = 3.7 Hz) corresponding to furane core. Meanwhile, the 1H NMR spectrum of the isatin-based azomethine-heterocyclic derivative 5c distinguished by four singlets signals at δ 2.36, 8.69, 9.30, and 10.78 ppm for the methyl group, azomethine proton, proton of the pyrazole H-5, and NH proton of the isatin ring, respectively. Besides, aromatic protons of the isatin ring appeared as two doublets at δ 6.83 and 7.90 ppm with coupling constant (J = 7.8 and 7.4 Hz) and two triplets signals at δ 6.88 and 7.40 ppm. Moreover, the other nine protons of the two aromatic rings (phenyl and 4-methylphenyl) emerged at δ 7.30–7.34, 7.56, 7.62, and 8.02 as one multiplet, one triplet, and two doublets (J = 8.0 and 8.1 Hz) integrated for 3H, 2H, and 4H. Further, the 13C NMR spectrum of azomethine derivative 5c exhibited two peaks at δ 21.00 and 164.94 ppm for the carbon atoms of the methyl (CH3) and carbonyl (CO) groups. In addition, peaks of the aromatic and azomethine carbon atoms (the twenty-three carbons) were marked between δ 110.53–157.25 ppm.
Bis-Schiff bases are famous for their antitumor, antioxidant, antiglycation, antibacterial, and antiviral activities.44–46 Also, act as inhibitors of enzymes such as α-glucosidase,47 butrylcholinesterase, and acetylcholinesterase.48 As shown in Scheme 3, the reaction was readily extended to synthesize a new bis-Schiff base by reaction of 3-hydrazonoindolin-2-one (1) with various bis-aldehyde. Spectral data (IR, 1H NMR, and 13C NMR) and elemental analysis confirmed the structures of all newly synthesized compounds 7a and 7b.
Regarding the IR spectra of bis-Schiff bases 7a and 7b displayed high similarity by displaying bands around ν 1605–1607, 1725–1730, and 3199–3204 cm−1 corresponding to CN, CO, and NH groups, respectively. The 1H NMR spectrum of compound 7a is characterized by a singlet signal at δ 6.03 ppm related to the methylene group, two signals at δ 8.58 and 8.61 ppm for two azomethine protons, and a signal at δ 10.81 ppm for two NH protons. Besides, the aromatic protons displayed between δ 6.85–7.96 ppm for sixteen protons. The 13C NMR spectrum of bis-Schiff base 7a exhibited two peaks at δ 89.38 and 164.67 ppm for the methylene carbon atom (1C, CH2) and carbonyl atoms (2C, 2CO), respectively. In addition, the twenty-six aromatic carbon with the two azomethine carbon atoms ranged from δ 110.79 to 160.94 ppm (the twenty-eight carbon atoms). Finally, all the newly synthesized Schiff bases were characterized by forming a new azomethine proton, as represented in the discussion and Experimental section.
2.2. In vitro biological activities and SAR analysis
| Isatin-based Schiff bases | Antioxidant activities | Free radical scavenging activities | ||
|---|---|---|---|---|
| IRP (μg mL−1) | TAC (mg gallic acid per g) | ABTS (%) | DPPH (IC50 μg mL−1) | |
| a Denotes the most effective compound, values were calculated from three replicates and expressed as mean ± SE. | ||||
| 2a | 19.93 ± 0.04 | 35.59 ± 0.08 | 28.78 ± 0.06 | 15.10 ± 0.03 |
| 2b | 20.12 ± 0.04 | 35.92 ± 0.08 | 29.05 ± 0.06 | 14.96 ± 0.03 |
| 3a | 21.13 ± 0.05 | 37.74 ± 0.08 | 30.51 ± 0.07 | 14.25 ± 0.03 |
| 3b | 47.87 ± 0.10a | 64.62 ± 0.14a | 51.28 ± 0.11a | 9.11 ± 0.02a |
| 3c | 20.93 ± 0.05 | 37.36 ± 0.08 | 30.21 ± 0.07 | 14.39 ± 0.03 |
| 3d | 22.81 ± 0.05 | 40.72 ± 0.09 | 32.93 ± 0.07 | 13.20 ± 0.03 |
| 4 | 18.63 ± 0.04 | 33.26 ± 0.07 | 26.90 ± 0.06 | 16.16 ± 0.04 |
| 5a | 50.39 ± 0.11a | 68.02 ± 0.15a | 53.98 ± 0.12a | 8.65 ± 0.02a |
| 5b | 48.92 ± 0.11a | 66.03 ± 0.14a | 52.41 ± 0.11a | 8.91 ± 0.02a |
| 5c | 49.41 ± 0.11a | 66.70 ± 0.15a | 52.93 ± 0.12a | 8.82 ± 0.0a |
| 6 | 17.40 ± 0.04 | 30.70 ± 0.07 | 23.27 ± 0.05 | 18.63 ± 0.04 |
| 7a | 20.74 ± 0.05 | 37.03 ± 0.08 | 30.76 ± 0.07 | 15.04 ± 0.03 |
| 7b | 20.94 ± 0.05 | 37.40 ± 0.08 | 31.07 ± 0.07 | 14.89 ± 0.03 |
| Ascorbic acid | — | — | 39.09 ± 0.09 | 4.05 ± 0.01 |
Firstly, the synthesized derivatives incorporating azomethine-isatin Schiff base demonstrated various degrees of antioxidant activity represented by iron-reducing power (IRP = μg mL−1) and total antioxidant capacity (TAC = mg gallic acid per g). The 3-(((5-chloro-3-methyl-1H-pyrazol-4-yl)methylene)hydrazono)indolin-2-one derivative 5a recorded the best antioxidant activity among all the tested derivatives with the strongest reducing power (IRP = 50.39 ± 0.11 μg mL−1) and highest antioxidant capacity (TAC = 68.02 ± 0.15 mg gallic acid per g) and that may be related to the presence of methyl group and chlorine atom at position three and five in pyrazole moiety (electron delocalized aromatic nucleus).
As can be seen in Table 1, the other two pyrazole derivatives 5b (IRP = 48.92 ± 0.11 μg mL−1 and TAC = 66.03 ± 0.14 mg gallic acid per g) and 5c (IRP = 49.41 ± 0.11 μg mL−1 and TAC = 66.70 ± 0.15 mg gallic acid per g), showed nearly equipotent with a slight difference in antioxidant activity. In addition, introducing a heterocyclic ring with one hetero-atom as furan instead of pyrazole doesn't enhance the activity and the antioxidant activities may be related to the presence of two hetero-atoms in five-ring as pyrazole ring that showed effected in the antioxidant activity. To our surprise, replacing of aryl group at C3 of pyrazole to benzo[b]furan as compound 6 causes dramatically decrease for (IRP = 17.40 ± 0.04 μg mL−1 and TAC = 30.70 ± 0.07 mg gallic acid per g) compared with 3-arylpyrazole derivatives 5b and 5c, indicating that the presence of furan fused to benzene ring to form benzo[b]furan decrease antioxidant activity by nearly from 0.25 to 0.50 of its activity.
Furthermore, compounds 2a and 2b, which are Schiff bases with piperidinyl and morpholinyl derivatives showed moderate antioxidant activity with IRP values of (19.93 ± 0.04 and 20.12 ± 0.04 μg mL−1) and TAC values of (35.59 ± 0.08 and 35.92 ± 0.08 mg gallic acid per g), respectively. Moreover, forming new Schiff bases that bear phenolic hydroxyl group as salicylamide derivatives 3a–d with a different substituent at the para position of the azoaryl group don't enhance the antioxidant activity compared to other derivatives, except with compound 3b that bears (4-fluorophenyl)azo group at C5 of salicylamide derivative 3b with IRP values of (47.87 ± 0.10 μg mL−1) and TAC values of (64.62 ± 0.14 mg gallic acid per g). The values of IRP and TAC of bis-compounds exhibited moderate activity, indicating an increase in the isatin nucleus and the presence of new ether linkage in the structure don't affect activity.
The scavenging activities using the ABTS and DPPH radicals were evaluated for isatin-based Schiff bases, and the results obtained ranged from 23.27 ± 0.05 to 53.98 ± 0.12% compared with ascorbic acid (39.09 ± 0.09%) for ABTS. Moreover, the interaction of the synthesized compounds with stable free radical DPPH indicated radical scavenging ability represented by IC50 values ranging from 8.65 ± 0.02 to 8.65 ± 0.02 μg mL−1 in comparison to ascorbic acid (4.05 ± 0.01 μg mL−1). It was found that four isatin-based Schiff bases have the best ABTS and DPPH values, where the salicylamide derivative 3b showed (ABTS = 51.28 ± 0.11% and DPPH = 9.11 ± 0.02 μg mL−1), and the other three Schiff bases with pyrazole heterocyclic moiety as 5a (ABTS = 53.98 ± 0.12% and DPPH = 8.65 ± 0.02 μg mL−1), 5b (ABTS = 52.41 ± 0.11% and DPPH = 8.91 ± 0.02 μg mL−1), and 5c (ABTS = 52.93 ± 0.12% and DPPH = 8.82 ± 0.02 μg mL−1), and the order of activity as the most potent represented in the range of 5a > 5c > 5b > 3b.
As represented in Table 1, Schiff bases containing in the side chain amine derivatives 2a, 2b, and azo-aryl group 3a–d displayed weak to moderate free radical scavenging activities (ABTS and DPPH). This low activity may be related to the disability of amine (piperidinyl and morpholinyl) and azo-aryl groups (4-methyl, 4-bromo, and 4-nitro) to catch radicals, except 3b derivatives that indicate the presence of fluorine atom as strong electron withdrawing group that helps the aromatic aryl group permits the trapping of free radical. Additionally, the introduction of heterocyclic rings to the azomethine group showed enhancement of the antioxidant activity, especially with the pyrazole nucleus that displayed importance for antioxidant activity as represented in compounds 5a–c. The 3-(((1H-pyrazol-4-yl)methylene)hydrazono)indolin-2-one derivatives 5a–c demonstrated excellent ABTS values between (52.41 ± 0.11 to 53.98 ± 0.12%) and good DPPH values IC50 ranging between (8.65 ± 0.02 to 8.91 ± 0.02 μg mL−1) compared with 3-(((5-methylfuran-2-yl)methylene)hydrazono)-indolin-2-one (4) with ABTS (% = 26.90 ± 0.06) and DPPH (IC50 = 16.16 ± 0.04 μg mL−1) and 3-(((3-(benzofuran-2-yl)-1H-pyrazol-4-yl)methylene)hydrazono)indolin-2-one 6 with ABTS (% = 23.27 ± 0.05) and DPPH (IC50 = 18.63 ± 0.04 μg mL−1). The structure–activity relationship (SAR) revealed that the pyrazole core has antioxidant activity higher than furan and pyrazole containing benzo[b]furan group and this better activity may be due to the efficiency of the trapping. Even though the Schiff base with heterocyclic core 4–6 exhibited good antioxidant activity compared with other synthesized derivatives, but ascorbic acid remained the best activity with ABTS (% = 39.09 ± 0.09) and DPPH (IC50 = 4.05 ± 0.01 μg mL−1).
Furthermore, the bis-Schiff bases 7a and 7b exhibited moderate activity, and the presence of ethane-1,2-dioxyl as a linker between two Schiff bases demonstrated slightly higher free radical scavenging activities with ABTS (% = 31.07 ± 0.07) and low DPPH (IC50 = 14.89 ± 0.03 μg mL−1) compared with methane-1,1-dioxyl derivative activities (ABTS = 30.76 ± 0.07% and DPPH IC50 = 15.04 ± 0.03 μg mL−1). Finally, it can be concluded that among the isatin-based Schiff bases 2a, 2b, 3a–d, 4, 5a–c, 6, 7a, and 7b introducing the pyrazole motif as 5a–c or strong electron-withdrawing group to azo-salicylamide moiety as 3b are more effective antioxidants than others cores or substituents.
As described in Table 2, all compounds exhibited moderate to good α-amylase inhibition (%), ranging from 26.97 ± 0.06 to 57.64 ± 0.13%. Among the tested compounds, the Schiff base with pyrazole motif 5a–c and salicylamide containing azo-aryl with para-fluoro atom 3b are relatively more potent than other derivatives. The result of the anti-diabetic activity (Table 2) raveled that the four isatin-based Schiff bases 3b (α-amylase inhibition = 54.76 ± 0.12%), 5a (α-amylase inhibition = 57.64 ± 0.13%), 5b (α-amylase inhibition = 55.96 ± 0.12%), and 5c (α-amylase inhibition = 56.52 ± 0.12%) had anti-diabetic activities nearly close to the acarbose (standard reference), and the order of activity can be represented as 5a > 5c > 5b > 3b.
| Isatin-based Schiff bases | Anti-diabetic activity | Anti-Alzheimer's activity | Anti-arthritic activity | |
|---|---|---|---|---|
| α-Amylase inhibition (%) | Acetylcholinesterase (AChE) inhibition (%) | Proteinase denaturation (%) | Inhibition of proteinase (%) | |
| a Denotes the most effective compound, values were calculated from three replicates and expressed as mean ± SE; ACA = acarbose; DIC = diclofenac sodium. | ||||
| 2a | 29.84 ± 0.06 | 17.14 ± 0.04 | 19.23 ± 0.04 | 17.03 ± 0.04 |
| 2b | 30.12 ± 0.07 | 17.30 ± 0.04 | 19.40 ± 0.04 | 17.19 ± 0.04 |
| 3a | 31.64 ± 0.07 | 18.17 ± 0.04 | 20.38 ± 0.04 | 18.06 ± 0.04 |
| 3b | 54.76 ± 0.12a | 34.56 ± 0.08a | 37.61 ± 0.08a | 34.57 ± 0.08a |
| 3c | 31.33 ± 0.07 | 17.99 ± 0.04 | 20.18 ± 0.04 | 17.88 ± 0.04 |
| 3d | 34.15 ± 0.07 | 19.61 ± 0.04 | 22.00 ± 0.05 | 19.45 ± 0.04 |
| 4 | 27.89 ± 0.06 | 16.02 ± 0.03 | 17.97 ± 0.04 | 15.92 ± 0.03 |
| 5a | 57.64 ± 0.13a | 36.38 ± 0.08a | 39.59 ± 0.09a | 36.39 ± 0.08a |
| 5b | 55.96 ± 0.12a | 35.32 ± 0.08a | 38.44 ± 0.08a | 35.33 ± 0.08a |
| 5c | 56.52 ± 0.12a | 35.68 ± 0.08a | 38.82 ± 0.08a | 35.69 ± 0.08a |
| 6 | 26.97 ± 0.06 | 15.75 ± 0.03 | 17.24 ± 0.04 | 15.01 ± 0.03 |
| 7a | 31.58 ± 0.07 | 17.99 ± 0.04 | 19.77 ± 0.04 | 17.27 ± 0.04 |
| 7b | 31.90 ± 0.07 | 18.17 ± 0.04 | 19.96 ± 0.04 | 17.44 ± 0.04 |
| ACA | 69.11 ± 0.15 | — | ||
| DIC | 49.33 ± 0.11 | 41.88 ± 0.09 | ||
Modification of benzylidene derivative in compounds 2a and 2b by introducing secondary amine to form Schiff base containing amine moiety doesn't improve the activity. Furthermore, our work planned to study the effect of 5-azo-aryl-salicylamide-Schiff base derivatives 3a–d on inhibition of α-amylase and the result demonstrated that the presence of this moiety was affected directly by position and different substituent on azo-aryl group. Additionally, these derivatives showed good to potent inhibition percentages with values ranging between (% = 31.33 ± 0.07 to 54.76 ± 0.12). In addition, the introduction of a high electronegative atom as fluoro observed the best inhibition among these derivatives (α-amylase inhibition = 54.76 ± 0.12%) with an inhibition percentage nearly of 79.23% compared to acarbose. Considering the bis-Schiff base 7a and 7b, it was noticed that the amylase percentage decreased, and that illustrated that repeated the isatin core as another heterocyclic core causes a decrease in the activity.
