
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D printing of PLA:CaP:GO scaffolds for bone tissue applications†
L. González-Rodríguez *ab,
S. Pérez-Davilaab,
R. Lamac,
M. López-Álvarezab,
J. Serraab,
B. Novoac,
A. Figuerasc and
P. Gonzálezab
*ab,
S. Pérez-Davilaab,
R. Lamac,
M. López-Álvarezab,
J. Serraab,
B. Novoac,
A. Figuerasc and
P. Gonzálezab
aCINTECX, Universidade de Vigo, Grupo de Novos Materiais, 36310 Vigo, Spain. E-mail: laugonzalez@uvigo.gal
bGalicia Sur Health Research Institute (IIS Galicia Sur), SERGAS-UVIGO, 36213 Vigo, Spain
cInstitute of Marine Reseach (IIM), CSIC, Eduardo Cabello 6, 36208, Vigo, Spain
First published on 30th May 2023
Abstract
Graphene oxide (GO) has attracted increasing interest for biomedical applications owing to its outstanding properties such as high specific surface area, ability to bind functional molecules for therapeutic purposes and solubility, together with mechanical resistance and good thermal conductivity. The combination of GO with other biomaterials, such as calcium phosphate (CaP) and biodegradable polymers, presents a promising strategy for bone tissue engineering. Presently, the development of these advanced biomaterials benefits from the use of additive manufacturing techniques, such as 3D printing. In this study, we develop a 3D printed PLA:CaP:GO scaffold for bone tissue engineering. First, GO was characterised alone by XPS to determine its main bond contributions and C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio. Secondly, we determined the GO dose which ensures the absence of toxicity, directly exposed in vitro (human osteoblast-like cells MG-63) and in vivo (zebrafish model). In addition, GO was microinjected in the zebrafish to evaluate its effect on immune cells, quantifying the genetic expression of the main markers. Results indicated that the GO tested (C:O of 2.14, 49.50% oxidised, main bonds: C–OH, C–O–C) in a dose ≤0.25 mg mL−1 promoted MG63 cells viability percentages above 70%, and in a dose ≤0.10 mg mL−1 resulted in the absence of toxicity in zebrafish embryos. The immune response evaluation reinforced this result. Finally, the optimised GO dose (0.10 mg mL−1) was combined with polylactic acid (PLA) and CaP to obtain a 3D printed PLA:CaP:GO scaffold. Physicochemical characterisation (SEM/EDS, XRD, FT-Raman, nano-indentation) was performed and in vivo tests confirmed its biocompatibility, enabling a novel approach for bone tissue-related applications.
:O ratio. Secondly, we determined the GO dose which ensures the absence of toxicity, directly exposed in vitro (human osteoblast-like cells MG-63) and in vivo (zebrafish model). In addition, GO was microinjected in the zebrafish to evaluate its effect on immune cells, quantifying the genetic expression of the main markers. Results indicated that the GO tested (C:O of 2.14, 49.50% oxidised, main bonds: C–OH, C–O–C) in a dose ≤0.25 mg mL−1 promoted MG63 cells viability percentages above 70%, and in a dose ≤0.10 mg mL−1 resulted in the absence of toxicity in zebrafish embryos. The immune response evaluation reinforced this result. Finally, the optimised GO dose (0.10 mg mL−1) was combined with polylactic acid (PLA) and CaP to obtain a 3D printed PLA:CaP:GO scaffold. Physicochemical characterisation (SEM/EDS, XRD, FT-Raman, nano-indentation) was performed and in vivo tests confirmed its biocompatibility, enabling a novel approach for bone tissue-related applications.
1. Introduction
Graphene oxide (GO) and its derivatives have attracted growing interest for biomedical applications owing to their outstanding properties, such as high specific surface area, mechanical resistance, and good thermal conductivity,1 along with a higher reactivity, greater solubility, and stability in biological fluids compared to graphene.2 The latter properties, achieved by the incorporation of different oxygenated functional groups, including hydroxyls, epoxides and carboxyl groups, make GO hydrophilic and therefore able to bind to functional molecules through covalent bonding or electrostatic interactions. This functional flexibility makes the 2D nano-layered material an excellent transporter of molecules for therapeutic purposes in the physiological environment.3 Moreover, several studies have shown that GO can significantly promote cell growth by improving mammalian cell attachment and proliferation, related with the wealthy oxygen-containing functional groups.4–7 However, the ability of graphene oxide to generate reactive oxygen species, implying a potential mechanism of cytotoxicity on eukaryotic cells, microorganisms and organisms, has also been reported, triggering metabolic reactions that end in cell death.8,9 Therefore, its use as antitumor and antimicrobial agent is likewise being investigated.From each of these potential applications, the enormous relevance of determining the toxicity threshold in terms of the GO dose, sheet size, oxidation state, and route of administration both in vitro and in vivo is evident. To date, numerous in vitro studies have been carried out, mostly using fibroblasts and different cancer cells to evaluate their response when exposed to different doses of GO. The degree of toxicity found was shown to be a function of the physicochemical properties of GO and experimental conditions.7 Different GO forms have been evaluated, including in solution versus immobilised GO, concluding the influence of the oxidation state and a protective role of GO solutions against immobilisation at certain concentrations.1
In vivo studies investigated the effects of different doses, generally quantified in mg of GO per kg of animal body weight, and routes of entry of GO nanoparticles. The most commonly used animals so far have been rodents, and the most tested route of administration has been via intravenous injection, although there are also results from GO exposed by intratracheal, intraperitoneal, or dermal routes. Several authors furthermore concluded that the diameter of GO nanosheets influences the distribution, with generally higher accumulation in different organs for diameters in the range 10–30 nm, although the results of inflammatory response in this case will also depend on the route of administration.10
Apart from the in vitro and in vivo evaluation of GO doses and routes of administration, the GO combination with other biomaterials is likewise of interest to complement its properties and develop advanced biomaterials. Biodegradable polymers (including collagen, chitosan, alginate, polyvinyl alcohol, and polylactic acid [PLA]) and calcium phosphates are among the most interesting for bone tissue applications.11 Several works have obtained 3D porous scaffolds combining PLA and CaP by traditional methodologies, such as solvent-casting or phase-separation, as in the research published by Charles-Harris et al. (2008),12 indicating their biocompatibility in vitro and the relevance of the final architecture on the cell's behaviour. In relation to this last aspect, the advantages provided by the additive manufacturing to fabricate customised 3D structures, controlling the geometry and inner architecture of the structure, has promoted several authors to investigate the obtaining of PLA scaffolds,13 CaP scaffolds14,15 or combination of both16 using this 3D printing technology. Thus, for instance, in the particular case of Nevado et al. (2020) a filament of PLA-CaP composite was obtained by hot-melt extrusion to be then used in a filament printer.17
In the case of GO, it has been recently combined with the biodegradable polymer chitosan and CaP to obtain composite membranes by vacuum filtration and biomimetic mineralisation fabrication methods.18 Moreover, its combination with fish gelatin has also been investigated and the in vivo effects when implanted ectopically.19 Scaffolds of polyethylene oxide with GO were also fabricated by electrospinning and their no inflammatory response was confirmed when implanted in animal model.20 The use of 3D printing to obtain scaffolds based on GO has been recently addressed by Z. Cheng et al. (2020) proposing a bio-ink composite of GO, collagen and chitosan to be used in 3D printing to fabricate cartilage scaffolds.21 However, to our knowledge, the direct combination of PLA, CaP and GO in a pellets printer to obtain at the time 3D scaffolds for bone tissue applications has not yet been investigated.
In present work, the development of a 3D printed PLA:CaP:GO scaffolds for bone tissue engineering is addressed. Physicochemical characterisation of GO alone (XPS) was first carried out to determine its main bond contributions. Secondly, its biological response was evaluated in vitro (MG-63 cell line) and in vivo in the zebrafish model (embryos and larvae of Danio rerio) when directly bioavailable from the scaffold degradation, in doses from 0.0005 to 0.5 mg mL−1. The inmune response was also evaluated in vivo after being locally microinjected. The resulting GO dose that guarantees the absence of toxicity from the biological evaluation was the one incorporated to the versatile 3D printer of pellets, together with the corresponding amounts of PLA and CaP. Physicochemical (SEM/EDS, XRD, FT-Raman), mechanical (nano-indentation) and preliminar biological characterisation of the 3D printed PLA:CaP:GO scaffolds is also presented.
