
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, DNA binding and biological evaluation of benzimidazole Schiff base ligands and their metal(II) complexes†
Khalid Mahmooda,
Zareen Akhter *a,
Fouzia Perveenb,
Aishab,
Muneeba Bibic,
Hammad Ismaild,
Nida Tabassume,
Sammer Yousufe,
Ahmad Raza Ashraff and
Muhammad Abdul Qayyumf
*a,
Fouzia Perveenb,
Aishab,
Muneeba Bibic,
Hammad Ismaild,
Nida Tabassume,
Sammer Yousufe,
Ahmad Raza Ashraff and
Muhammad Abdul Qayyumf
aDepartment of Chemistry, Quaid-i-Azam University Islamabad, Pakistan. E-mail: khalid42_edu@yahoo.com; zareenakhter@yahoo.com
bResearch Centre for Modeling and Simulations, National University of Sciences and Technology (NUST) Islamabad, Pakistan
cDepartment of Biochemistry, Quaid-i-Azam University Islamabad, Pakistan
dDepartment of Biochemistry and Biotechnology, University of Gujrat, Gujrat, Pakistan
eH.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan
fDepartment of Chemistry, Division of Science and Technology, University of Education, Lahore, Pakistan
First published on 17th April 2023
Abstract
Two novel benzimidazole ligands (E)-2-((4-(1H-benzo[d]imidazole-2-yl)phenylimino)methyl)-6-bromo-4-chlorophenol (L1) and (E)-1-((4-(1H-benzo[d]imidazole-2-yl)phenylimino)methyl)naphthalene-2-ol (L2) with their corresponding Cu(II), Ni(II), Pd(II) and Zn(II) complexes were designed and synthesized. The compounds were characterized by elemental, IR, and NMR (1H & 13C) spectral analyses. Molecular masses were determined by ESI-mass spectrometry, and the structure of ligand L1 was confirmed by single crystal X-ray diffraction analysis. Molecular docking was carried out for the theoretical investigation of DNA binding interactions. The results obtained were verified experimentally by UV/Visible absorption spectroscopy in conjunction with DNA thermal denaturation studies. It was observed that ligands (L1 and L2) and complexes (1–8) were moderate to strong DNA binders, as evident from the binding constants (Kb). The value was found to be highest for complex 2 (3.27 × 105 M−1) and lowest for 5 (6.40 × 103 M−1). A cell line study revealed that breast cancer cells were less viable to the synthesized compounds compared to that of standard drugs, cisplatin and doxorubicin, at the same concentration. The compounds were also screened for in vitro antibacterial activity for which complex 2 showed a promising broad-spectrum effect against all tested strains of bacteria, almost in the proximity of the reference drug kanamycin, while the rest of the compounds displayed activity against selected strains.
1. Introduction
Metal complexes are inherently endowed with extensive biological properties. Despite their scant implementation in the pharmaceutical industry as drugs, these compounds are widely used to unveil the structural behavior and functions of nucleic acids and promoters in various fascinating processes.1 Since the discovery of cisplatin in 1965 by serendipity,2,3 and its outstanding anticancer properties,4 a research interest has been evoked in the development of new metallo–organic complexes as chemotherapeutic agents. The most investigated metal complexes to date entail platinum and ruthenium central metal ions.5,6 However, owing to the high cost and adverse side effects associated with the aforementioned metal centers, first-row transition metal complexes are considered promising candidates for the design of potential anticancer agents.7–9 Definite coordination geometries and distinctive photophysical properties enhance the functionality of these binding agents.10 Most inorganic medicines implemented as antifungal, antibacterial, and antineoplastic drugs incorporate these metal ions.11–13Along with metal, ligands also play a pivotal role in defining potential activity, which ranges from the recognition of a target site to the activity of any free ligand.7 Thus, the organic sphere around the metal core dictates binding with the target biopolymer, i.e. nucleic acid. Owing to the significance of organic moieties in tuning the propensity of metal complexes to bind with DNA, the oxygen and nitrogen donor benzimidazole Schiff bases seemed to be the strong candidates as chelating agents according to the literature.14–16 These are planar heterocyclic ligands with a structural scaffold similar to that found in purine bases of nucleic acid,17,18 which have an effective DNA binding ability. It has also been reported that the azomethine functionality of Schiff bases is responsible for various biological activities, such as antitumor, antimicrobial, and herbicidal activities.19 Additionally, the complexes of benzimidazole Schiff base ligands with metal ions are efficient DNA binders and exhibit stronger binding affinity than those of free ligands.20–22
There are three primary ways in which anticancer compounds interact with DNA: (i) electrostatic interaction, (ii) intercalation between base pairs, and (iii) minor and major DNA groove binding interaction.6
During intercalation, the compounds stack between adjacent DNA base pairs, leading to significant π-electron overlap. The forces that sustain the stability of the DNA-intercalator complex, even more than DNA alone, are van der Waals, hydrogen bonding, hydrophobic, and/or charge transfer forces.21–25 DNA intercalators are used in chemotherapeutic treatment to inhibit DNA replication in rapidly growing cancer cells. The DNA-intercalator complex is stabilized by π–π stacking interaction and is thus less sensitive to ionic strength relative to the two other binding modes (groove and external binding). Structural changes are induced in DNA by intercalators. Intercalation stabilizes, lengthens, stiffens, and unwinds the DNA double helix.26 For an intercalator to fit between base pairs, the DNA must dynamically open a space between its base pairs by unwinding. The degree of unwinding varies depending on the intercalator. The ethidium cation unwinds DNA by about 26° and proflavine by about 17°. These structural modifications can lead to functional changes, often leading to the inhibition of transcription and replication and DNA repair processes, which make intercalators potent mutagens.27
Intercalation is usually independent of the DNA sequence context (a slight GC specificity has been observed). This mode of binding is usually favored by the presence of an extended fused aromatic ligand, such as PHEHAT or DPPZ. With less extended aromatic systems, intercalation is usually prevented through the clashing of ancillary ligands with the phosphodiester backbone so that only partial intercalation can occur.
Owing to the role of organic ligands and chelates in DNA binding interactions, we planned to synthesize ligands and metal complexes (intercalators) with benzimidazole and azomethine functionalities along with the varying number of aromatic rings in their structure. A comparative DNA interaction study of organic intercalators was carried out using UV-vis absorption spectroscopy and a thermal melting study of DNA. It is well known that the DNA binding ability of complexes can initiate cytotoxicity in cancerous cells.23,28 Therefore, it is important to study the effect of synthesized complexes on cancerous cells. In this regard, the breast cancer cell line was used, and anticancer activities were investigated. Furthermore, the antimicrobial and preliminary cytotoxic studies of these compounds in normal shrimp cells made them an intriguing dimension of the research presented in this study.
2. Results and discussion
The heterocyclic benzimidazole Schiff base ligands (L1 and L2) and their corresponding metal complexes (1–8) were successfully synthesized. The compounds were structurally characterized using important spectroscopic techniques, such as FTIR, NMR (1H and 13C), high-resolution mass spectrometry, and elemental and single crystal X-ray diffraction analysis, which indicated the high purity of compounds. The analytical and physical data of the compounds are presented in Table 1. Moreover, the compounds were assessed for biological activity by performing various biological assays (antibacterial, antifungal, antitumor, and cytotoxic) to check their potential as safe antibiotic drugs. DNA binding studies of the compounds were carried out by absorption titrations and thermal melting studies of DNA.| Compound | Formula | Molecular weight | Color | Yield (%) | Melting point (°C) |
|---|---|---|---|---|---|
| A1 | C13H12N3 | 210 | Light pink | 62 | 159 |
| L1 | C20H14N3OClBr | 426 | Orange | 86 | 174 |
| L2 | C24H18N3O | 364 | Red | 78 | 169 |
| 1 | C40H26N6O2CuCl2Br2 | 914 | Brown | 83 | >300 |
| 2 | C40H26N6O2NiCl2Br2 | 910 | Red | 67 | >300 |
| 3 | C40H26N6O2PdCl2Br2 | 957 | Brown | 62 | >300 |
| 4 | C40H26N6O2ZnCl2Br2 | 916 | Yellow | 77 | >300 |
| 5 | C48H34N6O2Cu | 789 | Green | 75 | 225 |
| 6 | C48H33N6O2Ni | 785 | Dark red | 68 | >300 |
| 7 | C48H33N6O2Pd | 831 | Brown | 61 | >300 |
| 8 | C48H33N6O2Zn | 791 | Yellow | 79 | 211 |
2.1 Infrared (IR) spectroscopy
The IR spectral data of precursor A1 (Table 2) revealed two absorption bands at 3436 and 3357 cm−1, which are attributed to the amino (NH2) group (Fig. S1†). The disappearance of these bands and the appearance of new strong bands at 1619 and 1613 cm−1 (for azomethine groups) in the spectrum of ligands L1 and L2, respectively, confirmed the successful conversion of amine functionality into an Schiff base moiety (Fig. S4a and c†). These absorption bands were shifted to the lower region (1619 to 1587 cm−1) in the spectrum of the corresponding metal complexes, which confirmed the coordination of the azomethine group through the nitrogen atom (N→M).24 This was further confirmed by the appearance of new bands in the region of 433–490 cm−1 due to ν(M–N) bond vibrations. Furthermore, the absence of broad bands at 3295 and 3203 cm−1 (owing to the OH moiety in the ligand structure) in the spectra of the complexes indicated the involvement of phenolic oxygen in bonding with a metal center. Further evidence regarding the binding of oxygen with metal ions was provided by the presence of bands around 540–576 cm−1 in the spectra of complexes25 (Fig. S4b, d, S2a–c, and S3a–c†).| Compound | N–H (ring) | ν NH2 amine | ν OH | ν C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N (ring) N (ring) |
ν (CN) azomethine |
ν C–N (ring) | ν M–O | ν M–N |
|---|---|---|---|---|---|---|---|---|
| A1 | 3436, 3357 | — | 1623 | — | 1439 | — | — | |
| L1 | 3625 | — | 3295 | — | 1619 | 1434 | — | — |
| L2 | 3618 | — | 3203 | — | 1613 | 1432 | — | — |
| 1 | 3620 | — | — | — | 1587 | 1447 | 569 | 490 |
| 2 | 3650 | — | — | — | 1587 | 1433 | 562 | 476 |
| 3 | 3656 | — | — | — | 1598 | 1435 | 576 | 461 |
| 4 | 3615 | — | — | — | 1594 | 1429 | 576 | 426 |
| 5 | 3622 | — | — | — | 1594 | 1435 | 540 | 433 |
| 6 | 3617 | — | — | — | 1587 | 1431 | 562 | 454 |
| 7 | 3615 | — | — | — | 1589 | 1429 | 554 | 433 |
| 8 | 3621 | — | — | — | 1594 | 1436 | 576 | 475 |
2.2 1H and 13C NMR spectroscopy
Purity and structure confirmation were further validated by NMR spectroscopy. 1H NMR spectrum showed characteristic signals in the form of one proton singlet resonating at δ 9.13 and δ 9.73, attributable to the azomethine protons (–NCH–) of L1 and L2, respectively. For the same ligands, the phenolic (–OH) and imine protons (–NH) were expectedly observed as two singlets at δ13.02 (L1) and δ 12.62 (L2) (–OH) as well as δ 14.49 (L1) and δ 15.74 (L2) (–NH) (Fig. 1a and S9a†).