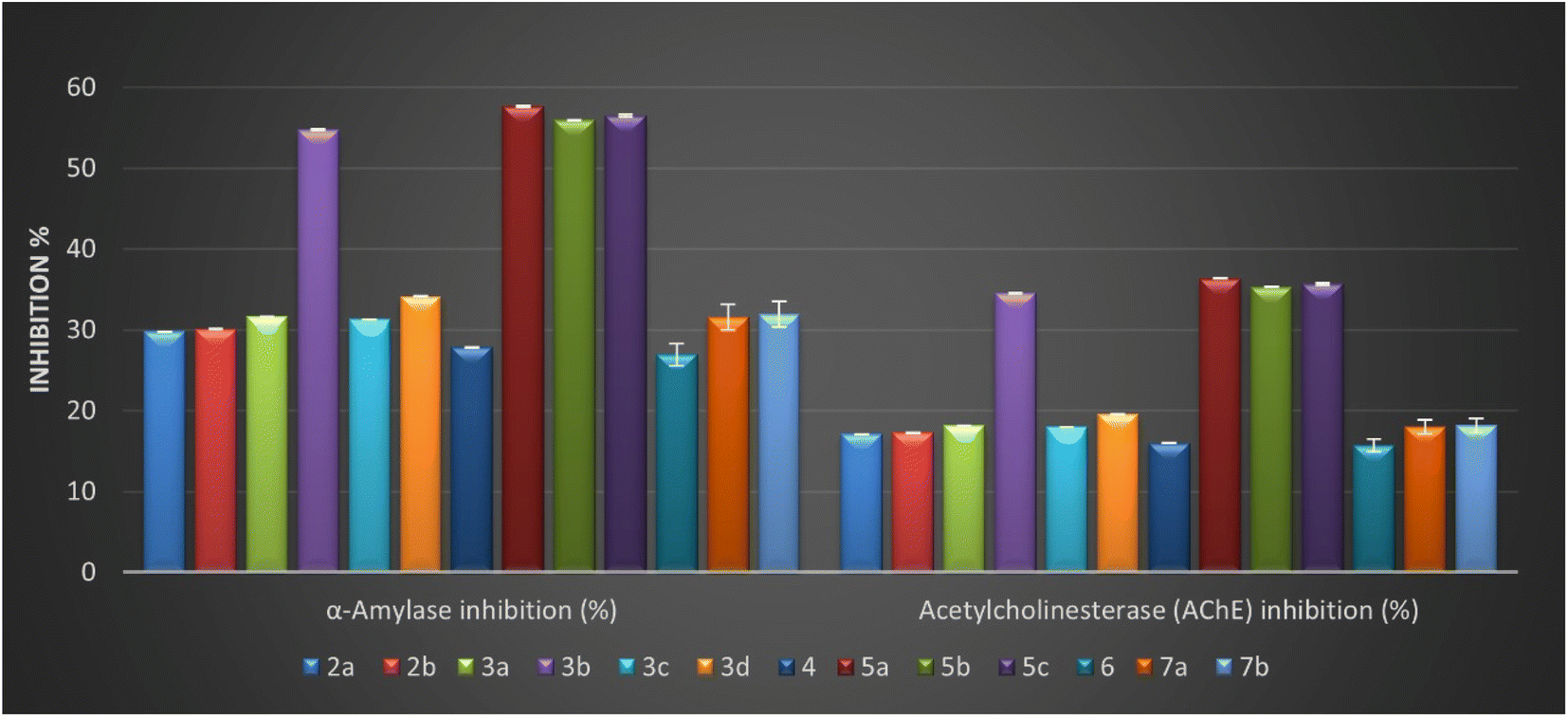 | ||
| Fig. 3 In vitro anti-diabetic activity represented as α-amylase inhibition (%) and anti-Alzheimer's activity represented as acetylcholinesterase (AChE) inhibition (%) of the newly designed derivatives 2–7. | ||
Generally, the inhibition percentage values toward the acetylcholinesterase for the designed isatin-Schiff bases 2–7 were determined and ranged from 15.75 ± 0.03 to 36.38 ± 0.08%. More importantly, four derivatives 3b, 5a, 5b, and 5c revealed the highest inhibitory percentage on the acetylcholinesterase enzyme with values equal to 34.56 ± 0.08, 36.38 ± 0.08, 35.32 ± 0.08, and 35.68 ± 0.08%, respectively. Interestingly, three of the four derivatives contain the same structure pattern with (1-phenyl-1H-pyrazol-4-yl) as a bioactive core side by side with 3-hydrazonoindoline pharmacophore with the new methylic group (CHN). Moreover, the values of inhibition to other derivatives 2a, 2b, 3a, 3c, 3d, 4, 6, 7a, and 7b showed inhibition values ranging from 15.75 ± 0.03 to 19.61 ± 0.04%, indicating a narrow range between them.
The anti-arthritic activity of the newly designed isatin-based Schiff bases derivatives 2a, 2b, 3a–d, 4, 5a–c, 6, 7a, and 7b was evaluated by determining the inhibition percentage of proteinase denaturation and proteinase enzyme. Firstly, the newly designed isatin-based Schiff bases showed proteinase denaturation inhibition (PDI) values ranging between (17.24 ± 0.04 to 39.59 ± 0.09%) and proteinase inhibition (PI) enzyme (15.01 ± 0.03 to 36.39 ± 0.08%) when compared with reference standard drug diclofenac sodium (proteinase denaturation % = 49.33 ± 0.11 and proteinase inhibition % = 41.88 ± 0.09) as represented in Table 2.
The present study observed the maximum percentage of proteinase denaturation inhibition (PDI) and proteinase inhibition (PI) by four Schiff bases 3b, 5a, 5b, and 5c. These derivatives revealed proteinase denaturation and proteinase inhibition in the range from 37.61 ± 0.08 to 39.59 ± 0.09% and from 34.57 ± 0.08 to 36.39 ± 0.08%, respectively, and close to the diclofenac sodium PDI = 49.33 ± 0.11% and PI = 41.88 ± 0.09% (Fig. 4 and Table 2). These good inhibitory activities can be referred to the presence of pyrazole moiety side by side with indolin-2-one in compounds 5a–c that causes a diminution in protein denaturation and proteinase inhibitory activity and an increase in enhanced protection. Moreover, it was observed that the lowest anti-arthritic activity was determined by isatin Schiff base 6 with 1-phenyl-1H-pyrazole containing the benzo[b]furan group as a substituent in position two of the pyrazole motif. Compound 6 exhibited proteinase denaturation inhibition (PDI = 17.24 ± 0.04%) and proteinase inhibition (PI = 15.01 ± 0.03%) and this decrease in anti-arthritic activity may be related to the presence of a benzofuran group.
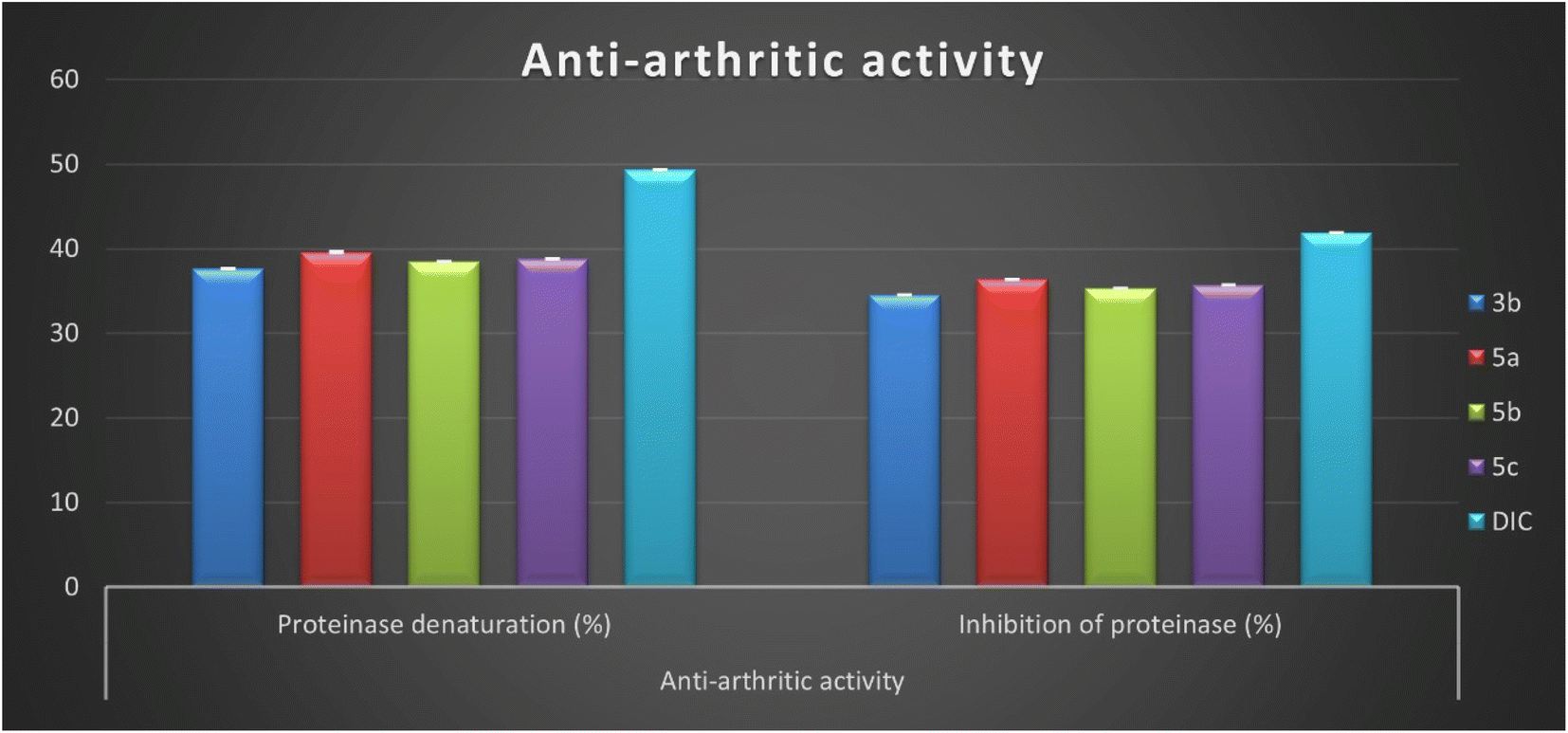 | ||
| Fig. 4 In vitro inhibition percentage of anti-arthritic activity (proteinase denaturation and proteinase) of most active isatin derivatives 3b, 5a, 5b, 5c, and diclofenac sodium. | ||
Furthermore, the bis-indolin-2-one derivatives 7a and 7b demonstrated low inhibitory activity against the proteinase denaturation inhibition (PDI = 19.77 ± 0.04 and 19.96 ± 0.04%) and proteinase inhibition (PI = 17.27 ± 0.04 and 17.44 ± 0.04%), respectively. In addition, compounds 2a and 2b that contain a hydrazone group with 4-(piperidin-1-yl or morpholin-1-yl)benzylidene moiety exhibited very close inhibitory activity to bis-indolin-2-one derivatives 7a and 7b with proteinase denaturation inhibition (PDI = 19.23 ± 0.04 and 19.40 ± 0.04%) and proteinase inhibition (PI = 17.03 ± 0.04 and 17.19 ± 0.04%), respectively.
Finally, it can be concluded that the newly developed isatin-based Schiff bases 2–7 reduce protein denaturation and proteinase inhibition, resulting in an anti-arthritic activity. In addition, among the synthesized derivatives, four Schiff bases 3b, 5a, 5b, and 5c displayed good inhibitory activity ranging between (76.2–80.25%) and (82.5–86.89%) compared to diclofenac.
As depicted in Table S1 (ESI file†), it was noticed that IRP, TAC, the scavenging activity against ABTs, the inhibitory effect against α-amylase and ACE in addition to the anti-arthritic activity are positively correlated with each other and negatively correlated with the concentration required to inhibit 50% of DPPH radicals (IC50). When these biological activities increase, the concentration required to inhibit 50% of the DPPH radical decreases.
N) to 3-hydrazonoindolin-2-one (1) improved biological evaluation Fig. 5. The SAR analysis based on the pharmacophore group introduced in the side chain of the main core can be illustrated as follows:
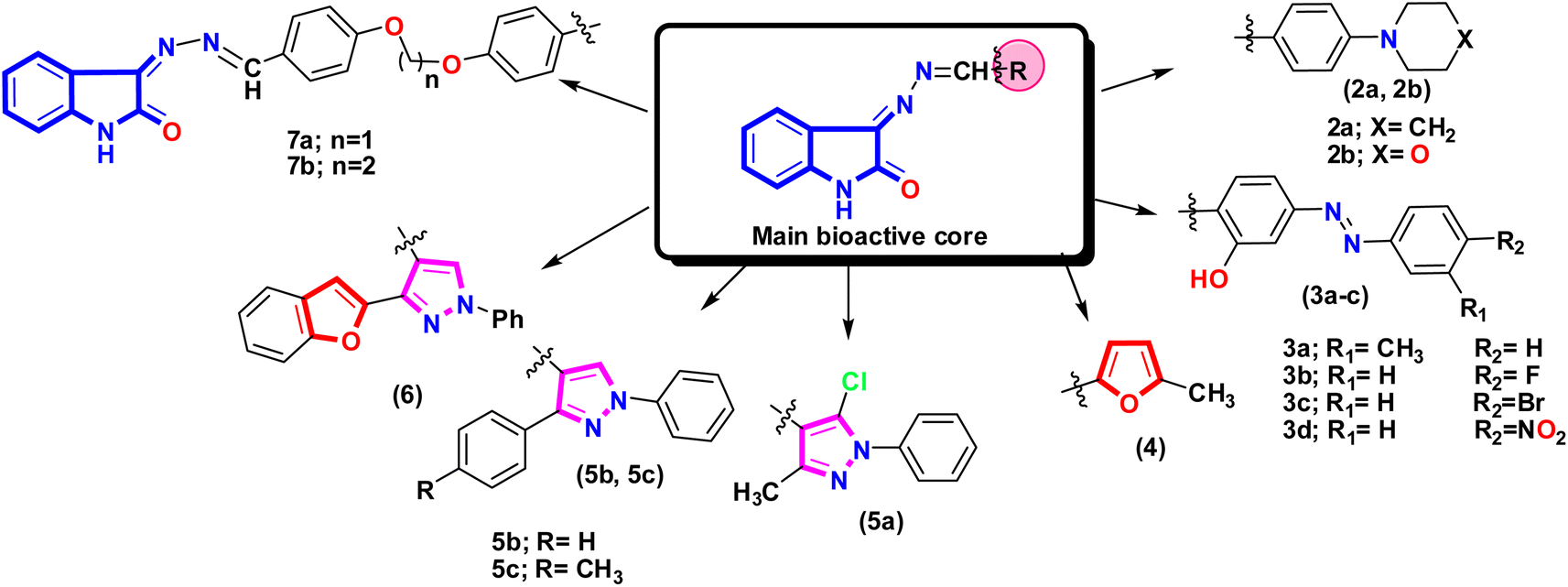 | ||
| Fig. 5 Showed the different fragment that attached to 3-hydrazonoindolin-2-one (1) and formed new azomethine group with different pharmacophore that affect the activity. | ||
[I] Introducing the benzylidene derivative with morpholinyl or piperidinyl moiety exhibited moderate activity with a slight difference between the two derivatives, and the morpholinyl moiety often exhibited slight potency, and that may be related to the presence of hetero atom oxygen.