2. Materials and methods
2.1. Raw materials
Natural polylactic acid, in pellets form (Luminy® LX175, CAS No. 9051-89-2), was acquired from Filament2print (Spain), which claim that it is a material with characteristics such as a density of 1.24 g cm−3, a tensile modulus of 3500 MPa and a glass transition temperature of 60 °C, among others. The calcium phosphate, in this case as hydroxyapatite powder (Captal®, CAS No. 12167-74-7), was acquired from Plasma-Biotal Limited (UK). Its particles are spherical (average particle size 3.29 μm) and consists in a high-purity (>95%) bio-resorbable porous bone substitute material, with a high surface area (BET) >30 m2 g−1 and nano-sized crystallites. An aqueous suspension of graphene oxide sheets (0.5 mg mL−1) with a 95% elemental content of C, H, N, S and O and CAS No. 1034343-98-0 was the GO raw material used (Graphenea, C1703/GOB151/D).2.2. 3D printing
To obtain the 3D printed PLA:CaP:GO scaffolds, GO sheets were first carefully and repeatedly mixed, at the desired dilution from the original aqueous suspension (0.5 mg mL−1), with the PLA and the CaP powder in a Petri dish to unite them three into a single mass. The amount of CaP varied from 3.4–13.2 wt% of the total mass of the scaffold and GO contribution varied from 0.02–0.002 wt%, including the ideal one optimised during the results section. This single mass was then dried in a laboratory stove at 45 °C for 12 h, mixed again and introduced into the 3D printer (TUMAKER Voladora NX Pellet). Scaffolds with dimensions of 8 mm diameter and 2.5 mm height were obtained in a rectilinear framework, defined by a filling patern of 45/−45° angle, according to the printer parameters. This 3D printer based on fused deposition modelling (FDM) technology works with two temperature control points, whose temperature is adjusted according to the material of the pellet to be extruded. In our case the temperatures were 160 and 230 °C, corresponding to extruders 1 and 2, respectively. The samples were printed using a bed temperature of 45 °C and a manufacturing process speed of 60 mm s−1. Furthermore, Fig. S1† (ESI) shows a simulation using Simplify3D Professional Software (Version 4.1.2) of the printing process (previously designed in an STL file by SolidWorks 2016 software). 3D printed scaffolds free of GO (PLA:CaP) were also obtained to be used as reference.2.3. Physicochemical and mechanical characterisation
The GO sheets morphology was analysed by transmission electron microscopy (TEM) with a JEOL JEM-1010 microscope model (CACTI, UVigo) and their elemental composition was characterised by X-ray photoelectron spectroscopy (XPS) using a Thermo Scientific K-Alpha ESCA instrument (CACTI, UVigo) equipped with a hemispherical electron analyser and micro-focused monochromatic aluminium Al Kα X-ray source (1486.6 eV). The morphological characterisation of 3D printed PLA:CaP:GO scaffolds was addressed with a Stereo Microscope Nikon SMZ1500 to obtain 3D optimal images and with a scanning electron microscope (SEM) JEOL JSM-6700F high-resolution. The elemental surface composition was detected by EDS using an Oxford Inca Energy 300 coupled to the SEM equipment. The crystalline structure of GO sheets alone and the one of the 3D printed PLA:CaP:GO scaffolds was evaluated in both by X-ray diffraction (XRD) in a X'Pert Pro Panalytical system (CACTI, UVigo). Fourier – transform Raman spectroscopy (FT-Raman) was also performed using a Horiba HR800 system with a 632.82 nm line from an HeNe laser, a power of 0.011 mW under a 100× magnifying objective of the microscope and a spectral resolution of ±1 cm−1. This made it possible to identify the main molecular vibrations, which were then assigned to the corresponding chemical bonds both for GO sheets alone and fabricated scaffolds. In order to give an insight into the mechanical properties of scaffolds, a nanoindenter XP model of MTS NanoSystem was used to measure hardness and Young's Modulus values with a 100 nm radius triangular pyramid indenter tip (Berkovich-type indenter) and the CSM (Continuous Stiffness Measurement) to perform dynamic measurements as a function of depth and XP head. A total of 30 indentations were carried out and the average of the valid results was calculated ±standard deviation.2.4. Cell culture and in vitro viability study: MG-63 cell line
Prior to the obtaining of the 3D printed PLA:CaP:GO scaffolds the GO toxicity threshold was determined in vitro with the osteoblast-like cell line MG-63. A known volume of the original 0.5 mg mL−1 aqueous GO suspension was first subjected to centrifugation with a Sigma 2-6 Sartorius laboratory centrifuge for a cycle of 20 min at 4000 rpm. Then, the aqueous supernatant was carefully removed and the GO sheets, deposited on the bottom, were resuspended in the same volume of cell growth medium. With this procedure, the original concentration of GO sheets is maintained, while the cells' exposure to water is avoided. The growth medium used was DMEM with glucose and L-glutamine (Lonza), supplemented with 10% foetal bovine serum (FBS, HyClone) and 1% of a combination of amphotericin B, penicillin, and streptomycin in a solution 100× (Sigma-Aldrich). Next, different dilutions were prepared from the resuspended 0.5 mg mL−1 GO in supplemented DMEM to obtain GO concentrations of: 0.25, 0.10, 0.05, 0.005, and 0.0005 mg mL−1.All the dilutions were tested with osteoblast-like cell line MG-63 (ECACC, catalogue no.: 86051601), including the 0.5 mg mL−1 suspension in growth medium. To this end, a volume of 100 μL of 7 × 104 cells per mL of supplemented DMEM was initially cultured in 96-microwells and incubated at 37 °C and 5% of CO2 in humidified atmosphere. After 3 days, when the desired confluence was obtained, the supplemented DMEM was replaced by the prepared GO dilutions and incubated for 24 h. A phenol solution at 6.4 mg mL−1 in supplemented DMEM and the supplemented DMEM itself were both incorporated as positive and negative controls of cytotoxicity, respectively. These were also used to validate the healthy stage of cells.
After the incubation time, cell viability was measured with the MTS colorimetric test, based on the reduction of the tetrazolium compound MTS (3-[4,5, dimethylthiazol-2-yl]-5-[3-carboxymethoxy-phenyl]-2-[4-sulfophenyl]-2H-tetrazolium) by NAD(P)H-dependent dehydrogenase enzymes in metabolically active mammalian cells. The formazan dye was quantified using a Bio-Rad Model 550 microplate spectrophotometer at 490 nm. The obtained absorbance values are proportional to the cell viability. Four replicates per concentration were used, and the experiment was repeated three times. Wells with the corresponding GO concentration without cells were also tested in same conditions and subjected to the MTS colorimetric test as blank control to avoid false positives. Finally, a quantitative evaluation of the viability percentages was carried out according to the scale of cytotoxicity previously described by Rodríguez-López et al., where no toxicity is considered to exist at >90% of cell viability and severe toxicity occurs at viabilities below 30%, with a mild to severe range in between.22
2.5. Toxicity studies: zebrafish model
The GO toxicity threshold and immune response was then determined with zebrafish model. The in vivo biocompatibility of extracts obtained from the 3D printed PLA:CaP:GO scaffolds was also carried out as described below.Embryos and larvae were obtained from IIM-CSIC experimental facilities, where the animals were maintained using established protocols.23,24 Different fish lines were used: wild type (WT) zebrafish and the transgenic lines Tg-(mpx:GFP) and Tg-(mpeg:mCherry), with neutrophils and macrophages labelled, respectively. All experimental procedures were reviewed and approved by the CSIC National Committee of Bioethics under approval number ES360570202001/21/FUN.01/INM06/BNG01.
A corresponding volume of the original 0.5 mg mL−1 aqueous GO suspension was diluted in the zebrafish water, whose composition was previously specified,25 or distilled water, depending on the experiment, to obtain desired final concentrations in the 0.25–0.0005 mg mL−1 range. In the case of PLA:CaP:GO scaffolds, the evaluated extracts were obtained after 30 days at 37 °C under agitation (60 rpm), according to a protocol following the UNE-EN-ISO 10993-13:2009 standard. These were also immersed, in this case, in zebrafish water. Different concentrations were used in each assay depending on the results of previous tests or according to the possibilities of the technique. Both the zebrafish water, necessary for the maintenance of young fish, and distilled water, which simulates the solvent in the GO solution, were incorporated alone as controls. Furthermore, for the extracts assays, a control obtained under the same conditions and from PLA alone was added. This control was already shown to be safe.
2.5.1.1 Bath exposure. In the bath experiments, each tested suspension was diluted in a final volume of 6 mL of zebrafish water and assays were performed in six-well plates. The available concentrations of GO were in the 0.25–0.0005 mg mL−1 range, while the extracts used for these tests were those obtained from various scaffolds with different compositions of both CaP (in proportion as indicated in Section 2.1) and GO (0.02–0.002 wt%). Embryos and larvae were maintained at 28 °C and both viability and hatching of embryos and larvae survival were monitored daily, up to 72 h for embryo assays and 7 days for larvae. Each exposure method was performed in three biological replicates (n = 30) for each condition and repeated twice. A control of distilled water diluted in a 1
:2 ratio in zebrafish water was added in all cases.
2.5.1.2 GO microinjection. To assess whether GO can cause fatal damage by spreading through the bloodstream of the larvae, various concentrations of GO were introduced into the systemic circulation by microinjection in the duct of Cuvier (DC) according to the following protocol: larvae were anesthetised in zebrafish water containing 160 μg mL−1 MS-222 (Sigma-Aldrich) and placed on an agarose plate to be individually microinjected with 2 nL of selected GO concentrations using a glass microneedle from the Narishige MN-151 micromanipulator and a FemtoJet 4x microinjector (Eppendorf). Only the two highest GO concentrations of 0.25 and 0.1 mg mL−1 were tested by this route. After microinjection, larvae were maintained at 28 °C, and their mortality was recorded. The resulting data were used to calculate the survival rate. Each assay was performed twice, on a total of 30 embryos per condition, and with the reference of a control based on distilled water.
The expression of specific genes involved in the immune response after GO exposition was analysed in the aforementioned WT fish. To this end, three biological replicates, five larvae per replicate, were microinjected with defined GO dilution of 0.1 mg mL−1. Total RNA was isolated after 24 hpe using the Maxwell® RSC simplyRNA Tissue kit (Promega) and cDNA synthesised following the supplier's instructions (NZY First-Strand cDNA Synthesis Kit, NZYtech). Subsequently, quantitative analysis was carried using a 7300 Real-Time PCR System (Applied Biosystems). The amplification of cDNA corresponding to interleukin 1-beta (il1β) and tumour necrosis factor alpha (tnfa), two of the most important genes during the inflammatory reaction, as well as tp53, an evolutionarily conserved tumour suppressor, which also regulates innate and adaptive immune responses, were examined based on their specific primers.26,27 Analyses were performed using 0.5 μg of cDNA and the SYBR Green PCR Master Mix (Thermo Fisher Scientific) in a total PCR volume of 25 μL. The temperature cycles were as follows: (1) initial denaturation at 95 °C for 10 min, (2) 40 cycles of denaturation at same temperature for 15 s each, and (3) one hybridisation-elongation at 60 °C for 1 min. The transcription levels of the target genes were normalised using the Pfaffl method,28 and 18S ribosomal RNA (18S) was selected as reference gene.