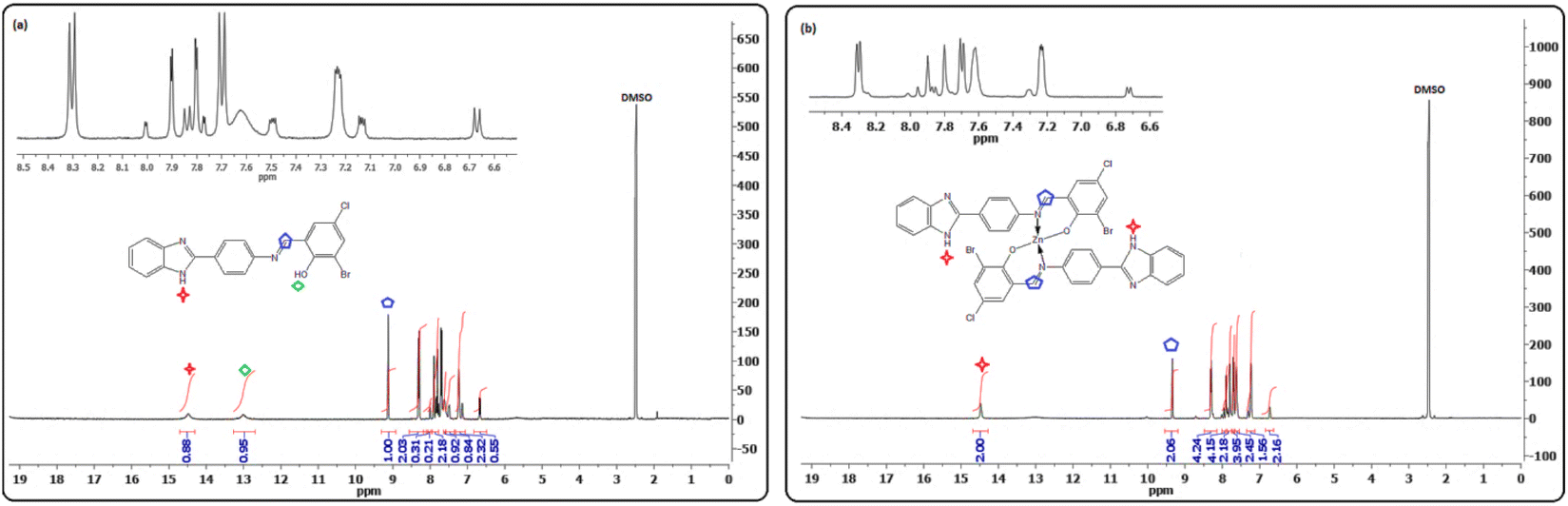 | ||
| Fig. 1 Representative 1H NMR spectrum of ligand L1 (a) and Zn(II) complex 4 (b) in DMSO-d6. | ||
The formation of Schiff base metal complexes was supported by observing a downfield shift of the azomethine proton from δ 9.11 (L1) and δ 9.73 (L2) to 9.52 (2), 9.50 (3), 9.46 (4), 9.76 (6), 10.03 (7), and 9.86 (8), as illustrated in Table 3. Strong evidence in favor of deprotonation and coordination of phenolic oxygen of ligands to metal ions was inferred from the disappearance of –OH proton singlets in the spectrum of complexes.26,27 Additionally, the number of protons obtained from the integrated spectral curves agreed well with those calculated from the CHN analyses.
| L1 | C20H14N3OClBr | 1H NMR (400 MHz, DMSO) δ 14.49 (s, 1H), 13.02 (s, 1H), 9.13 (s, 1H), 8.35 (d, J = 8.5 Hz, 2H), 8.01 (d, J = 2.5 Hz, 1H), 7.95–7.78 (m, 2H), 7.62 (s, 1H), 7.49 (d, J = 3.2 Hz, 1H), 7.18 (ddd, J = 38.6, 5.8, 3.0 Hz, 2H), 6.67 (d, J = 8.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 165.12, 158.81, 150.13, 142.31–140.17, 134.83, 129.02, 122.13, 119.79, 113.42, 110.82 |
| L2 | C24H18N3O | 1H NMR (300 MHz, DMSO) δ 15.74 (s, 1H), 12.62 (s, 1H) 9.73 (s, 1H), 8.55 (d, J = 8.4 Hz, 1H), 8.28 (d, J = 8.4, 2H), 8.95 (d, J = 9, 1H), 7.85 (m, 3H), 7.42 (d, J = 10.5 Hz, 2H), 7.36 (d, m, 2H), 7.02 (d, J = 9 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 171.98, 162.59, 155.67, 152.56, 145.34, 137.90 133.65, 129.54, 128.69, 128.18, 128.09, 127.16, 124.14, 121.43, 120.96, 110.59, 109.18 |
| 1 | C40H26N6O2CuCl2Br2 | No data for copper complexes |
| 2 | C40H26N6O2NiCl2Br2 | 1H NMR (400 MHz, DMSO) δ 14.52 (s, 1H), 9.52 (s, 1H), 8.24 (dd, J = 24.3, 6.1 Hz, 4H), 7.83 (t, J = 22.3 Hz, 2H), 7.65 (dd, J = 27.7, 18.9 Hz, 2H), 7.35 (s, 1H), 7.26 (s, 1H), 7.06 (s, 2H), 6.94 (d, J = 7.7 Hz, 1H), 6.65 (d, J = 8.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 170.51, 159.91, 151.65, 148.61–146.26, 142.19, 138.96, 137.03, 121.91–110.52 |
| 3 | C40H26N6O2PdCl2Br2 | 1H NMR (400 MHz, DMSO) δ 14.41 (s, 1H), 9.50 (s, 1H), 7.91 (d, J = 7.2 Hz, 4H), 7.73–7.65 (m, 6H), 7.51 (s, 2H), 7.05 (d, J = 7.1 Hz, 4H), 6.82 (d, J = 8.2, 4H). 13C NMR (101 MHz, DMSO) δ 166.15, 158.79, 151.75, 148.71, 147.19, 145.82, 144.36, 135.03, 128.61, 127.52, 123.58–11.15 |
| 4 | C40H26N6O2ZnCl2Br2 | 1H NMR (400 MHz, DMSO) δ 14.47 (s, 2H), 9.46 (s, 2H), 8.30 (d, J = 7.2 Hz, 4H), 8.01–7.87 (m, 4H), 7.81 (s, 2H), 7.70 (d, J = 7.1 Hz, 4H), 7.62 (s, 2H), 7.23 (dd, J = 17.2, 14.5 Hz, 2H), 6.74 (d, J = 7.1 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 169.21, 158.92, 151.42, 149.73, 146.11, 144.23, 138.82, 137.51, 135.02, 129.41, 128.12, 123.20–111.07 |
| 5 | C48H34N6O2Cu | No data for copper complexes |
| 6 | C48H33N6O2Ni | 1H NMR (400 MHz, DMSO) δ 15.78 (s, 2H), 9.76 (s, 2H), 8.54 (s, 2H), 8.35–8.17 (m, 4H), 8.12 (d, J = 7.1 Hz, 2H), 7.95 (s, 2H), 7.83 (d, J = 22.4 Hz, 2H), 7.69 (d, J = 6.7 Hz, 4H), 7.63–7.49 (m, 6H), 7.38–7.10 (m, 4H), 7.10–6.83 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 181.68, 164.21, 154.69, 150.07, 146.33, 144.0, 138.16, 132.30, 130.50, 129.50, 125.06, 124.03, 123.54–123.41, 122.03, 113.64–113.45, 110.02, 108.39 |
| 7 | C48H33N6O2Pd | 1H NMR (400 MHz, DMSO) δ 15.91 (s, 2H), 10.03 (s, 2H), 8.58 (d, J = 8.8 Hz, 2H), 8.31 (s, 4H), 8.12 (m, 2H), 7.95 (dd, J = 34.4, 25.3 Hz, 2H), 7.84 (d, J = 7.4 Hz, 4H), 7.65 (d, J = 36.5 Hz, 6H), 7.52 (m, 2H), 7.40 (m, 2H), 7.07 (d, J = 9.1 Hz, 4H). 13C NMR (101 MHz, DMSO) δ 182.05, 169.15, 155.54, 150.72, 145.03, 137.90, 133.65, 129.54, 128.69–128.08, 127.14, 124.28–124.13, 122.91, 121.37, 109.14 |
| 8 | C48H33N6O2Zn | 1H NMR (400 MHz, DMSO) δ 15.75 (s, 2H), 9.86 (s, 2H), 8.56 (d, J = 8.8 Hz, 2H), 8.32 (s, 4H), 8.13–8.10 (m, 2H), 8.05–7.95 (m, 2H), 7.84 (d, J = 7.4 Hz, 4H), 7.65–7.55 (m, 6H), 7.51–7.45 (m, 2H), 7.43–7.37 (m, 2H), 7.07 (d, J = 9.1 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 178.04, 165.74, 156.52, 152.69, 145.11, 142.51, 133.38, 131.26, 128.61, 122.99, 122.28, 121.28, 120.10, 119.20, 111.13, 109.96 |
Correspondingly, 13C NMR data (summarized in the experimental section and Table 3) also corroborated well with the formation of ligands L1 and L2 (Fig. S8a and S9b†). All assignments of the carbon atoms were found in their expected regions29 and well supported by their IR and 1H NMR spectral data. The azomethine and phenolic carbon of ligands L1 and L2 shifted downfield in complexes, i.e. from δ 172 (L2 and Fig. S9b†) to δ 182 (complex 7 and Fig. S11b†), indicating the involvement of azomethine-N and phenolic-O in coordination with the metal ion. Furthermore, the aromatic carbons found near the coordination sites showed a slight downfield shift (Table 3, Fig. S6b–S8b and S10b–S12b†), indicating the shift of electron density from ligand donor sites to the metal ion.30
2.3 Mass spectral studies
The molecular weight and stoichiometric composition of the ligands and complexes were determined using the respective m/z values in the mass spectrum of the compounds. The successful synthesis of ligands L1 and L2 was proved by the presence of base peak signals at m/z = 426 (M + H) and 364 (M + H), respectively (Fig. 2a and S14†). The molecular ion peaks at m/z = 916 (M + H+), 910 (M + H+), 959 (M + H+), 917 (M + H+), 789 (M + H+), 786 (M + H), 831 (M + H+) and 792 (M + H) confirmed the molecular weight of complexes 1–8, respectively, as shown in Fig. 2b and c and S15–S21 in the ESI.† | ||
| Fig. 2 Representative high-resolution ESI mass spectrum of ligand (L1) (a), Pd(II) complex (3) (b), and isotope model of Pd(II) complex (3) (c). | ||
2.4 Single-crystal X-ray diffraction analysis of L1
Single-crystal X-ray diffraction studies of ligand (L1) was carried out by mounting an orange color crystal of suitable size 0.02 × 0.09 × 0.30 mm3 on a narrow glass fiber fixed with wax on the copper pin for data collection. The collection of data was carried out using a Bruker D8 venture fitted with a CCD detector diffractometer and graphite monochromator with Cu Kα radiation (λ = 1.54178 Å) at T = 100(2) K. The integration and reduction of the collected data were done using the SAINT program. The structure was solved by applying the direct method and Fourier transformation techniques, and further refinement was performed using the SHELXL97 program with full-matrix least-square calculations on F2. PLATON and SHELXL programs were used for the final refinement of the structure. The ORTEP view in Fig. 3 represents the crystal structure of ligand (L1) and the MERCURY program used for drawing crystal packing and intermolecular interactions present in the crystal lattice.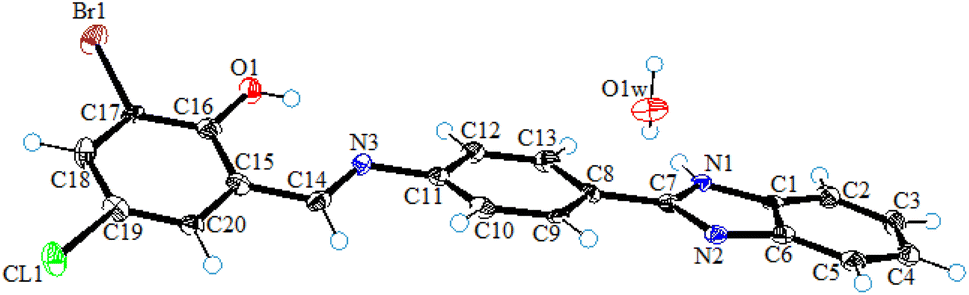 | ||
| Fig. 3 The ORTEP view of ligand (L1) drawn at a 50% probability level. | ||
A summary of crystal data collection and refinement is presented in Table 4.