[II] Looking at the series of isatin-azomethine-aryl azo derivatives 3a–d, the formation of salicylamide derivatives showed moderate activity in general, and the most potent derivative among this series is 3b which is linked to the fluorine atom. This activity may be due to the small size and highly electronegative of fluorine atom, which can improve pharmacokinetics and drug-likeness properties.59,60
[III] Introduction of a new heterocyclic to the azomethine group as 5-methyl furan-2-yl motif does not improve activity, and this can be illustrated by the electron may run and push in the furan core more than the azomethine group and an indole moiety due to high electronegativity of the oxygen atom than the nitrogen.
[IV] The activity of the three isatin-based Schiff bases 5a, 5b, and 5c may be related to the presence of the pyrazole scaffold with various substitutions. Additionally, the isatin-based Schiff base 5a, with different substituents on pyrazole moiety as 5-chloro-3-methyl exhibited the most potent compound, which may be due to increasing the hydrophobic properties in the pyrazole ring.61
[V] The two isatin-based Schiff bases 5b and 5c showed almost the same activities where the isatin-based Schiff base 5b linked with the phenyl ring at C3 of the pyrazole moiety while derivative 5c linked with the 4-methylphenyl ring at the same position (C3 of the pyrazole moiety) indicating that the methyl group in this position doesn't affect the activity. Additionally, replacing this aryl group with heteroatom-containing bicyclic moiety (benzofuran) at C-3 resulted in weaker activity.
[VI] It was observed that the incorporation of bis-formyl derivatives to 3-hydrazonoindolin-2-one (1) to form a bis-Schiff base with a dioxo-linker by introducing more lipophilic group (phenyl group) doesn't enhance the activity.
Finally, it can be concluded that the four compounds 3b, 5a, 5b, and 5c have high antioxidant, anti-diabetes, anti-Alzheimer's, and anti-arthritis activities. This result agrees with the result of Russo and co-workers, which state the relationship and well correlation between the biological activities of the compounds. Additionally, it was reported that the antioxidant activity improves and enhances some activities such as anti-diabetes, anti-Alzheimer's, and anti-arthritis.62
3. Computational studies
3.1. Quantum chemical calculations
In medicinal chemistry, the density functional theory (DFT) is most commonly used in quantum chemical studies to confirm the structural features essential for biological activity.63,64 Additionally, it was used widely because of its low cost, satisfactory performance, and understanding of molecules' behavior in the reactions.65,66 In this work, the structure of the most active Schiff bases 3b, 5a, 5b, and 5c was constructed and optimized using the popular hybrid functional B3LYP with the double zeta basis set 6–31 g (d) as described previously.67–69 The frontier molecular orbitals (FMO) are represented by the highest occupied orbital (HOMO) and lowest occupied orbital (LUMO), and, therefore, the difference between HOMO and LUMO defined as energy band gap and these descriptors are important to evaluate the stability of compounds and intermolecular interaction in the drug-receptor binding system.70As represented in Fig. 6, the salicylamide derivative 3b showed that the HOMO orbital and LUMO were localized overall molecule. However, the HOMO orbital is condensed over the isatin nucleus and salicylamide-azo group with EHOMO = −5.76 eV and ELUMO = −2.88 eV. Similarly, compounds 5a–c revealed that the HOMO orbitals are localized over all the molecules. At the same time, LUMO orbitals are distributed over all molecules except N-phenyl and chlorine atom attached to pyrazole (5a), two phenyl rings attached to pyrazole core (5b), and phenyl and tolyl bounded to pyrazole derivative (5c). Additionally, it was found that the hydrazone group (–CN–NC–) contributed to both HOMO and LUMO orbitals. Moreover, compound 3b exhibited the lowest energy band gap ΔE = 2.88 eV, while the other pyrazole derivatives 5a, 5b, and 5c showed very close energy band gaps with values ΔE = 3.42, 3.38, and 3.36 eV, respectively. Notably, the most active compound 5a showed the lowest total energy with a value of ETotal = −1541.58 Hartree compared to 5b (ETotal = −1273.73 Hartree), 5c (ETotal = −1313.04 Hartree), and salicylamide derivative 3b (ETotal = −1332.63 Hartree), and therefore, compound 5a is the most stable and least reactive and these results may illustrate the potency of biological activity (Table S2, ESI file†).
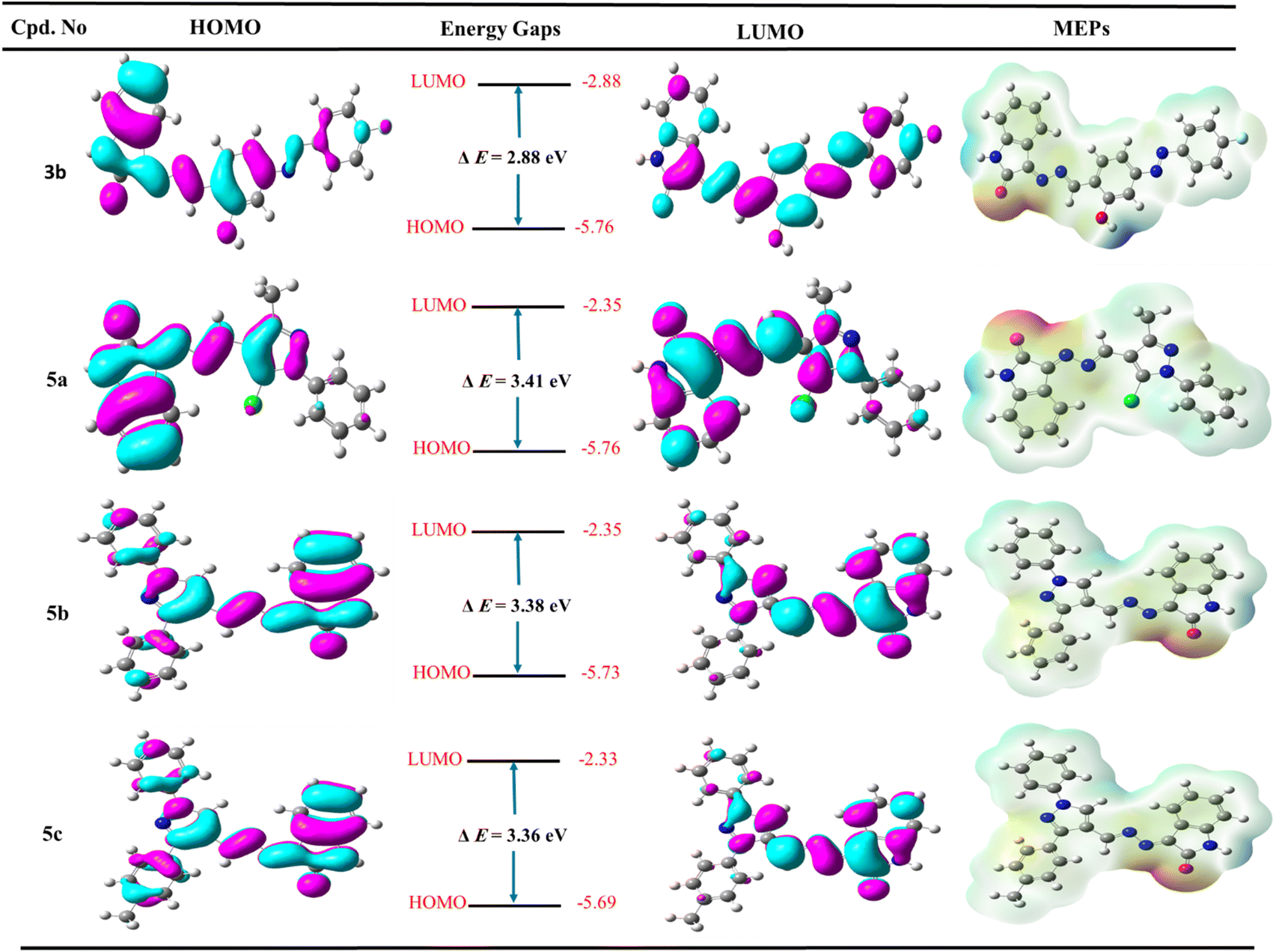 | ||
| Fig. 6 The frontier molecular orbitals (HOMO and LUMO) and molecular electrostatic potential of the most active derivatives 3b, 5a, 5b, and 5c. | ||
The molecular electrostatic potential (MEP Maps) is an important tool that used to recognize the reactive sites in a molecule by determine the positive and negative regions through the molecules. These regions help to form different types of interaction as hydrogen bonds, arene–arene “π–π”, arene–cation “π–cation”, and hydrophobic interactions inside the active site of pocket in the biological target.
The MEP maps diagram represents various electrostatic potentials on a molecule with different colors. Additionally, the electrostatic potential areas are colored based on their electrostatic potential and range from blue to red, the blue regions being the most positive, while the red regions being the most negative, and finally, the green regions being zero. The ascending order is expressed as the following: red > orange > yellow > green > blue. The red regions (negative charge) are localized over the oxygen of isatin, nitrogen of hydrazone, oxygen of hydroxyl group and nitrogen of azo group in the salicylamide derivative 3b, and nitrogen of pyrazole (mainly nitrogen number 2) at pyrazole-Schiff bases 5a–c. Moreover, the blue color is mainly related to the hydrogen atom of NH of isatin and hydrogen of the hydroxyl group in the salicylamide derivative 3b. The green color represented the neutral regions distributed over the whole molecule, especially the aromatic rings. These charged surface areas that represented as blue and red regions could form hydrogen bonds in molecular docking and, therefore, could bind to the active site and inhibit the target enzyme and appear the potency of the synthesized compound.
3.2. Molecular docking simulations
In developing and designing a new drug, molecular docking is a crucial approach used to predict how a synthesized compound is bound to a drug target protein and its affinity.71,72 The docking simulation for the most active Schiff bases 3b, 5a, 5b, and 5c was evaluated inside the active sites of two proteins to determine the binding mode pattern, binding conformation, and different types of interaction of the active derivatives with the active site in enzymes. The human pancreatic α-amylase complexed with co-crystallized ligand acarbose (α-amylase PDB: 2QV4) was selected for anti-diabetic activity. Further, the recombinant human acetylcholinesterase (AChE) (PDB: 4EY7) was used for anti-Alzheimer's activity. The crystal structures of these two proteins were downloaded from the protein data bank (https://www.rcsb.org/) and the active site was generated as described previously73 using Molecular Operating Environment (MOE) software version 2009.10.Interestingly, the Schiff base derivative 5a revealed binding energy S = −22.09 kcal mol−1 and interacted with the pocket via hydrogen bond acceptors with the side chain between the residue Arg195 and carbonyl of isatin with a distance of 2.67 Å and strength 17%. Besides, an arene–arene interaction was observed between Trp59 and phenyl of the N-phenyl pyrazole group. Moreover, the isatin bioactive core in compound 5b could make two hydrogen bonds with the active site of pocket through NH of isatin with Glu240 (2.13 Å and 12%) and carbonyl of isatin derivative with Lys200 (2.00 Å and 32%), as well as arene–arene interaction between the phenyl of isatin and Tyr151 (Fig. 7, and full size figures represented in ESI file†).