2.6. Data analysis
GraphPad and SPSS software were used for both data processing and statistical analysis. Most results were expressed as mean values ±standard error of the mean. Non-parametric and parametric tests were used to calculate statistical significance, depending on the characteristics of the variables (Mann–Whitney U for osteoblasts data exclusively and t-Student for remaining assays). For survival experiments, Kaplan–Meier cumulative survival curves were analysed using the log-rank (Mantel–Cox) test. Significant differences are displayed as *** (p < 0.001), ** (0.001 < p < 0.01) or * (0.01 < p < 0.05) in all cases.3. Results and discussion
3.1. Physicochemical characterisation of GO
The morphological characterisation of GO was performed by TEM microscopy and it is presented in the ESI (Fig. S2(a–c)),† revealing their sheet-like structure, with a thickness of the sheet folds of 9.53 ± 0.01 nm (Fig. S2(b)†). In addition, the crystallographic evaluation by the inverted SAED pattern is also provided, Fig. S2(d)† in the hexagonal system, where crystal planes where crystal planes (2,−1,−1,0), (1,−1,0,0), (0,−1,1,0) and (−1,−1,2,0) were identified in accordance with the study by Pandey, who proposed a GO structure with epoxy, hydroxyl and carbonyl groups randomly attached to the carbon structure.25 The quantitative analysis of the GO elemental composition was also evaluated in depth by XPS to confirm, the major expected contributions of carbon (C 1s, 67.24 at%), followed by oxygen (O 1s, 31.37 at%) with a C:O ratio of 2.14. The general XPS spectrum and high-resolution spectra for the transitions are also presented at Fig. S2(e–g),† where the two characteristic peaks at 532.15 eV and at 285.46 eV, corresponding to the oxygen (O 1s) and carbon (C 1s), were respectively detected. The high-resolution XPS spectra for the electronic transitions of C 1s and O 1s are also provided.
In order to identify the major type of bonds present at the tested GO sheets, both spectra were deconvoluted. Table 1 summarizes the main bond contributions attributed for binding energy after the deconvolutions of C 1s and O 1s transitions. Going into detail, C 1s spectrum was deconvoluted in two main contributions: an intense peak at 284.80 eV attributed to C–C, C–H bonds, another intense peak at 286.92 eV attributed to C–OH, C–O–C, and a third contribution at 288.6 eV from a deconvoluted peak at the shoulder region attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. In relative percentage the major contribution was for C–C and C–H bonds (50.5%), followed by C–OH and C–O–C (43.17%) and a minor contribution of double bonds CO (6.33%). According to the deconvolution of the O 1s high-resolution spectrum three contributions were also identified: the main one with an intense band at 532.80 eV attributed to C–OH, C–O–C bonds (representing the 82.89% in relative percentage) and two less intense ones at 532.10 and 531.38 eV attributed to CO bonds and oxidised sulphur, respectively.
O. In relative percentage the major contribution was for C–C and C–H bonds (50.5%), followed by C–OH and C–O–C (43.17%) and a minor contribution of double bonds CO (6.33%). According to the deconvolution of the O 1s high-resolution spectrum three contributions were also identified: the main one with an intense band at 532.80 eV attributed to C–OH, C–O–C bonds (representing the 82.89% in relative percentage) and two less intense ones at 532.10 and 531.38 eV attributed to CO bonds and oxidised sulphur, respectively.
| C 1s | |||
|---|---|---|---|
| C–C, C–H | C–OH, C–O–C | CO |
|
| Binding energy (eV) | 284.80 eV | 286.92 eV | 288.60 eV |
| Bond contribution (rel. %) | 50.50% | 43.17% | 6.33% |
|
|||
| O 1s | |||
| C–OH, C–O–C | CO |
Oxidised sulphur | |
| Binding energy | 532.80 eV | 532.10 eV | 531.38 eV |
| Bond contribution (rel. %) | 82.89% | 10.43% | 6.68% |
These XPS results indicated that almost the 50% of the whole carbon detected was combined with oxygen (49.50%), which confirms that the graphene is oxidised in a great extent. Moreover, carbon to oxygen single bonds (C–OH, C–O–C) were more abundant than carbon to oxygen double bonds (CO). This preference of the single bond conformation under mild oxidation, results in favour of hydroxyl (C–OH) and carbonyl groups (CO), and is in agreement with a previous work,29 where the increase of biocompatibility of highly oxidised graphene-based materials is likewise demonstrated. In particular hydroxyl (–OH) functional groups, when present on a surface represent a hydrophilic surface,30 particularly reactive and extensively studied for the functionalisation of GO.31 Despite the fact that hydrophilic functionality corresponds in general to a reduced protein bonding, it provides significant inhibition of leukocyte adhesion and macrophage fusion, resulting in decreased cytokine secretion and attenuated inflammatory reactions. Moreover, in relation to bone tissue related applications, –OH functionality has been published to increase the levels of mineralisation of osteoblasts, as opposed to other functional groups.32,33
The GO sheets were also subjected to XRD analysis for structural analysis and again provided as ESI in Fig. S3(a)† with prominent diffraction peak at 2θ = 9.80 ± 0.01°, corresponding to the crystal plane (002).34 Fig. S3(b)† also incorporates the FT-Raman spectrum, where the typical G (1581 cm−1) and D (1342 cm−1) bands are represented.35–37
Following the physicochemical characterisation of the GO nanoparticles, which confirmed their sheet-like appearance and a composition based on carbons mostly together with oxygen groups by single bonds, which brings a certain degree of amorphousness to the GO crystalline structure, its toxicity was also evaluated. For it, GO alone in different doses was directly exposed to both a cell line and zebrafish model to select the concentration which guarantees the biological safety of the PLA:CaP:GO scaffold, given that once the scaffold degrades GO will be fully bioavailable.
3.2. GO in vitro viability
In the context of biological characterisation, the first test conducted was a cell culture assay used to determine a biocompatibility range of the GO in contact with osteoblastic-like cells. Thus, cellular trials were carried out using cell line MG-63 in the presence of different GO dilutions. Fig. 1 presents the viability percentage, calculated with respect to the negative control. As shown, only cells treated with the highest concentration of GO (0.5 mg mL−1) decrease their viability to a value (64%) below that as “tolerable toxicity” according to UNE-EN-ISO 10993-5 standard, the limit of which is marked by a dotted line (70%). Additionally, the remaining doses (from 0.1 to 0.0005 mg mL−1) allow for an acceptable cell viability (above 73%). This is consistent with what has been found in a previous study, where the same concentrations of nano-sheet like GO resulted in the same viability percentages after 24 h exposure with MCF-7 tumour cells.38 Identical findings were obtained by another group who, using the same methodology but with carcinoma cells (A549 cell line), observed a viability ≥80% for concentrations of 0.125 mg mL−1 of GO in solution and below.39 A similar pattern of results was obtained by J. Wu et al., who found slightly higher viability at the highest concentrations of GO (0.5 and 0.1 mg mL−1) in contact with breast adenocarcinoma cells.40 In Table 2, the equivalences between cellular viability and values in cytotoxicity scale were summarised, being “0” the lowest toxicity value and “4” the highest one. In this sense, it was demonstrated that the cytotoxicity of GO in contact with MG-63 was dose-dependent, and there is a clear range of cellular biocompatibility to be considered.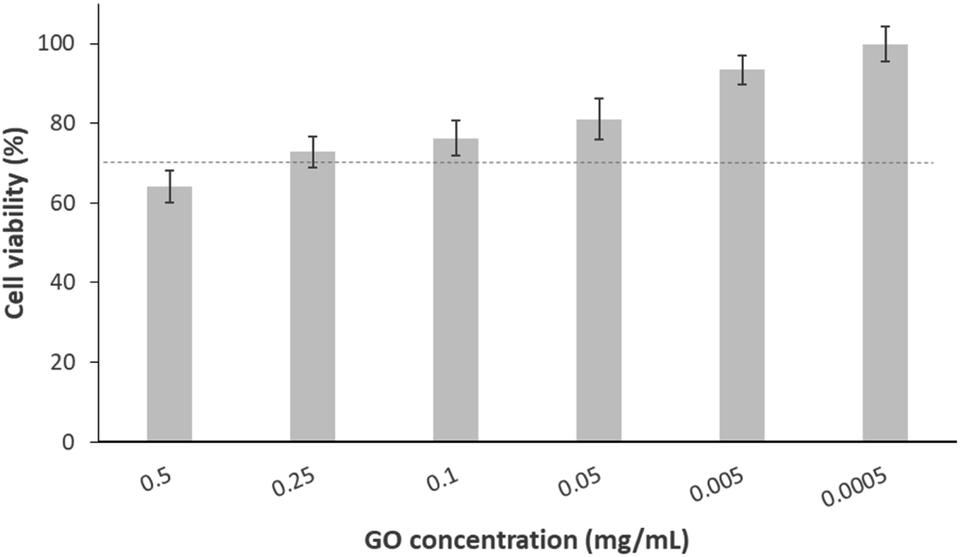 | ||