| Empirical formula | C40H30Br2Cl2N6O4 |
| Formula weight | 889.42 |
| Temperature | 100(2) K |
| Wavelength | 1.54178 Å |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| Unit cell dimensions | a = 8.3713(3) Å, a = 90° |
| b = 14.0145(4) Å, b = 90° | |
| c = 31.3792(9) Å, g = 90° | |
| Volume | 3681.4(2) Å3 |
| Z | 4 |
| Density (calculated) | 1.605 Mg m−3 |
| Absorption coefficient | 4.558 mm−1 |
| F(000) | 1792 |
| Crystal size | 0.300 × 0.090 × 0.020 mm3 |
| Theta range for data collection | 3.454 to 66.683° |
| Index ranges | −9 ≤ h ≤ 9, −16 ≤ k ≤ 16, −33 ≤ l ≤ 37 |
| Reflections collected | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 443 443 |
| Independent reflections | 6474 [R(int) = 0.0913] |
| Completeness to theta = 66.683° | 99.8% |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 6474/1/512 |
| Goodness-of-fit on F2 | 1.028 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0406, wR2 = 0.0762 |
| R indices (all data) | R1 = 0.0542, wR2 = 0.0805 |
| Absolute structure parameter | 0.47(2) |
| Extinction coefficient | n/a |
| Largest diff. peak and hole | 0.339 and −0.398 e Å−3 |
 | ||
| Fig. 4 Crystal packing diagram of ligand (L1) showing intermolecular interactions within the unit cell along the b-axis. | ||
| D | H | A | D–H | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|---|---|---|
| a Symmetric codes: 1/2 − X, −Y, 1/2 + Z, −X, 1/2 + Y, 1/2 − Z, 1/2 + X, 1/2 − Y, and −Z. | ||||||
| N(1) | H(1A) | O(1W) | 0.88 | 2.05 | 2.7810(1) | 140 |
| O(1W) | H(1W) | N(2) | 0.92 | 1.77 | 2.6900(1) | 171 |
| O(2W) | H(3W) | O(1W) | 0.78 | 2.02 | 2.7711(1) | 161 |
| O(2W) | H(4W) | N4 | 0.89 | 1.93 | 2.8130(1) | 168 |
| N5 | H(5A) | O(2W) | 0.88 | 1.88 | 2.7473(1) | 170 |
| C(5) | H(5) | N(4) | 0.95 | 2.61 | 3.4290(1) | 145 |
| C(10) | H(10) | CL(1) | 0.95 | 2.82 | 3.5941(1) | 140 |
| C29 | H(29) | O(2W) | 0.95 | 2.46 | 3.3823(1) | 165 |
| C40 | H(40) | O(1) | 0.95 | 2.45 | 3.3375(1) | 156 |
2.5 DNA binding studies
The DNA binding proclivity of the synthesized compounds was investigated theoretically by molecular docking and experimentally by UV-vis absorption titrations and thermal denaturation studies, as illustrated below.O oxygen of thymine DT-C16 and DT-A16, whereas two hydrogen bonds were built between the N atom of L1 and the H atom of cuanine and cytosine, respectively, i.e., DG-A14 and DC-G14 of DNA. The 3D and 2D pose images of L1–Ni complex 2 (Fig. 5 (3)) indicated the formation of four hydrogen bonds with –NH2 of cytosine (DC-A6 and DC-B8) as well as with a CO group of thiamine (DT-C11, DT-A11) of DNA base pairs. Compound 2 showed a greater DNA binding propensity owing to its capacity to develop a greater number of direct hydrogen bonds with nitrogenous bases of DNA. Thus, the docked results suggest an intercalative mode and groove binding of the studied compounds. Complex 1 was able to develop two hydrogen bonds with an –NH2 group of DC-D7 and DC-B7 DNA, suggesting a comparatively smaller value of binding for complex 1.
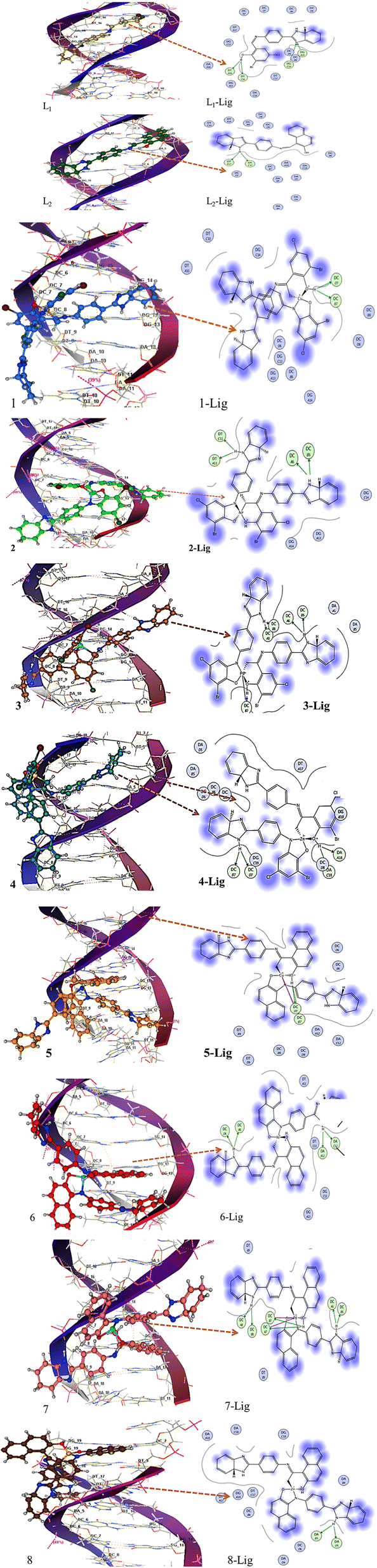 | ||
| Fig. 5 3D complex (left) and 2D lig plot (right) showing docking interaction map of ligands L1 and L2 and representative complexes 2–8. Hydrogen bonding is shown by the orange dotted line. | ||
Similar docking simulations were carried out for ligand L2 and metal complexes 5–8. From the 3D pose and 2D ligplot images (Fig. 5), it can be observed that ligand L2 formed two hydrogen bonds involving phenolic-H and the imidazole ring nitrogen atom as a binding site. The most effective binding interactions were shown by the Pd complex of L2 with the following six hydrogen bonds: DC-A8, DC-B8, DC-A7, DC-B7, DC-A6, and DC-B6. Complex 8 exhibited minimum binding, as indicated in Fig. 5 (right). It exhibited only two hydrogen bonds, which were formed with an –NH2 group of adenine DA-B5 and DA-D5. The intercalation of the planar aromatic part of L2 complexes is also evident along with the mixed mode of interactions.