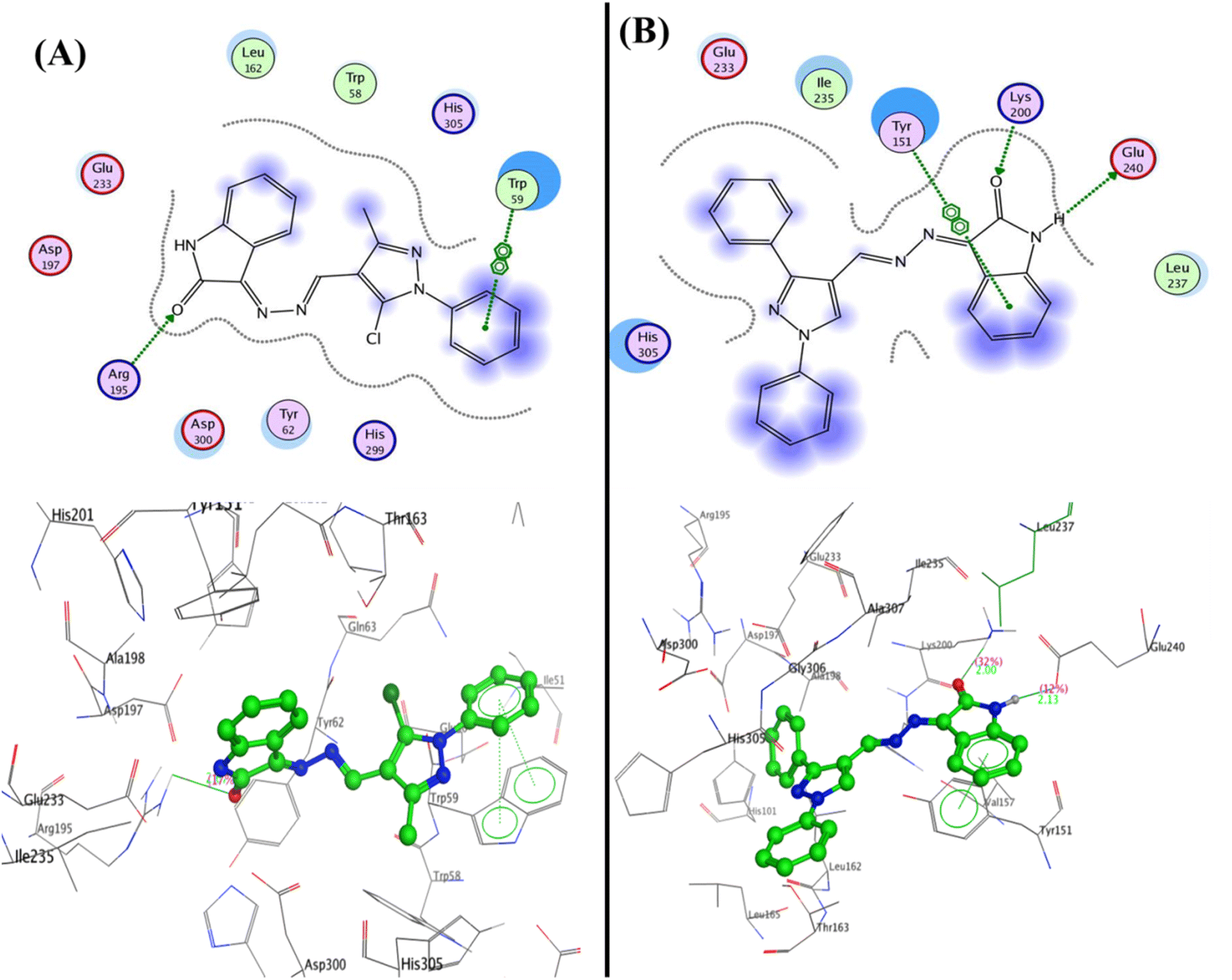 | ||
| Fig. 7 2D and 3D structure of compounds (A) 5a and (B) 5b inside the active site of α-amylase (PDB: 2QV4). | ||
Furthermore, compound 5c was able to interact with the residues in diverse sites in the pocket as Glu233 and formed a hydrogen bond sidechain donor with NH of isatin with a bond length of 2.17 Å and strength of 14%, moreover, it interacted with amino acids in the peripheral anionic siteTrp59 and His101 through arene–arene “π–π” interactions with phenyl of N-phenyl of pyrazole and phenyl of isatin, respectively. Additionally, compound 3b displayed two hydrogen bonds sidechain acceptors between the His201 and Lys200 with the hydroxyl group and carbonyl of isatin with bond lengths 2.18 Å (11%) and 1.93 Å (13%), respectively (Fig. 8).
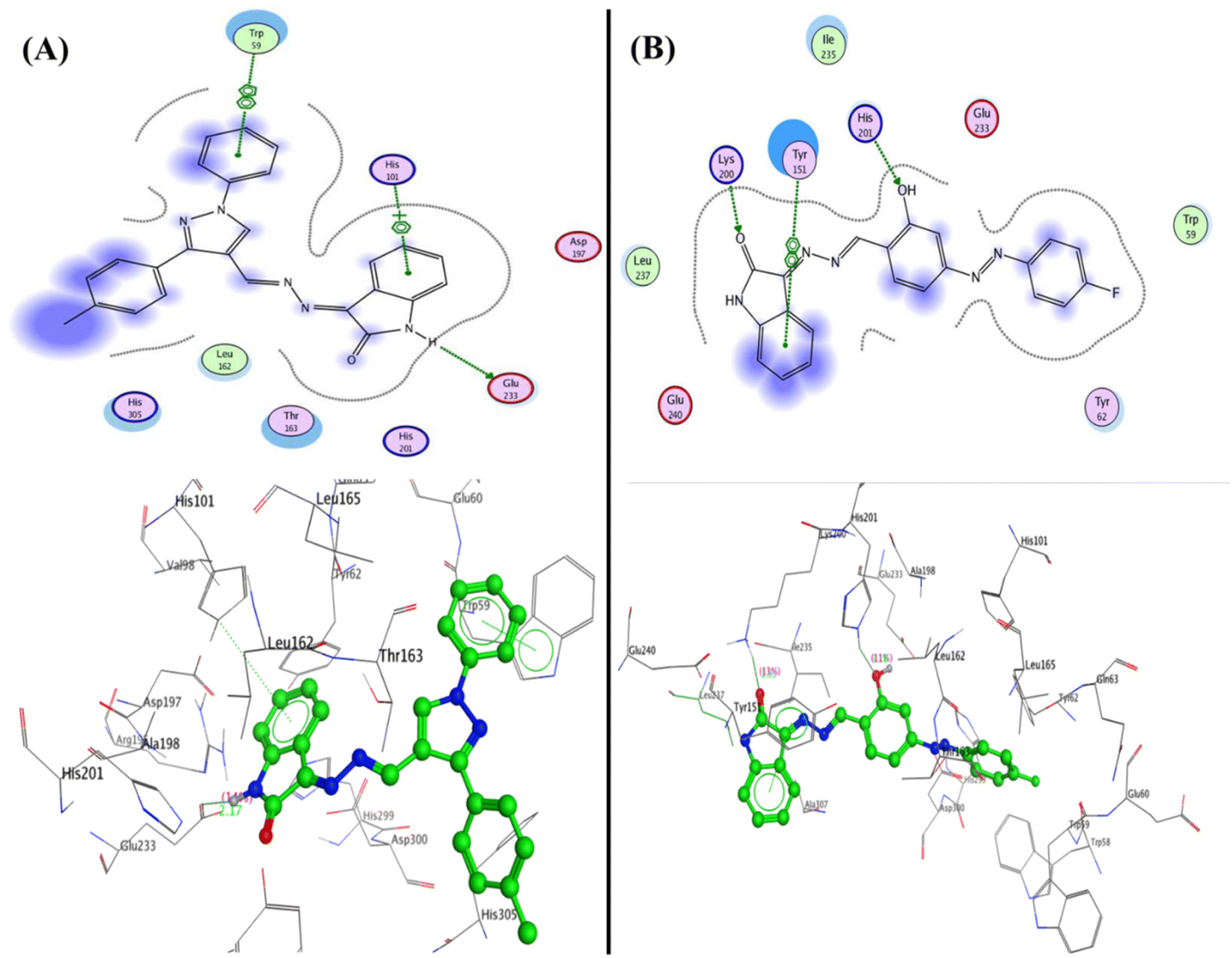 | ||
| Fig. 8 2D and 3D structure of compounds (A) 5c and (B) 3b inside the active site of α-amylase (PDB: 2QV4). | ||
 | ||
| Fig. 9 2D and 3D structure of compounds (A) co-crystallized ligand and (B) 5a inside the active site of acetylcholinesterase (AChE) (PDB: 4EY7). | ||
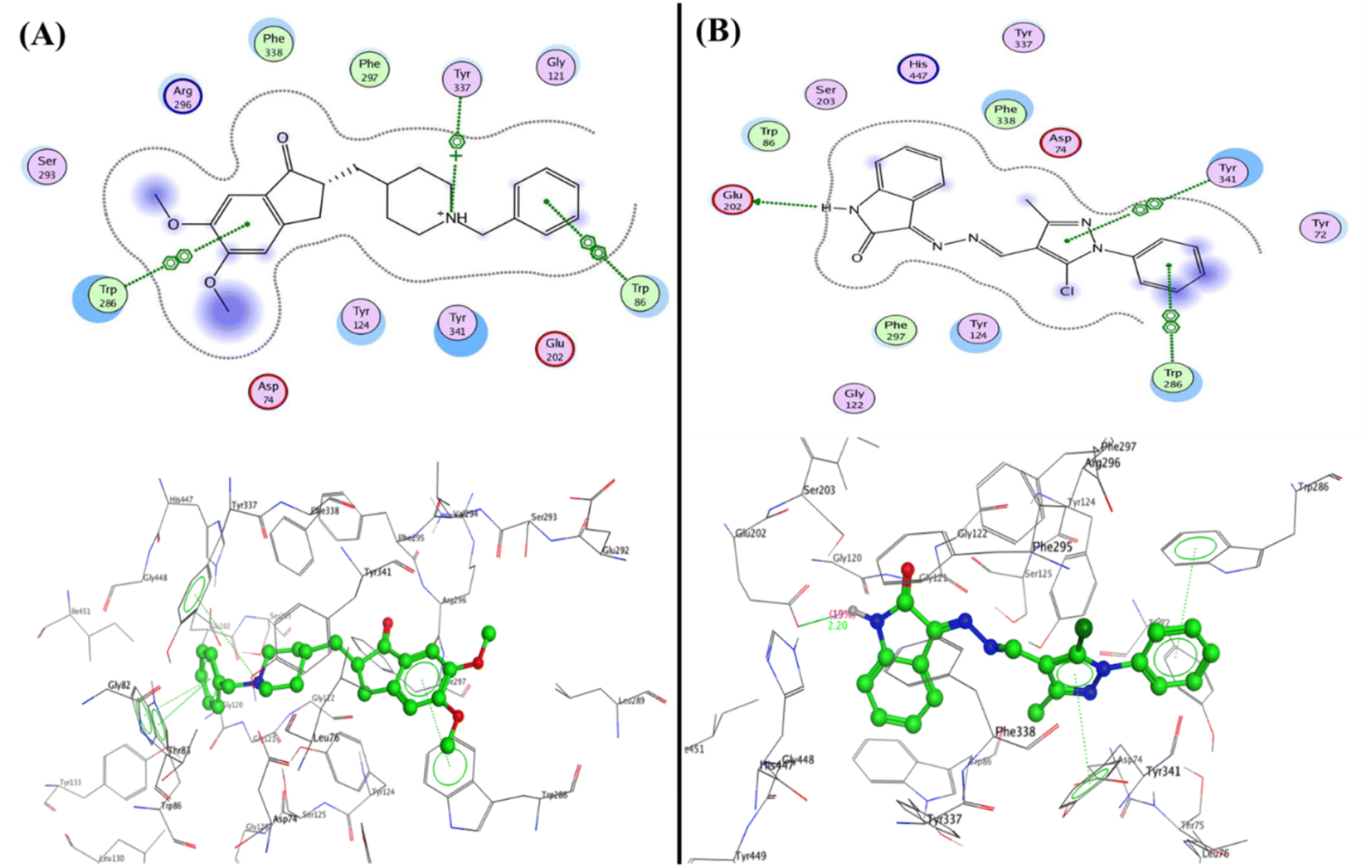
Furthermore, the compounds 5b and 5c demonstrated one type of interaction known as arene–arene interaction with three residues of amino acid, where compound 5b presented binding energy S = −27.22 kcal mol−1 with four arene–arene interactions between the residues Tyr124 and Tyr341 with phenyl of isatin and the Trp286 with N-phenyl of pyrazole and pyrazole bioactive core. At the same time, compound 5c showed binding energy S = −25.61 kcal mol−1 with the residues Trp286, Tyr337, and Tyr341 with NH of isatin, N-phenyl of pyrazole and pyrazole bioactive core, respectively. Additionally, the hydrophobic interaction was observed inside the pocket with phenyl groups substituted on pyrazole, and phenyl of isatin moiety (Fig. 10). Finally, the salicylamide derivative 3b revealed the lowest binding energy S = −24.66 kcal mol−1 through one hydrogen bond backbone acceptor between the Phe295 with the hydroxyl group with a distance of 1.99 and strength of 27%, as well as one arene–arene “π–π” interaction between the phenyl group of azo (p-fluorophenyl) moiety with the amino acid Tyr337.
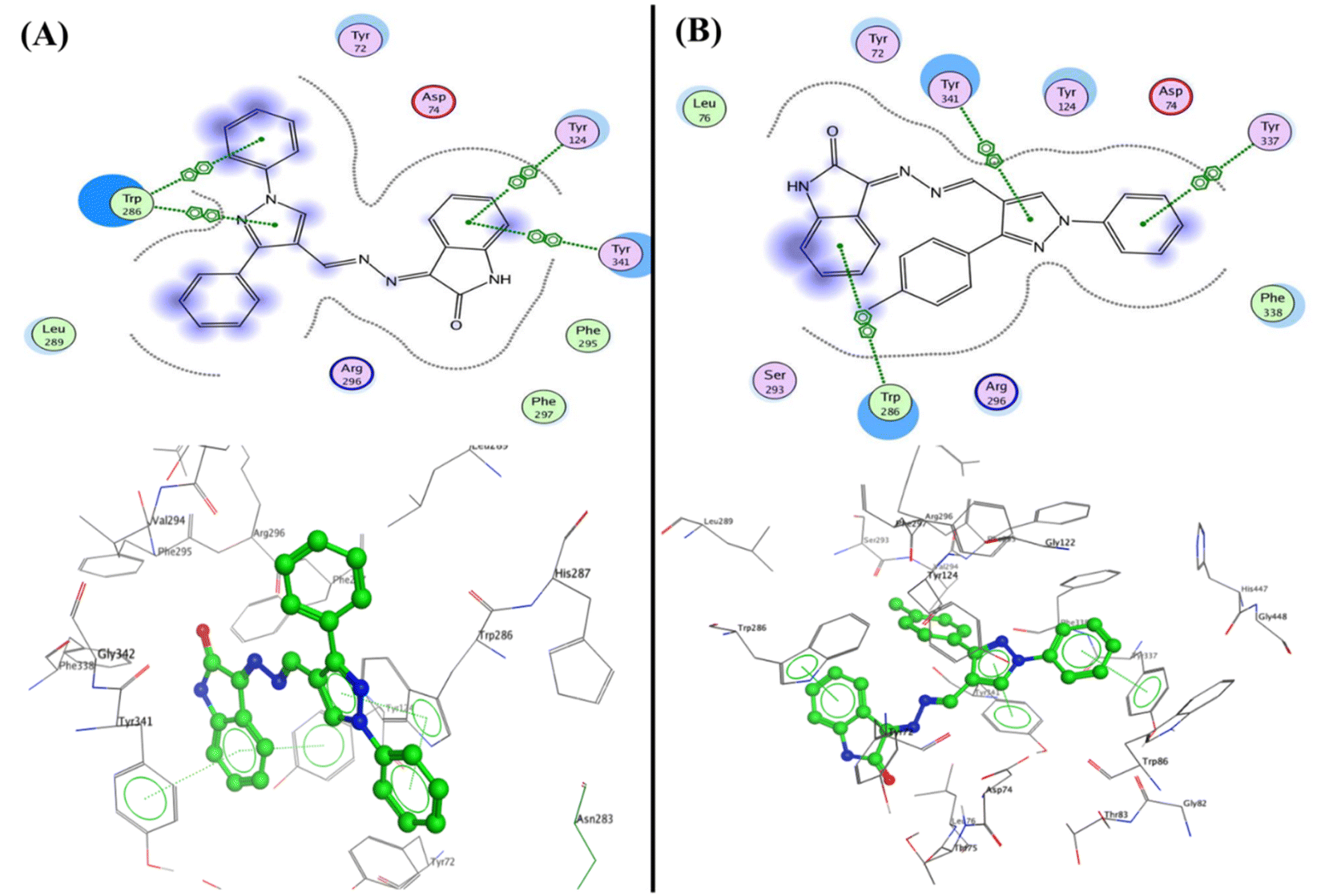 | ||
| Fig. 10 2D and 3D structure of compounds (A) 5b and (B) 5c inside the active site of acetylcholinesterase (AChE) (PDB: 4EY7). | ||
3.3. In silico drug-likeness and ADMET prediction
| Properties | 3b | 5a | 5b | 5c |
|---|---|---|---|---|
| Formula | C21H14FN5O2 | C19H14ClN5O | C24H17N5O | C25H19N5O |
| Molecular weight (MW) | 387.37 | 363.80 | 391.42 | 405.45 |
| Num. Heavy atoms (atoms) | 29 | 26 | 30 | 31 |
| Num. aromatic heavy atoms (arom-atoms) | 18 | 17 | 23 | 23 |
| Fraction Csp3 | 0.00 | 0.05 | 0.00 | 0.04 |
| Num. Rotatable bonds (R-bonds) | 4 | 3 | 4 | 4 |
| Num. H-bond acceptors (H-acc) | 7 | 4 | 4 | 4 |
| Num. H-bond donors (H-don) | 2 | 1 | 1 | 1 |
| Molar refractivity (MR) | 111 | 105.5 | 120.96 | 125.92 |
| Topological polar surface area (TPSA) | 98.77 | 71.64 | 71.64 | 71.64 |
M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP (solubility) logP (solubility) |
2.48 | 2.48 | 2.90 | 3.11 |
| WlogP (solubility) |
4.57 | 3.04 | 3.74 | 4.05 |
| XlogP (solubility) |
4.15 | 4.15 | 4.26 | 4.62 |
| Lipinski filter violations | 0 | 0 | 0 | 0 |
| Ghose filter violations | 0 | 0 | 0 | 0 |
| Veber (GSK) filter violations | 0 | 0 | 0 | 0 |
| Egan (pharmacia) filter violations | 0 | 0 | 0 | 0 |
| Drug-likeness model score | −0.14 | 0.05 | −0.05 | 0.31 |
The selection of new drugs and formulations are based on the physicochemical properties, which include the molecular weight (MW), solubility, molar refractivity (MR), topological polar surface area (TPSA), and hydrogen-bonding capacities; therefore, the physicochemical properties of isatin-based Schiff bases 3b and 5a–c (Table 3) are the essential factors for finding new drug candidates and should be an agreement with the different rule-based filters (drug-likeness rules).76,77
There are some drug-likeness rules78 which can describe as (i) Lipinski's rule (MW ≤ 500; MlogP ≤ 5; H-acc ≤ 10; H-don ≤ 5), (ii) Ghose's filter (160 ≤ MW ≤ 480, −0.4 ≤ WlogP ≤ 5.6, 40 ≤ MR ≤ 130, and 20 ≤ atoms ≤ 70), (iii) Veber's filter (R-bonds ≤ 10, and TPSA ≤ 140), and (iv) Egan's filter (WlogP ≤ 5.88, and TPSA ≤ 131.6). Based on these rules, we found that the four isatin-based Schiff bases (3b and 5a–c) agree with the four drug-likeness rules (Lipinski, Ghose, Veber, and Egan rules) and these derivatives are accepted as new drug candidates.