| Fig. 1 Cell viability of MG-63 after exposure to different concentrations of GO for 24 h. The dotted line indicates the acceptable limit of cytotoxicity according to the UNE-EN-ISO 10993-5:2009 standard. Data are presented as mean ±cumulative error. | ||
| GO concentration (mg mL−1) | 0.5 | 0.25 | 0.1 | 0.05 | 5 × 10−3 | 5 × 10−4 |
|---|---|---|---|---|---|---|
| Viability percentage | 64% | 73% | 76% | 81% | 93% | 99% |
| Value on cytotoxicity scale | 2 | 2 | 2 | 1 | 0 | 0 |
3.3. GO in vivo viability
The next step was the evaluation of the toxic potential of GO nanosheets in vivo using the zebrafish model. Fig. 2 reports an overview of the influence of different concentrations of GO, discarding the stock solution of 0.5 mg mL−1, which was highly toxic to the cells in the previous in vitro study. The effect was analysed by bath exposure in embryos in the first hours after fertilisation and in their hatchability, as well as in 3 dpf larvae, after exposure to GO by bath or microinjection.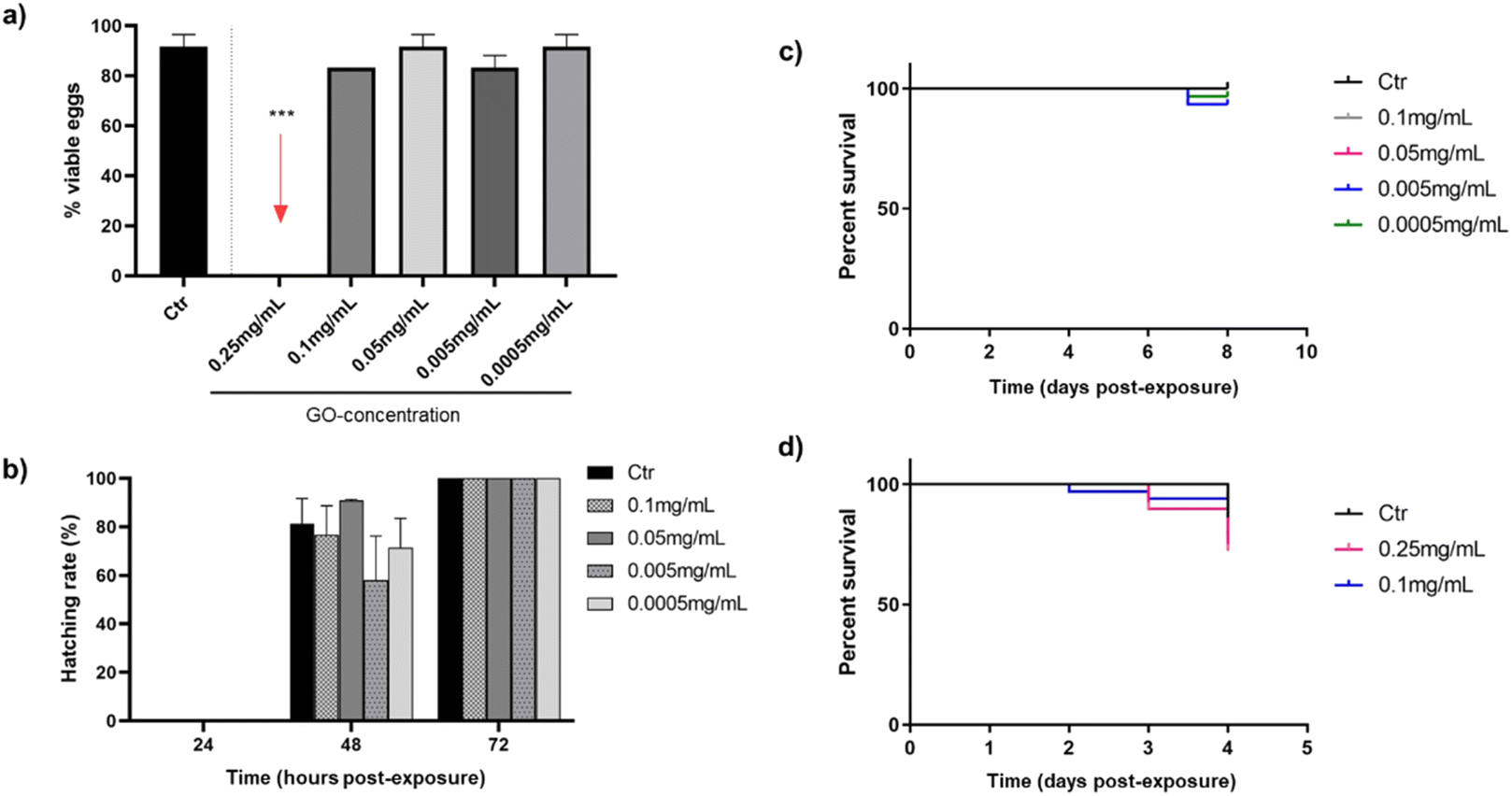 | ||
| Fig. 2 Toxicity response of zebrafish embryos and larvae at different concentrations of GO through two exposition routes: viability rate of embryos at 24 hpe by bath (a), hatching rate during 72 hpe (b), viability rate of larvae exposed to GO by bath for 7 dpe (c) and viability rate of larvae exposed to GO by microinjection for 72 hpe (d). In all cases the results are shown with respect to the control (Ctr). | ||
As shown in Fig. 2(a), only the highest concentration of GO (0.25 mg mL−1) was toxic after 24 h in contact with embryos, as none were viable. These lethal consequences of the highest tested GO concentration appear to be consistent with some authors that associate mitochondrial and metabolic damage caused by ROS in the embryonic development of zebrafish after translocation of GO from water to fish.42 Nevertheless, after 72 h of GO exposure by bath, all concentrations that were not toxic to the embryos in the first 24 h (0.1–0.0005 mg mL−1) allow them to hatch completely (Fig. 2(b)). In addition, mortality caused by bath exposure of zebrafish larvae to GO is represented in Fig. 2(c), showing no differences in survival rates between the control group and the GO-exposed groups after seven days of immersion. In this case, the effect of the 0.25 mg mL−1 concentration was no longer evaluated, because it had already been proved toxic to the embryos by bath, but it was considered necessary to assess the response of zebrafish larvae by microinjecting the GO nanosheets. In this respect, the response of larvae to the two highest GO concentrations (0.25 and 0.1 mg mL−1) after being microinjected into the DC is shown in Fig. 2(d), assuming that if both are non-toxic, concentrations below them will not be either. Although a discrete mortality started two days post-exposure (dpe), there were no significant differences in survival rates at the end of the experiment after 72 h, which is the time considered critical for assessing viability in this case, in which the GO was introduced directly into the systemic circulation.
Several researchers evaluated the toxicity ranges of GO after bath exposure in both embryos and larvae reinforcing the toxic effect of GO concentrations close to 0.25 mg mL−1 observed in the present study: 0.4 and 0.2 mg mL−1 affect to hatching rate and larvae viability after 72 hpe.43,44 However, M. D'Amora et al. found lower concentrations of GO to be toxic, between 0.05 and 0.1 mg mL−1.45 With regard to the microinjection method, present results are in line with Gollavelli and Ling, who did not observe toxic effects of GO at concentration 0.1 mg mL−1 or below on larval survivability.46 These findings were quite different from the ones by other authors, who documented similar GO concentrations causing developmental changes,47 although the different microinjection sites and modifications of the GO used must be taken into account.
To study the immune response of zebrafish to GO microinjections at 0.25 and 0.1 mg mL−1 concentrations, we analysed the changes in the number of neutrophils and macrophages and the migration of these cells to the microinjection site in the corresponding transgenic lines. The data shown in Fig. 3 correspond to the quantification of total cells and cells migrated to the anterior region after 2 and 24 h of exposure. Even though no significant differences in the quantity or mobilisation of the neutrophils were observed in the first few hpe (Fig. 3(a)), they did appear at 24 h after incorporating 0.25 mg mL−1 of GO directly via the DC (Fig. 3(b)). The macrophage response was more immediate, showing significant differences for the same GO dose (0.25 mg mL−1) 2 h after microinjection (Fig. 3(c)) that was restabilised after 24 h, returning to a homeostatic state (Fig. 3(d)). The results presented demonstrate a rapid increase of the macrophage and subsequent neutrophil induction caused by injection of the highest applied concentration of GO and, consequently, an influence of GO on the innate immune response of zebrafish during their early embryonic stages that must be considered.
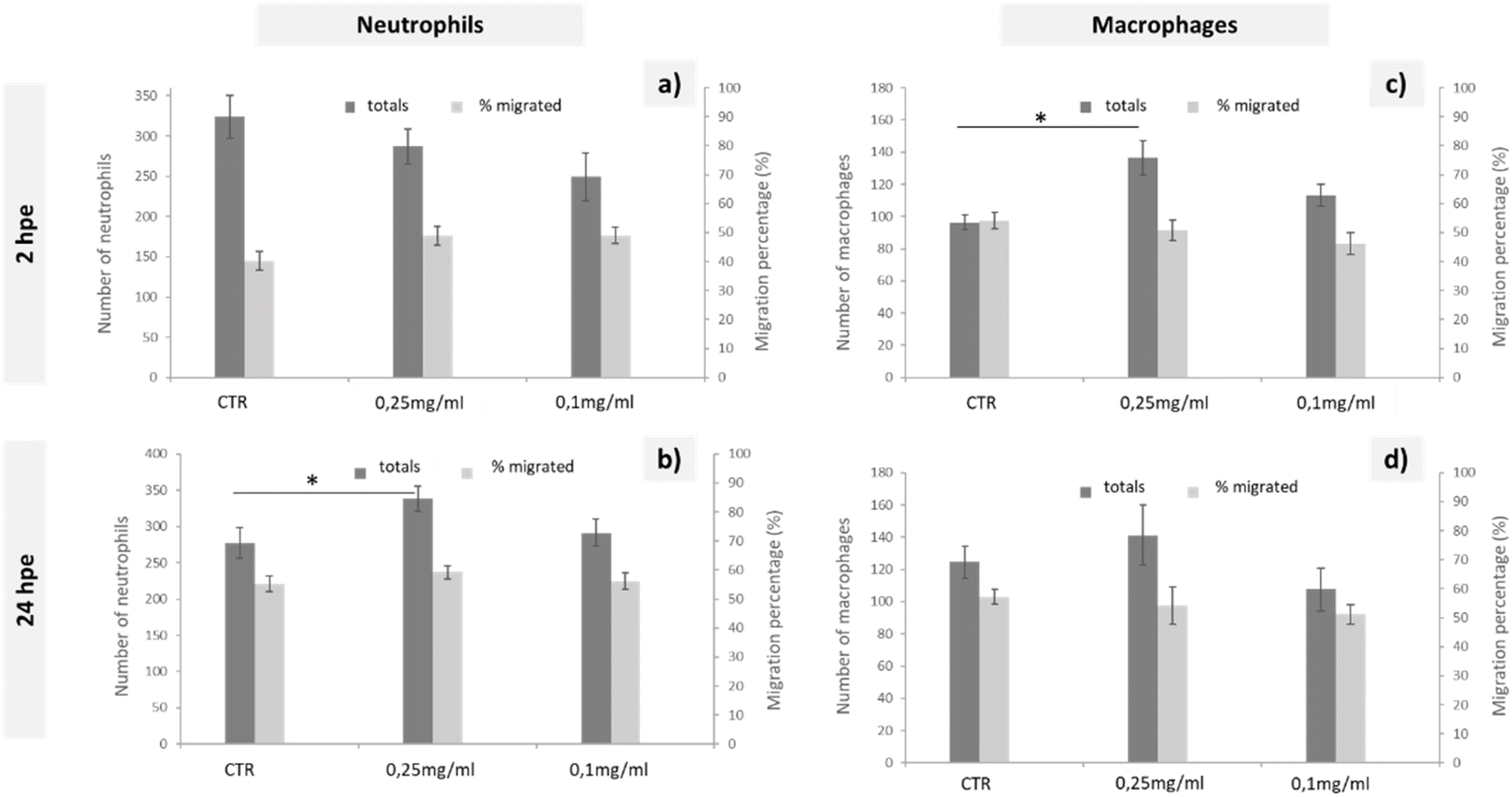 | ||
| Fig. 3 Quantification of neutrophil (a and b) and macrophage (c and d) behaviour in response to the injection of different solutions of GO in zebrafish transgenic embryos (3dpf) with fluorescently labelled immune cells, from 2 to 24 hpe. Data are presented as mean ±SEM. Statistically significant differences from the control group are indicated by *, p < 0.05. | ||
The natural response when a foreign substance is introduced into the body is the generation of an oxidative stress, leading to systemic inflammation. In the zebrafish immune system, neutrophils and macrophages are the essential immune cells that respond in this case.48 Considering that macrophages initiate the inflammatory response, secreting proinflammatory cytokines and chemokines that consequently alert the immune system and promote neutrophil recruitment,49 the findings presented were consistent with this theory, as there was first an increase of total macrophages that was subsequently solved and led to a total increase of neutrophils. Similarly, Chen et al. suggested the presence of an immunomodulatory effect after the exposure to GO, even at lower concentrations, such as 0.01 mg mL−1.48 This macrophage-neutrophil cooperation as an effector mechanism in the regulation of the immune system was already described by other authors.50 The lack of a migratory response towards the GO microinjection site could be explained by the rapid distribution of GO throughout the larval body. Chen et al. shows a broad GO distribution pattern when larvae are exposed by immersion,48 such that by microinjecting GO directly into the bloodstream, we could expect a rapid GO distribution resulting in an increase in the total number of immune cells, but without being recruited to a specific site. Further experimental assays would be needed to determine any type of bioaccumulation using the microinjection exposure, although we can conclude that if there was, it would not be fatal to the larvae according to the mortality tests carried out in this study.