The absorption spectra resulting from titrations are depicted in Fig. 6 and 7. As observed from spectral curves, the absorbance of test compounds decreased with the addition of SS-DNA aliquots (10–80 μM). This hypochromism accompanied by a red shift/blue shift (Table 7) suggests that these compounds bind to DNA through intercalation mode and groove binding.34,35 The hypochromic effect is considered to be due to the interaction between the electronic states of the intercalating chromophore (compounds) and the nitrogen bases of DNA.36 This resulted in structural changes in DNA that entailed the local unwinding of the double helix and the creation of a cavity to accommodate guest compound molecules.37 It is also obvious from previous knowledge that the electronic interaction between chromophores and DNA bases decreases as a cube of the distance of separation between them.35 The maximum hypochromism was observed in our case for compound 2, which was 73%, indicating the very close proximity of compound 2 chromophores to DNA bases. The other compounds, including ligands and complexes, showed hypochromism from 28–57%, suggesting their stronger binding interaction with DNA (Table 7).
 | ||
| Fig. 6 Changes in the UV-vis spectra of ligand L1 (a), Cu(II) complex 1 (b), Ni(II) complex 2 (c), Pd(II) complex 3 (d), and Zn(II) complex 4 (e), having a concentration of 1 × 10−4 M, as a function of increasing quantities of fish sperm DNA (10–80 μM) in 50 mM Tris–HCl/NaCl buffer (pH = 7.35) at 25 °C. The arrows indicate a decrease in absorbance with an increase in DNA concentration. Inset plots show intrinsic binding constant kb. | ||
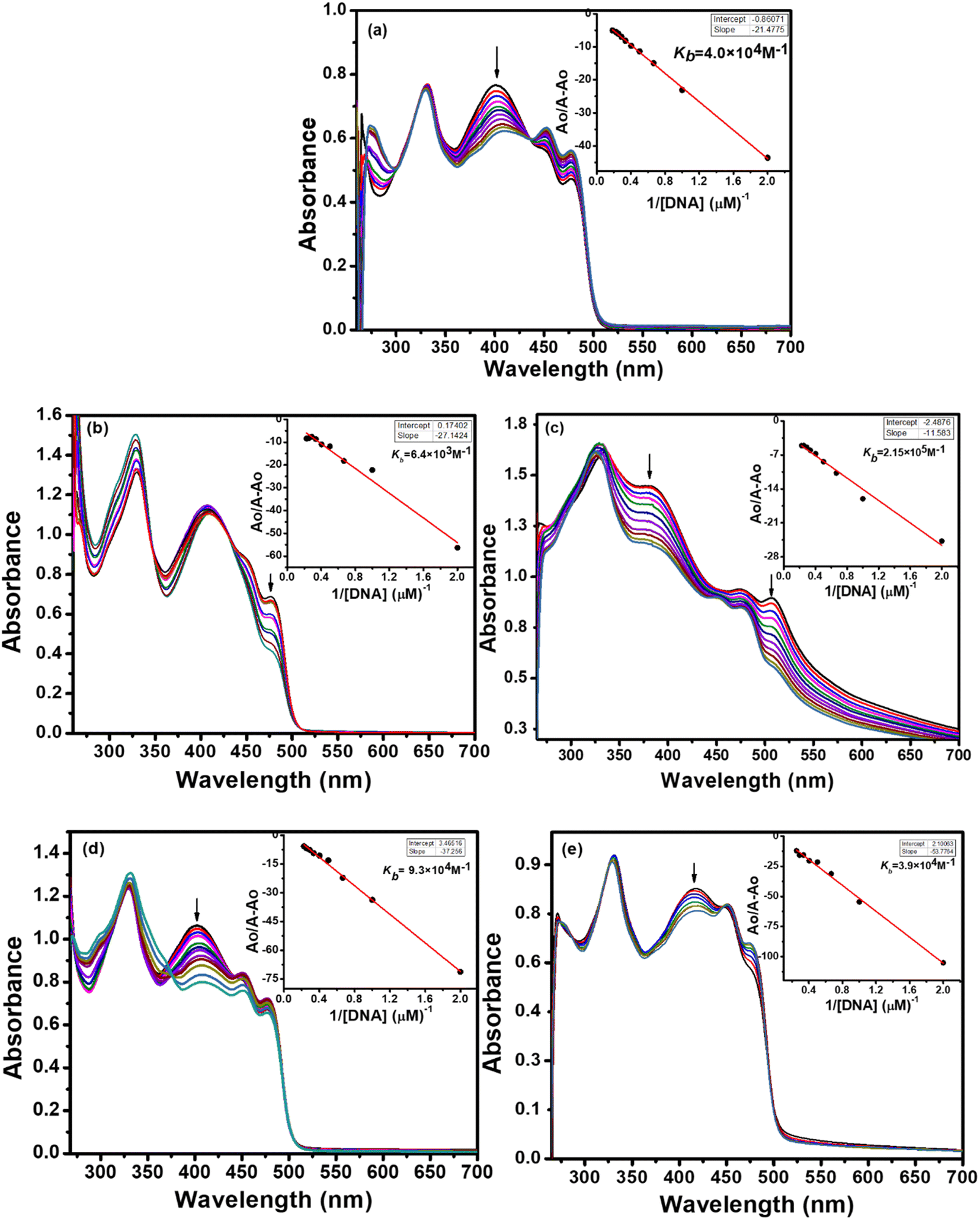 | ||
| Fig. 7 Changes in the UV-vis spectra of ligand L2 (a), Cu(II) complex 5 (b), Ni(II) complex 6 (c), Pd(II) complex 7 (d) and Zn(II) complex 8 (e) having a concentration of 1 × 10−4 M, as a function of increasing quantities of fish sperm DNA (10–80 μM) in 50 mM Tris–HCl/NaCl buffer (pH = 7.35) at 25 °C. The arrows indicate a decrease in absorbance with an increase in DNA concentration. Inset plots show intrinsic binding constant kb. | ||
To quantitatively compare the binding strength of the ligands and complexes with SS-DNA, the intrinsic binding constants (Kb) were calculated from the absorption data; their values are collated in Table 7. In the first series (L1 and its complexes), the highest binding constant was observed for Ni(II) complex 2 and the lowest for Pd(II) complex 3. By comparing the calculated binding constant values of test compounds with that of the known intercalator, proflavine,38 it was inferred that compounds 1 and 2 were stronger intercalators, while compounds L1, 3, and 4 bind to DNA via partial intercalation along with other modes, such as groove binding. This type of interaction of compounds 1 and 2 could be justified by the square planar geometry of Ni(II) and Cu(II) complexes (confirmed by the diamagnetic nature of Ni(II) and the d9 electronic configuration of Cu(II)39), facilitating them to diffuse between the DNA bases more effectively; hence, stronger stacking interactions are developed.
In the second series (L2 and complexes), the highest binding constant was observed for Ni(II) complex 6 and the lowest for Cu(II) complex 5. Thus, compound 6 interacted with DNA through intercalation, while compounds L2, 5, 7 and 8 interacted through partial intercalation and groove binding. The trend in the hypochromism between the ligands followed the order L1 > L2. The same trend was found with the binding constant values. Thus, the two results supported each other. The trend in hypochromism among complexes was of the following order: 2 > 6 > 1 > 4 > 7 > 3 > 8 > 5. The same order was found for the binding constant values, as illustrated in Table 7. Thus, the magnitude of the hypochromism reflects the strength of the stacking interactions between the compound and DNA.
In summary, among the synthesized compounds, Ni(II) complex 2 exhibited the strongest interaction with DNA. The corresponding absorption spectrum showed an isobestic point at 344 nm, indicating DNA-2 complex formation and a hypochromic effect along with bathochromic shift Δλ = 4 nm. Similarly, complexes 6 and 1 also manifested stronger DNA interaction proclivity (see Table 6). Hence, it was concluded that complexes 2, 6, and 1 bind with DNA through intercalation, and the rest of the compounds interact through partial intercalation and groove binding.
| Compound | Kb (M−1) | ΔG (kJ mol−1) |
|---|---|---|
| L1 | 1.55 × 105 | −29.68 |
| L2 | 5.11 × 104 | −26.88 |
| 1 | 2.33 × 105 | −30.62 |
| 2 | 45.8 × 105 | −38.01 |
| 3 | 7.46 × 104 | −27.80 |
| 4 | 2.34 × 105 | −30.64 |
| 5 | 1.14 × 104 | −23.16 |
| 6 | 1.52 × 105 | −29.57 |
| 7 | 2.85 × 105 | −31.12 |
| 8 | 6.56 × 104 | −27.48 |
It is established from the results of hypochromicity and intrinsic binding constant values that the synthesized compounds show binding interactions with DNA. Further experiments must be conducted to determine the energy parameters for compound–DNA complexes.
The Gibb's free energy (ΔG) of the compound–DNA complex explained the thermodynamic stability of the compound–DNA complex when DNA was added to the solution of the compound. Eqn (1) helps to calculate free energy by putting the values of suitable parameters:
|
ΔG = −RTlnKb.
| (1) |
A comparison of the free energy values (Table 7) indicates that the binding of Ni(II) complex 2 with DNA was most spontaneous and thermodynamically favorable under the given set of conditions. The remaining compounds also generated stable adducts with DNA and could be used as future therapeutic agents to cure various disorders in the living system.
| Compound | ΔTma (°C) | Kbb (M−1) | Hypochromism (%) | Δλmax | ΔG (kJ mol−1) | Isobestic points (nm) |
|---|---|---|---|---|---|---|
| a The increment in DNA thermal melting (ΔTm, °C) was measured in unspecific (st) DNA. Experiments were run in phosphate buffer (10 mM, pH 7). Thermal denaturation for natural (st) DNA = 68.5 °C.b Calculated from the A0/A − A0 vs. 1/[DNA] plots of the UV-vis DNA titration results. | ||||||
| L1 | 4.2 | 1.13 × 105 | 43 | 15 nm (red) | −28.81 | 301, 341, 422 |
| L2 | 3.1 | 4.00 × 104 | 35 | 9 nm (red) | −26.24 | 438 |
| 1 | 5.7 | 2.1 × 105 | 60 | 2 nm (blue) | −30.34 | 342 |
| 2 | 6.1 | 3.27 × 105 | 75 | 4 nm (blue) | −31.44 | 344 |
| 3 | 2.7 | 4.75 × 104 | 39 | 3 nm (red) | −26.66 | — |
| 4 | 3.6 | 1.07 × 105 | 55 | 8 nm (red) | −28.67 | 338, 371 |
| 5 | 2.2 | 6.40 × 103 | 28 | 2 nm (red) | −21.70 | 344, 432 |
| 6 | 4.7 | 2.15 × 105 | 73 | 7 nm (blue) | −30.40 | 452 |
| 7 | 3.2 | 9.3 × 104 | 42 | 6 nm (red) | −28.33 | 376 |
| 8 | 3.9 | 3.9 × 104 | 35 | 4 nm (blue) | −26.18 | 444, 464 |
The transition in the structure of DNA from double-stranded to single strands was monitored by measuring absorbance at 260 nm as a function of temperature. Fig. 8a and b indicate that in the absence of any compound, the Tm of ST-DNA was 68.5 °C under our experimental conditions. The melting point increased in the range of 71.2 to 74.6 °C for compounds of the first series (ligand L1 and complexes 1–4) and 70.7 to 73.2 °C for the second series (L2 and complexes 5–8), as indicated in Table 7. From the results, it was deduced that all compounds interacted efficiently with DNA via an intercalation mode. This type of stacking interaction stabilizes the double helix structure of DNA and increases Tm. The significantly increased Tm of DNA in the presence of complexes 2 and 6 revealed a very strong interaction between the compound and DNA, which was also reflected in the binding constant (Kb) values obtained spectroscopically.