The drug-likeness model score analysis is based on the positive and negative values of the target compounds, where the compound is like a drug if the drug-likeness score is valued positively. In contrast, the compound is non-drug if the drug-likeness score is valued negatively.79 Based on this rule, we found that (i) the two Schiff bases, isatin-azomethine-pyrazole derivatives 5a and 5c, showed drug-likeness scores equal to 0.05 and 0.31, respectively, and therefore considered drug-like. (ii) On the other hand, the other two compounds, isatin-azomethine-aryl azo derivative 3b, and isatin-pyrazole based Schiff base 5b, revealed negative drug-likeness scores equal to −0.14 and −0.05, respectively, and consequently, these compounds were non-drug like (Fig. 11).
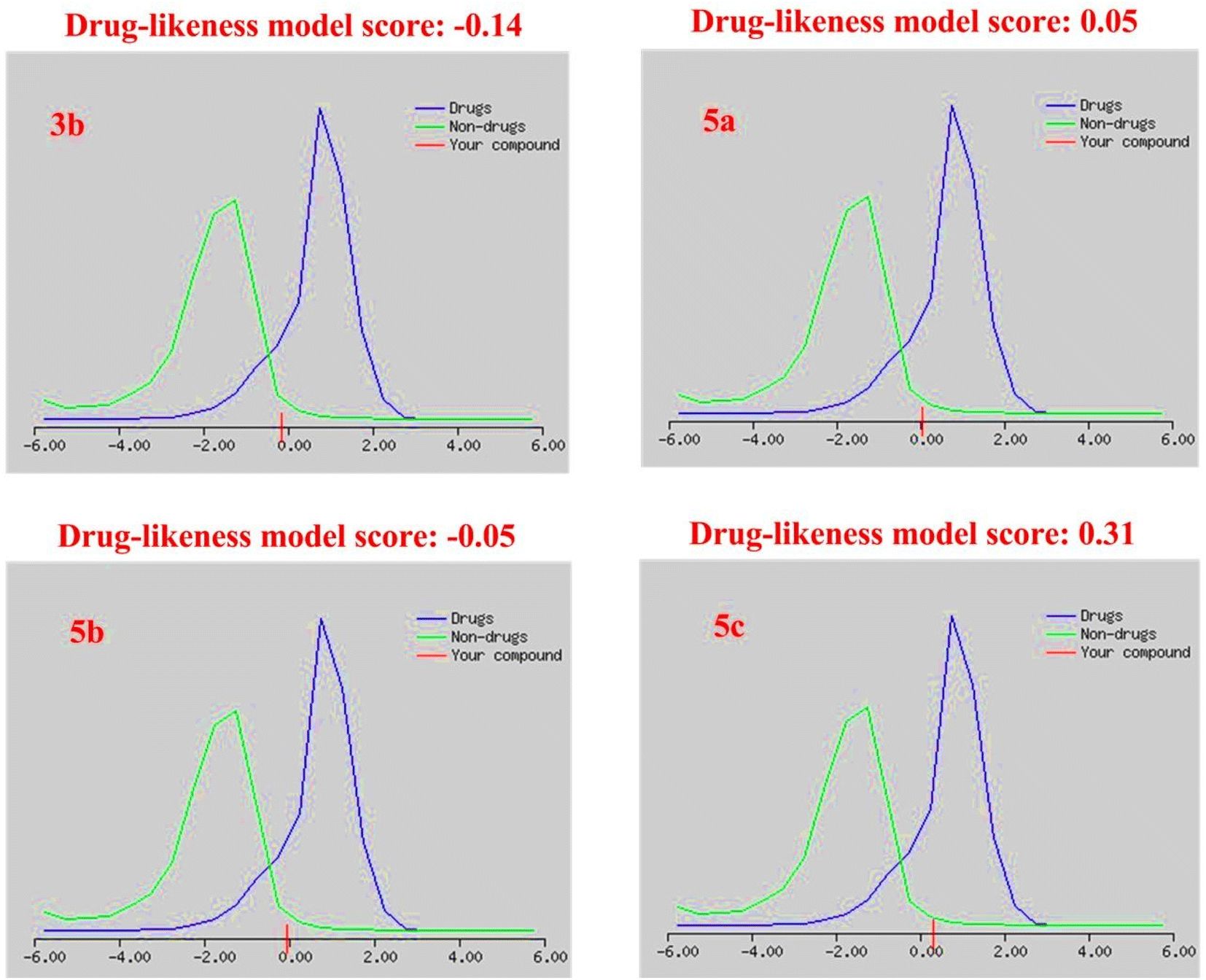 | ||
| Fig. 11 The drug-likeness model scores of the isatin-based Schiff bases 3b and 5a–c. | ||
| Properties | 3b | 5a | 5b | 5c |
|---|---|---|---|---|
| Toxicity | ||||
| Human hepatotoxicity (H-HT) | 0.974/ |
0.975/ |
0.951/ |
0.958/ |
| Carcinogens | 0.910/ |
0.597/ |
0.576/ |
0.616/ |
| AMES test | 0.969/ |
0.754/ |
0.447/ |
0.558/ |
| Skin sensitization (SS) | 0.600/ |
0.650/ |
0.173/ |
0.102/ |
| Eye irritation (EI) | 0.630/ |
0.479/ |
0.500/ |
0.408/ |
| Eye corrosion (EC) | 0.003/ |
0.008/ |
0.003/ |
0.003/ |
|
||||
| Metabolism | ||||
| CYP1A2 inhibitior | Yes/0.730 | Yes/0.892 | Yes/0.895 | Yes/0.702 |
| CYP2C19 inhibitior | Yes/0.578 | Yes/0.521 | Yes/0.840 | Yes/0.777 |
| CYP2C9 inhibitior | Yes/0.542 | Yes/0.583 | Yes/0.885 | Yes/0.920 |
| CYP2D6 inhibitior | No/0.022 | No/0.011 | No/0.020 | No/0.022 |
| CYP3A4 inhibitior | Yes/0.277 | Yes/0.202 | Yes/0.272 | Yes/0.450 |
Human hepatotoxicity (H-HT) is essential to risk inspection for new drugs. The destruction and injury of the liver because of the drug's effects is a major patient safety concern, and the drug may be withdrawn from the market. The new target compounds are either non-toxic for the liver, H-HT negative, with a value of 0 to 0.3 (green), or toxic for the liver, H-HT positive, with two categories; the first is moderate toxicity with a predicted value of 0.3 to 0.7 (yellow) and the second is strong toxicity with a value from 0.7 to 1.0 (red). All isatin-based Schiff bases 3b and 5a–c were predicted as liver toxic (H-HT positive) from the strong toxicity with high hazard (red color, the degree from 0.951 to 0.975).
Carcinogenic substances have the ability to damage the genome; as a result, they have serious effects on human health. Some drugs are withdrawn from the market when they are proven to be carcinogenic. The target compounds are marked as non-carcinogens with a predicted value of zero to 0.3 (green), moderately carcinogens with a predicted value of 0.3 to 0.7 (yellow) or strong carcinogens with a predicted value from 0.7 to 1.0 (red). Hence, compound 3b is labeled a strong carcinogen and exhibited a high predicted value of 0.910. At the same time, the three derivatives 5a, 5b, and 5c are characterized as moderate carcinogens with a predicted value of 0.597, 0.576, and 0.616, respectively.
Furthermore, the Ames test for mutagenicity is used for testing the compound's mutagenicity, where the mutagenic has a close relationship with carcinogenicity. A target compound is considered strongly mutagenic when it has a predicted value of 0.7 to 1.0 (red) but considered a medium mutagenic when it has a predicted value of 0.3 to 0.7 (yellow) and considered a non-mutagenic when it has a predicted value of 0 to 0.3 (green). The compounds 3b and 5a are considered strongly mutagenic, but the compounds 5b and 5c are mutagenic from the medium degree. Additionally, evaluating the skin sensitization (SS), eye irritation (EI), and eye corrosion (EC) of substances is a necessary part of risk assessment. The target substances were divided into non-sensitizer/non-irritants/non-corrosive substances (value 0–0.3, green) or sensitizer/irritants/corrosive substances (high-risk substances with value 0.7–1.0 shown red color or medium substances risk with value 0.3–0.7 shown yellow color). The two Schiff bases 3b and 5a are skin sensitizers, but the other compounds 5b and 5c are non-sensitizers with values of 0.173 and 0.102, respectively. All isatin-based Schiff bases 3b and 5a–c were predicted as eye irritants (medium risk) and are predicted as eye non-corrosive substances.
Nowadays, the metabolism prediction model had concentrated on the interaction between the target compounds with cytochromes P450 monooxygenase enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4) which catalyze the phase 1 metabolism of pharmaceuticals and concentrated in the liver. The inhibition of cytochromes enzymes is one of the principal mechanisms which cause pharmacokinetic drug–drug interactions.82,83 Table 4 demonstrates that all isatin-based Schiff bases 3b and 5a–c were predicted as inhibitors of the four enzymes (namely: CYP1A2, CYP2C19, CYP2C9, and CYP3A4) with the probability value range from 0.202 to 0.920. Also, predicted non-inhibition of the CYP2D6 enzyme of isatin-based Schiff bases 3b and 5a–c with a probability value ranging from 0.011 to 0.020.
| Properties | 3b | 5a | 5b | 5c |
|---|---|---|---|---|
| Absorption | ||||
| Caco-2 permeability (log cm s−1) | −4.781/ |
−4.699/ |
−4.842/ |
−4.827/ |
| MDCK permeability (cm s−1) | 1.7 × 10−5/ |
2.1 × 10−5/ |
1.6 × 10−5/ |
1.4 × 10−5/ |
| P-glycoprotein-inhibitor | 0.013/ |
0.001/ |
0.304/ |
0.799/ |
| P-glycoprotein-substrate | 0.017/ |
0.003/ |
0.001/ |
0.001/ |
|
||||
| Distribution | ||||
| Plasma protein binding (PPB, %) | 100.3/ |
98.68/ |
100.6/ |
100.8/ |
| Fraction unbound in plasma (FU, %) | 1.268/ |
2.537/ |
1.423/ |
1.586/ |
| Volume distribution (VD, L kg−1) | 0.987/ |
1.964/ |
0.476/ |
0.39/ |
The new drug must pass via intestinal cell membranes before it reaches systemic circulation. To evaluate drug permeability, we utilized the human colon adenocarcinoma cell lines (Caco-2). Accordingly, Caco-2 cell permeability has been an important index for the new drug. A target compound is considered to fit proper Caco-2 permeability when it has a predicted value > −5.15 log cm s−1 (green). Based on the rule of Caco-2 permeability, we found that the four isatin-based Schiff bases (3b and 5a–c) have a fit proper Caco-2 permeability with a range of −4.699 to −4.827 log cm s−1. The apparent permeability coefficient (Papp) is used for estimating the uptake efficiency of target compounds in the body. Hence, the development of the Madin–Darby Canine Kidney cells (MDCK) for employing it as a model for permeability screening. Also, for evaluating the effect of the blood–brain barrier (BBB), we used Papp values of MDCK cell lines. A target compound is considered to have a high passive MDCK permeability when a Papp is more than 20 × 10−6 cm s−1 (green). Table 5 illustrates that the target compounds 3b and 5a–c have a high passive MDCK permeability of more than 20 × 10−6 cm s−1 in a range from 1.4 × 10−5 to 2.1 × 10−5 cm s−1.
P-glycoprotein (P-gp), known as MDR1 or 2 ABCB1, is an efflux membrane transporter and is responsible for limiting cellular uptake. Furthermore, P-gp is one of the main barriers to delivering drugs properly. Thus, a suitable inhibitor or substrate will allow an increase in cellular uptake and predict the pharmacokinetics of drugs.85 A target compound is considered a fit proper P-glycoprotein inhibitor or substrate when it has a predicted value of zero to 0.3: excellent (green); 0.3–0.7: medium (yellow); 0.7–1.0: poor (red). Accordingly, the two Schiff bases 3b and 5a are inhibitors for P-glycoprotein with values of 0.013 and 0.001, respectively. Schiff base 5b is a moderate P-glycoprotein inhibitor, but 5c is a non-inhibitor of P-glycoprotein. On the other hand, the four isatin-based Schiff bases (3b and 5a–c) are P-glycoprotein substrates with predicted values a range from 0.001 to 0.017.