To better understand the influence of GO on immune response in zebrafish, we examined the expression of some genes involved in the response to inflammatory processes. Expression levels of il1b, tnfa and tp53, 24 h after injection of 0.1 mg mL−1 of GO, are shown in Fig. 4. The selected dose of GO, chosen as the maximum concentration with no detrimental effects in the previous in vivo experiments, also did not modify the expression levels of tnfa and tp53. In the case of il1b, although there was a downregulation trend, no significant differences in expression with respect to the control were observed. Illβ and TNF-alpha are cytokines primarily produced during the innate immune response. Although there is still not sufficient knowledge on GO-induced immune toxicity in zebrafish, our results are in contrast with those obtained by Chen et al., who reported a significantly elevated expression of these two molecules, indicating induction of an inflammatory response. This was observed after an exposure to GO by bath and for a prolonged time of 14 days.27 Moreover, with regard to the expression of tp53, an evolutionarily conserved tumour suppressor that also regulates innate and adaptive immune responses,51 Jia et al. found that it was significantly up-regulated after exposure to GO at different sizes.26 This is not in line with our results, which indicate that this gene is not affected, at least by the GO and under the conditions we have specifically set up.
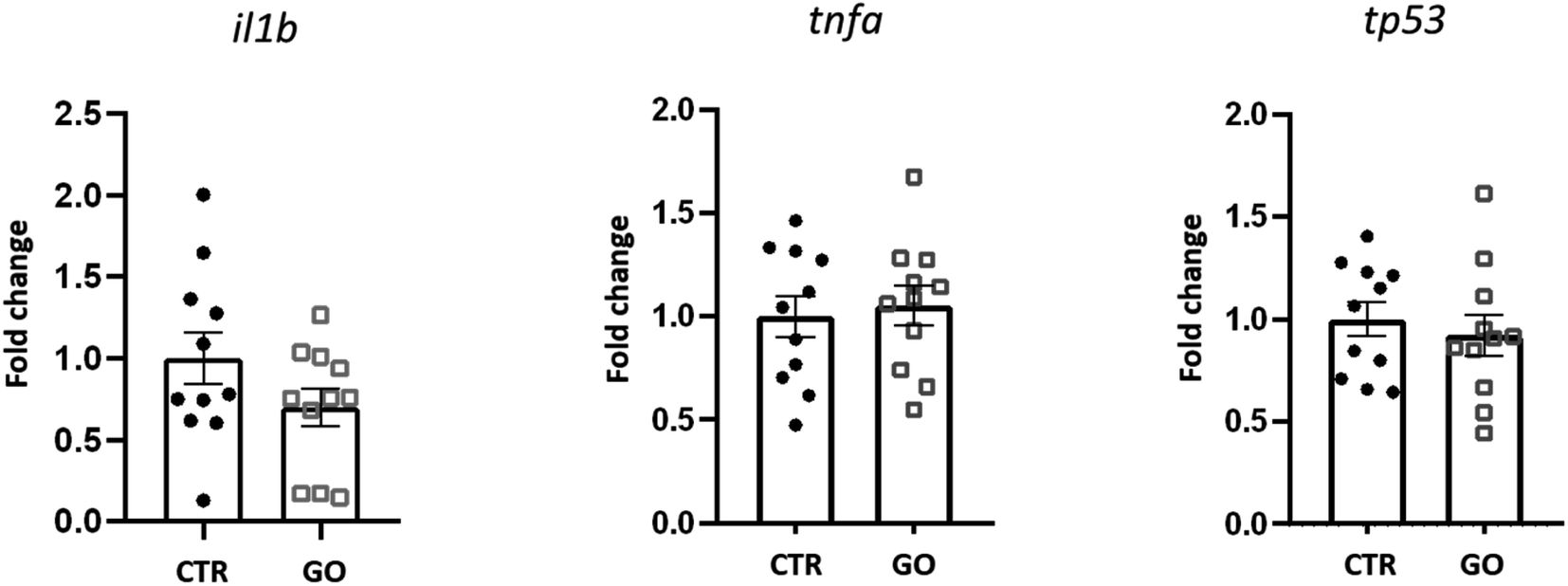 | ||
| Fig. 4 Expression profile of genes related to inflammatory/immune response in zebrafish larvae 24 h after being microinjected with 0.1 mg mL−1 of GO. Data are presented as mean of fold changes, calculated with respect to the basal expression of each gene (CTR), ±standard error of the mean. | ||
In summary, GO nanoparticles in the shape of nanoscale sheets, oxidised at 32% and with a C/O ratio close to 2, proved to be biocompatible in contact with both MG-63 cells after 24 h under the concentration ≤0.25 mg mL−1, and zebrafish model, after direct contact and being microinjected at the systemic level, at a concentration of 0.10 mg mL−1 and below.
3.4. Physicochemical and mechanical characterisation of 3D printed PLA:CaP:GO scaffolds
A GO concentration of 0.10 mg mL−1 was incorporated by 3D printing into the PLA:CaP scaffold, meaning the 0.004 wt% of the entire composition. The CaP contribution was varied in the range 3.4–13.2 wt% (confirmed by ICP-calcination methodology, not shown) in total composition. The highest contribution was limited to 13.2 wt% due to technical requirements, to ensure an effective and uniform incorporation.The porous structure of the resulting 3D printed PLA:CaP:GO scaffolds was first analysed using a stereo microscope and the obtained 3D optical images can be observed in Fig. 5. In this case a scaffold with pores of 1 × 1 mm2 and the 3.4 wt% of CaP and 0.004 wt% of GO is presented Fig. 5(a) and (b). In a general view, the homogeneous pore dimensions and symmetrical pore distribution are clearly visible. In addition, in the transparent interpores PLA-based walls the contributions of CaP granules in aggregates of up to ∼150 μm can be elucidated Fig. 5(b) and (c). Moreover, the morphology of the same scaffold was also evaluated by SEM, and a micrograph is presented in Fig. 5(d) together with the EDS analysis Fig. 5(e). The porosity and morphology of the pore walls can be observed. In addition, differences in the width along each deposited line are clearly detected, related to reological factors. These pore and extruded filament characteristics and deposition pattern, using 3D printing, were similar to those obtained by other authors who mixed PLA and GO52 or PLA and CaP.13,17 This therefore suggests that the introduction of the third material, GO, does not interfere with the structure of the PLA:CaP based polymeric/ceramic scaffolds. The elemental analysis by EDS Fig. 5(e) confirmed the composition of the scaffold with presence of C in an atomic percentage of 77.6%, O of 21.5%, P of 0.33% and Ca of 0.55%, with a Ca:P ratio of 1.67. These results proved the incorporation of CaP into the scaffolds. Furthermore, the Ca:P ratio of 1.67 has been proven to be the most effective in promoting bone regeneration.53
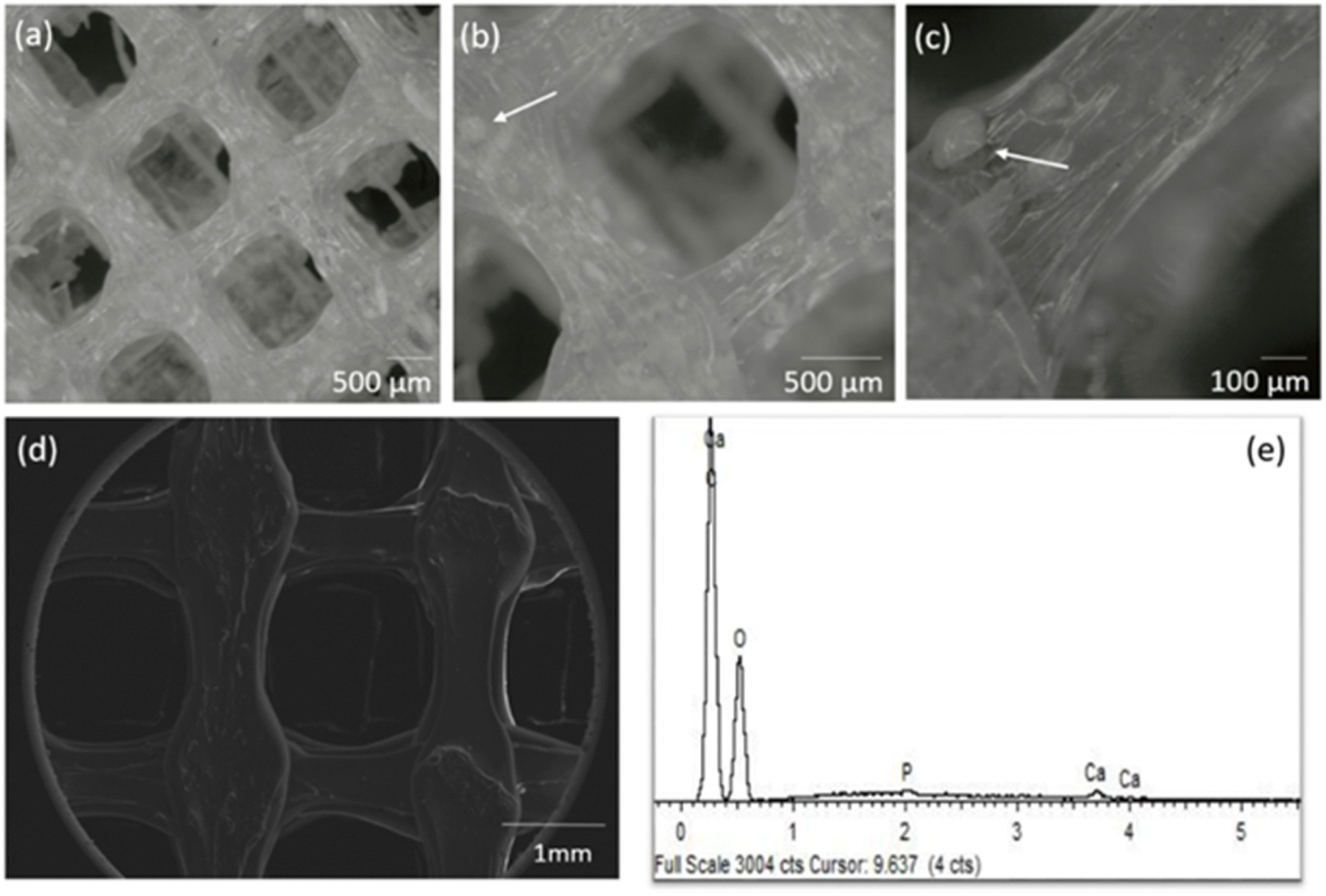 | ||
| Fig. 5 Stereo micrographs in different magnifications (a–c), SEM (d) and corresponding EDS spectrum (e) of the PLA:CaP:GO scaffold with 3.4 wt% of CaP and 0.10 mg mL−1 of GO (0.004 wt%) (d and e). | ||