 | ||
| Fig. 8 Melting curves for 100 μM of ST-DNA upon the addition of ligand L1 and complexes 1–4 (a), ligand L2 and complexes 5–8 (b). | ||
2.6 Biochemistry
| Compounds | Inhibition zone (mm) | MIC values (μg mL−1) | ||||||
|---|---|---|---|---|---|---|---|---|
| M. luteus | S. aureus | E. coli | E. aerogenes | M. luteus | S. aurus | E. coli | E. aerogenes | |
| a Kanamycin. | ||||||||
| L1 | — | — | 7.1 ± 0.3 | 6.3 ± 0.5 | — | — | 100 ± 3.1 | 100 ± 2.5 |
| L2 | — | — | 8.5 ± 0.5 | 5.2 ± 1.25 | — | — | 100 ± 2.02 | 100 ± 3.25 |
| 1 | — | 4.7 ± 1.03 | 13.1 ± 1.04 | 6.4 ± 0.4 | — | 100 ± 3.46 | 50 ± 0.5 | 100 ± 2.63 |
| 2 | 8.5 ± 0.4 | 10.3 ± 0.7 | 16.8 ± 2.7 | 14.5 ± 0.8 | 50 ± 3.5 | 25 ± 1.37 | 12.5 ± 0.5 | 25 ± 0.5 |
| 3 | — | — | 7.4 ± 0.5 | — | — | — | 100 ± 2.73 | — |
| 4 | 6.6 ± 0.5 | — | 11.4 ± 1.4 | 8.5 ± 1.52 | 100 ± 4.51 | — | 50 ± 1.41 | 50 ± 1.50 |
| 5 | — | — | — | — | — | — | — | — |
| 6 | 9.4 ± 0.3 | 7.20 ± 0.5 | 12.5 ± 1.6 | 6.5 ± 0.3 | 50 ± 2.1 | 100 ± 4.25 | 25 ± 0.5 | 100 ± 3.85 |
| 7 | — | — | 8.5 ± 0.9 | 6.8 ± 0.6 | — | — | 100 ± 2.5 | 100 ± 4.5 |
| 8 | 7.7 ± 0.5 | — | 10.8 ± 0.5 | — | 100 ± 4.07 | — | 50 ± 1.05 | — |
| Ka |
24.6 ± 0.1 | 26.3 ± 0.3 | 21.2 ± 01 | 22.4 ± 0.2 | 12.5 ± 0.5 | 6.25 ± 0.3 | 6.25 ± 0.5 | 12.5 ± 0.75 |
| Compounds | Brine shrimp cytotoxicity assay (LD50 = μg mL−1) |
|---|---|
| a Note: — represents inactivity. | |
| L1 | 127.61 ± 1.32 |
| L2 | >200 |
| 1 | >200 |
| 2 | >200 |
| 3 | >200 |
| 4 | — |
| 5 | — |
| 6 | >200 |
| 7 | 125 ± 2.43 |
| 8 | — |
| Doxorubicin | 3.22 ± 4.32 |
N) groups, respectively, which may be an important fragment for the formation of hydrogen bonding at the receptor site.50,51 Among these complexes, complexes 5 (L2-Cu), 2 (L1-Ni), and 7 (L2-Pd) exerted strong cytotoxic effects when compared with cisplatin and doxorubicin used as the standard, as illustrated in Fig. 9. This may partially be attributed to the lipophilic character of the central metal atom as explained based on Overton's concept and Tweedy's chelation theory.52,53 Complexes 5 (L2-Cu), 2 (L1-Ni), and 7 (L2-Pd) possess poor activity compared to those of the respective ligands, which could be attributed to their decreased solubility. A comparative study with cisplatin and doxorubicin based on % cell viability values obtained with the MCF7 cell line exhibited that the ligands L1, L2, and complexes 5 (L2-Cu), 2 (L1-Ni), and 7 (L2-Pd) showed percent cell viability values at 1.46, 4.12, 0.129, 1.89, and 3.09, respectively, while cisplatin and doxorubicin maintained 21.89 and 7.37 values correspondingly. The poor activity of Zn(II) complexes is attributed to the restriction of only tetrahedral structure, while square planar complexes of remaining metal ions help in the effective penetration of complexes between the grooves of DNA and show better binding effects. The Ni(II) and Pd(II) complexes induce apoptosis (programmed cell death) that may be a direct result of their action on the cell, while Cu(II) complexes on reduction to Cu(I) generate reactive oxygen species (ROS)54 such as hydroxyl radicals from H2O2 produced during normal cellular activities, leading to growth inhibition in tumor cells.55 The enhanced cytotoxic values of ligands L1 and L2 and complexes 5 (L2-Cu), 2 (L1-Ni), and 7 (L2-Pd) than cisplatin and doxorubicin support the use of the first-row transition metal complexes as effective anticancer agents, but their intrinsic side effects must be overcome before clinical trials.
 | ||
| Fig. 9 Percent cell viability of ligands L1 and L2 and their respective Zn(II), Cu(II), Ni(II) and Pd(II) complexes from 1 ppm to 5 ppm concentration against MCF7 cell line. | ||
3. Experimental
3.1 Materials and methods
All chemicals, including solvents and reactant materials, were purchased from Sigma Aldrich and were used without further purification. Salmon sperm DNA (st)DNA, was purchased from Sigma Aldrich (St Louis, MO, USA) with an extinction coefficient of ε260 = 6600 M−1 cm−1.3.2 Instrumentation and measurements
Elemental analysis was performed using a CHNS932 (Leco-USA) elemental analyzer. Melting points were recorded using an MPD Mitamura Riken Kogyo (Japan) electrothermal apparatus. Infrared (IR) spectra were recorded using a PerkinElmer Spectrum One Spectrometer in a 4000–400 cm−1 region. 1H and 13C{1H} NMR spectra were obtained using a Bruker Avance 400 and Bruker DPX 400 and were referenced to the residual 1H and 13C resonances of the solvent used. High-resolution mass spectrometry analysis was carried out using a Premier Waters Maldi-quadrupole time-of-flight (Q-TOF) mass spectrometer equipped with Z-spray electrospray ionization (ESI) and matrix-assisted laser desorption ionization (MALDI) sources.Single-crystal X-ray diffraction data were collected using Bruker APEX D8 Venture diffractometer fitted with PHOTON 100 detector (CMOS technology) and fine-focus sealed tube with an X-ray source [Cu Kα radiation, α = 1.54178 Å]. Reflection intensities were integrated using SAINT software. Absorption correction was done using the multi-scan method.
The structure was solved by direct methods using SHELXS97 (Sheldrick, 1997)56 and difmap synthesis using SHELXS96 (Sheldrick, 1996).57 All non-hydrogen atoms were refined anisotropically, and the hydrogen atoms were placed into the calculated positions.
3.3 Synthesis
The synthetic schemes for the ligands and corresponding metal complexes are shown in Scheme 1.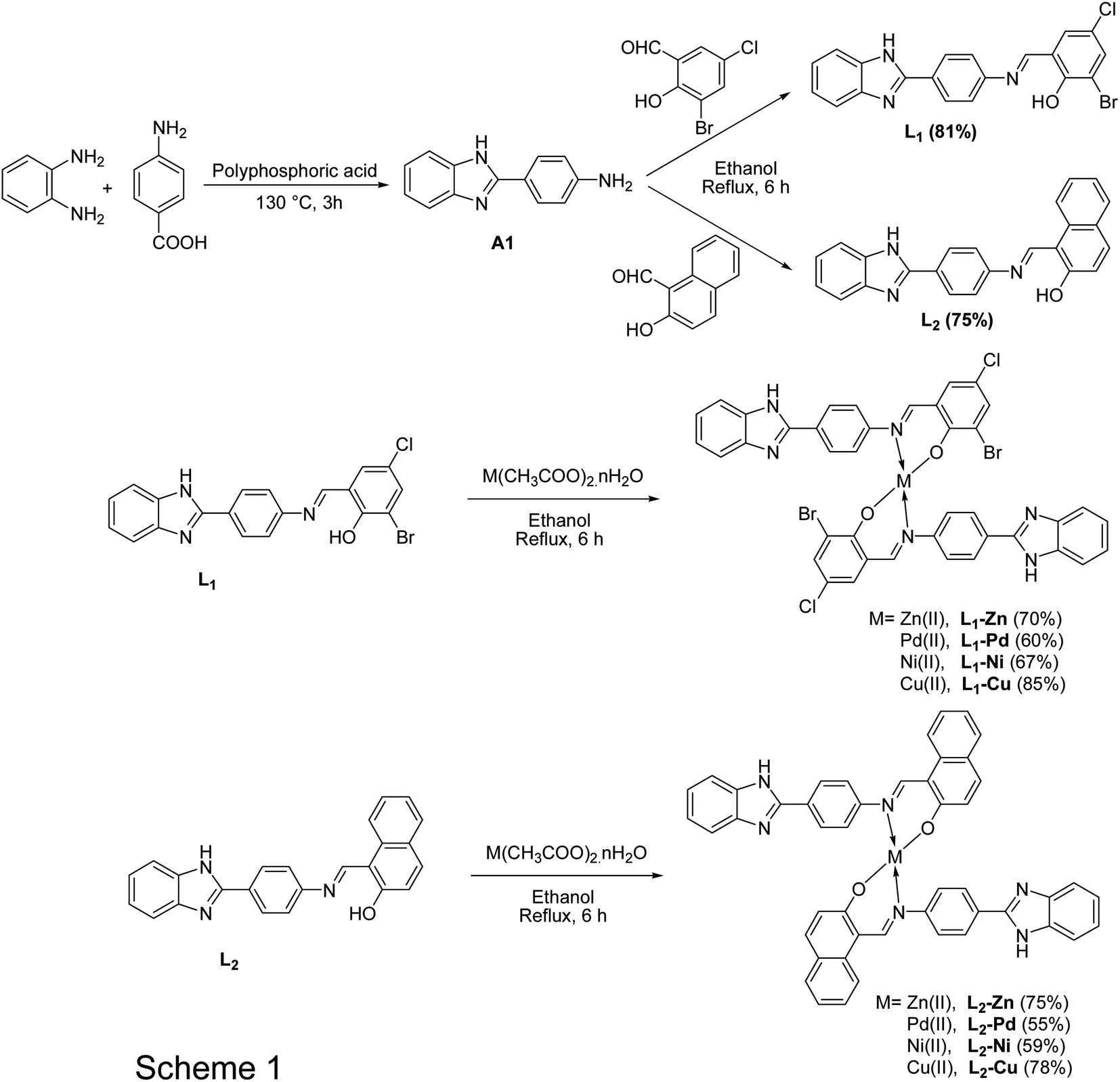 | ||
| Scheme 1 General synthetic chart of ligands and corresponding metal complexes. | ||
:1 molar ratio. The reaction mixture was heated at 130 °C for 3 h. After completion of the reaction, the thick liquid mixture was poured onto crushed ice and neutralized with a 4 N NaOH solution, and the precipitate formed was filtered and dried. Compound A1 was extracted with methanol and used immediately for the preparation of L1 and L2.N), 1434 (C–Nring). 1H NMR (400 MHz, DMSO) δ 14.49 (s, 1H), 13.02 (s, 1H), 9.13 (s, 1H), 8.35 (d, J = 8.5 Hz, 2H), 8.01 (d, J = 2.5 Hz, 1H), 7.95–7.78 (m, 2H), 7.62 (s, 1H), 7.49 (d, J = 3.2 Hz, 1H), 7.18 (ddd, J = 38.6, 5.8, 3.0 Hz, 2H), 6.67 (d, J = 8.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 165.12, 158.81, 150.13, 142.31–140.17, 134.83, 129.02, 122.13, 119.79, 113.42, 110.82. HRMS (m/z) calculated for C20H13N3OClBr [M + H+] 426.0009, found 426.0005.N), 1432 (C–Nring). 1H NMR (300 MHz, DMSO) δ 15.74 (s, 1H), 12.62 (s, 1H) 9.73 (s, 1H), 8.55 (d, J = 8.4 Hz, 1H), 8.28 (d, J = 8.4, 2H), 8.95 (d, J = 9, 1H), 7.85 (m, 3H), 7.42 (d, J = 10.5 Hz, 2H), 7.36 (d, m, 2H), 7.02 (d, J = 9 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 171.98, 162.59, 155.67, 152.56, 145.34, 137.90 133.65, 129.54, 128.69, 128.18, 128.09, 127.16, 124.14, 121.43, 120.96, 110.59, 109.18. HRMS (m/z) calculated for C24H17N3O [M + H+] 364.1450, found 364.1459.3.3.4.1 L1-Cu(II) complex (1). Yield: 83%. M.p.: >300 °C. Colour: brown. Anal. calc. (%) for C40H24N6O2CuCl2Br2 Calc. (%) C, 52.51; H, 2.64; N, 9.18. Found (%): C, 52.47; H, 2.63; N, 9.17. IR (ν, cm−1): 3620 (N–Hring), 1587 (azomethine, –C
N), 1447 (C–Nring), 569 (M–O), 490 (M–N). HRMS (m/z) calculated for C40H24N6O2CuCl2Br2 [M + H+] 914.9171, found 916.0054.