The pharmacodynamic behavior of the drugs is affected by their plasma protein binding (PPB) and the fraction unbound in plasma (FU). In general, drugs in plasma exist in an equilibrium state, unbound, or bound to serum proteins. Thus, the distribution of the drugs through the binding with plasma protein is one of the most important evaluations of drugs. A target compound is considered to have proper plasma protein binding if it has a predicted value of PPB of less than 90% (excellent, green) and a predicted value of FU of more than or equal to 5% (excellent, green). On the other hand, the drugs with a high binding with plasma protein of more than 90% (poor, red), and a predicted value for FU less than 5%, may have a low therapeutic index and decreased efficiency of traverse through cellular membranes. Based on the above, the four isatin-based Schiff bases (3b and 5a–c) have a high binding with plasma protein with predicted values of more than 90% (the range from 98.68% to 100.8%), also the predicted values of FU less than 5% (the range from 1.268% to 2.537%), and these compounds maybe have a low therapeutic index with slight efficiency in crossing the membranes.
The volume distribution (VD) is an important parameter to describe the in vivo distribution of the drug. Volume distribution (VD) is a theoretical concept that links the administered dose of the drug with the actual initial concentration presented in the circulation. If the target compound has a predicted value of volume distribution in the range of 0.04 to 20 L kg−1 (green), thus this compound has a good volume distribution. Table 5 depicts that all the target isatin-based Schiff bases 3b and 5a–c have a suitable volume distribution ranging from 0.39 to 1.964 L kg−1.
4. Conclusion
In summary, based on a new technique “one drug-multiple targets” for discovering novel therapeutics, novel derivatives of isatin-based Schiff bases (2a, 2b, 3a–d, 4, 5a–c, 6, 7a, and 7b) have been synthesized and were proved by physical and spectral analysis. The work was extended to evaluate multiple biological activities, such as antioxidant, anti-diabetic, anti-Alzheimer, and anti-arthritic activities in vitro of all isatin-based Schiff bases. The in vitro biological studies revealed that compound 5a possessed multiple–active and exhibited potency as an antioxidant, scavenging, anti-diabetic, and anti-Alzheimer among the four potent derivatives (3a and 5a–c). Also, the order of the potency is 5a > 5c > 5b > 3b. Furthermore, the computational studies of the most active Schiff bases 3b, 5a, 5b, and 5c were performed, and the quantum chemical calculation revealed that the most active compound 5a is the most stable and least reactive with the lowest total energy and a value of ETotal = −1541.58 Hartree. The docking simulation for 5a revealed binding energy S = −22.09 and −27.49 kcal mol−1 compared with S = −32.53 kcal mol−1 for acarbose and S = −32.42 kcal mol−1 for donepezil inside the active sites of the two enzymes α-amylase and acetylcholinesterase, respectively. Also, the drug-likeness prediction showed that the four active isatin-based Schiff bases (3b and 5a–c) agree with the four drug-likeness rules (Lipinski, Ghose, Veber, and Egan rules) and these derivatives are accepted as new drug candidates, especially the two derivatives 5a and 5c that possess drug-likeness scores equal to 0.05 and 0.31, respectively, and therefore considered drug-like molecules. Finally, the in silico ADMET properties results of the isatin-based Schiff bases (3b and 5a–c) indicate that (i) the Schiff bases (3b and 5a–c) are inhibitors of CYP1A2, CYP2C19, CYP2C9, and CYP3A4 enzymes with the high probability value from 0.202 to 0.920, (ii) are non-inhibitors of the CYP2D6 enzyme with a probability value from 0.011 to 0.020, (iii) some Schiff bases possess low toxicity and other with moderate or high toxicity, (iv) the two Schiff bases 3b and 5a have high absorption properties, but almost all the Schiff bases (3b and 5a–c) possess low distribution properties. From this contemporary manuscript, in the future, the four isatin-based Schiff bases 3b, 5a, 5b, and 5c, especially Schiff base 5a will be utilized for the design of further structurally optimized derivatives based on the structure–activity relationship study with further biological evaluation for producing a novel drug with multiple targets “one drug-multiple targets”.5. Materials and methods
5.1. Chemistry
All melting points were measured on a Gallenkamp melting point apparatus and are uncorrected. The IR spectra were recorded (KBr disk) on a PerkinElmer 1650 FTIR instrument. 1H-NMR (500 MHz) and 13C-NMR (126 MHz) spectra were recorded on a JEOL spectrometer using DMSO-d6 as solvent and tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ-values) are reported in ppm. Elemental analyses were performed at the Micro Analytical Center, Cairo University, Egypt. Additionally, the progress of the reactions was monitored by thin-layer chromatography (TLC) using aluminum sheets coated with silica gel F254 (Merck), viewing under a short-wavelength UV lamp effected detection. All evaporations were performed under reduced pressure at 40 °C.Glacial acetic acid, p-fluoroaniline, p-nitroaniline, m-methylaniline, p-bromoaniline, ethanol, 5-methyl furan-2-carbaldehyde, p-fluorobenzaldehyde, p-hydroxybenzaldehyde, o-hydroxybenzaldehyde, and hydrochloric acid were of Fluka. Isatin and hydrazine hydrate were supplied by Aldrich.
5.2. General producer for the synthesis of isatin-based Schiff bases 2a, 2b, 3a–d, 4, 5a–c, 6, 7a, and 7b
A mixture of 3-hydrazino-isatin (1) (0.01 mol, 1.66 g) and the appropriate aldehydes (0.01 mol) {namely; 4-(piperidin-1-yl) benzaldehyde, 4-morpholinobenzaldehyde, 2-hydroxy-4-((3-methylphenyl)azo)benzaldehyde, 4-((4-fluorophenyl)azo)-2-hydroxybenzaldehyde, 4-((4-bromophenyl)azo)-2-hydroxybenzaldehyde, 2-hydroxy-4-((4-nitrophenyl)azo)benzaldehyde, 5-methylfuran-2-carbaldehyde, 5-chloro-3-methyl-1-phenyl-1H-pyrazole-4-carbaldehyde, 1, 3-diphenyl-1H-pyrazole-4-carbaldehyde, 1-phenyl-3-(4-methylphenyl)-1H-pyrazole-4-carbaldehyde, 3-(benzofuran-2-yl)-1-phenyl-1H-pyrazole-4-carbaldehyde, 4,4′-methylenebis(oxy)dibenzaldehyde, and 4,4'-(ethane-1, 2-diylbis(oxy))dibenzaldehyde} with a catalytic amount of glacial acetic acid (0.0087 mol, 0.5 mL) in absolute ethanol (25 mL) was refluxed for four hours and then left to cool. The solid product was filtered of, dried, and finally recrystallized from ethanol to afford Schiff bases 2a, 2b, 3a–d, 4, 5a–c, 6, 7a, and 7b.O), 1605 (CN), 1511 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm): 1.55 (s, 6H, 3CH2, piperidine moiety), 3.35 (s, 4H, 2CH2, piperidine moiety), 6.86 (d, 1H, J = 8.0 Hz, aromatic-H), 6.99 (d, 2H, J = 8.5 Hz, aromatic-H), 7.03 (t, 1H, J = 7.5 Hz, aromatic-H), 7.34 (t, 1H, J = 7.5 Hz, aromatic-H), 7.78 (d, 2H, J = 9.0 Hz, aromatic-H), 8.16 (d, 1H, J = 7.5 Hz, aromatic-H), 8.57 (s, 1H, –CHN–, azomethine), 10.74 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm): 24.49, 25.45, 48.37 (5C, piperidine moiety), 111.12, 114.47, 114.85, 117.59, 122.26, 122.68, 129.29, 131.75, 133.46, 145.06, 150.77, 154.14, 164.75 (14C, 13C of aromatic + 1C of azomethine –NCH–), 165.66 (1C, CO); anal. calcd. (%) for C20H20N4O (332.40): C, 72.27; H, 6.06; N, 16.86. Found: C, 72.19; H, 6.10; N, 16.79%.O), 1611 (CN), 1521 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 3.27–3.29 (m, 4H, 2CH2, morpholine moiety), 3.71 (s, 4H, 2CH2, morpholine moiety), 6.86 (d, 1H, J = 6.5 Hz, aromatic-H), 7.00 (t, 2H, J = 7.75 Hz, aromatic-H), 7.05 (d, 2H, J = 9.0 Hz, aromatic-H), 7.34 (t, 1H, J = 7.75 Hz, aromatic-H), 7.82 (d, 2H, J = 9.0 Hz, aromatic-H), 8.11 (d, 1H, J = 7.5 Hz, aromatic-H), 8.58 (s, 1H, –CHN–, azomethine), 10.75 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 47.42, 66.40 (4C, morpholine moiety), 111.18, 114.54, 117.47, 122.75, 123.64, 129.34, 131.46, 133.65, 145.17, 150.98, 154.26, 164.17 (14C, 13C of aromatic + 1C of azomethine –NCH–), 165.54 (1C, CO); anal. calcd. (%) for C19H18N4O2 (334.37): C, 68.25; H, 5.43; N, 16.76. Found: C, 68.19; H, 5.50; N, 16.82%.O), 1621 (CN), 1536 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 2.42 (s, 3H, CH3), 6.92 (t, 1H, J = 5.8 Hz, aromatic-H), 7.08 (dd, 1H, J = 15.5, 7.8 Hz, aromatic-H), 7.20 (d, 1H, J = 10.0 Hz, aromatic-H), 7.37 (d, 1H, J = 6.5 Hz, aromatic-H), 7.44 (t, 1H, J = 8.0 Hz, aromatic-H), 7.49 (t, 1H, J = 6.5 Hz, aromatic-H), 7.70 (d, 2H, J = 8.0 Hz, aromatic-H), 7.95 (d, 1H, J = 7.0 Hz, aromatic-H), 8.01 (d, 1H, J = 8.5 Hz, aromatic-H), 8.50 (s, 1H, aromatic-H), 8.87 (s, 1H, –NCH–), 10.91 (s, 1H, OH exchangeable with D2O), 11.47 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 21.41 (1C, CH3), 118.77, 120.40, 120.56, 122.39, 122.90, 123.47, 129.54, 130.02, 134.17, 139.46, 145.75, 151.06, 152.56, 157.93, 161.78 (20C, 19C of aromatic + 1C of azomethine –NCH–), 165.07 (1C, CO); anal. calcd. (%) for C22H17N5O2 (383.40): C, 68.92; H, 4.47; N, 18.27. Found: C, 68.85; H, 4.51; N, 18.20%.O), 1620 (CN), 1535 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 6.91 (t, 1H, J = 8.0 Hz, aromatic-H), 7.05–7.10 (m, 1H, aromatic-H), 7.19 (dd, 1H, J = 9.5, 2.5 Hz, aromatic-H), 7.42 (d, 2H, J = 8.0 Hz, aromatic-H), 7.45 (d, 1H, J = 10.0 Hz, aromatic-H), 7.91–8.01 (m, 4H, aromatic-H), 8.50 (s, 1H, aromatic-H), 8.87 (s, 1H, –NCH–, azomethine), 10.90 (s, 1H, OH exchangeable with D2O), 11.03 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 111.51, 116.75, 116.93, 118.26, 120.40, 122.84, 123.21, 124.50, 125.01, 125.09, 125.17, 127.67, 128.82, 129.74, 145.72, 149.23, 149.25, 151.14, 158.17, 161.91 (20C, 19C of aromatic + 1C of azomethine –NCH–), 165.07 (1C, CO); anal. calcd. (%) for C21H14FN5O2 (387.37): C, 65.11; H, 3.64; N, 18.08. Found: C, 65.20; H, 3.58; N, 18.01%.O), 1618 (CN), 1574 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 6.93 (d, 1H, J = 7.5 Hz, aromatic-H), 7.07 (t, 1H, J = 7.5 Hz, aromatic-H), 7.19 (d, 1H, J = 8.5 Hz, aromatic-H), 7.43 (t, 1H, J = 8.3 Hz, aromatic-H), 7.79 (d, 2H, J = 9.0 Hz, aromatic-H), 7.83 (d, 2H, J = 8.5 Hz, aromatic-H), 7.93 (d, 1H, J = 7.0 Hz, aromatic-H), 7.99–8.02 (m, 1H, aromatic-H), 8.33–8.51 (m, 1H, aromatic-H), 8.86 (s, 1H, –NCH–, azomethine), 10.90 (s, 1H, OH exchangeable with D2O), 11.56 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 111.54, 116.93, 118.35, 118.89, 120.51, 122.89, 124.64, 124.81, 127.84, 128.86, 133.02, 134.43, 145.63, 145.74, 151.11, 151.41, 157.84, 162.15 (20C, 19C of aromatic + 1C of azomethine –NCH–), 165.06 (1C, CO); MS (m/z, %): 450 (M + 2, 10.81), 448 (M+, 13.25), 219 (100); anal. calcd. (%) for C21H14BrN5O2 (448.27): C, 56.27; H, 3.15; N, 15.62. Found: C, 56.20; H, 3.21; N, 15.56%.O), 1618 (CN), 1516 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 6.85 (t, 1H, J = 7.5 Hz, aromatic-H), 7.04 (t, 1H, J = 7.5 Hz, aromatic-H), 7.15 (d, 1H, J = 9.0 Hz, aromatic-H), 7.40 (d, 1H, J = 8.5 Hz, aromatic-H), 7.59 (d, 1H, J = 7.5 Hz, aromatic-H), 7.89 (d, 1H, J = 8.0 Hz, aromatic-H), 8.01 (d, 2H, J = 9.0 Hz, aromatic-H), 8.34 (d, 2H, J = 7.5 Hz, aromatic-H), 8.50 (s, 1H, aromatic-H), 8.81 (s, 1H, –NCH–, azomethine), 10.97 (s, 1H, OH exchangeable with D2O), 12.56 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 111.52, 116.88, 118.45, 120.66, 122.91, 123.74, 123.83, 125.30, 125.59, 128.20, 128.93, 134.46, 145.70, 148.57, 151.15, 155.75, 157.52, 162.96 (20C, 19C of aromatic + 1C of azomethine –NCH–), 165.04 (1C, CO); anal. calcd. (%) for C21H14N6O4 (414.37): C, 60.87; H, 3.41; N, 20.28. Found: C, 60.80; H, 3.48; N, 20.34%.O), 1617 (CN), 1536 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 2.38 (s, 3H, CH3), 6.50 (d, 1H, J = 3.7 Hz, furan H-4), 6.93 (d, 1H, J = 8.1 Hz, isatin), 7.08 (t, 1H, isatin), 7.30 (d, 1H, J = 3.7 Hz, furan H-3), 7.43 (t, 1H, isatin), 8.15 (d, 1H, J = 8.3 Hz, isatin), 8.53 (s, 1H, –CHN–, azomethine), 10.87 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 13.74 (1C, CH3), 109.98, 110.61, 116.76, 121.63, 122.16, 128.93, 133.38, 144.71, 147.74, 151.09, 158.13, 163.36 (12C, 11C of aromatic + 1C of azomethine –NCH–), 164.72 (1C, CO); anal. calcd. (%) for C14H11N3O2 (253.26): C, 66.40; H, 4.38; N, 16.59. Found: C, 66.45; H, 4.32; N, 16.64%.O), 1622 (CN), 1550 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 2.58 (s, 3H, CH3), 6.89 (d, 1H, J = 8.3 Hz, isatin), 7.03 (t, 1H, isatin), 7.39 (t, 1H, isatin), 7.52–7.65 (m, 5H, aromatic-H), 8.15 (d, 1H, J = 8.3 Hz, isatin), 8.65 (s, 1H, –CHN–, azomethine), 10.85 (s, 1H, NH-isatin exchangeable with D2O). 