The crystalline structure was assessed using XRD and presented at Fig. 6(a) where the diffraction patterns for different compositions of PLA:CaP:GO scaffolds are shown. The two characteristic diffraction peaks in the spectrum of 3D printed PLA alone (named as PLA:0CaP:0GO) were found at positions 16.5° and 21.5°, corresponding to (200)/(110) and (015) crystal planes, respectively.54 When low contributions of CaP and GO are present in the scaffold, differences at the diffraction patterns were observed, as a less intense diffraction peak for the (200)/(110) crystal plane, which suggest lower crystallinity degree. The presence of CaP is clearly detected in the diffraction pattern of the scaffold with the highest contribution, 13.2 at%, with the main peaks at positions 31.8°, 32.2°, 32.9°and 39.9°, which correspond to planes (121), (112), (300) and (310).55,56 The typical diffraction peak of GO is not detected by this technique given probably the low content incorporated. This non-appearance of the characteristic GO peaks in PLA:CaP:GO based structures is a fact previously found by authors such as M. Gong et al.57 Moreover, the PLA:CaP:GO scaffolds were also subjected to FT-Raman spectroscopy to evaluate their bonding configuration and results are presented in Fig. 6(b). The Raman spectrum obtained for the PLA:CaP:GO scaffold revealed the characteristic spectrum of poly(lactic acid)s58,59 with main strong bands registered, taking as reference the spectrum of PLA:0CaP:0GO scaffold, at: 872 cm−1 with an intense and sharp band attributed to C–COO stretching, 1769–1773 cm−1 assigned to CO asymmetric stretching, 1455–1457 cm−1 to asymmetric bending CH3, 1128 cm−1 to asymmetric rocking CH3 and, finally, 1043 cm−1 attributed to skeletal stretching C–CH3. It is also present the band near 960 cm−1 attributed to PO4−3 symmetric stretching mode of calcium phosphates. According to literature60 when this band appears at 970 cm−1 with a shoulder in 961 cm−1 is attributed to tricalcium phosphates (TCP) and other authors56 assigned this vibration at 962 and 956 cm−1 to hydroxyapatite and the one at 972, 958–968 and 948 cm−1 to β-TCP. The position of the peak in this case at 954 cm−1 indicates, as explained above, that we are dealing with a calcium phosphate of the apatitic type. Finally, the Raman spectrum presented two bands at 1596 cm−1 and at 1323 cm−1 which correspond to the typical G (1581 cm−1) and D (1342 cm−1) bands of GO (Fig. S3(b)†). The spectrum of GO alone (Ctr GO) is also incorporated as reference measured under the same conditions and related bands highlighted with pointed squares. These results confirm both the successful contribution of CaP and GO in the 3D printed PLA:CaP:GO scaffolds.
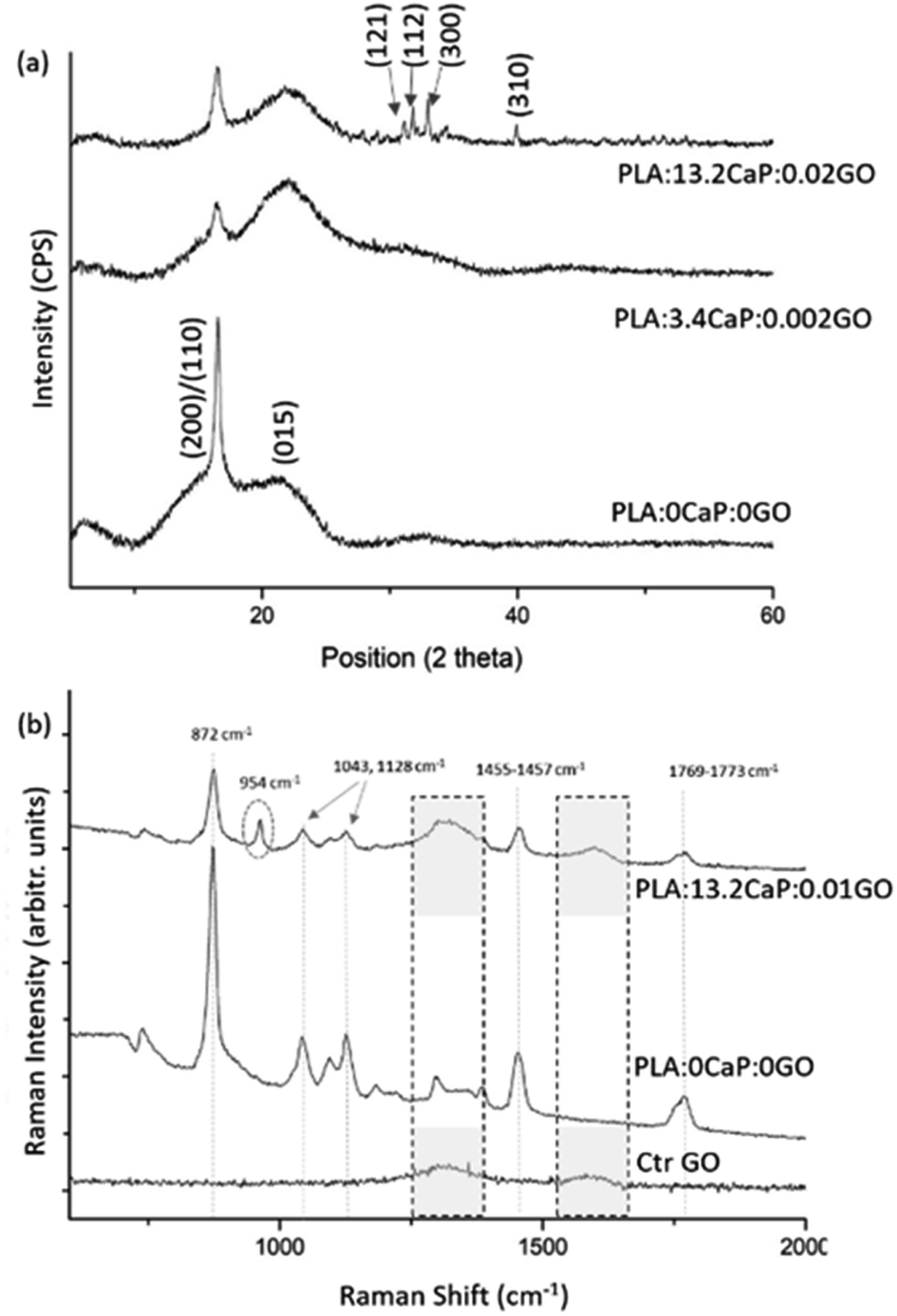 | ||
| Fig. 6 XRD diffraction pattern of the 3D printed PLA:CaP:GO scaffolds (a) and FT-Raman spectra showing the characteristic bands for each of the scaffold components (b). In both cases the sample PLA:0CaP:0GO is taken as reference, as well as the GO for FT-Raman. For Raman spectra, the regions associated with GO (1300–1600 cm−1) are highlighted with pointed square, while the band corresponding to CaP is enclosed by a dotted circle. | ||
Once the scaffolds were structurally and chemically characterised, the mechanical properties were also analysed. Fig. 7 shows the hardness and Young's Modulus measurements carried out on different PLA:CaP:GO scaffolds, for which the CaP content was kept constant in order to observe the changes associated with the incorporation of GO at different concentrations. For that reason, a PLA:CaP scaffold was also incorporated as control sample, with values of hardness (black bars) 0.19 ± 0.03 GPa and Young's modulus (grey bars) of 3.60 ± 0.30 GPa. In particular, the latter value is consistent with those of PLA:CaP scaffolds from other authors although with different CaP concentrations and demonstrates the increase with respect to PLA values alone.61 When GO is incorporated into the scaffolds (0.01 at% and 0.02 at%) the mean values of both hardness and Young's modulus increased in relation to the control, without GO. Going into detail with the Young's modulus, it was observed a maximum value of 4.24 ± 0.30 GPa obtained at the PLA:CaP:GO scaffold with the highest contribution in GO. This value represents an increase of 16% compared to the sample without GO, implying that the presence of GO improves the mechanical properties of the PLA:CaP scaffolds. This has already been affirmed by other authors such as Pinto et al. who obtained nanocomposites thin films of PLA after incorporating small amounts of GO, observing an increase in the modulus of elasticity value of 115% by adding 0.3 wt% of GO in a PLA matrix.62
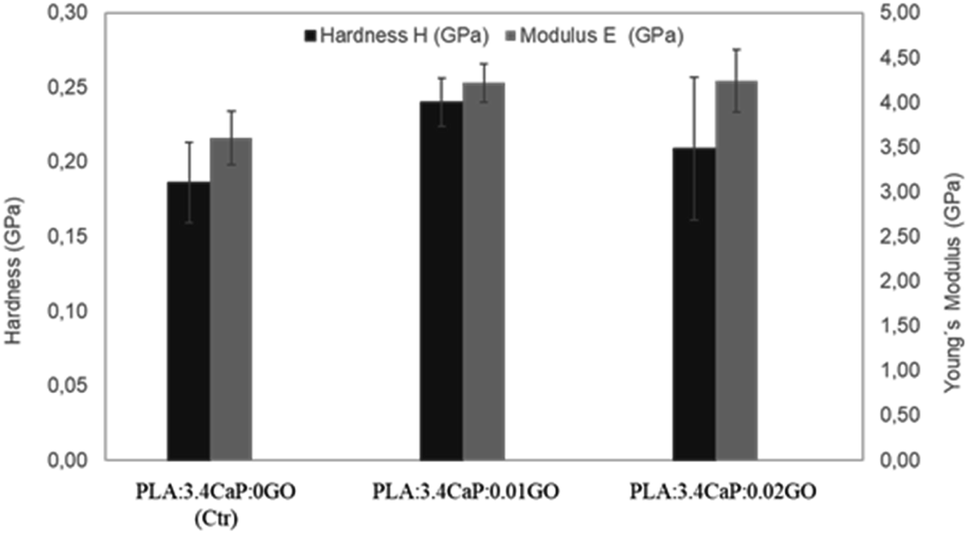 | ||
| Fig. 7 Young's modulus and hardness of different PLA:CaP:GO scaffolds. In the bar chart means ±standard errors are represented. | ||
After physicochemical characterisation of the PLA:CaP:GO scaffolds, which demonstrated the successful incorporation of GO into the composite and no influence on the homogeneity of the porous structure of the scaffolds, as well as improved mechanical properties compared to the control PLA:CaP scaffolds, the toxicity of the extracts obtained in contact with zebrafish was evaluated.