3.3.4.2 L1-Ni(II) complex (2). Yield: 67%. Colour: red. M.p.: >300 °C. Anal. calc. (%) for C40H24N6O2Cl2NiBr2: C, 52.79; H, 2.65; N, 9.23. Found: C, 52.80; H, 2.68; N, 9.17. IR (ν, cm−1): 3650 (N–Hring), 1587 (azomethine, –C
N), 1442 (C–Nring), 562 (M–O), 476 (M–N). 1H NMR (400 MHz, DMSO) δ 14.52 (s, 1H), 9.52 (s, 1H), 8.24 (dd, J = 24.3, 6.1 Hz, 4H), 7.83 (t, J = 22.3 Hz, 2H), 7.65 (dd, J = 27.7, 18.9 Hz, 2H), 7.35 (s, 1H), 7.26 (s, 1H), 7.06 (s, 2H), 6.94 (d, J = 7.7 Hz, 1H), 6.65 (d, J = 8.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 170.51, 159.91, 151.65, 148.61–146.26, 142.19, 138.96, 137.03, 121.91–110.52. HRMS (m/z) calculated for C40H24N6O2Cl2NiBr2 [M + H+] 910.1233, found 910.1239.
3.3.4.3 L1-Pd(II) complex (3). Yield: 62%. Colour: brown. M.p.: >300 °C. Anal. calc. (%) for C40H24N6O2Cl2PdBr2: calc. (%): C, 50.10; H, 2.51; N, 8.77. Found (%): C, 50.07; H, 2.48; N, 8.76. IR (ν, cm−1): 3656 (N–Hring), 1598 (azomethine, –C
N), 1442 (C–Nring), 576 (M–O), 461 (M–N). 1H NMR (400 MHz, DMSO) δ 14.41 (s, 1H), 9.50 (s, 1H), 7.91 (d, J = 7.2 Hz, 4H), 7.73–7.65 (m, 6H), 7.51 (s, 2H), 7.05 (d, J = 7.1 Hz, 4H), 6.82 (d, J = 8.2, 4H). 13C NMR (101 MHz, DMSO) δ 166.15, 158.79, 151.75, 148.71, 147.19, 145.82, 144.36, 135.03, 128.61, 127.52, 123.58–11.15. HRMS (m/z) calculated for C40H24N6O2Cl2PdBr2 [M + H+] 958.88, found 958.8786.
3.3.4.4 L1-Zn(II) complex (4). Yield: 77%. Colour: yellow. M.p.: >300 °C. Anal. calc. (%) for C40H24N6O2Cl2ZnBr2: C, 52.40; H, 2.63; N, 9.16. Found: C, 52.38; H, 2.58; N, 9.12. IR (ν, cm−1): 3615 (N–Hring), 1594 (azomethine, –C
N), 1429 (C–Nring), 576 (M–O), 426 (M–N). 1H NMR (400 MHz, DMSO) δ 14.47 (s, 2H), 9.46 (s, 2H), 8.30 (d, J = 7.2 Hz, 4H), 8.01–7.87 (m, 4H), 7.81 (s, 2H), 7.70 (d, J = 7.1 Hz, 4H), 7.62 (s, 2H), 7.23 (dd, J = 17.2, 14.5 Hz, 2H), 6.74 (d, J = 7.1 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 169.21, 158.92, 151.42, 149.73, 146.11, 144.23, 138.82, 137.51, 135.02, 129.41, 128.12, 123.20–111.07. HRMS (m/z) calculated for C40H24N6O2Cl2ZnBr2 [M + H+] 916.9764, found 916.9791.
3.3.5.1 L2-Cu(II) complex (5). Yield: 75%. Colour: green. M.p.: 225 °C. Anal. calc. for C48H32N6O2Cu: Calc. (%): C, 73.03; H, 4.09; N, 10.65. Found (%): C, 72.98H, 4.05; N, 10.61. IR (ν, cm−1): 3622 (N–Hring), 1594 (azomethine, –C
N), 1446 (C–Nring), 540 (M–O), 433 (M–N). HRMS (m/z) calculated for C48H32N6O2Cu [M + H+] 789.2039, found 789.2026.
3.3.5.2 L2-Ni(II) complex (6). Yield: 68%. Colour: dark red. M.p.: >300 °C. Anal. calc. for C48H32N6O2Ni: calc. (%): C, 73.35; H, 4.10; N, 10.69. Found (%): C, 73.37; H, 4.11; N, 10.71. IR (ν, cm−1): 3617 (N–Hring), 1587 (azomethine, –C
N), 1431 (C–Nring), 582 (M–O), 454 (M–N). 1H NMR (400 MHz, DMSO) δ 15.78 (s, 2H), 9.76 (s, 2H), 8.54 (s, 2H), 8.35–8.17 (m, 4H), 8.12 (d, J = 7.1 Hz, 2H), 7.95 (s, 2H), 7.83 (d, J = 22.4 Hz, 2H), 7.69 (d, J = 6.7 Hz, 4H), 7.63–7.49 (m, 6H), 7.38–7.10 (m, 4H), 7.10–6.83 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 181.68, 164.21, 154.69, 150.07, 146.33, 144.0, 138.16, 132.30, 130.50, 129.50, 125.06, 124.03, 123.54–123.41, 122.03, 113.64–113.45, 110.02, 108.39. HRMS (m/z) calculated for C48H32N6O2Ni [M + H+] 785.9635, found 785.9643.
3.3.5.3 L2-Pd(II) complex (7). Yield: 61%. Colour: brown. M.p.: >300 °C. Anal. calc. for C48H32N6O2Pd: calc. (%) C, 69.36; H, 3.88; N, 10.11. Found (%): C, 69.39; H, 3.89; N, 10.12. IR (ν, cm−1): 3615 (N–Hring), 1589 (azomethine, –C
N), 1429 (C–Nring), 554 (M–O), 433 (M–N). 1H NMR (400 MHz, DMSO) δ 15.91 (s, 2H), 10.03 (s, 2H), 8.58 (d, J = 8.8 Hz, 2H), 8.31 (s, 4H), 8.12 (m, 2H), 7.95 (dd, J = 34.4, 25.3 Hz, 2H), 7.84 (d, J = 7.4 Hz, 4H), 7.65 (d, J = 36.5 Hz, 6H), 7.52 (m, 2H), 7.40 (m, 2H), 7.07 (d, J = 9.1 Hz, 4H). 13C NMR (101 MHz, DMSO) δ 182.05, 169.15, 155.54, 150.72, 145.03, 137.90, 133.65, 129.54, 128.69–128.08, 127.14, 124.28–124.13, 122.91, 121.37, 109.14. HRMS (m/z) calculated for C48H32N6O2Pd [M + H+] 831.1700, found 831.1741.
3.3.5.4 L2-Zn(II) complex (8). Yield: 79%. Colour: yellow. M.p.: 211 °C. Anal. calc. (%) for C48H32N6O2Zn: C, 72.78; H, 4.07; N, 10.61. Found (%): C, 72.72; H, 4.00; N, 10.55. IR (ν, cm−1): 3621 (N–Hring), 1594 (azomethine, –C
N), 1436 (C–Nring), 576 (M–O), 475 (M–N). 1H NMR (400 MHz, DMSO) δ 15.75 (s, 2H), 9.86 (s, 2H), 8.56 (d, J = 8.8 Hz, 2H), 8.32 (s, 4H), 8.13–8.10 (m, 2H), 8.05–7.95 (m, 2H), 7.84 (d, J = 7.4 Hz, 4H), 7.65–7.55 (m, 6H), 7.51–7.45 (m, 2H), 7.43–7.37 (m, 2H), 7.07 (d, J = 9.1 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 178.04, 165.74, 156.52, 152.69, 145.11, 142.51, 133.38, 131.26, 128.61, 122.99, 122.28, 121.28, 120.10, 119.20, 111.13, 109.96. HRMS (m/z) calculated for C48H32N6O2Zn [M + H+] 792.0961, found 792.0940.