13C NMR (DMSO-d6, 126 MHz, δ ppm) 14.76 (1C, CH3), 111.34, 113.27, 117.11, 122.77, 125.76, 128.86, 129.68, 129.91, 130.44, 134.18, 137.56, 145.50, 150.70, 151.45, 156.06 (17C, 16C of aromatic + 1C of azomethine –NCH–), 165.34 (1C, CO); MS (m/z, %): 365 (M + 2, 2.75), 363 (M+, 6.97), 77 (100); anal. calcd. (%) for C19H14ClN5O (363.80): C, 62.73; H, 3.88; N, 19.25. Found: C, 62.67; H, 3.94; N, 19.19%.O), 1613 (CN), 1536 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 6.83 (d, 1H, J = 8.0 Hz, isatin), 6.90 (t, 1H, isatin), 7.34 (t, 1H, aromatic-H), 7.41 (t, 1H, isatin), 7.49–7.58 (m, 5H, aromatic-H), 7.74 (d, 2H, J = 8.1 Hz, aromatic-H), 7.90 (d, 1H, J = 8.0 Hz, isatin), 8.03 (d, 2H, J = 8.8 Hz, aromatic-H), 8.70 (s, 1H, –CHN–, azomethine), 9.32 (s, 1H, pyrazole H-5), 10.79 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 111.02, 117.02, 117.15, 119.87, 122.75, 128.03, 129.25, 129.32, 129.45, 129.98, 130.17, 131.36, 132.37, 133.94, 139.38, 145.32, 151.50, 153.78, 157.24 (23C, 22C of aromatic + 1C of azomethine –NCH–), 165.40 (1C, CO); anal. calcd. (%) for C24H17N5O (391.42): C, 73.64; H, 4.38; N, 17.89. Found: C, 73.70; H, 4.33; N, 17.95%.O), 1615 (CN), 1532 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 2.36 (s, 3H, CH3), 6.83 (d, 1H, J = 7.8 Hz, isatin), 6.88 (t, 1H, isatin), 7.34–7.30 (m, 3H, aromatic-H), 7.40 (t, 1H, isatin), 7.56 (t, 2H, aromatic-H), 7.62 (d, 2H, J = 8.0 Hz, aromatic-H), 7.90 (d, 1H, J = 7.4 Hz, isatin), 8.02 (d, 2H, J = 8.1 Hz, aromatic-H), 8.69 (s, 1H, –CHN–, azomethine), 9.30 (s, 1H, pyrazole H-5), 10.78 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 21.00 (1C, CH3), 110.53, 116.41, 116.65, 119.33, 122.23, 127.52, 128.48, 128.72, 128.96, 129.36, 129.70, 130.79, 133.50, 138.50, 138.86, 144.80, 151.06, 153.38, 157.25 (23C, 22C of aromatic + 1C of azomethine –NCH–), 164.94 (1C, CO); anal. calcd. (%) for C25H19N5O (405.45): C, 74.06; H, 4.72; N, 17.27. Found: C, 74.00; H, 4.80; N, 17.35%.O), 1610 (CN), 1566 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 6.88 (d, 1H, J = 8.2 Hz, isatin), 6.95 (t, 1H, isatin), 7.31 (t, 1H, aromatic-H), 7.38 (t, 2H, 1H of isatin and 1H of aromatic-H), 7.46 (t, 1H, aromatic-H), 7.59–7.63 (m, 3H, aromatic-H), 7.73 (d, 1H, J = 8.1 Hz, aromatic-H), 7.75 (d, 1H, J = 8.6 Hz, aromatic-H), 8.09 (d, 2H, J = 8.8 Hz, aromatic-H), 8.14 (d, 1H, J = 7.9 Hz, isatin), 9.06 (s, 1H, –CHN–, azomethine), 9.40 (s, 1H, pyrazole H-5), 10.86 (s, 1H, NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm) 106.75, 111.17, 112.02, 117.09, 117.60, 120.00, 120.19, 122.23, 122.92, 124.09, 125.92, 128.45, 130.03, 130.23, 130.32, 130.71, 134.15, 139.21, 144.06, 145.48, 149.18, 151.32, 155.04 (25C, 24C of aromatic + 1C of azomethine –NCH–), 164.94 (1C, CO). Anal. calcd. (%) for C26H17N5O2 (431.45): C, 72.38; H, 3.97; N, 16.23. Found: C, 72.45; H, 3.90; N, 16.30%.O), 1605 (CN), 1544 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 6.03 (d, 2H, –OCH2O–), 6.85 (d, 1H, J = 8.0 Hz, aromatic-H), 6.98–7.01 (t, 1H, J = 7.5 Hz, aromatic-H), 7.21 (t, 2H, J = 8.0 Hz, aromatic-H), 7.26–7.29 (m, 4H, aromatic-H), 7.37 (t, 2H, J = 7.5 Hz, aromatic-H), 7.78–7.83 (m, 2H, aromatic-H), 7.86–7.89 (m, 1H, aromatic-H), 7.94–7.96 (m, 3H, aromatic-H), 8.58, 8.61 (2s, 2H, 2-NCH– azomethine), 10.81 (s, 2H, 2NH-isatin exchangeable with D2O); 13C NMR (DMSO-d6, 126 MHz, δ ppm): 89.38 (1C, CH2), 110.79, 116.68, 122.38, 127.75, 128.92, 130.09, 130.97, 131.82, 133.62, 144.94, 150.68, 159.19, 160.94 (28C, 26C of aromatic + 2C of azomethine –NCH–), 164.67 (2C, 2CO); anal. calcd. (%) for C31H22N6O4 (542.54): C, 68.63; H, 4.09; N, 15.49. Found: C, 68.70; H, 4.03; N, 15.41%.O), 1607 (CN), 1553 (CC); 1H NMR (DMSO-d6, 500 MHz, δ ppm) 4.49 (s, 4H, –OCH2CH2O–), 6.90 (d, 2H, J = 8.3 Hz, aromatic-H), 7.04 (t, 2H, aromatic-H), 7.11 (t, 2H, aromatic-H), 7.21 (d, 2H, J = 8.5 Hz, aromatic-H), 7.39 (t, 2H, aromatic-H), 7.83 (d, 2H, J = 8.1 Hz, aromatic-H), 7.97 (d, 2H, J = 8.0 Hz, aromatic-H), 8.05 (d, 2H, J = 7.4 Hz, aromatic-H), 8.56 (s, 2H, –NCH–, azomethine), 10.85 (s, 2H, NH-isatin exchangeable with D2O); anal. calcd. (%) for C32H24N6O4 (556.57): C, 69.06; H, 4.35; N, 15.10. Found: C, 69.00; H, 4.40; N, 15.00%.5.3. In vitro biological activities
5.3.1.1. Total antioxidant capacity. Total antioxidant capacity (TAC) was determined by evaluating the green phosphate/Mo5+ complex at wavelength 695 nm using the method described by Prieto et al.86 It was expressed as mg gallic acid equivalent per gram dry weight (see ESI file†).
5.3.1.2. Iron-reducing power. Total iron-reducing power was assessed using the method suggested by Oyaizu87 as μg mL−1. A high absorbance of the reaction mixture at 700 nm indicates a higher reducing power (see ESI file†).
5.3.1.3. DPPH radical-scavenging activity. The scavenging activity against 1,1-diphenyl-2-picryl-hydrazyl (DPPH) radical was assayed by calculating the median inhibitory concentration (IC50) according to the method suggested88 (see ESI file†).
5.3.1.4. ABTS radical scavenging assay. The 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) assay was carried out to determine the percent of the ABTS radical inhibition (%) according to the method modified by Arnao et al.89 using ascorbic acid as standard (see ESI file†).
5.3.4.1. Protein denaturation. The percentage of protein denaturation inhibition was calculated according to the reported method.90 and modified by Soni and Sureshkumar.91 All results were compared with the standard (diclofenac sodium) that was prepared according to the method described.92 The control represents 100% protein denaturation (see ESI file†).
5.3.4.2. Proteinase inhibitory activity. This assay was carried out by calculating the percentage of the proteinase inhibitory activity was according to the method suggested93 (see ESI file†).
5.4. Statistical analysis
The statistical positive and negative correlations were evaluated among measurements of the biological activities using one-way analysis of variance (oneway ANOVA) by Statistical Package for Social Sciences (SPSS for windows, version 11.0). Values of P < 0.05 were considered significant correlation and those of P < 0.01 were considered highly significant one.5.5. Density function theory (DFT) calculation
The fully geometry optimized for the most active derivatives 3b, 5a, 5b, and 5c was performed using the Gaussview 6.0 and Gaussian 0994 at the DFT/B3LYP 6–31 g (d) basis set, in gas phase for all atoms as described previously.64,69
5.6. Molecular docking simulation
The molecular docking simulation was performed inside the active site of human pancreatic α-amylase complexed with co-crystallized ligand acarbose (α-amylase PDB: 2QV4) was selected for antidiabetic activity. Further, the recombinant human acetylcholinesterase (AChE) (PDB: 4EY7) was used for anti-Alzheimer's activity. The crystal structures of these two proteins were downloaded from the protein data bank (https://www.rcsb.org/) and the active site was generated as described previously 73 using Molecular Operating Environment (MOE) software version 2009 × 10.95–97 In addition, target compound structures were generated with ChembioDraw 2014, exported to MOE, where hydrogen atoms were added, and structures were minimized with MMFF94X forcefield using 0.001 RMS kcal mol−198,99 (all details are represented in ESI file†).
5.7. In silico studies
The physicochemical properties and drug-likeness of isatin-based Schiff bases 3b and 5a–c were calculated using SwissADME website http://www.swissadme.ch/index.php.74 While the Drug-likeness scores were calculated using Molsoft LLC website https://www.molsoft.com/mprop/.75 The toxic properties were calculated using ADMETlab 2.0 website https://admetmesh.scbdd.com/service/evaluation/cal.80 But the metabolism properties was computed using the pkCSM-pharmacokinetics website https://biosig.lab.uq.edu.au/pkcsm/prediction81 also the probability value (the range of zero to one) of the metabolism properties, the absorption, and distribution properties were calculated using ADMETlab 2.0 website https://admetmesh.scbdd.com/service/evaluation/cal.84Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors wish to thank the National Research Centre for the facilities provided.References
- J. Ahmad Malik, S. Ahmed, M. Shinde, M. H. S. Almermesh, S. Alghamdi, A. Hussain and S. Anwar, Saudi J. Biol. Sci., 2022, 29, 3586–3599 CrossRef CAS PubMed.
- J. Dowarah and V. P. Singh, Bioorg. Med. Chem., 2020, 28, 115263 CrossRef CAS PubMed.
- N. Oumata, K. Lu, Y. Teng, C. Cavé, Y. Peng, H. Galons and B. P. Roques, Eur. J. Med. Chem., 2022, 240, 114578 CrossRef CAS PubMed.
- D. J. Selkoe, Ann. N. Y. Acad. Sci., 2000, 924, 17–25 CrossRef CAS PubMed.
- Z. Breijyeh and R. Karaman, Molecules, 2020, 25, 5789 CrossRef CAS PubMed.
- Alzheimer’s Association, Alzheimer’s Assoc. Rep., 2019, vol. 15, pp. 321–387 Search PubMed.
- A. A. Alhazzani, M. Alqahtani, A. Alshbriqe, A. Awwadh, T. Alyami, M. Alshomrani and O. A. Mostafa, J. Neurol. Sci., 2017, 381, 315 Search PubMed.
- C. G. Lyketsos and J. Olin, Biol. Psychiatry, 2002, 52, 243–252 CrossRef PubMed.
- S. Gauthier, J. Cummings, C. Ballard, H. Brodaty, G. Grossberg, P. Robert and C. Lyketsos, Int. Psychogeriatr., 2010, 22, 346–372 CrossRef PubMed.
- D. Harman, Ann. N. Y. Acad. Sci., 2001, 928, 1–21 CrossRef CAS PubMed.
- L. Zhu, M. Luo, Y. Zhang, F. Fang, M. Li, F. An, D. Zhao and J. Zhang, Coord. Chem. Rev., 2023, 475, 214875 CrossRef CAS.
- B. Akbari, N. Baghaei-Yazdi, M. Bahmaie and F. Mahdavi Abhari, BioFactors, 2022, 48, 611–633 CrossRef CAS PubMed.
- E. Jaul and J. Barron, Front. Public Health, 2017, 5, 335 CrossRef PubMed.
- S. K. Beura, R. Dhapola, A. R. Panigrahi, P. Yadav, D. H. Reddy and S. K. Singh, Life Sci., 2022, 306, 120855 CrossRef CAS PubMed.
- C. Huang, J. Luo, X. Wen and K. Li, Biochem. Genet., 2022, 60, 1049–1075 CrossRef CAS PubMed.
- X. Xia, Q. Jiang, J. McDermott and J. D. J. Han, Aging Cell, 2018, 17, e12802 CrossRef PubMed.
- Z. Haghighijoo, L. Zamani, F. Moosavi and S. Emami, Eur. J. Med. Chem., 2022, 227, 113949 CrossRef CAS PubMed.
- S. Gupta, V. Singh, P. K. Varadwaj, N. Chakravartty, A. V. S. K. M. Katta, S. P. Lekkala, G. Thomas, S. Narasimhan, A. R. Reddy and V. B. Reddy Lachagari, J. Biomol. Struct. Dyn., 2022, 40, 2264–2283 CrossRef CAS PubMed.
- A. Kabir and A. Muth, Pharmacol. Res., 2022, 176, 106055 CrossRef CAS PubMed.
- X. H. Makhoba, C. Viegas, R. A. Mosa, F. P. D. Viegas and O. J. Pooe, Drug Des., Dev. Ther., 2020, 14, 3235–3249 CrossRef CAS PubMed.