3.5. In vivo zebrafish assays
The toxic potential of extracts obtained from the PLA:CaP:GO scaffolds (with 13.2 wt% of CaP and 0.004 wt% of GO) in zebrafish water for 30 days was evaluated using the zebrafish model. Extracts from PLA:0CaP:0GO scaffolds were also tested. Fig. 8 shows the effect on embryo viability by bath exposure. As expected, the GO added to the scaffolds and potentially released to the extracts after 30 days in zebrafish water at physiological temperature did not generate any significant toxic response in direct contact with zebrafish larvae after 7 days in the bath. | ||
| Fig. 8 Toxicity response of zebrafish larvae exposed to extracts obtained from PLA:CaP:GO scaffolds (with 3.4 wt% of CaP and 0.004 wt% of GO) for 30 days: viability rate of WT embryos 7 dpe by bath. | ||
In summary, the GO used in the present study at nanoscale size and 32% oxidisation, is not toxic to MG-63 bone cells at a concentration ≤0.25 mg mL−1 in contact for 24 h. These results are similar, in terms of the dosage and time, to those obtained by Bengtson et al. who confirmed the non-toxicity of up to 200 μg mL−1 GO in contact with an epithelial tissue cell line for 24 h; however, in this case the GO used was much less oxidised.63 However, for other cell lines (fibroblasts, neuronal or cancer cells), GO, also in the form of nanoscale sheets, is only biocompatible up to 20–80 μg mL−1, although the contact times with the material are longer, 4–5 days.7,64,65
With respect to GO administrated in vivo in suspension both by direct contact and after being systemically injected, the non-toxic concentration was 0.1 mg mL−1. These values agree with previous studies using the same animal model, which concluded no toxicity of GO up to 0.3 mg mL−1 via the same route of exposure, i.e., direct contact.66 However, other researchers observed damage in zebrafish from a dose of 0.05 mg mL−1 of GO administered intraperitoneally.67 Furthermore, taking into account the results obtained using other animal models, several authors demonstrated pathological effects in mice after the intratracheal administration of 1 mg mL−1 of GO.68
Therefore, on the basis of our own results that are supported by some previous comparable research, 0.1 mg L−1 is defined as the ideal GO concentration to incorporate in the PLA:CaP hybrid scaffold, thus ensuring the absence of toxicity. This is not only because it has been validated in an in vivo model, but also because of the gradual degradation of the scaffold that would occur under physiological conditions, implying that the threshold concentration would not appear at any time in the implanted area.
The proven sensitivity and similarity to human biological mechanisms of the in vivo model (Danio rerio) defines this threshold dose as a guarantee of safety of the device that could be developed from the scaffold model studied herein.
4. Conclusions
In this research, porous scaffolds consisting of PLA, CaP and GO were 3D manufactured and characterised. The in-depth observation, EDS, XRD and Raman analysis allowed to determine the successful incorporation of GO by the proposed procedure. While at the surface level, microscopy techniques allowed detailed observation of the final porous structure of the scaffolds, from a mechanical point of view, these scaffolds have improved their modulus of elasticity compared to those without GO. In terms of biological results, the previous characterisation of GO in its original nano-sheet form allowed us to define 0.10 mg mL−1 as the non-toxic concentration both in contact with cells and with the in vivo model. Further, the degradation products of the PLA:CaP:GO scaffolds showed a biocompatibility with the same concentration of GO, which is 0.004 wt%, after being in direct contact with zebrafish body. Furthermore, these biocompatible scaffolds offer an interesting morphological structure and physiochemical characteristics that could be highly beneficial both for cell adhesion and for its mechanical performance. These findings support the validation of GO-enriched composites for future applications in biomedical engineering.Author contributions
L. G-R., S. P-D. and R. L. carried out the experiments, L. G-R and M. L-A. wrote the manuscript with support from R. L. and B. N. B. N, A. F., P. G. and J. S. supervised the project. All authors provided critical feedback and helped shape the research, analysis and manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This research was financially supported by BIOHEAT project (PID 2020-115415RB-100, Ministerio Ciencia e Innovación España), Xunta de Galicia ED431C 2021/49 and IN607B 2022/13 Program for Consolidación e estructuración de unidades de investigación competitivas (GRC and GPC). Pérez-Davila, S. and Lama, R. are grateful for funding support from Xunta de Galicia pre-doctoral grants (ED481A 2019/314 and IN606A-2017/011, respectively). The technical staff from CACTI (University of Vigo) and from the IIM-CSIC (Judit Castro, Lucía Sánchez and the aquarium staff) are gratefully acknowledged. Universidade de Vigo is acknowledged for funding for the open access charge.References
- P. P. Wibroe, S. V. Petersen, N. Bovet, B. W. Laursen and S. M. Moghimi, Biomaterials, 2016, 78, 20–26 CrossRef CAS PubMed.
- E. Rozhina, S. Batasheva, R. Miftakhova, X. Yan, A. Vikulina, D. Volodkin and R. Fakhrullin, Appl. Clay Sci., 2021, 205, 106041 CrossRef CAS.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- O. N. Ruiz, K. A. S. Fernando, B. Wang, N. A. Brown, P. G. Luo, N. D. McNamara, M. Vangsness, Y. P. Sun and C. E. Bunker, ACS Nano, 2011, 5, 8100–8107 CrossRef CAS PubMed.
- G. Y. Chen, D. W. P. Pang, S. M. Hwang, H. Y. Tuan and Y. C. Hu, Biomaterials, 2012, 33, 418–427 CrossRef CAS PubMed.
- H. H. Yoon, S. H. Bhang, T. Kim, T. Yu, T. Hyeon and B. S. Kim, Adv. Funct. Mater., 2014, 24, 6455–6464 CrossRef CAS.
- A. Rhazouani, H. Gamrani, M. El Achaby, K. Aziz, L. Gebrati, M. S. Uddin and F. Aziz, BioMed Res. Int., 2021, 2021, 5518999 Search PubMed.
- M. Cicuéndez, V. S. Silva, M. J. Hortigüela, M. C. Matesanz, M. Vila and M. T. Portolés, Colloids Surf., B, 2017, 158, 33–40 CrossRef PubMed.
- T. A. Tabish, M. Z. I. Pranjol, H. Hayat, A. A. M. Rahat, T. M. Abdullah, J. L. Whatmore and S. Zhang, Nanotechnology, 2017, 28, 504001 CrossRef PubMed.
- J. Ma, R. Liu, X. Wang, Q. Liu, Y. Chen, R. P. Valle, Y. Y. Zuo, T. Xia and S. Liu, ACS Nano, 2015, 9, 10498–10515 CrossRef CAS PubMed.
- S. Gai, G. Yang, P. Yang, F. He, J. Lin, D. Jin and B. Xing, Nano Today, 2018, 19, 146–187 CrossRef CAS.
- M. Charles-Harris, M. A. Koch, M. Navarro, D. Lacroix, E. Engel and J. A. Planell, J. Mater. Sci.: Mater. Med., 2008, 19, 1503–1513 CrossRef CAS PubMed.
- T. Serra, M. A. Mateos-Timoneda, J. A. Planell and M. Navarro, Organogenesis, 2013, 9, 239–244 CrossRef PubMed.
- B. Zhang, H. Sun, L. Wu, L. Ma, F. Xing, Q. Kong, Y. Fan, C. Zhou and X. Zhang, Bio-Des. Manuf., 2019, 2, 161–171 CrossRef CAS.
- R. Trombetta, J. A. Inzana, E. M. Schwarz, S. L. Kates and H. A. Awad, Ann. Biomed. Eng., 2017, 45, 23–44 CrossRef PubMed.
- T. Petrovskaya and N. Toropkov, AIP Conf. Proc., 2020, 2310, 020253 CrossRef CAS.
- P. Nevado, A. Lopera, V. Bezzon, M. R. Fulla, J. Palacio, M. A. Zaghete, G. Biasotto, A. Montoya, J. Rivera, S. M. Robledo, H. Estupiñan, C. Paucar and C. Garcia, Mater. Sci. Eng., C, 2020, 114, 111013 CrossRef CAS PubMed.