3.4 DNA binding studies
3.4.2.1 UV-vis absorption spectroscopic studies. Absorption spectroscopic studies were carried out using a PerkinElmer Lambda 35 UV-vis spectrometer. Stock solutions (500 μM) of the compounds were prepared in DMSO, and for further dilutions (100 μM), deionized water was used. Fish sperm DNA (st)DNA was prepared in Tris–HCl buffer solution (0.6 M HCl) and 50 mM NaCl with pH = 7.33, and this buffered DNA was filtered using the syringe filtration method to make it protein-free. The solution of (st)DNA gave a ratio of UV absorbance at 260 and 280 nm, A260/A280 of 1.9, indicating that (st)DNA was sufficiently free of proteins.59 The concentration of (st)DNA was calculated from its absorption intensity at 260 nm with a molar extinction coefficient of 6600 cm−1.60 The solution of (st)DNA and drug (compounds) was incubated for 5 min before recording the absorption spectrum. A fixed quantity (3 mL) of the compound (100 μM) was titrated with an increasing quantity (10–80 μL) of DNA. The intrinsic binding constants (kb) were calculated using the following equation:
where [DNA] represents the (st)DNA concentration in base molarity, A0 represents the absorbance of the drug in the absence of DNA, and A represents the absorbance of the drug in the presence of DNA. εG and εH–G represent the absorption coefficients of the drug and drug–DNA complex, respectively. The intrinsic binding constant Kb can be calculated from the intercept to the slope ratio of A0/A − A0 vs. 1/[DNA] plots.
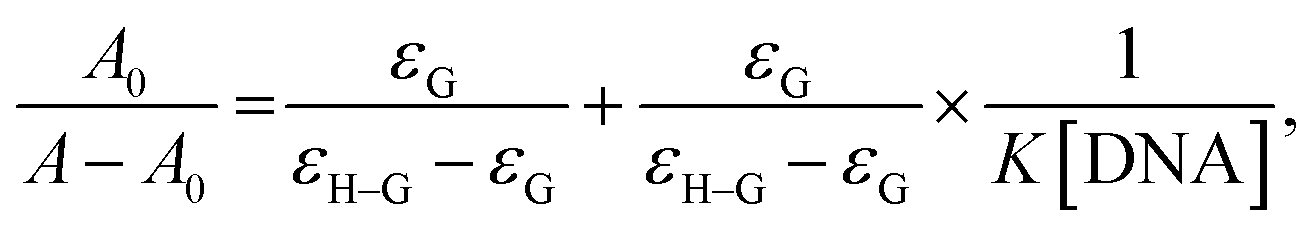
3.4.2.2 Thermal stability of DNA. DNA thermal stability as a function of temperature was investigated by absorbance versus temperature curve. The change in absorbance with the temperature at 260 nm was monitored from 30 °C to 90 °C at a 1 °C per minute scan rate and fixed [drug]/[DNA] concentration ratio. Accordingly, the DNA melting temperature (Tm) was recorded, and the difference between Tm values in the absence and presence of compound (ΔTm) was calculated.
3.5 Biochemistry
4. Conclusion
In search of DNA targeting antimicrobial agents, two novel benzimidazoles “O” and “N” donor Schiff base ligands and their complexes with various transition metals were successfully synthesized and characterized by various spectroscopic techniques. DNA binding affinity of compounds was measured by performing UV/Vis absorption titrations and thermal denaturation experiments. Based on the obtained results, it can be concluded that the ligands, and most of their complexes, can bind with DNA by intercalation and groove binding mode as evidenced by the binding constant value, ranging from 103 to 105 M−1. Among all the complexes, compound 2 was stronger (Kb, 3.27 × 105 M−1) and compound 5 was weaker (Kb, 6.4 × 103 M−1) DNA binder. The in vitro antibacterial studies indicated that various compounds have the potential to inhibit the growth of bacteria, and compound 2 showed broad-spectrum antibacterial potency against M. luteus, S.aureus, E. coli and E. aerogenes with inhibition zone values of 8.5, 10.3, 16.8 and 14.5 mm. The preliminary cytotoxic effect of these compounds was studied on shrimp larvae, and most of the compounds were declared to be non-toxic towards normal cells. Only two compounds, L1 and 7, showed slight toxicity with LD50 values of 127 and 125 μg mL−1, respectively. The compounds showed a marked cytotoxic effect against human breast cancer cells. This leads to the conclusion that the synthesized compounds possessed significant anticancer activity comparable to the activity of commonly used anticancer drugs, cisplatin and doxorubicin. According to the obtained results, the design and synthesis of benzimidazole Schiff base-transition metal complexes were of immense importance to produce novel antimicrobial drug libraries with fewer or no side effects and to expunge the concept of multidrug resistance. Through this study, we want to explain that heterocyclic Schiff base ligands and complexes have all the inherent properties that are required to act as DNA targeting antimicrobial drugs.Conflicts of interest
Authors have no conflict of interest for this research work.Acknowledgements
Khalid Mahmood is highly thankful to HEC Pakistan for providing financial support under the Indigenous 5000 PhD Fellowship Program and for International Research Support Initiative Program (IRSIP). The funding from Quaid-i-Azam University, Islamabad, Pakistan under the University Research Fund (URF) is also greatly acknowledged.References
- G. Barone, A. Terenzi, A. Lauria, A. M. Almerico, J. M. Leal, N. Busto and B. García, DNA-binding of nickel(II), copper(II) and zinc(II) complexes: Structure-affinity relationships, Coord. Chem. Rev., 2013, 257, 2848–2862 CrossRef CAS.
- B. Rosenberg, L. Van Camp and T. Krigas, Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- B. Rosenberg, L. Van Camp, E. B. Grimley and A. J. Thompson, The Inhibition of Growth or Cell Division in Escherichia by Different Ionic Species of Platinum(IV) Complexes, J. Biol. Chem., 1967, 242, 1347–1352 CrossRef CAS PubMed.
- H. A. Wee and P. J. Dyson, Classical and non-classical ruthenium-based anticancer drugs: Towards targeted chemotherapy, Eur. J. Inorg. Chem., 2006, 4003–4018 Search PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, In vitro and in vivo activity and cross resistance profiles of novel ruthenium(II) organometallic arene complexes in human ovarian cancer, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- S. Y. Park, S. J. Yoon, S. itiroh Hakomori, J. M. Kim, J. Y. Kim, B. Bernert, T. Ullman, S. H. Itzkowitz and J. H. Kim, Dimeric Le(a) (Le(a)-on-Le(a)) status of beta-haptoglobin in sera of colon cancer, chronic inflammatory disease and normal subjects, Int. J. Oncol., 2010, 36, 1291–1297 CAS.
- I. Romero-Canelón and P. J. Sadler, Next-generation metal anticancer complexes: Multitargeting via redox modulation, Inorg. Chem., 2013, 52, 12276–12291 CrossRef PubMed.
- L. S. Liao, Y. Chen, Z. Y. Mo, C. Hou, G. F. Su, H. Liang and Z. F. Chen, Ni(II), Cu(II) and Zn(II) complexes with the 1-trifluoroethoxyl-2,9,10-trimethoxy-7-oxoaporphine ligand simultaneously target microtubules and mitochondria for cancer therapy, Inorg. Chem. Front., 2021, 8, 2225–2247 RSC.
- D. Hernández-Romero, S. Rosete-Luna, A. López-Monteon, A. Chávez-Piña, N. Pérez-Hernández, J. Marroquín-Flores, A. Cruz-Navarro, G. Pesado-Gómez, D. Morales-Morales and R. Colorado-Peralta, First-row transition metal compounds containing benzimidazole ligands: An overview of their anticancer and antitumor activity, Coord. Chem. Rev., 2021, 439, 213930 CrossRef.
- T. J. M. N. Farrell, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty, Pergamum, Oxford, 2003, p. 809 Search PubMed.
- A. A. Holder, Inorganic pharmaceuticals, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2013, 109, 317–339 RSC.
- L. Ronconi and P. J. Sadler, Using coordination chemistry to design new medicines, Coord. Chem. Rev., 2007, 251, 1633–1648 CrossRef CAS.
- C. W. Schwietert and J. P. McCue, Coordination compounds in medicinal chemistry, Coord. Chem. Rev., 1999, 184, 67–89 CrossRef CAS.
- A. Hussain, M. F. AlAjmi, T. R. Md, A. A. Khan, P. A. Shaikh and R. A. Khan, Evaluation of transition metal complexes of benzimidazole-derived scaffold as promising anticancer chemotherapeutics, Molecules, 2018, 23, 1–18 Search PubMed.
- E. Alterhoni, A. Tavman, M. Hacioglu, O. Şahin and A. Seher Birteksöz Tan, Synthesis, structural characterization and antimicrobial activity of Schiff bases and benzimidazole derivatives and their complexes with CoCl2, PdCl2, CuCl2 and ZnCl2, J. Mol. Struct., 2021, 1229, 129498 CrossRef CAS.
- A. Aragón-Muriel, Y. Liscano, Y. Upegui, S. M. Robledo, M. Teresa Ramírez-Apan, D. Morales-Morales, J. Oñate-Garzón, D. Polo-Cerón and S. Sinicropi, In Vitro Evaluation of the Potential Pharmacological Activity and Molecular Targets of New Benzimidazole-Based Schiff Base Metal Complexes, Antibiotics, 2021, 10, 728 CrossRef PubMed.
- N. M. Goudgaon, Synthesis , characterization and biological evaluation of novel C-2 substituted benzimidazole heterocycles, J. Pharm. Res., 2015, 9, 643–649 CAS.
- F. Fei and Z. Zhou, New substituted benzimidazole derivatives: a patent review (2010 – 2012), Expert Opin. Ther. Pat., 2013, 23, 1157–1179 CrossRef CAS PubMed.
- N. Raman and A. Selvan, Investigation of DNA binding mechanism, photoinduced cleavage activity, electrochemical properties and biological functions of mixed ligand copper(II) complexes with benzimidazole derivatives: Synthesis and spectral characterization, J. Enzyme Inhib. Med. Chem., 2012, 27, 380–389 CrossRef CAS PubMed.
- A. Nykjaer, J. C. Fyfe, R. Kozyraki, J. Leheste, C. Jacobsen, M. S. Nielsen, P. J. Verroust, M. Aminoff, A. De, S. K. Moestrup, R. Ray, J. Gliemann, T. E. Willnow, I. Erik, P. J. Verroust, M. Aminoff, A. De Chapelle, S. K. Moestrup and R. Raytt, Induced-fit recognition of DNA by organometallic complexes with dynamic stereogenic centers, Proc. Natl. Acad. Sci., 2016, 25, 16281–16286 Search PubMed.