- E. G. Guzman-Lopez, M. Reina, A. Perez-Gonzalez, M. Francisco-Marquez, L. F. Hernandez-Ayala, R. Castañeda-Arriaga and A. Galano, Int. J. Mol. Sci., 2022, 23, 13246 CrossRef CAS PubMed.
- A. Pérez-González, R. Castañeda-Arriaga, E. G. Guzmán-López, L. F. Hernández-Ayala and A. Galano, ACS Omega, 2022, 7, 38254–38268 CrossRef PubMed.
- M. G. A. Al-Khuzaie, M. M. Fahad and A. J. Al-Safi, Biomed. Chem. Sci., 2022, 3, 193–206 CrossRef.
- R. S. Cheke, V. M. Patil, S. D. Firke, J. P. Ambhore, I. A. Ansari, H. M. Patel, S. D. Shinde, V. R. Pasupuleti, M. I. Hassan, M. Adnan, A. Kadri and M. Snoussi, Pharmaceuticals, 2022, 15, 272 CrossRef CAS PubMed.
- B. D. Varpe, A. A. Kulkarni, S. B. Jadhav, A. S. Mali and S. Y. Jadhav, Mini-Rev. Med. Chem., 2020, 21, 1182–1225 Search PubMed.
- I. Abbasi, H. Nadeem, A. Saeed, H. A. A. Kharl, M. N. Tahir and M. M. Naseer, Bioorg. Chem., 2021, 116, 105385 CrossRef CAS PubMed.
- B. Mavroidi, A. Kaminari, D. Matiadis, D. Hadjipavlou-Litina, M. Pelecanou, A. Tzinia and M. Sagnou, Brain Sci., 2022, 12, 806 CrossRef CAS PubMed.
- I. Tumosienė, I. Jonuškienė, K. Kantminienė, V. Mickevičius and V. Petrikaitė, Int. J. Mol. Sci., 2021, 22, 7799 CrossRef PubMed.
- A. M. S. El-Sharief, Y. A. Ammar, A. Belal, M. A. M. S. El-Sharief, Y. A. Mohamed, A. B. M. Mehany, G. A. M. Elhag Ali and A. Ragab, Bioorg. Chem., 2019, 85, 399–412 CrossRef CAS PubMed.
- M. A. Salem, A. Ragab, A. El-Khalafawy, A. H. Makhlouf, A. A. Askar and Y. A. Ammar, Bioorg. Chem., 2020, 96, 103619 CrossRef PubMed.
- Y. A. Ammar, A. M. Sh El-Sharief, A. Belal, S. Y. Abbas, Y. A. Mohamed, A. B. M. Mehany and A. Ragab, Eur. J. Med. Chem., 2018, 156, 918–932 CrossRef CAS PubMed.
- S. A. El-Kalyoubi, A. Ragab, O. A. Abu Ali, Y. A. Ammar, M. G. Seadawy, A. Ahmed and E. A. Fayed, Pharmaceuticals, 2022, 15, 376 CrossRef CAS PubMed.
- A. S. Hassan, G. O. Moustafa, H. M. Awad, E. S. Nossier and M. F. Mady, ACS Omega, 2021, 6, 12361–12374 CrossRef CAS PubMed.
- S. H. Sumrra, W. Zafar, M. L. Asghar, F. Mushtaq, M. A. Raza, M. F. Nazar, M. A. Nadeem, M. Imran and S. Mumtaz, J. Mol. Struct., 2021, 1238, 130382 CrossRef CAS.
- A. S. Hassan, A. A. Askar, A. M. Naglah, A. A. Almehizia and A. Ragab, Molecules, 2020, 25, 2593 CrossRef CAS PubMed.
- A. M. Abdelghany, T. K. Khatab and A. S. Hassan, Bull. Chem. Soc. Ethiop., 2021, 35, 185–196 CrossRef CAS.
- H. R. Elgiushy, S. H. Mohamed, H. Taha, H. Sawaf, Z. Hassan, N. A. Abou-Taleb, E. M. El-labbad, A. S. Hassan, K. A. M. Abouzid and S. F. Hammad, Bioorg. Chem., 2022, 120, 105646 CrossRef CAS PubMed.
- A. S. Hassan, Bull. Chem. Soc. Ethiop., 2020, 34, 533–541 CrossRef CAS.
- A. S. Hassan, D. M. Masoud, F. M. Sroor and A. A. Askar, Med. Chem. Res., 2017, 26, 2909–2919 CrossRef CAS.
- A. S. Hassan, S. A. Osman and T. S. Hafez, Egypt. J. Chem., 2015, 58, 113–139 CrossRef.
- M. Eldeeb, E. F. Sanad, A. Ragab, Y. A. Ammar, K. Mahmoud, M. M. Ali and N. M. Hamdy, Biomedicines, 2022, 10, 722 CrossRef CAS PubMed.
- M. M. Abdelgalil, Y. A. Ammar, G. A. M. Elhag Ali, A. K. Ali and A. Ragab, J. Mol. Struct., 2023, 1274, 134443 CrossRef CAS.
- A. El-Faham, A. A. Elzatahry, Z. A. Al-Othman and E. A. Elsayed, Int. J. Nanomed., 2014, 9, 1167–1174 CrossRef PubMed.
- N. M. Morsy, A. S. Hassan, T. S. Hafez, M. R. H. Mahran, I. A. Sadawe and A. M. Gbaj, J. Iran. Chem. Soc., 2021, 18, 47–59 CrossRef CAS.
- G. Yapar, N. Demir, A. Kiraz, G. Y. Özkat and M. Yıldız, J. Mol. Struct., 2022, 1266, 133530 CrossRef CAS.
- M. M. J. Al-Mudhafar, T. N.-A. Omar and S. L. Abdulhadi, Al Mustansiriyah J. Pharm. Sci., 2022, 22, 23–48 CrossRef.
- S. Daoud, S. Thiab, T. M. A. Jazzazi, T. M. A. Al-Shboul and S. Ullah, Acta Pharm., 2022, 72, 449–458 CrossRef CAS PubMed.
- S. H. Sumrra, S. Kausar, M. A. Raza, M. Zubair, M. N. Zafar, M. A. Nadeem, E. U. Mughal, Z. H. Chohan, F. Mushtaq and U. Rashid, J. Mol. Struct., 2018, 1168, 202–211 CrossRef CAS.
- Y. Liu, F. Wen, H. Yang, L. Bao, Z. Zhao and Z. Zhong, Heliyon, 2022, 8, e10624 CrossRef CAS PubMed.
- W. L. Henry, J. Natl. Med. Assoc., 1962, 54, 476–478 CAS.
- M. N. Wickramaratne, J. C. Punchihewa and D. B. M. Wickramaratne, BMC Complementary Altern. Med., 2016, 16, 466 CrossRef PubMed.
- M. Goschorska, I. Gutowska, I. Baranowska-Bosiacka, K. Piotrowska, E. Metryka, K. Safranow and D. Chlubek, Int. J. Environ. Res. Public Health, 2019, 16, 10 CrossRef CAS PubMed.
- G. L. Ellman, K. D. Courtney, V. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
- B. Balraj, N. Senthilkumar, I. Vetha Potheher and M. Arulmozhi, Mater. Sci. Eng., B, 2018, 231, 121–127 CrossRef CAS.
- N. I. Osman, N. J. Sidik, A. Awal, N. A. M. Adam and N. I. Rezali, J. Intercult. Ethnopharmacol., 2016, 5, 343–349 CrossRef CAS PubMed.
- V. S. Djova, A. M. Nyegue and F.-X. Etoa, J. Drug Delivery Ther., 2018, 8, 174–181 CrossRef.
- R. Ayman, A. M. Radwan, A. M. Elmetwally, Y. A. Ammar and A. Ragab, Arch. Pharm., 2023, 356, e2200395 CrossRef PubMed.
- R. Thirumalaisamy, F. Ameen, A. Subramanian, T. Selvankumar, S. S. Alwakeel and M. Govarthanan, Int. J. Pept. Res. Ther., 2020, 26, 2179–2189 CrossRef CAS.
- P. Shah and A. D. Westwell, J. Enzyme Inhib. Med. Chem., 2007, 22, 527–540 CrossRef CAS PubMed.
- B. M. Johnson, Y. Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- S. S. Mukhtar, A. S. Hassan, N. M. Morsy, T. S. Hafez, F. M. Saleh and H. M. Hassaneen, Synth. Commun., 2021, 51, 1564–1580 CAS.
- D. Russo, P. Valentão, P. B. Andrade, E. C. Fernandez and L. Milella, Int. J. Mol. Sci., 2015, 16, 17696–17718 CrossRef CAS PubMed.
- A. Y. Alzahrani, Y. A. Ammar, M. A. Salem, M. Abu-Elghait and A. Ragab, Arch. Pharm., 2022, 355, e2100266 CrossRef PubMed.
- A. Y. Alzahrani, Y. A. Ammar, M. Abu-Elghait, M. A. Salem, M. A. Assiri, T. E. Ali and A. Ragab, Bioorg. Chem., 2022, 119, 105571 CrossRef CAS PubMed.
- D. M. Elsisi, A. Ragab, A. A. Elhenawy, A. A. Farag, A. M. Ali and Y. A. Ammar, J. Mol. Struct., 2022, 1247, 131314 CrossRef CAS.
- R. Ayman, M. S. Abusaif, A. M. Radwan, A. M. Elmetwally and A. Ragab, Eur. J. Med. Chem., 2023, 249, 115138 CrossRef CAS PubMed.
- A. Ragab, M. S. Abusaif, N. A. Gohar, D. S. Aboul-Magd, E. A. Fayed and Y. A. Ammar, Bioorg. Chem., 2023, 131, 106307 CrossRef CAS PubMed.
- A. Ragab, M. S. Abusaif, D. S. Aboul-Magd, M. M. S. Wassel, G. A. M. Elhagali and Y. A. Ammar, Drug Dev. Res., 2022, 1305–1330 CrossRef CAS PubMed.
- K. E. Saadon, N. M. H. Taha, N. A. Mahmoud, G. A. M. Elhagali and A. Ragab, J. Iran. Chem. Soc., 2022, 19, 3899–3917 CrossRef CAS.
- A. Ragab, Y. A. Ammar, A. Ezzat, A. M. Mahmoud, M. B. I. Mohamed, A. S. El-Tabl and R. S. Farag, Comput. Biol. Med., 2022, 145, 105473 CrossRef CAS PubMed.
- E. S. A. E. H. Khattab, A. Ragab, M. A. Abol-Ftouh and A. A. Elhenawy, J. Biomol. Struct. Dyn., 2022, 40, 1–19 CrossRef PubMed.
- S. A. Ibrahim, A. Ragab and H. A. El-Ghamry, Appl. Organomet. Chem., 2022, 36, e6508 CAS.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, Drug Dev. Res., 2023, 84, 3–24 CrossRef CAS PubMed.
- A. M. Naglah, A. A. Askar, A. S. Hassan, T. K. Khatab, M. A. Al-Omar and M. A. Bhat, Molecules, 2020, 25, 1431 CrossRef CAS PubMed.
- A. S. Hassan, N. M. Morsy, H. M. Awad and A. Ragab, J. Iran. Chem. Soc., 2022, 19, 521–545 CrossRef CAS.
- F. Zhao, P. Wang, Y. Jiao, X. Zhang, D. Chen and H. Xu, Front. Pharmacol., 2020, 11, 579332 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- A. S. Al Wasidi, A. S. Hassan and A. M. Naglah, J. Appl. Pharm. Sci., 2020, 10, 142–148 CAS.
- E. M. Gad, M. S. Nafie, E. H. Eltamany, M. S. A. G. Hammad, A. Barakat, A. Barakat and A. T. A. Boraei, Molecules, 2020, 25, 2523 CrossRef CAS PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, Nucleic Acids Res., 2021, 49, W5–W14 CrossRef CAS PubMed.
- A. S. Hassan, J. Iran. Chem. Soc., 2022, 19, 3577–3589 CrossRef CAS.
- J. Hakkola, J. Hukkanen, M. Turpeinen and O. Pelkonen, Arch. Toxicol., 2020, 94, 3671–3722 CrossRef CAS PubMed.
- Y. Wang, J. Xing, Y. Xu, N. Zhou, J. Peng, Z. Xiong, X. Liu, X. Luo, C. Luo, K. Chen, M. Zheng and H. Jiang, Q. Rev. Biophys., 2015, 48, 488–515 CrossRef PubMed.
- N. N. Wang, J. Dong, Y. H. Deng, M. F. Zhu, M. Wen, Z. J. Yao, A. P. Lu, J. B. Wang and D. S. Cao, J. Chem. Inf. Model., 2016, 56, 763–773 CrossRef CAS PubMed.
- M. L. Amin, Drug Target Insights, 2013, 2013, 27–34 Search PubMed.
- P. Prieto, M. Pineda and M. Aguilar, Anal. Biochem., 1999, 269, 337–341 CrossRef CAS PubMed.
- M. Oyaizu, Jpn. J. Nutr. Diet., 1986, 44, 307–315 CrossRef CAS.
- M. M. Rahman, M. B. Islam, M. Biswas and A. H. M. Khurshid Alam, BMC Res. Notes, 2015, 8, 621 CrossRef PubMed.
- M. B. Arnao, A. Cano and M. Acosta, Food Chem., 2001, 73, 239–244 CrossRef CAS.
- P. Shivanand, Int. J. Pharm. Life Sci., 2010, 1, 38–43 Search PubMed.
- D. Soni and P. Sureshkumar, Res. J. Biotechnol., 2016, 11, 4 Search PubMed.
- S. Meera, N. Ramaiah and N. Kalidindi, Saudi Pharm. J., 2011, 19, 279–284 CrossRef PubMed.
- O. O. Oyedapo and A. J. Famurewa, Int. J. Pharmacogn., 1995, 33, 65–69 CrossRef.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- H. F. Rizk, M. A. El-Borai, A. Ragab, S. A. Ibrahim and M. E. Sadek, Polycyclic Aromat. Compd., 2023, 43, 500–522 CrossRef CAS.
- A. Ezzat, M. B. I. Mohamed, A. M. Mahmoud, R. S. Farag, A. S. El-Tabl and A. Ragab, J. Mol. Struct., 2022, 1251, 132004 CrossRef CAS.
- E. A. Fayed, M. Mohsen, S. M. A. El-Gilil, D. S. Aboul-Magd and A. Ragab, J. Mol. Struct., 2022, 1262, 133028 CrossRef CAS.
- Y. A. Ammar, J. A. Micky, D. S. Aboul-Magd, S. M. A. Abd El-Hafez, S. A. Hessein, A. M. Ali and A. Ragab, Chem. Biol. Drug Des., 2023, 101, 245–270 CrossRef CAS PubMed.
- H. Ali Mohamed, Y. A. Ammar, G. A. M. Elhagali, H. A. Eyada, D. S. Aboul-Magd and A. Ragab, ACS Omega, 2022, 7, 4970–4990 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00297g |
| This journal is © The Royal Society of Chemistry 2023 |