- S. Liu, Z. Li, Q. Wang, J. Han, W. Wang, S. Li, H. Liu, S. Guo, J. Zhang, K. Ge and G. Zhou, ACS Appl. Bio Mater., 2021, 4, 8049–8059 CrossRef CAS PubMed.
- A. Şelaru, H. Herman, G. M. Vlăsceanu, S. Dinescu, S. Gharbia, C. Baltă, M. Roşu, C. V. Mihali, M. Ioniţă, A. Serafim, H. Iovu, A. Hermenean and M. Costache, Int. J. Mol. Sci., 2022, 23, 491 CrossRef PubMed.
- A. Ivanoska-Dacikj, G. Bogoeva-Gaceva, A. Krumme, E. Tarasova, C. Scalera, V. Stojkovski, I. Gjorgoski and T. Ristoski, Int. J. Polym. Mater. Polym. Biomater., 2020, 69, 1101–1111 CrossRef CAS.
- Z. Cheng, L. Xigong, D. Weiyi, H. Jingen, W. Shuo, L. Xiangjin and W. Junsong, J. Nanobiotechnol., 2020, 18, 1–13 CrossRef PubMed.
- L. Rodríguez-López, A. López-Prieto, M. Lopez-Álvarez, S. Pérez-Davila, J. Serra, P. González, J. M. Cruz and A. B. Moldes, ACS Omega, 2020, 5, 31381–31390 CrossRef PubMed.
- Zebrafish: A Practical Approach, ed. C. Nüsslein-Volhard and R. Dahm, Oxford University Press, USA, 2002 Search PubMed.
- M. Westerfield, The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio), University of Oregon Press, Eugene, USA, 5th edn, 2007 Search PubMed.
- M. Sendra, P. Pereiro, M. P. Yeste, L. Mercado, A. Figueras and B. Novoa, Environ. Pollut., 2021, 268, 115769 CrossRef CAS PubMed.
- P. P. Jia, T. Sun, M. Junaid, L. Yang, Y. B. Ma, Z. S. Cui, D. P. Wei, H. F. Shi and D. S. Pei, Environ. Pollut., 2019, 247, 595–606 CrossRef CAS PubMed.
- M. Chen, J. Yin, Y. Liang, S. Yuan, F. Wang, M. Song and H. Wang, Aquat. Toxicol., 2016, 174, 54–60 CrossRef CAS PubMed.
- M. W. Pfaffl, Nucleic Acids Res., 2001, 29(9), e45 CrossRef CAS PubMed.
- A. M. Pinto, C. Goncąlves, D. M. Sousa, A. R. Ferreira, J. A. Moreira, I. C. Goncąlves and F. D. Magalhães, Carbon, 2016, 99, 318–329 CrossRef CAS.
- P. Thevenot, W. Hu and L. Tang, Curr. Top. Med. Chem., 2008, 8, 270–280 CrossRef CAS PubMed.
- W. Yu, L. Sisi, Y. Haiyan and L. Jie, RSC Adv., 2020, 10, 15328–15345 RSC.
- B. G. Keselowsky, D. M. Collard and A. J. García, Biomaterials, 2004, 25, 5947–5954 CrossRef CAS PubMed.
- B. G. Keselowsky, D. M. Collard and A. J. García, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5953–5957 CrossRef CAS PubMed.
- S. Abraham, V. Ciobota, S. Srivastava, S. K. Srivastava, R. K. Singh, J. Dellith, B. D. Malhotra, M. Schmitt, J. Popp and A. Srivastava, Anal. Methods, 2014, 6, 6711–6720 RSC.
- Y. W. Cheng, S. H. Wang, C. M. Liu, M. Y. Chien, C. C. Hsu and T. Y. Liu, Surf. Coat. Technol., 2020, 385, 125441 CrossRef CAS.
- N. Mauro, C. Scialabba, S. Agnello, G. Cavallaro and G. Giammona, Mater. Sci. Eng., C, 2020, 107, 110201 CrossRef CAS PubMed.
- D. Zaharie-Butucel, M. Potara, S. Suarasan, E. Licarete and S. Astilean, J. Colloid Interface Sci., 2019, 552, 218–229 CrossRef CAS PubMed.
- C. Li, X. Chen, Z. Zhang and G. Jiang, J. Cluster Sci., 2020, 31, 1097–1102 CrossRef CAS.
- H. Ali-Boucetta, D. Bitounis, R. Raveendran-Nair, A. Servant, J. Van den Bossche and K. Kostarelos, Adv. Healthcare Mater., 2013, 2, 433–441 CrossRef CAS PubMed.
- J. Wu, R. Yang, L. Zhang, Z. Fan and S. Liu, Toxicol. Mech. Methods, 2015, 25, 312–319 CrossRef CAS PubMed.
- W. Xian, in A Laboratory Course in Biomaterials, CRC Press-Taylor & Francis, Boca Raton, Florida, 2009, pp. 99–128 Search PubMed.
- W. Zou, Q. Zhou, X. Zhang, L. Mu and X. Hu, Ecotoxicol. Environ. Saf., 2018, 159, 221–231 CrossRef CAS PubMed.
- Z. G. Wang, R. Zhou, D. Jiang, J. E. Song, Q. Xu, J. Si, Y. P. Chen, X. Zhou, L. Gan, J. Z. Li, H. Zhang and B. Liu, Biomed. Environ. Sci., 2015, 28, 341–351 Search PubMed.
- B. Manjunatha, E. Seo, S. H. Park, R. R. Kundapur and S. J. Lee, Environ. Sci. Pollut. Res., 2021, 28, 34664–34675 CrossRef CAS PubMed.
- M. D’Amora, A. Camisasca, S. Lettieri and S. Giordani, Nanomaterials, 2017, 7(12), 414 CrossRef PubMed.
- G. Gollavelli and Y. C. Ling, Biomaterials, 2012, 33, 2532–2545 CrossRef CAS PubMed.
- J. Jeong, H. J. Cho, M. Choi, W. S. Lee, B. H. Chung and J. S. Lee, Carbon, 2015, 93, 431–440 CrossRef CAS.
- Z. Chen, C. Yu, I. A. Khan, Y. Tang, S. Liu and M. Yang, Ecotoxicol. Environ. Saf., 2020, 197, 110608 CrossRef CAS PubMed.
- B. Amulic, C. Cazalet, G. L. Hayes, K. D. Metzler and A. Zychlinsky, Annu. Rev. Immunol., 2012, 30, 459–489 CrossRef CAS PubMed.
- M. T. Silva, J. Leukocyte Biol., 2010, 87, 93–106 CrossRef CAS PubMed.
- D. Shi and P. Jiang, Front. Cell Dev. Biol., 2021, 9, 1–10 Search PubMed.
- H. Belaid, S. Nagarajan, C. Teyssier, C. Barou, J. Barés, S. Balme, H. Garay, V. Huon, D. Cornu, V. Cavaillès and M. Bechelany, Mater. Sci. Eng., C, 2020, 110, 110595 CrossRef CAS PubMed.
- M. Akram, R. Ahmed, I. Shakir, W. A. W. Ibrahim and R. Hussain, J. Mater. Sci., 2014, 49, 1461–1475 CrossRef CAS.
- S. Pérez-Davila, L. González-Rodríguez, R. Lama, M. López-Álvarez, A. L. Oliveira, J. Serra, B. Novoa, A. Figueras and P. González, Polymers, 2022, 14, 4117 CrossRef PubMed.
- I. Fernández-Cervantes, M. A. Morales, R. Agustín-Serrano, M. Cardenas-García, P. V. Pérez-Luna, B. L. Arroyo-Reyes and A. Maldonado-García, J. Mater. Sci., 2019, 9478–9496 CrossRef.
- H. Aguiar, S. Chiussi, M. López-Álvarez, P. González and J. Serra, Ceram. Int., 2018, 44, 495–504 CrossRef CAS.
- M. Gong, Q. Zhao, L. Dai, Y. Li and T. Jiang, J. Asian Ceram. Soc., 2017, 5, 160–168 CrossRef.
- G. Kister, G. Cassanas and M. Vert, Polymer, 1998, 39, 267–273 CrossRef CAS.
- M. G. Gandolfi, F. Zamparini, M. Degli Esposti, F. Chiellini, C. Aparicio, F. Fava, P. Fabbri, P. Taddei and C. Prati, Mater. Sci. Eng., C, 2018, 82, 163–181 CrossRef CAS PubMed.
- S. Koutsopoulos, J. Biomed. Mater. Res., 2002, 62, 600–612 CrossRef CAS PubMed.
- G. E. Dubinenko, A. L. Zinoviev, E. N. Bolbasov, V. T. Novikov and S. I. Tverdokhlebov, Mater. Today: Proc., 2020, 22, 228–234 CAS.
- A. M. Pinto, J. Cabral, D. A. P. Tanaka, A. M. Mendes and F. D. Magalhães, Polym. Int., 2013, 62, 33–40 CrossRef CAS.
- S. Bengtson, K. Kling, A. M. Madsen, A. W. Noergaard, N. R. Jacobsen, P. A. Clausen, B. Alonso, A. Pesquera, A. Zurutuza, R. Ramos, H. Okuno, J. Dijon, H. Wallin and U. Vogel, Environ. Mol. Mutagen., 2016, 57, 469–482 CrossRef CAS PubMed.
- M. Lv, Y. Zhang, L. Liang, M. Wei, W. Hu, X. Li and Q. Huang, Nanoscale, 2012, 4, 3861–3866 RSC.
- K. Wang, K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Nanoscale Res. Lett., 2010, 6, 8 CrossRef PubMed.
- M. Bangeppagari, S. H. Park, R. R. Kundapur and S. J. Lee, Sci. Total Environ., 2019, 673, 810–820 CrossRef CAS PubMed.
- A. L. Fernandes, J. P. Nascimento, A. P. Santos, C. A. Furtado, L. A. Romano, C. Eduardo da Rosa, J. M. Monserrat and J. Ventura-Lima, Chemosphere, 2018, 210, 458–466 CrossRef CAS PubMed.
- M. C. Duch, G. R. S. Budinger, Y. T. Liang, S. Soberanes, D. Urich, S. E. Chiarella, L. A. Campochiaro, A. Gonzalez, N. S. Chandel, M. C. Hersam and G. M. Mutlu, Nano Lett., 2011, 11, 5201–5207 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra00981e |
| This journal is © The Royal Society of Chemistry 2023 |