- G. Barone, N. Gambino, A. Ruggirello, A. Silvestri, A. Terenzi and V. T. Liveri, Spectroscopic study of the interaction of NiII-5-triethyl ammonium methyl salicylidene ortho-phenylendiiminate with native DNA, J. Inorg. Biochem., 2009, 103, 731–737 CrossRef CAS PubMed.
- A. Silvestri, G. Barone, G. Ruisi, D. Anselmo, S. Riela and V. T. Liveri, The interaction of native DNA with Zn(II) and Cu(II) complexes of 5-triethyl ammonium methyl salicylidene ortho-phenylendiimine, J. Inorg. Biochem., 2007, 101, 841–848 CrossRef CAS PubMed.
- A. M. Mansour and O. R. Shehab, Lysozyme and DNA binding affinity of Pd(II) and Pt(II) complexes bearing charged N,N pyridylbenzimidazole bidentate ligands, Dalton Trans., 2018, 3459–3468 RSC.
- M. A. Ibrahim and K. M. El-Mahdy, Synthesis and antimicrobial activity of some new heterocyclic schiff bases derived from 2-Amino-3-formylchromone, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 2945–2958 CrossRef CAS.
- K. R. Reddy, K. M. Reddy and K. N. Mahendra, Synthesis, characterization, antibacterial and anthelmentic activities of copper (II) complexes with benzofuran Schiff bases, Indian J. Chem., 2006, 45, 377–381 Search PubMed.
- Z. H. Chohan, H. A. Shad and C. T. Supuran, Synthesis, characterization and biological studies of sulfonamide Schiff's bases and some of their metal derivatives, J. Enzyme Inhib. Med. Chem., 2012, 27, 58–68 CrossRef CAS PubMed.
- S. Sathiyaraj, G. Ayyannan and C. Jayabalakrishnan, Synthesis, spectral, DNA binding and cleavage properties of ruthenium(II) Schiff base complexes containing PPh3/AsPh3 as co-ligands, J. Serb. Chem. Soc., 2014, 79, 151–165 CrossRef CAS.
- L. Ma, L. Li and G. Zhu, Platinum-containing heterometallic complexes in cancer therapy: advances and perspectives, Inorg. Chem. Front., 2022, 9, 2424–2453 RSC.
- S. W. T. Sadtler, Handbook of Proton NMR Spectra, 1978 Search PubMed.
- D. J. Pasto, Organic structure determination, 1969 Search PubMed.
- C. N. Cavasotto, Ligand docking and virtual screening in structure-based drug discovery, AIP Conf. Proc., 2006, 851, 34–49 CrossRef CAS.
- F. Caruso, M. Rossi, A. Benson, C. Opazo, D. Freedman, E. Monti, M. B. Gariboldi, J. Shaulky, F. Marchetti, R. Pettinari and C. Pettinari, Ruthenium-arene complexes of curcumin: X-ray and density functional theory structure, synthesis, and spectroscopic characterization, in vitro antitumor activity, and DNA docking studies of (p-cymene)Ru(curcuminato)chloro, J. Med. Chem., 2012, 55, 1072–1081 CrossRef CAS PubMed.
- L. G. Ferreira, R. N. Santos, G. Oliva and A. D. Andricopulo, Dms43_Vitae[1].Pdf, 2015 Search PubMed.
- A. Kosiha, C. Parthiban, S. Ciattini, L. Chelazzi and K. P. Elango, Metal complexes of naphthoquinone based ligand: synthesis, characterization, protein binding, DNA binding/cleavage and cytotoxicity studies, J. Biomol. Struct. Dyn., 2017, 1102, 1–12 Search PubMed.
- M. Parveen, A. M. Malla, Z. Yaseen, A. Ali and M. Alam, Synthesis, characterization, DNA-binding studies and acetylcholinesterase inhibition activity of new 3-formyl chromone derivatives, J. Photochem. Photobiol., B, 2014, 130, 179–187 CrossRef CAS PubMed.
- R. Fukuda, S. Takenaka and M. Takagi, Metal ion assisted DNA-intercalation of crown ether-linked acridine derivatives, J. Chem. Soc., Chem. Commun., 1990, 2, 1028–1030 RSC.
- M. Sakthi and A. Ramu, Synthesis, structure, DNA/BSA binding and antibacterial studies of NNO tridentate Schiff base metal complexes, J. Mol. Struct., 2017, 1149, 727–735 CrossRef CAS.
- A. Basu and G. Suresh Kumar, Thermodynamic characterization of proflavine-DNA binding through microcalorimetric studies, J. Chem. Thermodyn., 2015, 87, 1–7 CrossRef CAS.
- A. F. Wells, Structural Inorganic Chemistry, Clarendon Press, Oxford, 5th edn, 1984 Search PubMed.
- S. Xiao, W. Lin, C. Wang and M. Yang, Synthesis and biological evaluation of DNA targeting flexible side-chain substituted β-carboline derivatives, Bioorg. Med. Chem. Lett., 2001, 11, 437–441 CrossRef CAS PubMed.
- M. Cory, D. D. Mckee, J. Kagan, D. W. Henry and J. A. Milled, Bifunctional Intercalators. Comparison of Polymethylene, J. Am. Chem. Soc., 1985, 2528–2536 CrossRef CAS.
- C. V Kumar and E. H. Asuncion, DNA Binding Studies and Site Selective Fluorescence Sensitization of an Anthryl Probe, J. Am. Chem. Soc., 1993, 8547–8553 CrossRef.
- B. Selvakumar, V. Rajendiran, P. Uma Maheswari, H. Stoeckli-Evans and M. Palaniandavar, Structures, spectra, and DNA-binding properties of mixed ligand copper(II) complexes of iminodiacetic acid: The novel role of diimine co-ligands on DNA conformation and hydrolytic and oxidative double strand DNA cleavage, J. Inorg. Biochem., 2006, 100, 316–330 CrossRef CAS PubMed.
- Y. An, S. D. Liu, S. Y. Deng, L. N. Ji and Z. W. Mao, Cleavage of double-strand DNA by linear and triangular trinuclear copper complexes, J. Inorg. Biochem., 2006, 100, 1586–1593 CrossRef CAS PubMed.
- G. A. Neyhart, N. Grover, S. R. Smith, W. A. Kalsbeck, T. A. Fairley, M. Cory and H. H. Thorp, Binding and kinetics studies of oxidation of DNA by Oxoruthenium(IV), J. Am. Chem. Soc., 1993, 115, 4423–4428 CrossRef CAS.
- A. A. Osowole, R. Kempe and R. Schobert, Synthesis , Spectral , Thermal , In-vitro Antibacterial and Anticancer Activities of some Metal (II) Complexes of 3- (1- (4-methoxy-6-), Int. Res. J. Pure Appl. Chem., 2012, 2, 105–129 CrossRef CAS.
- G. Ceyhan, C. Çelik, S. Uruş, I. Demirtaş, M. Elmastaş and M. Tümer, Antioxidant, electrochemical, thermal, antimicrobial and alkane oxidation properties of tridentate Schiff base ligands and their metal complexes, Spectrochim. Acta, Part A, 2011, 81, 184–198 CrossRef CAS PubMed.
- H. S. I. Bertini and A. Sigel, Handbook on Metalloproteins, Marcel Dekker, New York, 2001 Search PubMed.
- S. N. Gajbhiye and R. Hirota, Toxicity of Heavy Metals To Brine Shrimp Artemia, J. Indian Fish. Assoc., 1990, 20, 43–50 Search PubMed.
- J. Lacroix, A. Patenaude and J. L. C. Rousseau, N -Phenyl-N0-(2-chloroethyl)ureas (CEUs) as potential antineoplastic agents. Part 3: Role of carbonyl groups in the covalent binding to the colchicine-binding site, Bioorg. Med. Chem., 2008, 16, 1206–1217 CrossRef PubMed.
- B. Thati, A. Noble, B. S. Creaven, M. Walsh, M. Mccann, K. Kavanagh and M. Devereux, In vitro anti-tumour and cyto-selective effects of coumarin-3-carboxylic acid and three of its hydroxylated derivatives , along with their silver-based complexes, using human epithelial carcinoma cell lines, Cancer Lett., 2007, 248, 321–331 CrossRef CAS PubMed.
- B. S. Creaven, B. Duff, D. A. Egan, K. Kavanagh, G. Rosair, V. Reddy and M. Walsh, Inorganica Chimica Acta Anticancer and antifungal activity of copper (II) complexes of quinolin-2 (1H) -one-derived Schiff bases, Inorg. Chim. Acta, 2010, 363, 4048–4058 CrossRef CAS.
- P. Taylor, Synthesis, Characterization , and Antiproliferative Activity of Cu , V (IV) O , Co , Mn , and Ni Complexes with 3- ( 2- ( 4- Methoxyphenylcarbamothioyl)Hydrazinyl)-3-OXO-N-(Thiazol-2-yl), Phosphorus, Sulfur Silicon Relat. Elem., 2014, 37–41 Search PubMed.
- K. Mahmood, W. Hashmi, H. Ismail and B. Mirza, Synthesis , DNA binding and antibacterial activity of metal (II) complexes of a benzimidazole Schiff base, Polyhedron, 2018, 157, 326–334 CrossRef.
- D. H. Petering, R. W. Byrnes and W. E. Antholine, Oxidation-reduction reactions in ehrlich cells treated with copper-neocuproine, Free Radical Biol. Med., 1992, 13, 469–478 CrossRef PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- https://www.rcsb.org/structure/1w0t.
- I. M. El-Deen, A. F. Shoair and M. A. El-Bindary, Synthesis, structural characterization, molecular docking and DNA binding studies of copper complexes, J. Mol. Liq., 2018, 249, 533–545 CrossRef CAS.
- M. E. Reichmann, C. A. Rice, C. A. Thomas and P. Doty, A further examination of the molecular weight and size of deoxypentose nucleic acid, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- M. Zaheer, A. Shah, Z. Akhter, R. Qureshi, B. Mirza, M. Tauseef and M. Bolte, Synthesis, characterization, electrochemistry and evaluation of biological activities of some ferrocenyl Schiff bases, Appl. Organomet. Chem., 2011, 25, 61–69 CrossRef CAS.
- I. Ul-Haq, N. Ullah, G. Bibi and S. Kanwal, Antioxidant and cytotoxic activities and phytochemical analysis of Euphorbia wallichii root extract and its fractions, Pharm. Res., 2012, 11, 241–249 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR, IR and HRMS spectra of the compounds/complexes presented in this article. CCDC 2047147 (L1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra00982c |
| This journal is © The Royal Society of Chemistry 2023 |